

Abstract
Altered oxidative stress has long been observed in cancer cells, and this biochemical property of cancer cells represents a specific vulnerability that can be exploited for therapeutic benefit. The major role of an elevated oxidative stress for the efficacy of molecular targeted drugs is under investigation. Menadione is considered an attractive model for the study of oxidative stress, which can induce apoptosis in human leukemia HL-60 cell lines. Prostaglandin E2 (PGE2) via its receptors not only promotes cell survival but also reverses apoptosis and promotes cancer progression. Here, we present evidence for the biological role of PGE2 as a protective agent of oxidative stress-induced apoptosis in monocytic cells. Pretreatment of HL-60 cells with PGE2 markedly ameliorated the menadione-induced apoptosis and inhibited the degradation of PARP and lamin B. The EP2 receptor antagonist AH6809 abrogated the inhibitory effect of PGE2, suggesting the role of the EP2/cAMP system. The PKA inhibitor H89 also reversed apoptosis and decreased the PKA activity that was elevated 10-fold by PGE2. The treatment of HL-60 cells with NAC or zinc chloride showed a similar protective effect as with PGE2 on menadione-treated cells. Furthermore, PGE2 activated the Ras/Raf/MEK pathway, which in turn initiated ERK activation, and ultimately protected menadione-induced apoptosis. These results imply that PGE2 via cell survival pathways may protect oxidative stress-induced apoptosis in monocytic cells. This study warrants further pre-clinical investigation as well as application towards leukemia clinics.
Keywords: apoptosis, menadione, oxidative stress, prostaglandin
INTRODUCTION
Oxidative stress is like two sides of a coin and used with caution can destroy with cancer cells, since cancer cells exhibit increased level of reactive oxygen species (ROS) compared to normal cells (Trachootham et al., 2009). Mounting evidence suggest that excessive ROS generation occurs in hypoxic environments and it is possible to enhance the ROS level to such extent that it can damage proteins, lipids, DNA, and ultimately lead to cell death (Coffey et al., 2000). Hence, glutathione (GSH) depletion along with ROS generation can effectively be used to induce cell death in cancer cells irrespective of their sensitivity toward anticancer drugs. Among the anti-cancer agents, menadione is a polycyclic aromatic ketone (2-methyl-1,4-naphthoquinone), which has the ability to generate intracellular ROS at multiple cellular sites through redox cycling reactions (Brière et al., 2004). Menadione induced apoptosis through the downregulation of ERK and JNK, followed by the activation of caspase 3 and poly ADP-ribose polymerase (PARP) cleavage (Osada et al., 2008).
Prostaglandin E2 (PGE2) is generated by the enzymes phospholipase A2 (PLA2), cyclooxygenase (COX), and prostaglandin E synthase (PGES), which stimulates key downstream signaling cascades and have roles in the maintenance of normal physiology as well as in the various pathological conditions including different cancers (Smith et al., 2000). The PGE2 differentially mediates its physiological role via receptors, due to the binding affinity of these receptors with PGE2 (Sampey et al., 2005; Smith et al., 2000). It has been reported that prostaglandin E receptors 1 and 3 (EP1 and EP3) mingle with Gαq to activate Ca+ signaling and with Gαi to inhibit adenyl cyclase, respectively. The other two receptors EP2 and EP4 couple to Gαs and in turn stimulate adenyl cyclase (Chun et al., 2010). Ras is a small GTP-binding protein, which is the common upstream molecule of several signaling pathways including Raf-MEKERK. PGE2 activates a Ras-Raf-ERK pathway through the EP2 receptor, which in turn upregulates COX-2 expression and stimulates cancer cell proliferation (Sung et al., 2006). In addition, EP2 contributed to tumor formation through the activation of protein kinase A (PKA), epidermal growth factor receptor (EGFR), and several effectors such as cyclic adenosine monophosphate (cAMP) response element-binding protein (CREB), H-Ras, Src, protein kinase B (AKT) and ERK (Chun et al., 2009).
Leukemia is cancer of the blood or bone marrow, which is characterized by unusual proliferation of blood cells. The human promyelocytic leukemia cell line (HL-60) is considered a useful model to study various genetic and biochemical factors regulating the development of the myeloid feature (Fontana et al., 1981). Therefore, the aim of this study was to elucidate the mechanisms of cell death induced by menadione as well as the anti-apoptotic role of PGE2. Menadione induced apoptotic cell death through oxidative stress, which is inhibited by PGE2 possibly through the scavenging of ROS and EP2 receptor in HL-60 cells. Furthermore, oxidative stress-induced signaling pathways were investigated by examining the role of PGE2 with hydrogen peroxide (H2O2), taxol, etoposide, and menadione. It was observed that PGE2 reverses menadione-induced apoptosis through the modulation of the Ras/Raf/ERK signaling pathway. We hope that the findings of this study will be a potential step forward in the understanding of the PGE2 role in apoptosis and further insights into the clinical applications of menadione for leukemia.
MATERIALS AND METHODS
Reagents and cell culture
Menadione was obtained from the Sigma-Aldrich and dissolved in dimethyl sulfoxide (DMSO) to produce a 100 mM stock solution. PGE2, butaprost, AH6809, and EP2 antagonist, were purchased from the Cayman Chemical Company. PD98059 was purchased from BioMol Plymouth Meeting. Paclitaxel (Taxol) was purchased from Bristol-Myers Squibb Princeton. HL-60 cells obtained from ATCC, CCL-240™ were cultured at a density of 5 × 105 in RPMI-1640 medium powdered with L-glutamine, containing 10% heat inactivated fetal bovine serum (FBS) and supplemented with 1% (v/v) antibiotic-penicillin streptomycin (Gibco, Invitrogen Corporation) at 37°C in a 5% CO2-humidified atmosphere. When the cells reached 70% confluence, the growth medium was removed from the cells and the cells were washed with phosphate-buffered saline (PBS) to remove all traces of serum. Trypsin/EDTA solution dispensed into culture dish completely covered the cell layer and incubated the cells for 2 min at 37°C to ensure that the cells completely detached from the base of the dish. Cells were centrifuged at 1,000 × g for 5 min and after the cell counting through the standard tryphan blue method and hematocytometer, the cell pellet was resuspend in fresh growth media with 10% FBS. Then the cells were seeded onto a new plate.
Cell cycle analysis
Cells were trypsinized and collected by centrifugation at 1,000 × g for 5 min. The collected cells were washed with PBS and resuspened at 1 × 106 cells/ml. For fixation, the cell suspension was kept overnight at 4°C, after the addition of 70% ethanol. Then, the cells were stained with propidium iodide solution (50 μg/ml propidium iodide, 0.1% Triton X-100, 0.1 mM EDTA, and 50 μg/ml RNase) for 20 min at 4°C. Stained DNA was analyzed by fluorescence activated cell sorting (FACS Calibur; Beckton Dickinson). For the detection of propidium iodide, cells were exited at 488 nm and their emissions were determined between 515 and 545 nm (Thayyullathil et al., 2008).
Determination of apoptosis
Cell viability was determined by a trypan blue exclusion test. A morphological analysis of apoptosis was performed after staining with Hoechst 33342 dye. Cells were fixed in 4% paraformaldehyde and permeabilized with 1X PBS and 0.5% Triton X-100 the nuclei were stained for 20 min with Hoechst dye. The coverslips were then washed, mounted onto slides, and viewed with a fluorescence microscope.
Apoptotic DNA fragments were isolated according to a previously described method and DNA was separated by electrophoresis on 1% agarose gels, stained with ethidium bromide, and visualized under ultraviolet light (Herrmann et al., 1994).
Cell morphology studies
After treating the cells with 1 μM PGE2 for 2 h followed by 10 μM of menadione. The HL-60 cells were then incubated at 37°C with 5 μM 2′,7′-dichlorfluorescein-diacetate (DCFHDA) (Calbiochem, USA) for 10 min. After indicated time, we gathered the cells and wash with 1× PBS for 3 times. The cell morphology was then observed by a fluorescence microscope (Axiovert 40 CFL, Carl Zeiss, USA) and the camera used was the Axiocam MRC 5 (Park et al., 2002).
Measurement of ROS production
Interacellular ROS concentrations were measured with the oxidant-sensitive fluorescent probe DCFHDA with inverted microscopy. Cells were grown to a density of 1 × 106 cells per 35 mm culture dish and maintained in the growth medium for 24 h. The cells were exposed to 5 μM DCFHDA for 30 min, and treated with 10 μM menadione for 12 h. The cells were washed with PBS and cover glass was put on the dish. DCF fluorescence (excitation, 485 nm; emission, 530 nm) was imaged on an inverted microscope (Yu and Kim, 2011).
Measurement of lipid peroxidation
Lipid peroxidation was estimated with the fluorescent probe diphenyl-1-pyrenylphosphine (DPPP) as peviously described (Park et al., 2002). After HL-60 cells were incubated with 5 μM DPPP for 15 min in the dark, the cells were exposed to 10 μM menadione or 1 μM PGE2 for 1 h. The images of DPPP fluorescence by reactive species were analyzed by the Zeiss Axiovert 200 inverted microscope at the fluorescence DAPI region (excitation, 351 nm; emission, 380 nm).
Protein kinase assay
cAMP-dependent protein kinase activity was determined by measuring the transfer 32P-ATP to a peptide substrate that was then captured on P81 phosphocellulose papers. Aliquots of cell lysates were incubated with 50 mM Tris at pH 7.5, 200 μM kemptite, 40 mM MgCl2, and 400 μM ATP. After 5 min at 30°C, the reactions were terminated by adding EDTA to a final concentration of 20 mM. Relative PKA activity was quantified with a liquid scintillation counter as described previously (Blum et al., 1999)
Western blotting
Gathered cells were washed with 1× PBS and lysed in a RIPA lysis buffer, which contain 50 mM Tris HCl (pH 7.4), 1% NP-40, 40 mM sodium fluoride (NaF), 10 mM sodium chloride (NaCl), 10 mM sodium orthovenodate (Na3VO4), 1 mM phenylmethyl-sufonyl fluoride (PMSF), 10 mM dithiothreitol (DTT), and protease inhibitor. The cell lysates were centrifuged at 12,000 × g for 15 min and protein was quantified through the Bio-Rad protein assay, was mixed with 4X loading buffer and boiled for 5 min. Samples were then resolved by SDS-PAGE and the separated proteins were transferred onto nitrocellulose membranes by the transfer method using transfer buffer (CAPS and methanol). The blots were blocked with 5% non-fat milk in Tris buffer saline containing 0.1% Tween 20 (1x TBST) and then incubated with primary antibodies followed by secondary antibodies. Western blot data presented here are representative of three independent experiments. The signals were detected by ECL Western blotting detection kit from Amersham Biosciences (USA) by using chemiluminescence system (Thayyullathil et al., 2008).
Ras activity assay
Ras activity was measured with a Ras activity assay kit (Upstate). The assay was performed according to the manufacturer’s instructions. Briefly, cells were washed twice with ice-cold PBS, lysed in lysis buffer (25 mM HEPES, pH 7.5, 150 mM NaCl, 1% Igepal CA-630, 10 mM MgCl2, 1 mM EDTA, 25 mM NaF, 10% glycerol, 10 μg/ml aprotinin, and 10 μg/ml leupeptin) and centrifuged. An equal volume of lysate was incubated with 7 μg of the Ras-binding domain for Raf-1 at 4°C for 2 h, and the beads were washed three times with lysis buffer. Bound Ras proteins were then detected by Western blotting using mouse monoclonal Ras antibody with the ECL system.
Statistical analysis
All the data presented as the mean values ± SD (standard deviation) were from 3 separate experiments. The data were evaluated by SPSS software with the dependent student t-test and where applicable (multiple group comparisons), the data were subjected to one-way analysis of variance (ANOVA). The significance of the differences between the mean was determined by the Tukey range test. Statistical significance was at P ≤ 0.05.
RESULTS
PGE2 prevents oxidative- and menadione-induced apoptosis in HL-60 cells
Menadione has been shown to stimulate ROS generation through activation of NADPH/quinone oxidase in various cells. As shown by the gel electrophoresis (Fig. 1A), menadione induced DNA fragmentation with rapid kinetics, which are considered the hallmark of apoptosis. Lamin B and PARP were cleaved with similar kinetics as the DNA fragmentation, yielding cleavage bands (Fig. 1B) that were detected after 4 h of menadione treatment. Since PARP and lamin B have been shown to be cleaved by caspase 3, therefore the menadione treated whole cell lysates were analyzed by Western blot with anti-caspase 3. The disappearance of a 32 KDa caspase zymogen was used to show the activation of caspase 3 (Fig. 1B). These data suggest that the caspase 3 dependent pathway might be responsible for the menadione-induced apoptosis.
Fig. 1.

Menadione induced apoptosis of HL-60 cells. Cells (1 × 106 cells/ml) were cultured in the absence or presence of 10 μM menadione. (A) Analysis of menadione-induced DNA fragmentation of cells by agarose gel electrophoresis. (B) Lamin B (upper panel) and PARP (middle panel) are cleaved as a result of capase-3 activation (lower panel) in apoptotic cells after treatment with menadione. At the indicated times, cell lysates were separated by SDS-PAGE and proteins were detected by immunoblotting. (C) Inhibition of menadione-induced apoptosis. HL-60 cells were pretreated with 1 μM PGE2 or 100 μM dibutyryl cAMP (dbcAMP). After 15 min, cells were incubated in the absence (control) or presence of menadione for 12 h. Cells were harvested and stained with propidium iodide, and their DNA content was analyzed by flow cytometry. Apoptotic cells are shown by the accumulation of cells with a DNA content of below 2n. Very similar results were obtained from three independent experiments.
To investigate the underlying mechanism of cAMP-dependent inhibition of apoptosis, we first examined the effect of cell-permeable cAMP analog dibutyryl cAMP on the menadione-induced apoptosis in HL-60 cells. To quantify the degree of apoptosis, the amount of sub-G1 DNA from the fixed nuclei was analyzed by flow cytometry (Fig. 1C). HL-60 cells were exprosed to menadione, which resulted in the increased accumulation of the sub-G1 phase. The addition of dibutyryl cAMP blocked menadione-induce apoptotic cell death. As expected, treatment of HL-60 cells with 1 μM PGE2 also effectively suppressed menadione-induced apoptosis.
To determine whether cAMP and PGE2 could also inhibit the induction of apoptosis by agents other than menadione, HL-60 cells were treated with H2O2, etopside, and taxol, and then allowed to recover in either the absence or presence of dibutyryl cAMP or PGE2. The flow cytometry results showed that PGE2 and dibutyryl cAMP also attenuated the generation of apoptosis through these cytotoxic oxidative agents (Table 1). The anti-apoptotic actions of PGE2 on drug-induced apoptosis were more effective than dibutyryl cAMP. To confirm this scenario, the anti-apoptotic effect of PGE2 was corroborated in U937 myeloid cells, indicating that it is not a cell line-specific phenomenon. The treatment of HL-60 cells with N-acetyl-cysteine (NAC) or zinc chloride showed a similar protective effect on menadione-treated cells (Table 1).
Table 1.
Anti-apoptotic action of prostaglandin E2 in human leukemia cell lines
| Cell type | Treatment | % Apoptosis | Apoptosis inhibition |
|---|---|---|---|
| HL-60 | Menadione | 20.57 ± 3.55 | - |
| + PGE2 | 9.47 ± 2.41 | 54.05 ± 7.60 | |
| + dibutylryl cAMP | 12.16 ± 3.48 | 41.51 ± 6.66 | |
| + N-acetylcysteine | 15.24 ± 2.31 | 30.65 ± 4.65 | |
| + Zinc chloride | 12.19 ± 1.15 | 40.42 ± 1.04 | |
| Hydrogen peroxide | 35.15 ± 2.39 | - | |
| + PGE2 | 22.10 ± 4.75 | 37.45 ± 9.79 | |
| + dibutylyl cAMP | 24.77 ± 4.85 | 30.95 ± 1.74 | |
| + N-acetylcysteine | 24.03 ± 6.05 | 33.01 ± 7.60 | |
| + Zinc chloride | 17.66 ± 4.96 | 51.42 ± 6.27 | |
| Etoposide | 21.02 ± 4.84 | - | |
| + PGE2 | 12.17 ± 1.59 | 40.87 ± 8.65 | |
| Taxol | 18.75 ± 3.12 | - | |
| + PGE2 | 13.01 ± 2.85 | 30.74 ± 8.58 | |
| U937 | Menadione | 27.03 ± 6.85 | - |
| + PGE2 | 13.83 ± 1.75 | 47.98 ± 6.70 | |
| Etoposide | 28.55 ± 8.05 | - | |
| + PGE2 | 21.65 ± 6.60 | 24.57 ± 2.87 |
Cells were pretreated with PGE2 (1 μM), dibutylryl cAMP (100 μM), N-acetylcysteine (15 mM), or zinc chloride (45 μM) for 10 min and then incubated with menadione, H2O2 (100 μM), etoposide (8.5 μM), or taxol (200 nM) for 12 h. Apoptotic cells were determined by flow cytometry. The treatment of HL-60 cells with NAC or zinc chloride and U937 cells with etoposide showed a similar protective effect as with PGE2 on menadione-treated cells. All results are the means ± SD of at least three independent experiments.
PGE2 may act as a scavenger of ROS
Menadione is known to catalyze the formation of H2O2 and hydroxyl radical (OH−), and these ROS are thought to mediate, at least in part, cellular toxicity. To confirm whether menadione-induce apoptosis in HL-60 cells is associated with the generation of cellular oxidative stress, the levels of intracellular peroxides were evaluated with DCFHDA. It is assumed that menadione-induce apoptosis in HL-60 cells occur through increases in DCF-detectable ROS (Fig. 2). Exposure of the HL-60 cells to radical scavenger such as PGE2 or NAC caused a clear decrease in the DCF fluorescence, consistent with the level of cleaved PARP (Fig. 2). These data strengthen the conclusion that PGE2 provides protection from the apoptotic actions of menadione by decreasing the steady-state level of intracellular oxidants.
Fig. 2.

Menadione-induced apoptosis is mediated by ROS production. (A) After cells were pretreated with 20 mM N-acetylcysteine (NAC) for 1 h, they were exposed to menadione. The cells were further incubated with DCFHDA, a cell-permeable dichlorofluorescein that can be used as an intracellular probe for oxidative stress for 30 min. Accumulation of the probe in the cells was measured by an increase at 530 nm when the sample was excited at 485 nm. Fluorescence images of DCF-loaded cells were obtained under a microscope. (B) PARP cleavage product was analyzed by Western blotting with anti-PARP antibody.
Glutathione (GSH) is one of the most abundant intracellular antioxidants, and measuring the changes in its concentration provides an alternative method to monitor oxidative stress in the cells. GSH-sensitive fluorescent dye t-butoxycarbonyl-Leu-Met-7-amino-4-chloromethylcoumarin (CMAC) can be used as a useful probe to evaluate the level of intracellular GSH. Cellular GSH levels were decreased in menadione treated HL-60 cells (Fig. 3A). However, the depletion of GSH was significantly inhibited by the pretreatment with 1 μM PGE2. To establish the extent to which oxidation of phospholipids was associated with the execution of the apoptotic program induced by menadione, DPPP was used as a probe to assess lipid peroxidation in monocytes. The DPPP fluorescent intensity was decreased markedly in PGE2-treated cells, whereas it increased in menadione-treated cells (Fig. 3B).
Fig. 3.
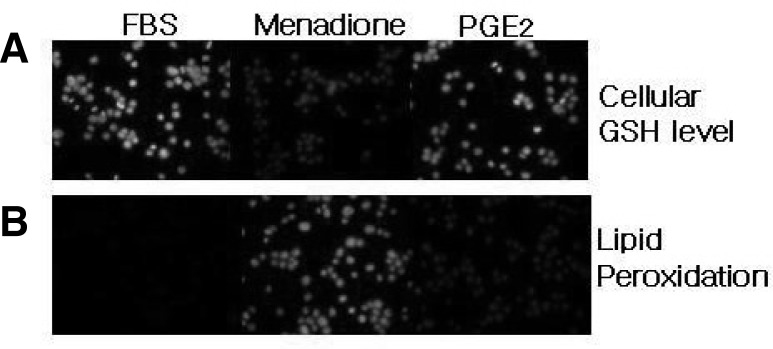
Measurement of cellular GSH and lipid peroxidation in menadione and PGE2 treated cells. (A) HL-60 cells (1 × 106 cells/ml) were stained with 5 μM CMAC for 15 min in the dark and then exposed to 10 μM or 1 μM of PGE2. Intracellular GSH levels were expressed relative to the FBS treated cells. Menadione treatment decreased cellular GSH levels, which was reversed by pretreatment of PGE2. (B) DPPP fluorescent intensity (lipid peroxidation) was decreased markedly in PGE2-treated cells, whereas menadione treatment increased the lipid peroxidation evident fluorescent intensity. Fluorescence images were obtained under microscopy.
PGE2-induced anti-apoptotic actions is mediated through EP2 receptor
Once the role of PGE2 in menadione-induced apoptosis in HL-60 cells was established, we next evaluated its functional relevance in anti-apoptotic actions. Above our results suggest that PGE2-induced anti-apoptotic effects might be mediated through a mechanism involving the cAMP system. Therefore, it seems that the activation of adenylate cyclase coupled with the activation of EP2 and EP4 receptors, plays an important role in the signal transduction mechanism associated with the PGE2-mediated protection from menadione-induced apoptosis.
To further support our conclusion for the role of PGE2 receptors, we compare their expression in HL-60 cells with the specific agonist for each receptor in order to identify EP receptors involved in the PGE2-mediated protection from menadione-induced apoptosis. Stimulation of the EP2 receptor with butaprost inhibited apoptotic cell death in HL-60 cells, whereas stimulation of EP3 or EP4 with sulprostone or PGE1 alcohol had no significant effect on cell survival (Fig. 4). Additionally, the EP2 antagonist AH-6809 abrogated the inhibitory effects of PGE2 on menadione-induced apoptosis (Fig. 4). Therefore, the EP2 receptor appeared to play a major role in mediating the PGE2-stimulated anti-apoptosis in HL-60 cells.
Fig. 4.

PGE2 protects apoptosis through EP2. HL-60 cells were incubated with butaprost, AH6809 and hydroxy PGE2. After treatment with PGE2 or menadione, cells were stained with propidium iodide and their DNA content was analyzed by flow cytometry. Stimulation of the EP2 receptor with butaprost or antagonist AH6809 inhibited apoptotic cell death in HL-60 cells, whereas stimulation of EP3 or EP4 with sulprostone or PGE1 alcohol had no significant effect on cell survival.
EP2 receptor activates PKA signaling pathway
To elucidate further the mechanism by which PGE2 regulated HL-60 apoptosis, we examined its downstream signaling pathways. It has been proposed that PGE2 stimulation of the EP2 receptor leads to the activation of PKA through the elevation of intracellular cAMP levels. To characterize the EP2 receptor mediated signaling pathways involved in the anti-apoptotic activity of PGE2, the effects of PGE2 on PKA activity were determined. PKA was measured in HL-60 cells with a radioisotope assay that utilizes the phosphorylation of the synthetic substrate kemptide (Fig. 5A). The PKA activity increased more than four-fold after treatment with PGE2. The ability of PGE2 to activate PKA was completely blocked by the addition of H-89, a specific PKA inhibitor. Prior to the incubation of HL-60 cells with H-89 for 15 min also suppressed anti-apoptotic activity of PGE2 by 85%, compared to the single treated PGE2 cells (Fig. 5B). These results indicate that PGE2-mediated cell survival in response to oxidative stress requires PKA activity.
Fig. 5.

PGE2 activate protein kinase A. (A) HL-60 cells were pretreated with vehicle alone, PGE2, or H89 for 15 min prior to being exposed to menadione. Cells were harvested at 2 h and measured PKA activity by in vitro kinase assay detailed in the “Materials and Methods”. (B) The cells were incubated with H89, PGE2 and then treated with menadione. After, stained with propi-dium iodide and their DNA content was analyzed by flow cytometry.
Studies have shown that PKA is a cytosolic, tetrameric holoenzymes that consists of two regulatory subunits associated with two catalytic subunits. The dissociated, active catalytic subunits can then affect the cell physiology through the phosphorylation of a wide variety of protein substrates in the cytoplasm and nucleus. cAMP-mediated transcriptional responses involve the ability of PKA to phosphorylate cAMP response element binding protein (CREB). To explore the functional significance of PKA activation by PGE2, we first tested translocation PKA catalytic subunits to nucleus in PGE2-challenged HL-60 cells was first tested. As shown in Fig. 6A, PGE2 treatment induced translocation of PKA catalytic subunits to the neucleus. However, in PGE2 stimulated cells the activated PKA did not phosphorylate cAMP response elementbinding (CREB) (Fig. 6B) and other cAMP-responsive transcription factors such as activating transcription factors (ATF-1 or ATF-2) (data not shown).
Fig. 6.

Effect of PGE2 on the expression of PKA, COX2, and transcription factors. (A) HL-60 cells were treated with 1 μM PGE2 and PKA catalytic subunit and COX2 expression was detected after indicated time. Immunoblotting analysis revealed that PGE2 enhanced the expression of PKA and COX2. (B) Cells were pretreated with or without PGE2, and incubated with 10 μM of menadione for 8, 12 and 24 h. PGE2 pretreatment inhibited the menadione-induced apoptosis via CREB and AKT.
Another possible signaling pathway for the effects of PGE2 on apoptosis is the phosphorylation of Akt. Since phosphorylation of Akt at Ser473 is required for its full activation, the phosphorylation of endogenous AKt was examined with an antibody that specifically recognizes Akt phosphorylation at the Serine residue. We did not observe any activation of endogenous Akt when the cells were treated with PGE2 (Fig. 6B). Thus, it did not seem likely that CREB and Akt could be involved in anti-apoptosis mediated by PGE2. Taken together, these results demonstrate that i) activation of PKA plays an important role in the regulation of PGE2-mediated anti-apoptotic effects, and ii) activation of CREB and Akt is not required for the protection of apoptosis in the monocytic HL-60 cells.
Activation of ERK is a critical event in PGE2-induced anti-apoptosis in HL-60 cells
Since Ras-Raf is implicated in the triggering of several different signal transduction pathways, we have verified that the integrity of the downstream EP2 receptor-targeted pathway was indeed required for the anti-apoptotic effects of PGE2. We directly measured the Raf activity in response to PGE2. Figure 7 shows that treatment of HL-60 cells with PGE2 induced an increase in Raf activity, which was assessed by immunoblotting samples of Raf immunoprecipitated from lysates using Raf-1-RBD. It seems that anti-apoptotic effect of EP2 leads to reinforcement of the activated Ras signal pathway due to the regulation of the cAMP levels in HL-60 cells simulated with PGE2. Western blot analysis also revealed that PGE2 treatment increased the levels of phosphorylated Raf and phosphorylated ERK in HL-60 cells (Fig. 7). To further insights into the signaling pathway leading to the protection of apoptosis in PGE2-induced HL-60 cells, a specific MEK1 inhibitor, PD98059, was used (Fig. 8). Pretreatment of HL-60 cells with PD98059 inhibited the PGE2 induced stimulation of Raf and ERK and reversed the anti-apoptotic effect in menadione treated HL-60 cells. Inhibition of the MEK pathway with PD98059 abolished the protective effect upon HL-60 cells, suggesting the role of the Ras-Raf-MEK-ERK pathway on this effect (Fig. 8). Taken together, these findings suggest that the activation of ERK is a critical event in PGE2-induced anti-apoptosis in HL-60 cells.
Fig. 7.

PGE2 induce Raf and ERK phosphorylation. (A) Ras activity was assessed by immunoblotting samples for Ras immunoprecipitated form lysates using Raf-1 RBD. Densitometer measurements of Ras activity (units as pixels) are shown below the blot. (B) Phosphorylated Raf-1 and Erk were detected by immunoblotting analysis with antibody specific for phosphorylated Raf-1 and Erk (pRaf and pErk, respectively). Protein loading was determined by stripping and reprobing the same nitrocellulose membrane for total Raf-1 and Erk protein level. Results are representative of three independent experiments.
Fig. 8.

The PGE2 effect is mediated by the ERK signaling pathway. (A) HL-60 cells were pretreated with specific MEK inhibitor, PD98095 for 1 h before treatment with PGE2 and menadione. Cells were harvested and stained with propidium iodide, and their DNA content was analyzed by flow cytometry. PD98059 abolished the protective effects of PGE2 on menadione-induced apoptosis. Apoptotic cell death is shown by the accumulation of cells with a DNA content of below 2n. Results were obtained from three independent experiments. (B) Cells were treated with PGE2 or PD98059 and ERK was detected by immunoblotting analysis with antibody specific for phosphorylated ERK(upper panel). PD98059 inhibited PGE2-induced phosphorylation of ERK in HL-60 cells. Actin was used as a control (lower panel).
DISCUSSION
Despite the recent advances in the understanding of signaling pathways, the role of PGE2 in mediating the effects of drugs has not been well publicized, and many questions have yet to be answered. PGE2 facilitates tumor progression by binding to its cognate receptors EP1–4 with the subsequent enhancement of cellular proliferation and angiogenesis, inhibiting apoptosis, enhancing cellular invasiveness, and modulating of immunosuppression (Narumiya et al., 1999). Our results demonstrate that PGE2 has a major role in promoting the survival of monocytes under oxidative-stress, and this effect might be mediated through its transmembrane receptor EP2. It was observed that treatment of HL-60 cells with menadione leads to the DNA fragmenataion and PARP and lamin B cleavage (Caricchio et al., 1999; Suzuki and Ono, 1999). PGE2 markedly increased the intracellular concentration of cAMP and reversed the menadione-induced apoptosis through the decreased degradation of PARP and lamin B (Figs. 1B and 1C). Several reports have demonstrated that the second messenger cAMP has anti-apoptotic actions in monocytic cell lines (García-Bermejo et al., 1998; Jun et al., 1998). cAMP also prevented apoptosis in a variety of other cell types including human neutrophil (Rossi et al., 1995), but the mechanism by which cAMP influences apoptosis is still unknown. The underlying mechanism was confirmed by the addition of cell-permeable cAMP analog dibutyryl and by the quantified amount of sub-G1 DNA, which showed that the addition of dibutyryl cAMP blocked menadione induced apoptotic cell death. The flow cytometry analysis revealed that PGE2 and dibutyryl cAMP inhibited the apoptosis induce by oxidative agents such as H2O2, etoposide and taxol in HL-60 cells and also attenuated the generation of apoptosis in U937 cells (Table 1).
Additionally, apoptotic cell death induced by menadione is thought to be mediated by intracellular ROS generation as shown previously (Criddle et al., 2006). PGE2 inhibited the menadione-induced apoptosis through the scavenging of ROS, observed by the decrease in the DCF fluorescence (Fig. 2). In accordance with this assumption, the GSH cellular level was significantly decreased in menadione treated cells (Checker et al., 2011). As shown in Fig. 3B, the fluorescent intensity of CMAC was decreased markedly in PGE2-treated cells, whereas it was increased in menadione-treated cells.
PGE2 is generated from arachidonic acid by cyclooxygenases, which binds to G protein-coupled receptors and increases the intracellular cAMP level (Coleman et al., 1994). It has been shown that HL-60 cells expressed three subtypes of PGE receptors including EP2, EP3, and EP4 (Ishiguro et al., 1998). Different studies have demonstrated the role of EP2 receptor in the enhanced effect of PGE2 on cellular and physiological processes (Ishiguro et al., 1998). Using pharmacological agonists for all four EP receptors, we noticed a significant increase in cAMP only with the EP2 specific agonist butaprost, suggesting that the other receptors do not mediate cell survival role of PGE2. Additionally, the EP2 antagonist AH-6809 abrogated the inhibitory effects of PGE2 on menadione-induced apoptosis (Fig. 4). These results are consistent with the previously published lines regarding the functional role of EP2-PGE2 in enhancing tumor promotion or preventing apoptosis (Aronoff et al., 2004; Kuo et al., 2009). Furthermore, Fig. 5A very clearly shows that the EP2 receptor activated PKA through an elevation of the intracellular cAMP level. PKA activity increased fourfold in PGE2 treated cells. The specific PKA inhibitor H89, completely prevented the PGE2 anti-apoptotic activity (85%) as shown in Fig. 5B, which suggest that PKA is an essential component of PGE2-mediated cell survival in response to oxidative stress.
To further insight into the survival role of PGE2, we explored the efficient mechanism of PKA and AKT activation by PGE2. Studies have shown that cAMP-mediated transcriptional responses required PKA to phosphorylate CREB (Seino and Shibasaki, 2005). It was observed that in PGE2 stimulated cells the translocated active PKA catalytic subunit did not phosphorylate CREB as well as other cAMP-responsive transcription factors such as ATF-1 and ATF-2. On the other hand, signaling through the PGE2 receptor (EP2) was coupled to the activation of Akt. Akt phosphorylation mediates anti-apoptotic and pro-survival events (Chang et al., 2003a). When the cells were treated with PGE2, no endogenous Akt was observed, which suggest that activation of Akt and CREB is not required for the protection of apoptosis in the monocytic HL-60 cells. Although, it has been observed that the PGE2 induced increase of CREB transcriptional activity is PKA dependent, however, this may be a cell specific role of PGE2 (Bidwell et al., 2010).
PGE2 exerted the majority of its actions through transmembrane receptors and the physiological role of PGE2via EP2 is diverse. It has been reported that PGE2 stimulates cell proliferation through the activation of ERK, suggesting the Ras signaling pathway (Yu et al., 2009). Overexpression of the activated Ras/Raf/ERK signaling pathway is associated with cell proliferation and inhibition of apoptosis (Chang et al., 2003b). Since Ras is implicated in the triggering of ERK, we verified that the integrity of the downstream EP2 receptor-targeted pathway was indeed required for the anti-apoptotic effects of PGE2. PGE2 treatment increased the phosphorylation of Raf and the ERK levels in HL-60 cells (Fig. 7), which suggest that the ERK signaling pathway is involved in the protection of apoptosis in PGE2-induced HL-60 cells. The activation of ERK1/2 is critical for PGE2-induced cell proliferation and is located upstream of COX-2 gene expression (Shen et al., 2004). PD98059, a specific inhibitor of MEK1, was used to block ERK1/2 activation, and PGE2-induced apoptosis was measured. It is noted that PD98059 inhibited the anti-apoptotic effect of PGE2, followed by the inhibition of ERK1/2 phosphorylation induced by PGE2 in HL-60 cells (Fig. 8). The functional outcome of ERK activation in HL-60 cells does not always mean a cell will go into a proliferative response. Rather, in certain cell types and under certain environmental circumstances, inhibition of ERK protects cells from apoptotic cell death (Goulet et al., 2005). However, in the present study, PGE2 might induce ERK phosphorylation at concentrations that stimulate HL-60 cell proliferation, which can be supported through the previously published work (Yu et al., 2009). Our results show that PGE2 activates the Ras pathway, which in turn initiates ERK activation, and ultimately protects oxidative-induced apoptosis in HL-60 monocytic cells.
In the current study, we present evidence for the biological role of PGE2 in promoting the survival of monocytes under oxidative-stress. Pretreatment of HL-60 cells with PGE2 markedly inhibited the menadione-induced apoptosis through the scavenging of ROS generation followed by an increase in the cellular GSH level and a decrease in lipid peroxidation. Challenging the HL-60 cells with PGE2 triggers an intracellular signaling pathway that leads to the activation of cAMP transcriptional activity. The selective EP2 receptor mediates anti-apoptotic and pro-survival events in PGE2 treated HL-60 cells. Additionally, the specific inhibition of ERK revealed that the anti-apoptotic effects of PGE2 might be related to the Ras/Raf/ERK signaling pathway. Our preliminary results will shed light on the hypothesis that PGE2 is directly responsible for the protection of monocytes under oxidative-stress. The importance of non-steroidal anti-inflammatory drugs (NSAIDs) to inhibit PGE2 remains a clinically relevant agent for the treatment of inflammation. The PGE2 inhibitors will play a pivotal role in the treatment and prevention not only for leukemia but also for other types of cancers. Further studies are warranted to investigate the modulating effects of PGE2 on the apoptotic signaling pathways of HL-60 cells as well as other cancer types either in vitro or in vivo.
Acknowledgments
This Research was supported by Kyungpook National University Research Fund, 2010.
REFERENCES
- Aronoff D.M., Canetti C., Peters-Golden M. Prostaglandin E2 inhibits alveolar macrophage phagocytosis through an E-prostanoid 2 receptor-mediated increase in intracellular cyclic AMP. Immunology. 2004;173:559–565. doi: 10.4049/jimmunol.173.1.559. [DOI] [PubMed] [Google Scholar]
- Bidwell P., Joh K., Leaver H.A., Rizzo M.T. Prostaglandin E2 activates cAMP response element-binding protein in glioma cells via a signaling pathway involving PKA-dependent inhibition of ERK. Prostaglandins Other Lipid Mediat. 2010;91:18–29. doi: 10.1016/j.prostaglandins.2009.12.002. [DOI] [PubMed] [Google Scholar]
- Blum S., Moore A.N., Adams F., Dash P.K. A mitogen-activated protein kinase cascade in the CA1/CA2 subfield of the dorsal hippocampus is essential for long-term spatial memory. J. Neurosci. 1999;19:3535–3544. doi: 10.1523/JNEUROSCI.19-09-03535.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brière J.J., Schlemmer D., Chretien D., Rustin P. Quinone analogues regulate mitochondrial substrate competitive oxidation. Biochem. Biophys. Res. Commun. 2004;316:1138–1142. doi: 10.1016/j.bbrc.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Caricchio R., Kovalenko D., Kaufmann W.K., Cohen P.L. Apoptosis provoked by the oxidative stress inducer menadione (Vitamin K3) is mediated by the Fas/Fas ligand system. Clin. Immunol. 1999;93:65–74. doi: 10.1006/clim.1999.4757. [DOI] [PubMed] [Google Scholar]
- Chang F., Lee J.T., Navolanic P.M., Steelman L.S., Shelton J.G., Blalock W.L., Franklin R.A., McCubrey J.A. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation a target for cancer chemotherapy. Leukemia. 2003a;17:590–603. doi: 10.1038/sj.leu.2402824. [DOI] [PubMed] [Google Scholar]
- Chang F., Steelman L.S., Lee J.T., Shelton J.G., Navolanic P.M., Blalock W.L., Franklin R.A., McCubrey J.A. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors potential targeting for therapeutic intervention. Leukemia. 2003b;17:1263–1293. doi: 10.1038/sj.leu.2402945. [DOI] [PubMed] [Google Scholar]
- Checker R., Sharma D., Sandur S.K., Khan N.M., Patwardhan R.S., Kohli V., Sainis K.B. Vitamin K3 suppressed inflammatory and immune responses in a redox-dependent manner. Free Radic. Res. 2011;45:975–985. doi: 10.3109/10715762.2011.585647. [DOI] [PubMed] [Google Scholar]
- Chun K.S., Lao H.C., Trempus C.S., Okada M., Langenbach R. The prostaglandin receptor EP2 activates multiple signaling pathways and beta-arrestin1 complex formation during mouse skin papilloma development. Carcinogenesis. 2009;30:1620–1627. doi: 10.1093/carcin/bgp168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun K.S., Lao H.C., Langenbach R. The prostaglandin E2 receptor, EP2, stimulates keratinocyte proliferation in mouse skin by G protein-dependent and β-arrestin1-dependent signaling pathways. J. Biol. Chem. 2010;285:39672–39681. doi: 10.1074/jbc.M110.117689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffey R.N., Watson R.W., Hegarty N.J., O’Neill A., Gibbons N., Brady H.R., Fitzpatrick J.M. Thiol-mediated apoptosis in prostate carcinoma cells. Cancer. 2000;88:2092–2104. doi: 10.1002/(sici)1097-0142(20000501)88:9<2092::aid-cncr15>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Coleman R.A., Smith W.L., Narumiya S. International Union of Pharmacology classification of prostanoid receptors: properties, distribution and structure of the receptors and their subtypes. Pharmacol. Rev. 1994;46:205–229. [PubMed] [Google Scholar]
- Criddle D.N., Gillies S., Baumgartner-Wilson H.K., Jaffar M., Chinje E.C., Passmore S., Chvanov M., Barrow S., Gerasimenko O.V., Tepikin A.V., et al. Menadione-induced reactive oxygen species generation via redox cycling promotes apoptosis of murine pancreatic acinar cells. J. Biol. Chem. 2006;281:40485–40492. doi: 10.1074/jbc.M607704200. [DOI] [PubMed] [Google Scholar]
- Fontana J.A., Colbert D.A., Deisseroth A.B. Identification of a population of bipotent stem cells in the HL-60 humman promyelocytic leukemia cell line. Proc. Natl. Acad. Sci. USA. 1981;78:3863–3866. doi: 10.1073/pnas.78.6.3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Bermejo L., Pérez C., Vilaboa N.E., de Blas E., Aller P. cAMP increasing agents attenuate the generation of apoptosis by etoposide in promonocytic leukemia cells. J. Cell Sci. 1998;111:637–644. doi: 10.1242/jcs.111.5.637. [DOI] [PubMed] [Google Scholar]
- Goulet A.C., Chigbrow M., Frisk P., Nelson M.A. Selenomethionine induces sustained ERK phosphorylation leading to cell-cycle arrest in human colon cancer cells. Carcinogenesis. 2005;26:109–117. doi: 10.1093/carcin/bgh306. [DOI] [PubMed] [Google Scholar]
- Herrmann M., Lorenz H.M., Voll R., Grunke M., Woith W., Kalden J.R. A rapid and simple method for the isolation of apoptotic DNA fragments. Nucleic Acids Res. 1994;22:5506–5507. doi: 10.1093/nar/22.24.5506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiguro S., Takahashi N., Nemoto K., Negishi M., Ichikawa A. Potentiation of retinoic acid-induced differentiation of HL-60 cells by prostaglandin EP2 receptor. Prostaglandins Other Lipid Mediat. 1998;56:145–153. doi: 10.1016/s0090-6980(98)00051-3. [DOI] [PubMed] [Google Scholar]
- Kuo K.T., Wang H.W., Chou T.Y., Hsu W.H., Hsu H.S., Lin C.H., Wang L.S. Prognostic role of PGE2 receptor EP2 in esophageal squamous cell carcinoma. Ann. Surg. Oncol. 2009;16:352–360. doi: 10.1245/s10434-008-0242-2. [DOI] [PubMed] [Google Scholar]
- Narumiya S., Sugimoto Y., Ushikubi F. Prostanoid receptors: structures, properties and functions. Physiol. Rev. 1999;79:1193–1226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- Osada S., Tomita H., Tanaka Y., Tokuyama Y., Tanaka H., Sakashita F., Takahashi T. The utility of vitamin K3 (menadione) against pancreatic cancer. Anticancer Res. 2008;28:45–50. [PubMed] [Google Scholar]
- Park J.E., Yang J.H., Yoon S.J., Lee J.H., Yang E.S., Park J.W. Lipid peroxidation-mediated cytotoxicity and DNA damage in U937 cells. Biochimie. 2002;84:1199–1205. doi: 10.1016/s0300-9084(02)00039-1. [DOI] [PubMed] [Google Scholar]
- Rossi A.G., Cousin J.M., Dransfield I., Lawson M.F., Chilvers E.R., Haslett C. Agents that elevate cAMP inhibit human neutrophil apoptosis. Biochem. Biophys. Res. Commun. 1995;217:892–899. doi: 10.1006/bbrc.1995.2855. [DOI] [PubMed] [Google Scholar]
- Sampey A.V., Monrad S., Crofford L.J. Microsomal prostaglandin E synthase-1: the inducible synthase for prostaglandin E2. Arthritis Res. Ther. 2005;7:114–117. doi: 10.1186/ar1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seino S., Shibasaki T. PKA-dependent and PKA-independent pathways for cAMP-regulated exocytosis. Physiol. Rev. 2005;85:1303–1342. doi: 10.1152/physrev.00001.2005. [DOI] [PubMed] [Google Scholar]
- Shen S.C., Ko C.H., Hsu K.C., Chen Y.C. 3-OH flavone inhibition of epidermal growth factor-induced proliferaton through blocking prostaglandin E2 production. Int. J. Cancer. 2004;108:502–510. doi: 10.1002/ijc.11581. [DOI] [PubMed] [Google Scholar]
- Smith W.L., DeWitt D.L., Garavito R.M. Cyclooxygenases: structural, cellular and molecular biology. Annu. Rev. Biochem. 2000;69:145–182. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- Sung Y.M., He G., Hwang D.H., Fischer S.M. Overexpression of the prostaglandin E2 receptor EP2 results in enhanced skin tumor development. Oncogene. 2006;25:5507–5516. doi: 10.1038/sj.onc.1209538. [DOI] [PubMed] [Google Scholar]
- Suzuki Y., Ono Y. Involvement of reactive oxygen species produced via NADPH oxidase in tyrosine phosphorylation in human B- and T-lineage lymphoid cells. Biochem. Biophys. Res. Commun. 1999;255:262–267. doi: 10.1006/bbrc.1999.0188. [DOI] [PubMed] [Google Scholar]
- Thayyullathil F., Chathoth S., Hago A., Patel M., Galadari S. Rapid reactive oxygen species (ROS) generation induced by curcumin leads to caspase-dependent and -independent apoptosis in L929 cells. Free Radic. Biol. Med. 2008;45:1403–1412. doi: 10.1016/j.freeradbiomed.2008.08.014. [DOI] [PubMed] [Google Scholar]
- Trachootham D., Alexandre J., Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat. Rev. Drug. Discov. 2009;8:579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- Yu J.S., Kim A.K. Wogonin induces apoptosis by activation of ERK and p38 MAPKs signaling pathways and generation of reactive oxygen species in human breast cancer cells. Mol. Cells. 2011;31:327–335. doi: 10.1007/s10059-011-0041-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L., Wu W.K., Li Z.J., Li H.T., Wu Y.C., Cho C.H. Prostaglandin E(2) promotes cell proliferation via protein kinase C/extracellular signal regulated kinase pathway-dependent induction of c-Myc expression in human esophageal squamous cell carcinoma cells. Int. J. Cancer. 2009;125:2540–2546. doi: 10.1002/ijc.24607. [DOI] [PubMed] [Google Scholar]