

Abstract
MyoD and myogenin (Myog) recognize sets of distinct but overlapping target genes and play different roles in skeletal muscle differentiation. MyoD is sufficient for near-full expression of early targets, while Myog can only partially enhance expression of MyoD-initiated late muscle genes. However, the way in which Myog enhances the expression of MyoD-initiated late muscle genes remains unclear. Here, we examine the effects of Myog on chromatin remodeling at late muscle gene promoters and their activation within chromatin environment. Chromatin immunoprecipitation (ChIP) assay showed that Myog selectively bound to the regulatory sequences of late muscle genes. Overexpression of Myog was found to overcome sodium butyrate-inhibited chromatin at late muscle genes in differentiating C2C12 myoblasts, shifting the transcriptional activation of these genes to an earlier time period. Furthermore, overexpression of Myog led to increased hyperacetylation of core histone H4 in differentiating C2C12 myoblasts but not NIH3T3 fibroblasts, and hyperacetylated H4 was associated directly with the late muscle genes in differentiating C2C12, indicating that Myog can induce chromatin remodeling in the presence of MyoD. In addition, co-immunoprecipitation (CoIP) revealed that Myog was associated with the nuclear protein Brd4 in differentiating C2C12 myoblasts. Together, these results suggest that Myog enhances the expression of MyoD-initiated late muscle genes through MyoD-dependent ability of Myog to induce chromatin remodeling, in which Myog-Brd4 interaction may be involved.
Keywords: chromatin remodeling, gene regulation, myogenin, myogenesis, MyoD
INTRODUCTION
Initial muscle development is controlled by the myogenic basic helix-loop-helix (bHLH) family of transcription factors (also referred to as myogenic regulatory factors, MRFs) in conjunction with the MEF2 MADS-box factors (Buckingham, 2001; Tapscott, 2005). The MRFs, including MyoD, Myf5, myogenin (Myog), and MRF4/Myf6/herculin, activate the expression of muscle-specific genes by dimerizing E proteins to bind the E-box sequences (CANNTG) in the promoters of muscle genes (Berkes and Tapscott, 2005; Blackwell and Weintraub, 1990). MRFs can also activate their own and one another’s expression (Braun et al., 1989; Brennan et al., 1990; Thayer et al., 1989). This stimulates a complex regulatory circuit that controls the expression of muscle-specific genes during myogenesis (Edmondson and Olson, 1989; Olson and Klein, 1994; Tapscott, 2005; Weintraub, 1993).
MyoD is regarded as the MRF most relevant to the regulation of myogenic differentiation. MyoD-deficient myoblasts are incapable of completing the differentiation program successfully (Tapscott, 2005). Genetic studies indicate that Myf5 and MRF4 act upstream of MyoD to specify the myogenic lineage (Kassar-Duchossoy et al., 2004; Rudnicki et al., 1993). However, Myog has a crucial role in the terminal differentiation of committed muscle cells (Hasty et al., 1993; Nabeshima et al., 1993). Protein motifs conserved in MyoD and Myf5 are necessary to initiate the expression of a subset of genes critical to the myogenic program, including transcription of the Myog gene (de la Serna et al., 2005; Edmondson et al., 1992; Rawls et al., 1995; Venuti et al., 1995). Myog functions downstream of Myf5 and MyoD to activate muscle gene expression. The spatial and temporal expression of MRFs is critical to myogenesis during embryogenesis (Tapscott, 2005).
MyoD and Myog recognize sets of distinct but overlapping target genes and play different roles in skeletal muscle differentiation (Blais et al., 2005; Ohkawa et al., 2006). Investigations of the way in which MyoD targets to the loci in inactive chromatin have shown that MyoD binds stably to the promoters of a restricted subset of muscle specific genes, such as Myog, by interacting with an adjacent protein complex containing Pbx homeodomain protein that is constitutively bound at these sites (Berkes et al., 2004). MyoD then recruits the histone acetyl-transferase (HAT) p300 to acetylate histones, and the p300/CBP-associated factor (PCAF) to acetylate MyoD (Tapscott, 2005). Both activities are necessary for the full transcriptional activity of MyoD on chromatin-associated templates (Dilworth et al., 2004). In addition, the SWI/SNF chromatin-remodeling enzymes containing the Brg1 ATPase were found to be involved in MyoD-initiated activation of muscle-specific genes, including the Myog gene (de la Serna et al., 2005; Simone et al., 2004). MyoD is sufficient for near full expression of early targets but not for late gene expression. The transcriptional activation of late genes requires the combined activities of MyoD and Myog. Part of the role of Myog is to enhance expression of a subset of genes previously initiated by MyoD (Cao et al., 2006). A study of the relationship between muscle-specific transcription factors and chromatin-remodeling enzymes demonstrated that Myog and MEF2D cooperate to determine the skeletal muscle phenotype through recruiting SWI/SNF chromatin-remodeling enzymes (Ohkawa et al., 2006). This indicates that chromatin remodeling is involved in the action of Myog in MyoD-initiated late muscle gene expression. However, the way in which MyoD and Myog coordinately work at a common set of genes remains unclear.
Bromodomain-containing protein 4 (Brd4) is a nuclear protein that binds preferentially to acetylated histone H3 and H4 (Dey et al., 2003). It has been shown that Brd4 mediates the transcriptional activation through recruitment of P-TEFb into a transcriptionally active complex (Ai et al., 2011; Jang et al., 2005; Wu and Chiang, 2007; Yang et al., 2005). Most recently, P-TEFb has been found to interact with the helix-loop-helix protein c-Myc in c-Myc-dependent transcription (Rahl et al., 2010). Given these findings, we suppose that Myog might interact with Brd4, which may play a role in regulating muscle gene expression.
In this study, we examine the effect of Myog on chromatin remodeling at late muscle genes and their activation within chromatin environment. Our results suggest that Myog enhances the expression of MyoD-initiated late muscle genes through MyoD-dependent ability of Myog to induce chromatin remodeling, in which Myog-Brd4 interaction may be involved.
MATERIALS AND METHODS
Plasmids
pBABE-MyoD-ER and pBABE-neo were provided by Stephen J Tapscott (Fred Hutchinson Cancer Research Center, USA); pSuper-Myog was provided by Thomas Braun (Max-Planck-Institute, Germany); pSuper empty vector was provided by Xia Yi (Peking University Health Science Center, China); shRNA708 MyoD was provided by Vivek Mittal (Cold Spring Harbor Laboratory, USA); pcDNA3.1(+)-flag and pcDNA3.1(+)-HA vector were provided by Yong-Ming Ren (Tsinghua University, Beijing, China). pcDNA3.1 (+)-MyoD-flag and pcDNA3.1 (+)-Myog-HA were generated by PCR from C2C12 cDNA and subcloned into the expression vector. The primers were 5′-GCT GGA TCC AGG GCC GCC ACC ATG GAG CTT CTA TCG CCG CCA CT-3′ and 5′-CGA TCT CGA GAA GCA CCT GAT AAA TCG-3′ for MyoD coding region, and 5′-ACA AAG CTT TGC CGC CAC CAT GGA GCT GTA TGA G-3′ and 5′-GAC ACT CGA GGT TGG GCA TGG TTT CGT C-3′ for Myog coding region. For convenience, both forward primers included the BamHI and HindIII restriction sites, and both reverse primers included an XhoI restriction site. Both forward primers also included a Kazak sequence. All constructs were sequence verified.
Cell culture and transfection
NIH3T3, C3H10T1/2 fibroblasts, and C2C12 myoblasts were grown in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% bovine calf serum (Biochrom, Germany), penicillin, and streptomycin. Differentiation medium was DMEM supplemented with 2% horse serum and 10 μg insulin per ml (Lu et al., 2004). Transfection was performed using transfection reagent Vigofect (Vigorus, China). Cells were seeded in 6 cm culture dishes (USA) at a density of approximately 105 cells/ml on the day prior to transfection. For RNA interference (RNAi), 0.9 μg pcDNA3.1 (+)-MyoD-flag and 5.4 μg pSuper-Myog or 3.2 μg pcDNA3.1 (+)-Myog-HA plasmid and 3.2 μg shRNA708 MyoD plasmid were transfected into the cells in 35 mm culture plates. To determine the role of Myog in myogenesis, 3.2 μg pBABE-MyoD-ER and 3.2 μg pcDNA3.1 (+)-Myog-HA were transfected into the cells in 35 mm culture plates. Then 10−7 M β-estradiol (Sigma, USA) was added into the medium 12 h after transfection. They were removed at the specific times. The cells were grown in growth medium (GM) for 24 h and then transferred into differentiation medium (DM). RNA was extracted after 24–48 h of differentiation for RT-PCR and RT-qPCR. For stable knockdown of Myog expression, C2C12 myoblasts were transfected with 3.2 μg pSuper-Myog and 0.32 μg pcDNA3.1 (+)-HA. Forty-eight hours later, cells were subcultured in the presence of 800 μg/ml G418 (Invitrogen, USA). Cells were then maintained in selection medium for about 2 weeks until G418-resistant colonies appeared. Single colonies were picked and transferred to 24 well plates. Individual clones were then maintained in the medium containing 400 μg/ml of G418.
RT-PCR and real-time quantitative PCR (RT-qPCR)
Total RNA was isolated with TRIzol (Invitrogen) and reverse-transcribed with M-MLV Reverse transcriptase (Promega, USA). PCR was performed using Taq DNA polymerase (Promega, China) and primers as follows: GAPDH, 5′-CAG CCT CGT CCC GTA GAC A-3′ and 5′-CGC TCC TGG AAG ATG GTG AT-3′; MyoD, 5′-GCA ACG CCA TCC GCT ACA-3′ and 5′-GGG TCT GGG TTC CCT GTT CT-3′; MyoD (endogenous), 5′-CTC TGA TGG CAT GAT GGA TT-3′ and 5′-CGA AAG GAC AGT TGG GAA GA-3′; Myog, 5′-GAA GCG CAG GCT CAA GAA AGT-3′ and 5′-GAT TGT GGG CGT CTG TAG GGT-3′; Myog (endogenous), 5′-CTT GCT CAG CTC CCT CAA CC-3′ and 5′-GGT GTT AGC CTT ATG TGA ATG G-3′; Myf5, 5′-AGG AAT GCC ATC CGC TAC-3′ and 5′-CGT GAT AGA TAA GTC TGG AGC-3′; Cdh-15, 5′-GGA CCT TCG GGA CAA CAT-3′ and 5′-CAG CCT CCA AGC CAT CAC-3′; Mck, 5′-CAT AAG ACC GAC CTC AAC CAC G-3′ and 5′-AAC CTC CTT CAT ATT GCC TCC CT-3′; Mylf3, 5′-TCC AAC AAC AAG GAC CAG-3′ and 5′-CAG CAA CGC TTC TAC CTC-3′; Tncs, 5′-AGG AGA CAA CCC ACA GCG-3′ and 5′-AAG ATG CGG AAG CAC TCA-3′; Mhc (embryonic), 5′-GCA AAG ACC CGT GAC TTC ACC TCT AG-3′ and 5′-GCA TGT GGA AAA GTG ATA CGT GG-3′. For real-time thermal cycling, triplicate aliquots of RT sample were used in a reaction mixture containing 250 nM of each primer and 20 μl of SYBR Green Real-time PCR Master Mix (Toyobo, Japan). ABI Prism 7300 SDS software was used for these experiments. The same primer sets of GAPDH used in RT-PCR were also used in the RT-qPCR. qCdh-15, 5′-GCA TGT GGA AAA GTG ATA CGT GG-3′ and 5′-GAG CCA TCT CCC TCG TAG TCA-3′; qMyog, 5′-AGT GGG GCA ATG CAC TGG AGT-3′ and 5′-ATG CAC ACC CAG CCT GAC AGA-3′; qMhc, 5′-CAA TAA ACT GCG GGC AAA GAC-3′ and 5′-CTT GCT CAC TCC TCG CTT TCA-3′. The thermal cycling was performed as previously described (Ji et al., 2009). GAPDH mRNA was used as an internal control. The expression of mRNA is represented as fold increase (2−ΔΔCt).
Chromatin immunoprecipitation (ChIP)
ChIP was performed as previously described (Ji et al., 2009). Briefly, sonicated chromatin was immunoprecipitated using anti-Myog Ab (F5D, NeoMarker, USA), anti-acetyl-histone H4 Ab (Upstate, USA) or IgG (Santa Cruz Biotechnology Inc.). PCR primers used for detecting Cdh15, Tncs, and IgH loci were described (Bergstrom et al., 2002). Primers for MyoD proximal regulatory region (PRR): 5′-CTA ACT CCT ATC CTT TGC CTG G-3′ and 5′-GGA GTG TAG TAG GGC GGA G-3′, distal regulatory region-3′ (DRR): 5′-TGA GTT GGG AAA GGA AAG TCT AG-3′ and 5′-GGC CTC TGG GGT GTT GC-3′, and core enhancer 5′-TCT TCC TAT AAA CTT CTG AGA C-3′ and 5′-GAG AAG ACC CAG GAA CAG C-3′. Primers for Mhc promoter: 5′-GCC CCC GAG GAG TTA CAG AG-3′ and 5′-CAG AAA ATG GCA GAG ACA GCT TTG-3′.
Histone extraction, TAU SDS-PAGE, and Western blot
Nuclei were isolated and digested with MNase (Worthington) at 100 U/ml for 30 min at 37°C. Then histones were extracted (Gong et al., 2002). Briefly, after centrifugation the supernatant (S1) was saved. The nuclear pellet was lysed in 10 vol of lysis buffer (10 mM Hepes pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 1.5 mM PMSF), added H2SO4 to a final concentration of 0.4 N and incubated on ice for 30 min. Then the supernatant (S2) was extracted by centrifugation. The S1 and S2 were pooled in one container, and TCA was added to a final concentration of 20%. This mixture was incubated at 4°C for 4 h. Pellet was obtained by centrifugation and washed with acid, dried and dissolved in sample buffer. For hyperacetylated histone analyses, TAU SDS-PAGE was performed as described with tritonacetic acid-urea minislab gels (10 × 15 × 0.1 cm) for the first direction and (10 × 15 × 0.15 cm) SDS minislab gels for the second direction (Gong et al., 2002). For Western blot analysis of acetylated H3 and H4, the isolated histones were separated by 15% SDS-PAGE, transferred onto nitrocellulose membranes, and incubated with antibodies against acetylated N-terminal sequences of H3 and H4 (Upstate Biotechnology, USA) at 44°C overnight. After washes in TBS-T, membranes were incubated with chemiluminescence reagents (Vigorus, China).
Co-immunoprecipitation (CoIP)
For analysis of Myog-Brd4 interaction, CoIP was performed as described previously (Zhang et al., 2010) with modification. The C2C12 myoblasts were grown in GM for 24 h, and maintained in DM for 48 h. Differentiating C2C12 cells were then lysed with RIPA buffer, and nuclear extracts were prepared. The supernatants were cleared by centrifugation. Equal amounts of protein (400 μg) were precleared using protein G PLUS agarose (Santa Cruz Biotechnology, USA) beads and proteins were immunoprecipitated with anti-Brd4 (H-250) or anti-Myog Ab-1 (NeoMarkers, USA); the associated partners in the precipitates were probed by anti-Myog Ab-1 and anti-Brd4 antibodies, respectively.
RESULTS
Knockdown of Myog decreases expression of late muscle genes in differentiating C2C12 myoblasts
To determine the effect of Myog on MyoD-targeted gene expression during myogenic differentiation, we knocked down the Myog gene in C2C12 myoblasts using RNAi and analyzed the expression of muscle specific genes that have previously been identified as MyoD targets by RT-PCR (Bergstrom et al., 2002). As shown in Fig. 1, the expression of late muscle genes Mhc, Mck, and Tncs in differentiating C2C12 myoblasts was dramatically decreased after knockdown of Myog, demonstrating that the Mhc, Mck, and Tncs genes are targets for Myog. In contrast, the expression of the early genes such as Cdh15 and Desmin and the late muscle gene Mylf3 was unaffected by Myog knockdown. Notably, following Myog knockdown, MyoD expression was also significantly reduced, indicating that MyoD can be regulated by Myog during myogenic differentiation. Unsurprisingly, the expression of Myf5 was increased, which is in agreement with previous observation that the expression profiles of MyoD and Myf5 become mutually exclusive during myoblast differentiation (Kitzmann et al., 1998). Together, these results suggest that Myog is involved in regulating late muscle gene but not early gene expression.
Fig. 1.

Knockdown of Myog decreases expression of late muscle genes in differentiating C2C12 myoblasts. Stable transfected cells were obtained after transfection of pSuper-Myog into C2C12 myoblasts and selection with G418. Total RNAs were extracted from the transfected cells and RT-PCR was performed to estimate the transcripts of Myog, MyoD, Myf5, early genes (Cdh15, Desmin), and late genes (Mhc, Mck, Tncs, Mylf3). GAPDH was used as an internal control. C2-V: C2C12 cells transfected with pSuper empty vector (control); C2-G1, 2 and 3: C2C12 cells transfected with pSuper-Myog.
Overexpression of Myog enhances late muscle gene expression in fibroblasts in the presence of MyoD
Because knockdown of Myog led to downregulation of MyoD in differentiating C2C12 cells (Fig. 1), and MyoD and Myog work in concert at a common set of genes (Cao et al., 2006), we cannot differentiate the effects of Myog and MyoD on late gene expression. It has demonstrated that exogenous MyoD can activate endogenous Myog but not endogenous MyoD in NIH3T3 fibroblasts (Thayer et al., 1989). To further demonstrate the role of Myog in regulating late muscle genes, we introduced exogenous MyoD and Myog into NIH3T3 fibroblasts by transfection of pBABE-MyoD-ER, pcDNA3.1-Myog-HA or both (Fig. 2A), and the expression of exogenous MyoD-ER and myogenin-HA fusion proteins were detected by Western blot with anti-MyoD and anti-HA antibodies, respectively (Fig. 2B). It is reported that the nuclear import of MyoD-ER fusion protein can be induced by β-estradiol (B-e) (Bergstrom et al., 2002). To mimic MyoD function in C2C12 cells, B-e was added to the transfected NIH3T3 fibroblasts at 12 h before low serum induced differentiation. After 24 h of low serum induction, mRNAs were analyzed by RT-qPCR. The expression of endogenous Myog gene (left panel in Fig. 2A) and the late gene Mhc (middle panel in Fig. 2A) was markedly higher in MyoD- and Myog-cotransfected NIH3T3 fibroblasts than in MyoD-transfected fibroblasts. However, Myog transfection alone could not activate the Mhc gene. Unlike the Myog and Mhc genes, the expression of early muscle gene Cdh15 in MyoD-transfected NIH3T3 cells reached a high level, similar to that in MyoD- and Myog-cotransfected NIH3T3 cells; however, Myog transfection rendered cells unable to activate the early gene Cdh15 (right panel in Fig. 2A). RT-PCR showed that the late Mhc gene was expressed only in MyoD-ER-transfected NIH3T3 fibroblasts (lane 3 in Fig. 2C) as well as differentiating C2C12 (lane 4, positive control), but not in untransfected (lane 1) and empty vector-transfected (lane 2) fibroblasts. These results indicate that the expression of the late Mhc gene might be associated to exogenous MyoD and possibly endogenous Myog, because MyoD transfection activated Myog expression (left panel in Fig. 2A). To further demonstrate the effects of Myog on MyoD-activated late gene expression, we performed RNAi to block MyoD-ER-activated Myog expression in NIH3T3 cells (Figs. 2D and 2E). RT-qPCR and RT-PCR showed that knock-down of the expression of Myog (endo) in MyoD-transfected NIH3T3 (upper, Fig. 2D) led to a significant decrease in Mhc transcript (bottom, Fig. 2D and upper, Fig. 2E), although exogenous MyoD was expressed there (Fig. 2F). Like Mhc gene, the expression of late genes Mck and Tncs was also decreased after Myog knockdown (Fig. 2E).
Fig. 2.

Myog maintains full expression of the late muscle genes in the presence of MyoD. (A) Enhancement of the expression of endogenous Myog and Mhc but not that of the Cdh15 gene by overexpression of Myog in the presence of MyoD. NIH3T3 fibroblasts were transfected with pBABE-MyoD-ER, pcDNA3.1-Myog-HA, or both. β-Estradiol (B-e) was administered 12 h before initial differentiation. The mRNAs for Myog (endo), Mhc, and Cdh15 genes were analyzed by RT-qPCR. GAPDH was used as an internal control. Relative levels of vector-transfected cells were normalized to 1. Data shown include the mean ± SD derived from three independent experiments performed in triplicate. *P < 0.05. **P > 0.05. (B) Protein expression of the overexpressed constructs in (A) was monitored by Western blot, in which exogenous MyoD-ER and Myog-HA were blotted by anti-MyoD and anti-HA antibodies, respectively. Tubulin was used as a loading control. (C) Activation of Mhc but not endogenous MyoD by overexpressed MyoD in NIH3T3 cells. NIH3T3 cells were transfected with pcDNA3.1-MyoD-flag. The mRNA of endogenous MyoD and Mhc genes was examined by RT-PCR. C2C12 myoblasts were used as positive controls. (D) Suppression of late gene Mhc by knockdown of Myog in NIH3T3 cells. The cells were transfected with pBABE-MyoD-ER (MyoD), pSuper-Myog (pSuper-G, i.e., RNAi of Myog), or RNAi empty vector (pSuper-V). B-e administration and RT-qPCR analyses of Myog (endo) and Mhc mRNA were performed. GAPDH was used as an internal control. Data represent one of two independent experiments performed in triplicate. *P < 0.05. (E) Downregulation of late but not early genes by knockdown of Myog in NIH3T3 cells. Gene transfection and cell treatment were performed as described in (D). mRNAs were analyzed by RT-PCR. GAPDH was here used as an internal control. (F) Protein expression of transfected constructs in (D) and E was monitored by Western blot using anti-MyoD antibody. Tubulin was used as a loading control. +MyoD-V: NIH3T3 cells transfected with pBABE-MyoD-ER and pSuper-vector (RNAi empty vector); +MyoD-Myog: NIH3T3 cells transfected with pBABE-MyoD-ER and pSuper-Myog.
Exogenous Myog can activate MyoD in C3H10T1/2 (Thayer et al., 1989) but not NIH3T3 (Ohkawa et al., 2006) fibroblasts. To demonstrate that MyoD is required for Myog-enhanced late gene expression, we performed RNAi to block MyoD expression in Myog-transfected C3H10T1/2 fibroblasts. RT-PCR (Fig. 3A) and RT-qPCR (Fig. 3B) showed that knock-down of MyoD expression caused down-regulation of all tested genes including Myog(endo), Desmin, Cdh15, and Mhc in Myog-HA transfected C3H10T1/2 cells, although exogenous Myog was expressed (Fig. 3C). Taking the data from Figs. 2 and 3 together, Myog may enhance late muscle gene and its own expression in the presence of MyoD but does not affect the early genes.
Fig. 3.

Knockdown of MyoD suppresses both early and late muscle gene expression. C3H10T1/2 fibroblasts were transfected with pcDNA3.1-Myog-HA (+Myog) and shRNA708MyoD (−MyoD) or empty vector (−V). The mRNAs were analyzed by (A) RT-PCR and (B) RT-qPCR. GAPDH was used as internal control. In (B), relative level in Myog-HA and sh-vector transfected cells was normalized to 1; data present mean ± SD derived from three independent experiments performed with triplicate. *P < 0.05. (C) The protein expression of Myog-HA in A and B was tested by Western blot using anti-HA antibody. Tubulin was used as a loading control.
Myog is associated with the regulatory sequences of late muscle genes in differentiating C2C12 myoblasts
To explore the mechanism by which Myog enhances late muscle gene expression, we performed ChIP to examine the binding of Myog with the regulatory sequences of the late genes in differentiating C2C12 myoblasts. After differentiation for 48 h, the chromatins were immunoprecipitated with anti-Myog antibody, and the genomic DNA encompassing the regulatory sequences of MyoD, Mhc, Tncs, and Cdh15 genes was amplified by a pair of primers. The results showed a strong binding of Myog to the Mhc promoter and the Tncs intron1 enhancer (Fig. 4). However, no binding of Myog to the promoter of Cdh15 gene, the proximal regulatory region (PRR) or core enhancer of MyoD could be detected in the chromatin precipitate; the exception was the MyoD distal regulatory region (DRR), which was found to bind to Myog in differentiating C2C12 cells. This is consistent with the observation that MyoD expression was reduced in Myog-knocked down C2C12 cells (Fig. 1). These results suggest that Myog can bind directly to the late gene promoters and distal regulatory region (DRR) of MyoD in differentiating C2C12 cells. This may contribute to enhancement of MyoD-initiated late gene expression.
Fig. 4.

Myog is associated with the regulatory sequences of the late muscle genes in ChIP assay. After C2C12 differentiation, the chromatins were immunoprecipitated using anti-Myog antibody. The specific sequences of the promoters of muscle specific genes were amplified by PCR as described in the Materials and Methods section. MyoD PRR: the proximal regulatory region of MyoD; MyoD DRR: the distal regulatory region of MyoD.
Overexpressed Myog may overcome sodium butyrate-inhibited chromatin remodeling at late muscle genes
It is well known that chromatin remodeling is involved in gene regulation. Sodium butyrate, an inhibitor of HDACs (histone deacetylases), has been used as a tool to induce histone hyperacetylation to study the effects of hyperacetylated histone on chromatin structure and function. In mammalian cells, however, butyrate does not cause an increase in histone hyperacetylation, and in fact just the opposite is observed (Covault et al., 1982; Plesko et al., 1983). Butyrate blocks MyoD-mediated chromatin reorganization and transcription initiation, but cannot block transcription when added after chromatin remodeling had occurred (Gerber et al., 1997; Johnston et al., 1992). We considered that Myog enhances late muscle gene expression through promoting chromatin remodeling. To assess this idea, we exposed C2C12 myoblasts to butyrate that was added at 0, 6, 12, 24, and 36 h after initial differentiation, respectively. After 72 h differentiation, cells were harvested, and RT-PCR was performed to examine gene expression. This allowed us to ascertain the chromatin status of muscle-specific genes. As shown in Fig. 5, the transcription of the early muscle genes Cdh15 and Desmin as well as MyoD was unaffected by butyrate added at all times, indicating that the chromatin at those loci has been remodeled after initial differentiation. However, the levels of Myog transcription were increased progressively when butyrate was added within 0–24 h of differentiation, and peaked when added at 36 h. In other word, adding butyrate at 36 h had not any inhibitory effect on Myog transcription, which was equal to that of without adding butyrate. These results suggest that, at the beginning of differentiation, the chromatin at Myog locus is minimally remodeled and progressively developed within 6–36 h of differentiation. The transcription of late muscle genes Tncs, Mck, and Mhc was almost inhibited when butyrate was added within 0–12 h of differentiation, but it was significantly increased when the agent was added within 24–36 h of differentiation. These data indicate that the addition of butyrate within 0–24 h of initial differentiation may completely or partially inhibit chromatin remodeling at the late genes. Namely, chromatin remodeling at those loci is established within 24–36 h of differentiation. We noted that the expression of the late muscle gene Mylf3 was not inhibited by butyrate, indicating that the chromatin at Mylf3 locus has been remodeled early during differentiation. It might be that MyoD mediates the chromatin remodeling at the Mylf3 gene, because the Mylf3 promoter has both MyoD- and Myog-specific binding sites (Asakura et al., 1993). This can also explain that Mylf3 gene expression is unaffected by Myog knockdown (Fig. 1).
Fig. 5.

The expression of MyoD-initiated late muscle genes but not early genes is affected by sodium butyrate in a time-dependent manner. C2C12 myoblasts were grown in GM and maintained in DM prior to harvest at 72 h. Butyrate was added at 0, 6, 12, 24, and 36 h after initial differentiation, respectively. Total RNAs were extracted and RT-PCR was performed to analyze the transcripts of Myog, MyoD, and their targeted genes, including early and late muscle genes. No: C2C12 myoblasts cultured in DM for 72 h without adding sodium butyrate. GAPDH was used as an internal control.
It is known that differentiating C2C12 myoblasts express both MyoD and Myog, but NIH3T3 fibroblasts do not. To ascertain the role of Myog in chromatin remodeling of late muscle genes, we transfected MyoD-ER, Myog, or both into NIH3T3 fibroblasts and exposed the cells to sodium butyrate to determine the chromatin status at muscle-specific genes. RT-PCR showed that addition of sodium butyrate 12 h after low-serum induction of differentiation inhibited expression of the Mhc, Tncs, and Mck genes but did not affect the early gene Cdh15 in MyoD-ER-transfected NIH3T3 (lane 1 in Fig. 6A). However, addition of butyrate at 12 h was not found to inhibit these gene expressions in MyoD-ER- and Myog-cotransfected NIH3T3 cells (lane 3 in Fig. 6A) relative to Myog-transfected cells (lane 2 in Fig. 6A). Similar results could be seen in the RT-qPCR assays for the expression of the endogenous Myog (Fig. 6E) and Mhc (Fig. 6F) genes in co-transfected cells relative to the early gene Cdh15 (Fig. 6D). These results indicate that within 12 h of the addition of butyrate, the chromatin remodeling at the early genes is established, but remodeling at late genes is not. These data also suggest that early overexpression of Myog can overcome butyrate-inhibited chromatin remodeling at the late genes, shifting their expression to an earlier stage in the presence of MyoD.
Fig. 6.

Overexpression of Myog can overcome sodium butyrate-inhibited chromatin remodeling at late muscle genes in the presence of MyoD. NIH3T3 fibroblasts were transfected with pBABE-MyoD-ER and/or pcDNA 3.1-Myog-HA. The β-Estradiol (B-e) was administrated 12 h before low serum (DM) induction, and sodium butyrate was added at 12 h after induction. (A) RT-PCR was performed to examine the transcripts of Cdh15, Mhc, Tncs and Mck. (B) Protein expression of transfected constructs in A was monitored by Western blot using anti-MyoD and anti-HA antibodies. Tubulin was used as a loading control. (C) Protein expression of transfected constructs in (D)-(F) was determined by Western blot, and RT-qPCR was performed to analyze Cdh15 (D), endogenous Myog (E) and Mhc mRNAs (F). Relative level in MyoD-transfected cells was normalized to 1. GAPDH was used as internal control. In (D)-(F), data represent mean ± SD derived from three independent experiments performed with triplicate. *P < 0.05.
Overexpressed Myog increases core histone H4 hyperacetylation in differentiating C2C12 myoblasts but not NIH3T3 fibroblasts
Core histone acetylation is implicated in chromatin remodeling and transcriptional regulation. To determine whether the ability of Myog to overcome butyrate-inhibited chromatin remodeling at the late genes is MyoD-dependent, we compared acetylated core histones in Myog-transfected C2C12 myoblasts to those in Myog-transfected NIH3T3 fibroblasts. After transfection of the cells with pEMSV-Myog and low serum induction, histones were extracted from isolated nuclei. Following 15% SDS-gel electrophoresis of histones (Fig. 7A), acetylated core histones H3 and H4 were analyzed using antibodies against acetylated N-terminal sequences of H3 and H4. Western blot analysis showed more acetylation of histone H4 but not H3 in differentiating C2C12 cells transfected with Myog than in control (empty-vector transfected) C2C12 (left panel in Fig. 7B). However, no increase in acetylated H4 could be seen in Myog-transfected NIH3T3 fibroblasts (right panel in Fig. 7B). The increased acetylation of histone H4 was also observed in TAU SDS-PAGE, which facilitates identification of hyperacetylated histone. More different forms of mono- and multi-acetylated (hyperacetylated) H4 were observed in Myog-transfected C2C12 cells than in control C2C12. In control cells, only monoacetylated H4 was observed. Myog-increased hyperacetylation of H4 could not be seen in Myog-transfected NIH3T3 (Fig. 7C). These results suggest that overexpressed Myog can induce hyperacetylation of core histones H4 in C2C12 rather than in NIH3T3 cells. This might be attributed to MyoD expression in differentiating C2C12 but not NIH3T3 cells. Namely, Myog-induced chromatin remodeling is MyoD-dependent. To examine whether Myog-induced hyperacetylated H4 was associated with the late muscle genes, chromatin immunoprecipitation was performed and showed that acetylated-histone H4 was recruited to the promoter regions of late muscle genes Mhc and Tncs when Myog was overexpressed in C2C12 myoblasts (Fig. 7D). These data strongly suggest that Myog may promote chromatin remodeling around the late muscle genes through increasing of hyperacetylated histone H4, which may contribute to Myog-enhanced expression of MyoD-initiated late muscle genes.
Fig. 7.

Overexpression of Myog increases hyperacetylation of histone H4 in differentiating C2C12 myoblasts but not NIH3T3 fibroblasts. NIH3T3 fibroblasts and C2C12 myoblasts were transfected with pEMSV-Myog or empty vector. After 24 h of transfection, cells were induced to differenttiate for 48 h. After harvest the nuclei were isolated, and histones were extracted as described in the “Materials and Methods.” (A) Analysis of histones by 15% SDS-PAGE. The gel was stained with Coomassie brilli-ant blue. (B) Western blot analysis of acetylated histone H3 and H4. Histones (60 μg) were separated by 15% SDS-PAGE and probed with antibodies against acetylated N-terminal sequences of H3 and H4. (C) Hyperacetylation of histones in 2-D gel electrophoresis. Histones (60 μg) were separated on TAU/SDS (2-D) gel using triton-acetic acid-urea (TAU) minislab gels for the first direction and SDS minislab gels for the second direction. The gel was stained with Coomassie brilliant blue. The arrows indicate mono- and multi-acetylated histones. (D) Recruitment of acetylated-histone H4 to the promoter regions of late muscle genes in control or Myogtransfected C2C12 myoblasts was analyzed by ChIP assay. After transfection, the chromatins were immuno-precipitated using anti-acetylated–histone H4 antibody or control IgG. The specific promoter sequences of late muscle genes were amplified by PCR. The IgH enhancer containing E boxes was used as negative control. S, standard histones (calf thymus histones); C, control (empty vector transfection); T, transfection; N, no transfection.
Myog directly interacts with Brd4 in differentiating C2C12 myoblasts
It has been shown that Brd4, a nuclear protein, is associated with diacetylated histone H3 and di- and tetra-acetylated histone H4 (Dey et al., 2003). It has also been shown that Brd4 mediates the transcriptional activation through recruitment of P-TEFb into a transcriptionally active complex (Ai et al., 2011; Jiang et al., 2005; Yang et al., 2005). To demonstrate whether Myog was associated with Brd4 in differentiating C2C12 myoblasts, we first examined the expression of Myog and Brd4 in the cells. Brd4 was constitutively expressed before differentiation induction (0 h) and seemed to be increased after 48 h differentiation. Importantly, Myog was found to be expressed in differentiating C2C12 cells (Fig. 8A). Co-immunoprecipitation and Western blot analysis showed that both Myog and Brd4 were detectable in the precipitates from differentiating C2C12 myoblasts (Fig. 8B). These data indicate that Myog can interact with Brd4 in differentiating C2C12 cells, and Brd4 may be involved in Myog-mediated chromatin remodeling and transcription of the late muscle genes.
Fig. 8.
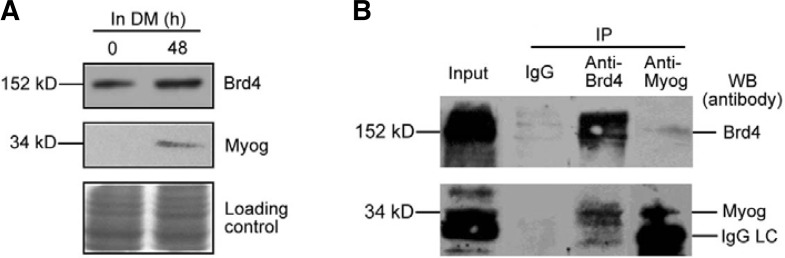
Endogenous Myog interacts with Brd4 in differentiating C2C12 myoblasts. (A) Western blot was performed to determine the expression of Brd4 and Myog in differentiating C2C12 myoblasts. The SDS-gel stained by Coomassie brilliant blue was used as loading control. (B) Endogenous Myog is associated with Brd4 in differentiating C2C12 cells. The nuclear extracts from differentiating C2C12 cells were co-immunoprecipitated with anti-Brd4 and anti-HA antibodies, respectively. Associated Brd4 and endogenous Myog in precipitates were probed with anti-Brd4 and anti-Myog antibodies, respectively. Precipitation with IgG was used as negative control. IgG LC, light chain of IgG.
DISCUSSION
In this study, we examine the effects of Myog on chromatin remodeling at late muscle genes and their transcription initiation within chromatin environment during muscle cell differentiation. Our findings demonstrate that Myog enhances the expression of MyoD-initiated late muscle genes through the MyoD-dependent ability of Myog to induce chromatin remodeling, in which hyperacetylated histone H4, and possibly, Myog-Brd4 interaction are involved.
The link between chromatin remodeling and myogenic factors is first revealed by deletion mutagenesis, demonstrating that MyoD can induce chromatin remodeling at muscle genes and activate transcription at previously silent loci. This is dependent on two motifs conserved in MyoD and Myf5, the H/C and Helix 3 domains (Tapscott et al., 2005). Through these domains, MyoD interacts with the Pbx/Meis homeodomain protein complex present in a subset of MyoD-regulated genes and localizes to its target promoters (Berkes et al., 2004). Although the ability of Myog to initiate chromatin remodeling is much weaker than that of MyoD and Myf5, overexpression of Myog in C3H10T1/2 fibroblasts may convert the cells to determined myoblasts (Bergstrom et al., 2002; Edmondson and Olson, 1989; Penn et al., 2004; Wright et al., 1989). However, Myog cannot induce muscle-specific genes in NIH3T3 fibroblasts efficiently (Gerber et al., 1997). This is because exogenous Myog can activate MyoD in C3H10T1/2 but not NIH3T3 (Ohkawa et al., 2006; Thayer et al., 1989). We found that addition of sodium butyrate, an inhibitor of chromatin reorganization, within 6–12 h after initial differentiation could almost completely inhibit the expression of late muscle genes in differentiating C2C12 myoblasts (Fig. 5), suggesting that chromatin remodeling at the MyoD-initiated late genes have not been established at that time. Furthermore, overexpression of Myog in the presence of MyoD could overcome butyrate-inhibited chromatin remodeling at the late genes, shifting the transcription initiation of these genes to an earlier time period (Fig. 6). These data indicate that Myog can induce chromatin remodeling in the presence of MyoD. Consistently, Myog was able to induce hyperacetylation of histone H4 in C2C12 cells but not in NIH3T3 cells (Fig. 7). In addition, we found that Myog-stimulated chromatin reorganization was at least partly selective to its targeted sequences (Figs. 4 and 7D). These data suggest that Myog has a MyoD-dependent ability to induce chromatin remodeling, which may contribute to enhancing MyoD-initiated late gene expression.
MyoD can bind p300 and PCAF, forming a MyoD complex with two distinct HAT activities. p300 acetylates histones, and PCAF acetylates MyoD. Both activities are necessary for the full transcriptional activity of MyoD on chromatin-associated templates (Dilworth et al., 2004). In addition, the SWI/SNF chromatin-remodeling enzymes containing the Brg1 ATPase are involved in MyoD-mediated activation of muscle-specific genes, including Myog, in which histone H4 hyperacetylation precedes Brg1 binding to MyoD-induced genes (de la Serna et al., 2005). Myog, even in the absence of MyoD, can also recruit the Brg1 ATPase to modify the chromatin structure at its own promoter in mature myofibers (Ohkawa et al., 2007). We observed H4 hyperacetylation in Myog-transfected C2C12 but not NIH3T3 cells (Fig. 7). Because C2C12 cells express MyoD (Fig. 2C) and Myog can regulate MyoD expression in differentiating C2C12 cells (Fig. 1) through binding the DRR of MyoD (Fig. 4), the hyperacetylation of histone H4 in Myog-transfected C2C12 cells (Fig. 7) should be MyoD-dependent. It has been reported that Brd4 associates with diacetylated histone H3 and di- and tetra-acetylated histone H4 (Dey et al., 2003). Interestingly, we observed the interaction of endogenous Myog with Brd4 in differentiating C2C12 cells (Fig. 8B). Based on previous reports (de la Serna et al., 2005; Ohkawa et al., 2007) and our own findings, we speculate that one of the functions of Myog in MyoD-initiated late gene expression could be that following MyoD-induced chromatin remodeling (including hyperacetylation of H4 by p300) and Brd4 association with hyperacetylated H4, Myog may recruit Brg1 to the promoters of MyoD-initiated late muscle genes by recognizing and binding Brd4. Consequently, the recruited Brg1 ATPase modifies promoter chromatin structure, facilitating MyoD-initiated late gene expression.
Myog could bind the DRR of MyoD in differentiating C2C12 cells (Fig. 4). This may help in maintaining the levels of MyoD, which is rapidly degraded before its binding to the target sites (Hatoum et al., 1998). Consistently, we found that knockdown of Myog led to suppression of MyoD in differentiating C2C12 cells (Fig. 1). We therefore suggest that another function of Myog in regulating late muscle genes is to support MyoD expression, strengthening MyoD-mediated chromatin remodeling at MyoD-initiated late genes and enhancing their expression.
Brd4 has been shown to mediate transcriptional activation, in which Brd4 binds the positive transcriptional elongation factor P-TEFb and transforms P-TEFb into a transcriptionally active complex (Ai et al., 2011; Jang et al., 2005; Yang et al., 2005). This complex can phosphorylate the largest subunit of RNA polymerase II (RNAPII) and induce a productive transcriptional elongation (Peterlin and Price, 2006; Saunders et al., 2006). Most recently, it was reported that the Brd4 extraterminal (ET) domain regulates transcriptional activity through recruitment of the specific effectors. These effectors regulate transcription by modifying the chromatin microenvironment at Brd4 target genes (Rahman et al., 2011). P-TEFb has also been shown to interact with MEF2 and c-Myc in MEF2- or c-Myc-dependent transcription (Nojima et al., 2008; Rahl et al., 2010). Our observation of the interaction of Myog with Brd4 provides evidence supporting the link between myogenic regulatory factors (MRFs) and Brd4, which may be related to not only MRF-mediated chromatin remodeling but also transcriptional elongation of muscle genes.
We have reported that Myog and MEF2C cooperate in activating the MLP gene (Ji et al., 2009). We have also demonstrated that MLP may enhance the expression of the AChR subunit genes in differentiated C2C12 cells through interaction with the Myog-E12 complex (Lu et al., 2004). These data suggest that Myog may have MyoD-independent functions in differentiated myotubes. Here, we focus on the role of Myog in promoting MyoD-initiated late muscle genes during myogenic differentiation and suggest that the ability of Myog to induce chromatin remodeling is MyoD-dependent. This conclusion does not exclude the observation that Myog, in the absence of MyoD, is able to modify chromatin structure in mature myofibers (Ohkawa et al., 2007).
In summary, Myog can enhance the expression of MyoD-initiated late muscle genes by virtue of its abilities (1) to support MyoD expression, (2) to stimulate hyperacetylation of histone H4 in the presence of MyoD, and (3) to interact with Brd4, possibly strengthening MyoD-mediated chromatin remodeling and transcriptional elongation of muscle genes. The precise nature of the role of Myog-Brd4 association in regulating muscle genes needs to be clarified.
Acknowledgments
We thank Stephen J. Tapscott (Fred Hutchinson Cancer Research Center, Seattle, USA) for the plasmids pBABE-MyoD-ER and pBABE-neo, Eric Olson (M.D. Anderson Cancer Center, USA) for the plasmid pEMSV-Myog, Xia Yi (Peking University Health Science Center, China) for the pSuper empty vector, Thomas Braun (Max-Planck-Institute, Germany) for the plasmid pSuper-Myog, Vivek Mittal (Cold Spring Harbor Laboratory, USA) for the plasmid shRNA708MyoD, and Yong-Ming Ren (Tsinghua University, China) for the plasmids pcDNA3.1(+)-flag-His and pcDNA3.1(+)-HA-His empty vector. This work was funded by National Natural Science Foundation of PR China grants 30471975, 30671062, 30770464, 30971449 and Education Committee of Beijing Intensifying Teaching Plan.
REFERENCES
- Ai N.P., Hu X.M., Ding F., Yu B.F., Wang H.P., Lu X.D., Zhang K., Li Y.N., Han A.D., Lin W., et al. Signal-induced Brd4 release from chromatin is essential for its role transition from chromatin targeting to transcriptional regulation. Nucleic Acids Res. 2011;39:9592–9604. doi: 10.1093/nar/gkr698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asakura A., Fujisawa-Sehara A., Komiya T., Nabeshima Y., Nabeshima Y. MyoD and myogenin act on the chicken myosin light chain 1 gene as distinct transcriptional factors. Mol. Cell. Biol. 1993;13:7153–7162. doi: 10.1128/mcb.13.11.7153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergstrom D.A., Penn B.H., Strand A., Perry R.L., Rudnicki M.A., Tapscott S.J. Promoter-specific regulation of MyoD binding and signal transduction cooperate to pattern gene expression. Mol Cell. 2002;9:587–600. doi: 10.1016/s1097-2765(02)00481-1. [DOI] [PubMed] [Google Scholar]
- Berkes C.A., Tapscott S.J. MyoD and the transcriptional control of myogenesis. Semin. Cell Dev. Biol. 2005;16:585–595. doi: 10.1016/j.semcdb.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Berkes C.A., Bergstrom D.A., Penn B.H., Seaver K.J., Knoepfler P.S., Tapscott S.J. Pbx marks genes for activation by MyoD indicating a role for a homeodomain protein in establishing myogenic potential. Mol Cell. 2004;14:465–477. doi: 10.1016/s1097-2765(04)00260-6. [DOI] [PubMed] [Google Scholar]
- Blackwell T.K., Weintraub H. Differences and similarities in DNA-binding preferences of MyoD and E2A protein complexes revealed by binding site selection. Science. 1990;250:1104–1110. doi: 10.1126/science.2174572. [DOI] [PubMed] [Google Scholar]
- Blais A., Tsikitis M., Acosta-Alvear D., Sharan R., Kluger Y., Dynlacht B.D. An initial blueprint for myogenic differenttiation. Genes Dev. 2005;19:553–569. doi: 10.1101/gad.1281105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun T., Buschhausen-Denkerm G., Bober E., Tannich E., Arnold H.H. A novel human muscle factor related to but distinct from MyoD1 induces myogenic conversion in 10T1/2 fibroblasts. EMBO J. 1989;8:701–709. doi: 10.1002/j.1460-2075.1989.tb03429.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan T.J., Edmondson D.G., Olson E.N. Aberrant regulation of MyoD1 contributes to the partially defective myogenic phenotype of the BC3H1 muscle cell line. J. Cell Biol. 1990;110:929–937. doi: 10.1083/jcb.110.4.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckingham M. Skeletal muscle formation in vertebrates. Curr. Opin. Genet. Dev. 2001;11:440–448. doi: 10.1016/s0959-437x(00)00215-x. [DOI] [PubMed] [Google Scholar]
- Cao Y., Kumar R.M., Penn B.H., Berkes C.A., Kooperberg C., Boyer L.A., Young R.A., Tapscott S.J. Global and gene-specific analyses show distinct roles for Myod and Myog at a common set of promoters. EMBO J. 2006;25:502–511. doi: 10.1038/sj.emboj.7600958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covault J., Sealy L., Schnell R., Shires A., Chalkley R. Histone hypoacetylation following release of HTC cells from butyrate. J. Biol. Chem. 1982;257:5809–5815. [PubMed] [Google Scholar]
- de la Serna I.L., Ohkawa Y., Berkes C.A., Bergstrom D.A., Dacwag C.S., Tapscott S.J., Imbalzano A.N. MyoD targets chromatin complexes to the myogenin locus prior to forming a stable DNA bound complex. Mol. Cell. Biol. 2005;25:3997–4009. doi: 10.1128/MCB.25.10.3997-4009.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey A., Chitsaz F., Abbasi A., Misteli T., Ozato K. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc. Natl. Acad. Sci USA. 2003;100:8758–8763. doi: 10.1073/pnas.1433065100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilworth F.J., Seaver K.J., Fishburn A.L., Htet S.L., Tapscott S.J. In vitro transcription system delineates the distinct roles of the coactivators pCAF and p300 during MyoD/E47-dependent transactivation. Proc. Natl. Acad. Sci USA. 2004;101:11593–11598. doi: 10.1073/pnas.0404192101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmondson D.G., Olson E.N. A gene with homology to the myc similarity region of MyoD1 is expressed during myogenesis and is sufficient to activate the muscle differentiation program. Genes Dev. 1989;3:628–640. doi: 10.1101/gad.3.5.628. [DOI] [PubMed] [Google Scholar]
- Edmondson D.G., Cheng T.C., Cserjesi P., Chakraborty T., Olson E.N. Analysis of the myogenin promoter reveals an indirect pathway for positive autoregulation mediated by the muscle-specific enhancer factor MEF-2. Mol. Cell. Biol. 1992;12:3665–3677. doi: 10.1128/mcb.12.9.3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber A.N., Klesert T.R., Bergstrom D.A., Tapscott S.J. Two domains of MyoD mediate transcriptional activation of genes in repressive chromatin: a mechanism for lineage determination in myogenesis. Genes Dev. 1997;11:436–450. doi: 10.1101/gad.11.4.436. [DOI] [PubMed] [Google Scholar]
- Gong M., Ni J.H., Jia H.T. Increased exchange rate of histone H1 on chromatin by exogenous myogenin expression. Cell Res. 2002;12:395–400. doi: 10.1038/sj.cr.7290141. [DOI] [PubMed] [Google Scholar]
- Hasty P., Bradley A., Morris J.H., Edmondson D.G., Venuti J.M., Olson E.N., Klein W.H. Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature. 1993;364:501–506. doi: 10.1038/364501a0. [DOI] [PubMed] [Google Scholar]
- Hatoum O.A., Gross-Mesilaty S., Breitschopf K., Hoffman A., Gonen H., Ciechanover A., Bengal E. Degradation of the myogenic transcription factor MyoD by the ubiquitin pathway in vivo and in vitro: regulation by specific DNA binding. Mol. Cell. Biol. 1998;18:5670–5677. doi: 10.1128/mcb.18.10.5670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang M.K., Mochizuki K., Zhou M., Jeong H.S., Brady J.N., Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell. 2005;19:523–534. doi: 10.1016/j.molcel.2005.06.027. [DOI] [PubMed] [Google Scholar]
- Ji Z.X., Du C., Wu G.S., Li S.Y., An G.S., Yang Y.X., Jia R., Jia H.T., Ni J.H. Synergistic up-regulation of muscle LIM protein expression in C2C12 and NIH3T3 cells by myogenin and MEF2C. Mol. Genet Genomics. 2009;281:1–10. doi: 10.1007/s00438-008-0393-7. [DOI] [PubMed] [Google Scholar]
- Johnston L.A., Tapscott S.J., Eisen H. Sodium butyrate inhibits myogenesis by interfering with the transcriptional activation function of MyoD and myogenin. Mol. Cell. Biol. 1992;12:5123–5130. doi: 10.1128/mcb.12.11.5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassar-Duchossoy L., Gayraud-Morel B., Gomes D., Rocancourt D., Buckingham M., Shinin V., Tajbakhsh S. Mrf4 determines skeletal muscle identitiy in Myf5: MyoD double-mutant mice. Nature. 2004;431:466–471. doi: 10.1038/nature02876. [DOI] [PubMed] [Google Scholar]
- Kitzmann M., Carnac G., Vandromme M., Primig M., Lamb N.J.C., Fernandez A. The muscle regulatory factors MyoD and Myf-5 undergo distinct cell cycle-specific expression in muscle cells. J. Cell Biol. 1998;142:1447–1459. doi: 10.1083/jcb.142.6.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P.Y., Taylor M., Jia H.T., Ni J.H. Muscle LIM protein promotes expression of the acetylcholine receptor γ-subunit gene cooperatively with the myogenin-E12 complex. Cell. Mol. Life Sci. 2004;61:2386–2392. doi: 10.1007/s00018-004-4213-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabeshima Y., Hanaoka K., Hayasaka M., Esuml E., Li S., Nonaka I., Nabeshima Y. Myogenin gene disruption results in perinatal lethality because of severe muscle defect. Nature. 1993;364:532–535. doi: 10.1038/364532a0. [DOI] [PubMed] [Google Scholar]
- Nojima M., Huang Y., Tyagi M., Kao H.Y., Fujinaga K. The positive transcription elongation factor b is an essential co-factor for the activation of transcription by myocyte enhancer factor 2. J. Mol. Biol. 2008;382:275–287. doi: 10.1016/j.jmb.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkawa Y., Marfella C.G., Imbalzano A.N. Skeletal muscle specification by myogenin and Mef2D via the SWI/SNF ATPase Brg1. EMBO J. 2006;25:490–501. doi: 10.1038/sj.emboj.7600943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson E.N., Klein W.H. bHLH factors in muscle development: dead lines and commitments, what to leave in and what to leave out. Genes Dev. 1994;8:1–8. doi: 10.1101/gad.8.1.1. [DOI] [PubMed] [Google Scholar]
- Penn B.H., Bergstrom D.A., Dilworth F.J., Bengal E., Tapscott S.J. A MyoD-generated feed-forward circuit temporally patterns gene expression during skeletal muscle differentiation. Genes Dev. 2004;18:2348–2353. doi: 10.1101/gad.1234304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterlin B.M., Price D.H. Controlling the elongation phase of transcription with P-TEFb. Mol Cell. 2006;23:297–305. doi: 10.1016/j.molcel.2006.06.014. [DOI] [PubMed] [Google Scholar]
- Plesko M.M., Hargrove J.L., Granner D.K., Chalkley R. Inhibition by sodium butyrate of enzyme induction by glucocorticoids and dibutyryl cyclic AMP. J. Biol. Chem. 1983;258:13738–13744. [PubMed] [Google Scholar]
- Rahl P.B., Lin C.Y., Seila A.C., Flynn R.A., McCuine S., Burge C.B., Sharp P.A., Young R.A. c-Myc regulates transcriptional pause release. Cell. 2010;141:432–445. doi: 10.1016/j.cell.2010.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman S., Sowa M.E., Ottinger M., Smith J.A., Shi Y., Harper J.W., Howley P.M. The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol. Cell. Biol. 2011;31:2641–2652. doi: 10.1128/MCB.01341-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawls A., Morris J.H., Rudnicki M., Braun T., Arnold H.H., Klein W.H., Olson E.N. Myogenin’s functions do not overlap with those of MyoD or Myf-5 during mouse embryogenesis. Dev. Biol. 1995;172:37–50. doi: 10.1006/dbio.1995.0004. [DOI] [PubMed] [Google Scholar]
- Rudnicki M.A., Schnegelsberg P.N., Stead R.H., Braun T., Arnold H.H., Jaenisch R. MyoD or Myf-5 is required for the formation of skeletal muscle. Cell. 1993;75:1351–1359. doi: 10.1016/0092-8674(93)90621-v. [DOI] [PubMed] [Google Scholar]
- Saunders A., Core L.J., Lis J.T. Breaking barriers to transcription elongation. Nat. Rev. Mol. Cell. Biol. 2006;7:557–567. doi: 10.1038/nrm1981. [DOI] [PubMed] [Google Scholar]
- Simone C., Forcales S.V., Hill D.A., Imbalzano A.N., Latella L., Puri P.L. p38 pathway targets SWI-SNF chromatin-remodeling complex to muscle-specific loci. Nat. Genet. 2004;36:738–743. doi: 10.1038/ng1378. [DOI] [PubMed] [Google Scholar]
- Tapscott S.J. The circuitry of a master switch: Myod and the regulation of skeletal muscle gene transcription. Development. 2005;132:2685–2695. doi: 10.1242/dev.01874. [DOI] [PubMed] [Google Scholar]
- Thayer M.J., Tapscott S.J., Davis R.L., Wright W.E., Lassar A.B., Weintraub H. Positive autoregulation of the myogenic determination gene MyoD1. Cell. 1989;58:241–248. doi: 10.1016/0092-8674(89)90838-6. [DOI] [PubMed] [Google Scholar]
- Venuti J.M., Morris J.H., Vivian J.L., Olson E.N., Klein W.H. Myogenin is required for late but not early aspects of myogenesis during mouse development. J. Cell Biol. 1995;128:563–576. doi: 10.1083/jcb.128.4.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub H. The MyoD family and myogenesis: redundancy, networks, and thresholds. Cell. 1993;75:1241–1244. doi: 10.1016/0092-8674(93)90610-3. [DOI] [PubMed] [Google Scholar]
- Wright W.E., Sassoon D.A., Lin V.K. Myogenin, a factor regulating myogenesis, has a domain homologous to MyoD. Cell. 1989;56:607–617. doi: 10.1016/0092-8674(89)90583-7. [DOI] [PubMed] [Google Scholar]
- Wu S.Y., Chiang C.M. The double bromodomain containing chromatin adaptor Brd4 and transcriptional regulation. J. Biol. Chem. 2007;282:13141–13145. doi: 10.1074/jbc.R700001200. [DOI] [PubMed] [Google Scholar]
- Yang Z., Yik J.H., Chen R., He N., Jang M.K., Ozato K., Zhou Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell. 2005;19:535–545. doi: 10.1016/j.molcel.2005.06.029. [DOI] [PubMed] [Google Scholar]
- Zhang H.J., Li W.J., Gu Y.Y., Li S.Y., An G.S., Ni J.H., Jia H.T. p14ARF interacts with E2F factors to form p14ARF-E2F/partner-DNA complexes repressing E2F-dependent transcription. J. Cell Biochem. 2010;109:693–701. doi: 10.1002/jcb.22446. [DOI] [PubMed] [Google Scholar]