

Abstract
Lung cancer remains a global health problem with a high mortality rate. CpG island methylation is a common aberration frequently associated with gene silencing in multiple tumor types, emerging as a highly promising biomarker. The transmembrane protein with a single EGF-like and two follistatin domains (TMEFF2) is epigenetically silenced in numerous tumor types, suggesting a potential role as a potential tumor suppressor. However, the role of TMEFF2 in lung cancer remains to be fully elucidated. We explored the methylation status of TMEFF2 gene in 139 patients with non-small cell lung cancer (NSCLC) and the feasibility of detecting circulating methylated DNA as a screening tool for NSCLC using methylation-specific PCR in 316 patients and 50 age-matched health controls. TMEFF2 methylation in tumor tissues was found in 73 of the 139 NSCLCs (52.5%) and was related to gene expression. The frequency of TMEFF2 methylation was higher in females and never-smokers than in males and smokers with borderline significance (65.8% vs 47.8%, p = 0.06; 65.7% vs 48.1%, p = 0.07). Notably, in adenocarcinomas, TMEFF2 methylation was significantly more frequent in tumors without EGFR mutation than those with EGFR mutation (adjusted odds ratio = 7.13, 95% confidence interval = 2.05–24.83, P = 0.002). Furthermore, TMEFF2 methylation was exclusively detected in the serum of NSCLC patients at a frequency of 9.2% (29/316). These findings suggest that methylation-associated down-regulation of TMEFF2 gene may be involved in lung tumorigenesis and TMEFF2 methylation can serve as a specific blood-based biomarker for NSCLC.
Keywords: methylation, methylation-specific PCR, non-small cell lung cancer, serum, TMEFF2
INTRODUCTION
Lung cancer is the leading cause of cancer deaths worldwide, with over 1 million deaths each year (Jemal et al., 2008). The poor prognosis is largely attributable to lack of efficient diagnostic methods for early detection (Minna et al., 2002). Thus, there is an increasing demand for molecular markers useful in the early diagnosis of lung cancer. DNA methylation within gene promoter regions results in aberrant gene silencing, which can make an important contribution to the emergence of a neoplasm (Jones and Baylin, 2002). Moreover, aberrant promoter methylation often occurs as very early events during cancer development (Baylin and Ohm, 2006). To date, a number of genes can now be methylated in lung cancer, which shows great promise for early detection of lung cancer (Anglim et al., 2008; Belinsky, 2004; Heller et al., 2010; Lee et al., 2011).
Transmembrane protein with epidermal growth factor (EGF)-like and two follistatin-like domains (TMEFF2), also known as TPEF, TENB2, and HPP1, was initially identified as a novel gene frequently methylated in human tumor cells (Liang et al., 2000). Although structural modules suggest that it may play multiple roles in cell growth, maturation, and adhesion (Young et al., 2001), the biological function of TMEFF2 in carcinogenesis remains elusive with conflicting reports (Ali and Knaüper, 2007; Elahi et al., 2008; Gery et al., 2002; Glynne-Jones et al., 2001). It has been reported that an elevated TMEFF2 expression and its extracellular portion promote cell growth (Ali and Knaüper, 2007; Glynne-Jones et al., 2001), whereas TMEFF2 plays a role in suppressing the growth and invasive potential of human cancer cells (Elahi et al., 2008; Gery et al., 2002). Moreover, TMEFF2 is mapped to human chromosome 2q32.3, where frequent loss of heterozygosity is seen in various human tumors (Beder et al., 2003; Otsuka et al., 1996; Takita et al., 2001) and is frequently hypermethylated in various human cancers (Lin et al., 2011; Suzuki et al., 2005; Young et al., 2001). In particular, chip-based analysis has shown that TMEFF2 gene is identified to have a distinct methylation pattern in nonsmall cell lung cancers (NSCLCs) compared to normal tissues (Field et al., 2005), and TMEFF2 methylation has been reported to frequently occur in the precancerous lesion and early stage of lung adenocarcinoma (AC), such as atypical adenomatous hyperplasia and bronchioloaveolar cell carcinoma (Chung et al., 2011; Selamat et al., 2011). Importantly, aberrantly methylated markers identified in lung tissue samples can be detected in peripheral blood, providing a valuable approach to non-invasive screening for early detection of lung cancer (Bremnes et al., 2005). We investigated the methylation status of the promoter region of the TMEFF2 gene in NSCLCs and correlated the results with clinicopathological characteristics. Its methylation was also evaluated in the blood of NSCLC patients and in controls.
MATERIALS AND METHODS
Patients and samples collection
Tumor and corresponding non-malignant lung tissue specimens (n = 139) were provided by the National Biobank of Korea - Kyungpook National University Hospital (KNUH), which is supported by the Ministry of Health, Welfare and Family Affairs. All materials derived from the National Biobank of Korea - KNUH were obtained by institutional review board approved protocols. All of the tumor and macroscopically normal lung tissue samples were obtained at the time of surgery, and were rapidly frozen in liquid nitrogen and stored at −80°C until genomic DNA preparation. Only tumors with > 80% tumor component were sent for DNA extraction and methylation analysis. The macroscopically normal lung tissues were confirmed to be normal by hematoxylineosin staining. Genomic DNA was extracted using the QIAamp DNA Mini Kit (QIAGEN, USA). Meanwhile, blood samples were obtained from 316 unselected informed NSCLC patients diagnosed at KNUH with written informed consent. The clinicopathological characteristics of the patients are summarized in Tables 1 and 2. Similarly, blood was drawn from 50 healthy donor volunteers in the same age group. The serum (2 ml) was isolated by centrifugation at 3000 rpm for 10 min and stored at −80°C until use. Serum DNA was extracted with the QIAamp DNA Blood Midi Kit (QIAGEN) according to the manufacturer’s instruction.
Table 1.
Associations of TMEFF2 methylation with the clinicopathological features of NSCLC patients
| Feature | Methylation frequency (%) | Crude
|
Adjusted
|
||||
|---|---|---|---|---|---|---|---|
| OR | 95% CI | pa | OR | 95% CI | pb | ||
| All subjects (n = 139) | 73 (52.5) | ||||||
| Age (years) | |||||||
| ≤ 62 (n = 60) | 34 (56.7) | 1.00 | 1.00 | ||||
| → 62 (n = 79) | 39 (49.4) | 0.75 | 0.38–1.46 | 0.40 | 0.77 | 0.38–1.56 | 0.46 |
| Gender | |||||||
| Male (n = 101) | 48 (47.5) | 1.00 | 1.00 | ||||
| Female (n = 38) | 25 (65.8) | 2.12 | 0.98–4.61 | 0.06 | 1.93 | 0.47–7.82 | 0.36 |
| Smoking status | |||||||
| Ever (n = 104) | 50 (48.1) | 1.00 | 1.00 | ||||
| Never (n = 35) | 23 (65.7) | 2.07 | 0.93–4.59 | 0.07 | 1.48 | 0.34–6.41 | 0.60 |
| Histology types | |||||||
| SCC (n = 60) | 32 (53.3) | 1.00 | 1.00 | ||||
| AC (n = 79) | 41 (51.9) | 0.944 | 0.48–1.85 | 0.87 | 0.62 | 0.28–1.34 | 0.23 |
| Pathologic stages | |||||||
| Stage I (n = 85) | 46 (54.1) | 1.00 | 1.00 | ||||
| Stage II–IIIA (n = 54) | 27 (50.0) | 0.85 | 0.43–1.68 | 0.64 | 0.79 | 0.38–1.61 | 0.51 |
| EGFR mutation in ACs | |||||||
| Present (n = 33) | 12 (36.4) | 1.00 | 1.00 | ||||
| Absent (n = 46) | 29 (63.0) | 2.99 | 1.18–7.55 | 0.02 | 7.13 | 2.05–24.83 | 0.002 |
P-value was calculated by the logic regression test.
P-value was calculated by the logic regression test adjusted for other variables.
SCC, squamous cell carcinoma; AC, adenocarcinoma; OR, odds ratio; CI, confidence interval
Table 2.
Associations of TMEFF2 methylation in serum DNA with the clinicopathological features of NSCLC patients
| Feature | Methylation frequency (%) | p |
|---|---|---|
| All subjects (n = 316) | 29 (9.2) | |
| Age (years) | ||
| ≤ 66 (n = 131) | 10 (7.6) | 0.424 |
| > 66 (n = 185) | 19 (10.3) | |
| Gender | ||
| Male (n = 243) | 20 (8.2) | 0.288 |
| Female (n = 73) | 9 (12.3) | |
| Smoking status | ||
| Ever (n = 249) | 19 (7.7) | 0.066 |
| Never (n = 67) | 10 (14.9) | |
| Histologic types* | ||
| SCC (n = 104) | 8 (7.7) | 0.579 |
| AC (n = 144) | 14 (9.7) | |
| Pathologic stages | ||
| Stage I (n = 67) | 5 (7.5) | 0.584 |
| Stage II–IV (n = 249) | 24 (9.6) |
Sixty eight patients including small cell lung cancers, large cell carcinomas, and unidentified non-small cell lung cancers were excluded from the analysis.
SCC, squamous cell carcinoma; AC, adenocarcinoma
Methylation analysis
The methylation status of the TMEFF2 gene in tissues and serum DNA was determined using a nested methylation-specific PCR (MSP). Bisulfite-treated DNA was amplified with the flanking primers 5′-GAGTTTAGTTTTTGGATGTTG-3′ and 5′-TACAACTCTACAACAACAAAC-3′. The amplicons from step one were diluted at 1:250 and then subjected to the second step of MSP that incorporated unmethylated or methylated primers; methylated, 5′-AAATTTTCGAGATTATGCGC-3′ (forward) and 5′-CCGAAAAACACAAAATCGCG-3′ (reverse); and unmethylated, 5′-AAATTTTTGAGATTATGTGT-3′ (forward) and 5′-CCAAAAAACACAAAATCACA-3′ (reverse). All PCR amplifications were carried out using reagents supplied by the GeneAmp DNA Amplification Kit with AmpliTaq Gold as the polymerase on a PTC-100 thermal cycler (MJ Research, USA). CpGenome™ Universal methylated and unmethylated DNA (Chemicon, USA) was used as a positive control for the methylated and unmethylated genes, respectively. Negative control samples without DNA were included for each set of PCR reactions. PCR products were analyzed on 2% agarose gels, stained with ethidium bromide, and visualized under UV light. Each MSP was repeated at least once to confirm the results.
Cell culture and 5-aza-2′-deoxycytidine (5-AzadC) treatment
Eight human NSCLC cancer cell lines, 4 adenocarcinoma (AC) (H522, A549, H1793, and H2009), 3 squamous cell carcinoma (SCC) (H157, H226, and H1703) and 1 large cell carcinoma (H1299), were obtained from the American Type Culture Collection (ATCC, USA). All cells were propagated following the instructions from ATCC. A549, H226, and H2009 cell lines were treated with 20 μM 5-AzadC for 3 days and the culture media was changed daily.
Total RNA isolation and reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was extracted from cultured NSCLC cell lines using TRIzol (Invitrogen, Australia) according to the manufacturer’s instructions. The structural integrity of the total RNA was confirmed by electrophoresis on 1.2% agarose-formaldehyde gels. Residual genomic DNA was digested with RNase-free DNase (Invitrogen). First strand cDNA was reverse-transcribed from 2 μg total RNA in a total volume of 20 μl using oligo(dT) and the SuperScript preamplification kit (Invitrogen). The resulting cDNA was amplified with primers specific to the TMEFF2 gene with AmpliTaq Gold (PE Applied Biosystems, USA). The primer sequences and annealing temperatures were described previously (Young et al., 2001). PCR was performed in a PTC100 and amplified products were separated on 2% agarose gels, visualized using ethidium bromide, and photographed.
Statistical analysis
The relationship between methylation and clinicopathological characteristics was analyzed with the Chi-square test or Fisher’s exact test for categorical variables. A P value < 0.05 was considered to be statistically significant. A logistic regression test was done to estimate the relationship between methylation and the covariates of age, gender, smoking history, tumor histology, and pathologic stage. The overall survival times of the NSCLC patients with or without methylation of the TMEFF2 gene were compared with the Kaplan-Meier method and the log-rank test. All analyses were performed using the Statistical Analysis System for Windows, version 9.1 (SAS Institute, USA).
RESULTS
Methylation status of the TMEFF2 gene in tissues and inverse correlation with its expression
The methylation status of the TMEFF2 gene was determined in 139 surgically resected NSCLCs and their corresponding nonmalignant lung tissues using nested MSP. The MSP primers were located at −197 to −11 bp relative to the translation start site, within the CpG island (CGI) described previously (Young et al., 2001). Each primer set yielded a single band of the expected size, and representative examples of the MSP analysis are shown in Fig. 1. Due to contamination of the tumor specimens with a small portion of normal tissues, unmethylated bands were present in most of all the malignant tissues. Bisulfite sequencing of the representative PCR products confirmed their methylation status and showed that all cytosines at non-CpG sites were converted to thymine (data not shown). Methylation of the TMEFF2 gene was found in 73 (52.5%) of the 139 tumor tissues and in 22 (15.8%) of their corresponding nonmalignant lung tissues (P < 0.0005), suggesting that TMEFF2 methylation may be a tumor-associated, de novo event. In addition, a high concordance of the methylation status between the tumor and adjacent normal tissues indicates the possibility that even phenotypically normal lung tissues can harbor epigenetic alterations already because the entire field of the lung was exposed to the carcinogenic insult. It has been reported that aberrant methylation frequently occurs in histologically normal-appearing lung tissues, representing a field defect of wide-spread epigenetic change in lung tissues (Belinsky et al., 2002; Guo et al., 2004). Accordingly, Ivanauskas et al. have found a substantial frequency of TMEFF2 methylation in the surrounding tumor-free mucosa of gastric ACs patients (Ivanauskas et al., 2008), supporting the field effect theory.
Fig. 1.
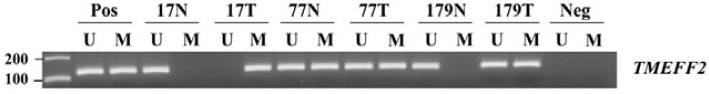
Representative data of the MSP analysis of TMEFF2 gene in NSCLC lung tissues. The methylation status of the TMEFF2 gene was analyzed in tumor and non-malignant lung tissues. MSP was carried out using unmethylation-specific (U) and methylation-specific (M) primers. CpGenome™ Universal methylated and unmethylated DNA (Chemicon) was used as a positive control for the M and U forms. Water was used as a negative control. Lane U, amplified product obtained using U primers; Lane M, amplified product obtained using M primers; Pos, positive control; Neg, negative control.
To determine whether promoter methylation was associated with transcriptional silencing of the TMEFF2 gene, the mRNA level and methylation status were investigated in NSCLC cell lines using RT-PCR and MSP analysis. TMEFF2 mRNA was expressed at high levels in the cell lines (H522, H1793, H2009, and H1703) with unmethylated alleles, while its expression was remarkably reduced in the cell lines (A549, H157, H226, and H1299) with a methylated allele (Fig. 2). Moreover, DNA demethylation with 5-AzadC treatment was able to significantly restore TMEFF2 mRNA expression. These data indicate that there is an inverse correlation between TMEFF2 promoter methylation and mRNA expression.
Fig. 2.

RT-PCR and MSP analysis of TMEFF2 gene in NSCLC cell lines. Expression TMEFF2 mRNA (A) and its methylation status (B) were determined in various human NSCLC cell lines by RT-PCR and MSP, respectively. Amplified products were run on 2% agarose gel and were visible at 178-bp for TMEFF2. Amplification of GAPDH was used as an internal loading control.
Relationship between promoter methylation and clinicopathological characteristics
TMEFF2 methylation was more frequent in the females than in males and in never-smokers than in ever-smokers with borderline significance (P = 0.06 and 0.07, respectively). Notably, the frequency of TMEFF2 methylation was significantly higher in EGFR mutation-negative ACs than EGFR mutation-positive ACs in univariate analysis (P = 0.02) (Table 1). This association was found to be independent in multivariate analysis adjusted for other variables (adjusted odds ratio = 7.13, 95% confidence interval = 2.05–24.83, P = 0.002). However, TMEFF2 methylation was not significantly associated with other factors (Table 1). Moreover, there was no significant difference in overall survival of the total patients according to TMEFF2 methylation status (data not shown).
Methylation status of TMEFF2 gene in the serum DNA of NSCLC patients
In the peripheral blood serum, TMEFF2 methylation was detected in 29 (9.2%) of the 316 tested NSCLC patients, while no sample carried its methylation in 50 age-matched, healthy controls, indicating that the sensitivity and specificity were 9.2% (29/316) and 100% (50/50), respectively. A representative of TMEFF2 methylation detection in serum is shown in Fig. 3. Unmethylated bands were observed in most of the tested tumor samples (data not shown), indicating the presence of circulating DNA in the patients’ serum. Notably, TMEFF2 methylation was detected more frequent in never-smokers than in ever-smokers with marginal significance (Table 2), being similar to the methylation pattern observed in lung tissues. However, it was not significantly correlated with other clinicopathological parameters of NSCLC patients (Table 2). Next, to confirm that serum TMEFF2 methylation are derived from tumor tissues, 20 matched samples including tissue and serum DNAs were analyzed. Although the prevalence of TMEFF2 methylation was significantly greater in tissue than in serum from NSCLCs, all three patients with serum DNA methylation showed the same alteration in the corresponding tumor tissues and no patients with only serum DNA methylation was identified (data not shown), indicating a significant concordance regarding the methylation pattern of the TMEFF2 gene in serum with tumor tissues. Taken together, these results suggest that TMEFF2 methylation can serve as a reliable and specific blood-based biomarker for NSCLC.
Fig. 3.
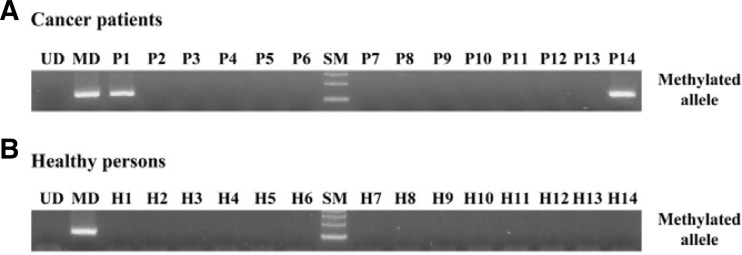
Representative data of the MSP analysis of TMEFF2 gene in serum DNA. The methylation status of the TMEFF2 gene was analyzed in serum DNA derived from NSCLC patients and healthy volunteers. MSP was carried out using unmethylation-specific (U) and methylation-specific (M) primers. CpGenome™ Universal methylated and unmethylated DNA (Chemicon) was used as a positive control for the M and U forms. Water was used as a negative control. Lane U, amplified product obtained using U primers; lane M, amplified product obtained using M primers; Pos, positive control; Neg, negative control.
DISCUSSION
We observed that TMEFF2 methylation frequently occurred in NSCLCs and that it was negatively correlated with its mRNA expression. In addition, TMEFF2 methylation in serum DNA appeared to be tumor-specific, representing the demonstration of the aberrant methylation of the TMEFF2 gene in serum DNA from NSCLC patients by MSP.
An interesting finding of the present study is that TMEFF2 methylation was more common in ACs without EGFR mutations than in those with EGFR mutations. In addition, TMEFF2 methylation was more frequent in females and never-smokers than in males and ever-smokers although the results were borderline significant. Interestingly, EGFR mutations highly occur in females and never-smokers (Shigematsu and Gazdar, 2006). Taken together, although the precise mechanisms are unknown, it is tempting to speculate a potential interplay between TMEFF2 and EGFR signaling pathways through aberrant methylation and mutation in lung ACs. In addition, it is likely that TMEFF2 methylation could be related to other environmental factors rather than tobacco smoking.
Notably, the prevalence of TMEFF2 methylation in NSCLCs is approximately an intermediate value compared with the results of the two groups showing a 72% to 30% frequency in lung ACs (Chung et al., 2011; Suzuki et al., 2005). These discrepancies may be attributable to methodological or geographical differences. The sensitivity of the nested MSP analysis used in the current study is at a medium level between the MethyLight assay and conventional MSP. Moreover, Toyook et al. (2003) have demonstrated geography-related differences in the methylation profiles of NSCLC. Alternatively, these divergent results might depend on the position of the examined CpG sites or the chance as a result of a relatively small number of study subjects examined.
The diagnosis of lung cancer at an early stage is critical for the effectiveness of treatment. Since increased levels of circulating DNA have frequently been found in the blood of cancer patients (Jahr et al., 2001), detection of methylation of the tumor suppressor gene (TSG) promoter in blood DNA may facilitate early detection of lung cancer. The methylation of several TSGs in blood is also detected on average in 42% (range 5–73%) of the NSCLC patients (Bremnes et al., 2005). Interestingly, TMEFF2 has been identified to be detected in peripheral blood as a frequent tumor-specific aberrantly methylated marker (Sabbioni et al., 2003). In addition, recent data have demonstrated that TMEFF2 methylation is detected in 65–71.2% of the plasma and fecal DNA from colorectal cancer patients and its methylation in plasma or fecal DNA and tumor is closely correlated (Huang et al., 2007; Lofton-Day et al., 2008). By contrast, our results indicate that TMEFF2 methylation in serum DNA is not a sensitive marker for NSCLC diagnosis even with excellent specificity, being similar to previous report that p16, DAPK, and MGMT methylation are significantly lower in the blood samples compared to the corresponding tumor tissues (Russo et al., 2005). We have no explanation for this difference yet, but the low incidence appears not to be based on the sensitivity of our assay. Generally, despite the low frequency of methylation in plasma or serum for each of the genes, using all for markers, they could detect 50% of the patients with methylation in the body fluids. Thus, further studies to enhance the sensitivity of TMEFF2 gene in combination with additional CpG sites frequently methylated in serum should be further explored.
Acknowledgments
This work was supported by the Korea Research Foundation Grant funded by the Korean Government (Ministry of Education & Human Resources Development, Basic Research Promotion Fund) (2010-0010000).
REFERENCES
- Ali N., Knaüper V. Phorbol ester-induced shedding of the prostate cancer marker transmembrane protein with epidermal growth factor and two follistatin motifs 2 is mediated by the disintegrin and metalloproteinase-17. J. Biol. Chem. 2007;282:37378–37388. doi: 10.1074/jbc.M702170200. [DOI] [PubMed] [Google Scholar]
- Anglim P.P., Alonzo T.A., Laird-Offringa I.A. DNA methylation-based biomarkers for early detection of non-small cell lung cancer: an update. Mol. Cancer. 2008;7:81–94. doi: 10.1186/1476-4598-7-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylin S.B., Ohm J.E. Epigenetic gene silencing in cancer - an early oncogenic pathway addiction? Nat. Rev. Cancer. 2006;6:107–116. doi: 10.1038/nrc1799. [DOI] [PubMed] [Google Scholar]
- Beder L.B., Gunduz M., Ouchida M., Fukushima K., Gunduz E., Ito S., Sakai A., Nagai N., Nishizaki K., Shimizu K. Genome-wide analyses on loss of heterozygosity in head and neck squamous cell carcinomas. Lab. Invest. 2003;83:99–105. doi: 10.1097/01.lab.0000047489.26246.e1. [DOI] [PubMed] [Google Scholar]
- Belinsky S.A. Gene-promoter hypermethylation as a biomarker in lung cancer. Nat. Rev. Cancer. 2004;4:1–11. [Google Scholar]
- Belinsky S.A., Palmisano W.A., Gilliland E.D., Crooks L.A., Divine K.K., Winters S.A., Grimes M.J., Harms H.J., Tellez C.S., Smith T.M., et al. Aberrant promoter methylation in bronchial epithelium and sputum from current and former smokers. Cancer Res. 2002;62:2370–2377. [PubMed] [Google Scholar]
- Bremnes R.M., Sirera R., Camps C. Circulating tumour-derived DNA and RNA markers in blood: a tool for early detection, diagnostics, and follow-up? Lung Cancer. 2005;49:1–12. doi: 10.1016/j.lungcan.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Chung J.H., Lee H.J., Kim B.H., Cho N.Y., Kang G.H. DNA methylation profile during multistage progression of pulmonary adenocarcinomas. Virchows Arch. 2011;459:201–211. doi: 10.1007/s00428-011-1079-9. [DOI] [PubMed] [Google Scholar]
- Elahi A., Zhang L., Yeatman T.J., Gery S., Sebti S., Shibata D. HPP1-mediated tumor suppression requires activation of STAT1 pathways. Int. J. Cancer. 2008;122:1567–1572. doi: 10.1002/ijc.23202. [DOI] [PubMed] [Google Scholar]
- Field J.K., Liloglou T., Warrak S., Burger M., Becker E., Berlin K., Nimmrich I., Maier S. Methylation discriminators in NSCLC identified by a microarray based approach. Int. J. Oncol. 2005;27:105–111. doi: 10.3892/ijo.27.1.105. [DOI] [PubMed] [Google Scholar]
- Gery S., Sawyers C.L., Agus D.B., Said J.W., Koeffler H.P. TMEFF2 is an androgen-regulated gene exhibiting antiproliferative effects in prostate cancer cells. Oncogene. 2002;21:4739–4746. doi: 10.1038/sj.onc.1205142. [DOI] [PubMed] [Google Scholar]
- Glynne-Jones E., Harper M.E., Seery L.T., James R., Anglin I., Morgan H.E., Taylor K.M., Gee J.M., Nicholson R.I. TENB2, a proteoglycan identified in prostate cancer that is associated with disease progression and androgen independence. Int. J. Cancer. 2001;94:178–184. doi: 10.1002/ijc.1450. [DOI] [PubMed] [Google Scholar]
- Guo M., House M.G., Hooker C., Han Y., Heath E., Gabrielson E., Yang S.C., Baylin S.B., Herman J.G., Brock M.V. Promoter hypermethylation of resected bronchial margins: a field defect of changes? Clin. Cancer Res. 2004;10:5131–5136. doi: 10.1158/1078-0432.CCR-03-0763. [DOI] [PubMed] [Google Scholar]
- Heller G., Zielinski C.C., Zochbauer-Muller S. Lung cancer: from single-gene methylation to methylome profiling. Cancer Metastasis Rev. 2010;29:95–107. doi: 10.1007/s10555-010-9203-x. [DOI] [PubMed] [Google Scholar]
- Huang Z.H., Li L.H., Yang F., Wang J.F. Detection of aberrant methylation in fecal DNA as a molecular screening tool for colorectal cancer and precancerous lesions. World J. Gastroenterol. 2007;13:950–954. doi: 10.3748/wjg.v13.i6.950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanauskas A., Hoffmann J., Jonaitis L.V., Markelis R., Juozaityte E., Kupcinskas L., Lofton-Day C., Rocken C., Malfertheiner P. Distinct TPEF/HPP1 gene methylation patterns in gastric cancer indicate a field effect in gastric carcinogenesis. Dig. Liver Dis. 2008;40:920–926. doi: 10.1016/j.dld.2008.05.004. [DOI] [PubMed] [Google Scholar]
- Jahr S., Hentze H., Englisch S., Hardt D., Fackelmayer F.O., Hesch R.D., Knippers R. DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001;61:1659–1665. [PubMed] [Google Scholar]
- Jemal A., Siegel R., Ward E., Hao Y., Xu J., Murray T., Thun M.J. Cancer statistics, 2008. CA Cancer J. Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- Jones P.A., Baylin S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- Lee S.M., Na Y.K., Hong H.S., Jang E.J., Yoon G.S., Park J.Y., Kim D.S. Hypomethylation of the thymosin 10 gene is not associated with its overexpression in non-small cell lung cancer. Mol. Cells. 2011;32:343–348. doi: 10.1007/s10059-011-0073-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang G., Robertson K.D., Talmadge C., Sumegi J., Jones P.A. The gene for a novel transmembrane protein containing epidermal growth factor and follistatin domains is frequently hypermethylated in human tumor cells. Cancer Res. 2000;60:4907–4912. [PubMed] [Google Scholar]
- Lin K., Taylor J.R., Wu T.D., Gutierrez J., Elliott J.M., Vernes J.M., Koeppen H., Phillips H.S., de Sauvage F.J., Meng Y.G. TMEFF2 is a PDGF-AA binding protein with methyla-tion-associated gene silencing in multiple cancer types including glioma. PLoS One. 2011;6:e18608. doi: 10.1371/journal.pone.0018608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lofton-Day C., Model F., DeVos T., Tetzner R., Distler J., Schuster M., Song X., Lesche R., Liebenberg V., Ebert M., et al. DNA methylation biomarkers for blood-based colorectal cancer screening. Clin. Chem. 2008;54:414–423. doi: 10.1373/clinchem.2007.095992. [DOI] [PubMed] [Google Scholar]
- Minna J.D., Roth J.A., Gazdar A.F. Focus on lung cancer. Cancer Cell. 2002;1:49–52. doi: 10.1016/s1535-6108(02)00027-2. [DOI] [PubMed] [Google Scholar]
- Otsuka T., Kohno T., Mori M., Noguchi M., Hirohashi S., Yokota J. Deletion mapping of chromosome 2 in human lung carcinoma. Genes Chromosomes Cancer. 1996;16:113–119. doi: 10.1002/(SICI)1098-2264(199606)16:2<113::AID-GCC5>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Russo A.L., Thiagalingam A., Pan H., Califano J., Cheng K., Ponte J.F., Chinnapan D., Nemani P., Sidransky D., Thiagalingam S. Differential DNA hypermethylation of critical genes mediates the stage-specific tobacco smoke-induced neoplastic progress of lung cancer. Clin. Cancer Res. 2005;11:2466–2470. doi: 10.1158/1078-0432.CCR-04-1962. [DOI] [PubMed] [Google Scholar]
- Sabbioni S., Miotto E., Veronese A., Sattin E., Gramantieri L., Bolondi L., Calin G.A., Gafà R., Lanza G., Carli G., et al. Multigene methylation analysis of gastrointestinal tumors: TPEF emerges as a frequent tumor-specific aberrantly methylated marker that can be detected in peripheral blood. Mol. Diagn. 2003;7:201–207. doi: 10.1007/BF03260039. [DOI] [PubMed] [Google Scholar]
- Selamat S.A., Galler J.S., Joshi A.D., Fyfe M.N., Campan M., Siegmund K.D., Kerr K.M., Laird-Offringa I.A. DNA methylation changes in atypical adenomatous hyperplasia, adenocarcinoma in situ, and lung adenocarcinoma. PLoS One. 2011;6:e21443. doi: 10.1371/journal.pone.0021443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigematsu H., Gazdar A.F. Somatic mutations of epidermal growth factor receptor signaling pathway in lung cancers. Int. J. Cancer. 2006;118:257–262. doi: 10.1002/ijc.21496. [DOI] [PubMed] [Google Scholar]
- Suzuki M., Shigematsu H., Shames D.S., Sunaga N., Takahashi T., Shivapurkar N., Lizasa T., Frenkel E.P., Minna J.D, Fujisawa T., et al. DNA methylation-associated inactivation of TGFβ-related genes DRM/Gremlin, RUNX3, and HPP1 in human cancers. Oncogene. 2005;93:1029–1037. doi: 10.1038/sj.bjc.6602837. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Takita J., Yang H.W., Chen Y.Y., Hanada R., Yamamoto K., Teitz T., Kidd V., Hayashi Y. Allelic imbalance on chromosome 2q and alterations of the caspase 8 gene in neuroblastoma. Oncogene. 2001;20:4424–4432. doi: 10.1038/sj.onc.1204521. [DOI] [PubMed] [Google Scholar]
- Toyooka S., Maruyama R., Toyooka K.O., McLerran D., Feng Z., Fukuyama Y., Virmani A.K., Zochbauer-Muller S., Tsukuda K., Sugio K., et al. Smoke exposure, histologic type and geography-related differences in the methylation profiles of non-small cell lung cancer. Int. J. Cancer. 2003;103:153–160. doi: 10.1002/ijc.10787. [DOI] [PubMed] [Google Scholar]
- Young J., Biden K.G., Simms I.A., Huggard P., Karamatic R., Eyre H.J., Sutherland G.R., Herath N., Barker M., Anderson G.J., et al. HPP1: a transmembrane protein-encoding gene commonly methylated in colorectal polyps and cancers. Proc. Natl. Acad. Sci. USA. 2001;98:265–270. doi: 10.1073/pnas.98.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]