

Abstract
Chronic inflammation plays an important role in atherogenesis. Experimental studies have demonstrated the accumulation of monocytes/macrophages in atherosclerotic plaques caused by inflammation. Here, we report the inhibitory effects of lipoteichoic acid (LTA) from Lactobacillus plantarum (pLTA) on atherosclerotic inflammation. pLTA inhibited the production of proinflammatory cytokines and nitric oxide in lipopolysaccharide (LPS)-stimulated cells and alleviated THP-1 cell adhesion to HUVEC by down-regulation of adhesion molecules such as intracellular adhesion molecule-1 (ICAM-I), vascular cell adhesion molecule-1 (VCAM-1), and E-selectin. The inhibitory effect of pLTA was mediated by inhibition of NF-κB and activation of MAP kinases. Inhibition of monocyte/macrophage infiltration to the arterial lumen was shown in pLTA-injected ApoE−/− mice, which was concurrent with inhibition of MMP-9 and preservation of CD31 production. The anti-inflammatory effect mediated by pLTA decreased expression of atherosclerotic markers such as COX-2, Bax, and HSP27 and also cell surface receptors such as TLR4 and CCR7. Together, these results underscore the role of pLTA in suppressing atherosclerotic plaque inflammation and will help in identifying targets with therapeutic potential against pathogen-mediated atherogenesis.
Keywords: ApoE, atherosclerosis, cytokine, Lactobacillus, lipoteichoic acid, inflammation
INTRODUCTION
Atherosclerosis is a disease in which artery walls thicken as a result of an accumulation of cholesterol and immune cells. Infiltration of serum cholesterol into endothelial cells of blood vessel induces inflammatory cytokines, which leads to the accumulation of macrophages and promotes the formation of multiple plaques within the arteries (Glass and Witztum, 2011; Lusis, 2000). Since atherosclerosis develops from chronic inflammation related to the accumulation of lipids on arterial walls, many studies have attempted to inhibit athero-inflammation and regulate reversible cholesterol transport from macrophages (Barlic et al., 2009; Chen et al., 2010; Han et al., 2010; Liu et al., 2005; Patel et al., 2009; Westerterp et al., 2007). Atherosclerotic lesions (atheromata) are asymmetric focal thickenings of the innermost layer of the artery, the intima. They consist of cells, connective-tissue elements, lipids, and debris. The atheroma is preceded by a fatty streak, an accumulation of lipid-laden cells beneath the endothelium. Most of these cells in the fatty streak are macrophages, together with some T cells. In the center of an atheroma, foam cells and extracellular lipid droplets form a core region, which is surrounded by a cap of smooth-muscle cells and a collagen-rich matrix. T cells, macrophages, and mast cells infiltrate the lesion and are particularly abundant in the shoulder region where the atheroma grows. Many of the immune cells exhibit signs of activation and produce inflammatory cytokines (Hansson, 2005). In particular, macrophages are involved in the uptake of modified lipoproteins, accumulation cholesterol esters, and recruitment of leukocytes.
The normal endothelium does not support binding of white blood cells. However, early after initiation of an atherogenic diet, patches of arterial endothelial cells begin to express selective adhesion molecules on their surface that bind to various classes of leukocytes. In particular, vascular cell adhesion molecule-1 (VCAM-1) binds precisely to the types of leukocytes found in early human and experimental atheroma, the monocyte and T lymphocyte (Libby et al., 2002). The recruited leukocytes penetrate into the intima and predispose the vessel wall to lipid deposits, triggering inflammatory responses (Prescott et al., 2002; Ross, 1999). The activated leukocytes and intrinsic arterial cells release fibrogenic mediators, including a variety of peptide growth factors that promote replication of arterial smooth muscle cells and contribute to the expansion of these cells into a dense extracellular matrix characteristic of more advanced atherosclerosis lesions (Ross, 1999).
Cytokines or growth factors produced in the inflamed intima and macrophage colony-stimulating factor induce monocytes entering the plaque to differentiate into macrophages. This step is critical for the development of atherosclerosis and is associated with up-regulation of pattern-recognition receptors for innate immunity, including toll-like receptors (TLRs). Although TLRs play a role in protection against infection, they also cause inflammatory diseases such as sepsis, autoimmune disease, cancer, and atherosclerosis (O’Neill et al., 2009). TLRs activate NF-κB and mitogen-activated protein kinase (MAPK) signaling pathways in a MyD88-dependent and/or -independent manner. Through this pathway, some pathogen-related ligands such as lipopolysaccharide (LPS), lipoteichoic acid (LTA), and peptidoglycan (PGN) induce excessive production of pro-inflammatory cytokines and interferons (Loiarro et al., 2010). TLRs also play an important role in the formation of pathogenassociated atherosclerosis through the induction of inflammation and formation of foam cells (Erridge, 2009).
Probiotics including L. plantarum have shown variety of health benefits such as improvement of atopy, activation of the intestinal immune system, protection and treatment of septic shock, regulation of cholesterol levels in the blood, and antioxidation effects (Gupta and Garg, 2009; Kim et al., 2008). However, since most studies have been performed with whole cell bacteria, it is difficult to define the precise mechanisms underlying the benefits of probiotics. In this study, we evaluated the role of LTA from L. plantarum in the formation of atherosclerosis. For this, we examined inhibition of LPS-induced inflammatory responses, which trigger the progression of atherosclerosis, and alleviation of inflammatory cell accumulation in the arterial intima using LTA-injected ApoE knockout mice. Together, our results suggest that pLTA can be considered as a novel therapeutic agent for the prevention and treatment of atherosclerosis.
MATERIALS AND METHODS
Cell lines
THP-1 and RAW264.7 cells were maintained in RPMI 1640 or MEM medium supplemented with 10% heat-inactivated FBS, 100 U/ml of penicillin, and 100 μg/ml of streptomycin. All cell culture media and reagents were purchased from Thermo Scientific HyClone (USA). Primary human umbilical vein endothelial cells (HUVEC, Modern Tissue Technology, Korea) were maintained in EGM-2 BulletKit (Lonza, Switzerland) supplemented with 10% heat-inactivated FBS in a humidified incubator of 5% CO2 at 37°C.
Preparation of LTA
Highly purified LTA was isolated from L. plantarum K8 (KCTC-10887BP) by n-butanol extraction, as previously described (Kim et al., 2008). The purity of the purified LTA was determined by measuring protein and endotoxin content through conventional silver staining after polyacrylamide gel electrophoresis and through the Limulus amebocyte lysate (LAL) assay (Thermo Fisher Scientific, USA), respectively. DNA or RNA contamination was assessed by measuring UV absorption at 260 and 280 nm.
RT-PCR and quantitative real-time PCR
Total cellular RNA was extracted using the guanidium thiocyanate-acid phenol-chloroform method. For each RT-PCR, 1 μg of total RNA was used with the One-Step RT-PCR Kit (Merck, USA). To quantify the target genes, real-time PCR amplification was carried out using the ABI prism 7000 sequence detection system (Applied BioSystems, USA), and the PCR products were detected using SYBR Green. The primers used for RT-PCR or real-time PCR are listed in Tables 1 and 2. The expression of mRNA was normalized with gly-ceraldehyde-3-phospate dehydrogenase (GAPDH) or β-actin.
Table 1.
Oligomer sequences used in RT-PCR
| Product | Forward primer (5′ to 3′) | Reverse primer (5′ to 3′) |
|---|---|---|
| TNF-α | TGCTGCACTTTGGAGTGAT | AGGCCCCAGTTTGAATTCTT |
| IL-8 | TCCTGATTTCTGCAGCTCTG | AAAGGAGAAACCAAGGCACA |
| E-selectin | ACTCAAGGGCAGTGGACACA | TTGGCAGGAAGGAACCTCTT |
| ICAM 1 | GAAGGGACCGAGGTGACAGTC | CCTGCAGTGCCCATTATGAC |
| VCAM 1 | TACCCATTTGACAGGCTGGA | TTGCATTTCCAGAAAGGTGC |
| β-actin | GAAAACAAGATGAGATTGGC | AGACCAAAAGCCTTCATACA |
| TLR2 | ACCCTAGGGGAA ACATCTCT | GCATCCTCACAGGCTGAAT |
| TLR4 | CTAGAGAACTTCCCC ATTGGAC | GCTGGTTGTCCCAAAATCAC |
| CCR2 | CCACATCTCGTTCTCGGTTTATCAG | CGTGGAAAATAAGGGCCACAG |
| CCR7 | CACAGTGCTCTCCATCCCAG | CCGATGAAGGCGTACAAGAA |
| CX3CR1 | TCCGCAATGTGGAAACAAAT | ACTTCCATTGCCTGCTCCTTT |
| GAPDH | GGGGCTCTCCAGAACATCAT | AGCGTCAAAGGTGGAGGAGT |
Table 2.
Oligomer sequences used in quantitative real-time PCR
| Product | Forward primer (5′ to 3′) | Reverse primer (5′ to 3′) |
|---|---|---|
| TNF-α | CTCTTCTGGCTCAAAAAGAGAATT | AGGCCCCAGTTTGAATTCTT |
| IL-8 | CAAACCTTTCCACCCCAAAT | ACCCTCTGCACCCAGTTTTC |
| E-selectin | AAGAGGTTCCTTCCTGCCAA | TTGCACACAGTGCCAAACAC |
| ICAM-1 | AGCACTCAAGGGGAGGTCAC | CCTGCAGTGCCCATTATGAC |
| VCAM-1 | CATTGACTTGCAGCACCACA | GTCCCCTCATTCGTCACCTT |
| β-actin | TGGCACCCAGCACAATGAA | CTAAGTCATCFTCCGCCTGAAGCA |
| GAPDH | GTCTTCACCACCATGGAGAA | AGGAGGCATTGCTGATGAT |
Measurement of cytokines and nitric oxide metabolites
Cell supernatants were collected and assayed for cytokine and nitric oxide production. TNF-α and IL-8 production were determined using Human TNF-alpha DuoSet, Mouse TNF-alpha DuoSet, or Human CXCL8/IL-8 QuantiGlo ELISA Kit (R & D Systems, USA), according to the manufacturer’s instructions. Nitrite, a stable oxidized product of NO, was measured by the Griess reagents according to the manufacturer’s instruction (Sigma-Aldrich, USA).
Western blot analysis
Total cellular protein was added to Laemmli sample buffer, boiled for 5 min, resolved by 12% SDS-PAGE in Tris/glycine/SDS buffer (25 mM Tris, 250 mM glycine, 0.1% SDS), and blotted onto PVDF membranes (100 V, 1.5 h, 4°C). After blocking for 1 h in TBS-T (20 mM Tris-HCl, 150 mM NaCl, 0.1% Tween-20) containing 3% BSA, membranes were washed three times in TBS-T and probed for 2 h with anti-COX-2 antibody, anti-Bax antibody, anti-HSP27 antibody, anti-phospho-HSP27 antibody, anti-phospho-MAP kinase antibodies (R & D Systems) or anti-IκB-α antibody (Santa Cruz Biotechnology, USA) in TBS-T/1% BSA. After washing three times in TBS-T, membranes were incubated with secondary HRP-conjugated anti-rabbit IgG or anti-mouse IgG for 1 h and then washed three times in TBS-T. Bands were detected using enhanced chemiluminescence (ECL) reagents (Invitrogen, USA), according to the manufacturer’s instructions.
Immunofluorescence staining
pLTA- or LPS-treated THP-1 cells were spread onto cover slips and fixed with 4% paraformaldehyde. Cells were incubated with Alexa-conjugated anti-NF-κB (clone; sc-8008 AF488, Santa Cruz Biotechnology) for 120 min and then examined by confocal microscopy.
Cell adhesion assay
THP-1 cells were incubated with [C14]-acetate (1 μCi/ml) in the presence or absence of pLTA (10 μg/ml) for 12 h. After incubation, cells were washed twice with phosphate-buffered saline (PBS) and then incubated in media containing LPS (0.1 μg/ml) for an additional 12 h. HUVEC grown in 96-well plates were treated with or without TNF-α (2 ng/ml) and then cultured for 6 h. The medium was removed from the wells and [C14]-labeled THP-1 cells (2 × 105 cells/ml) in 0.2 ml of the medium were added to each well. After 1 h incubation at 37°C in 5% CO2, wells were washed twice with 0.2 ml of PBS and the numbers of adherent cells were determined by measuring the radioactivity with a liquid scintillation counter (PerkinElmer, USA). THP-1 cell adhesion upon stimulation of pLTA with LPS or TNF-α was calculated relative to the basal adhesion of THP-1 cells to nonstimulated cells.
In vivo studies
All animal studies conformed to the Institutional Animal Care and Use Committee (IACUC) of Samsung Biomedical Research Institute (SBRI). ApoE knockout (ApoE−/−) mice were purchased from the Jackson Laboratory (USA) and bred at the SBRI under specific pathogen-free conditions. Eight-week-old ApoE−/− mice were randomly divided into two groups including high-fat high-cholesterol (HFHC) diet and placebo (n = 5) and HFHC diet and pLTA-treated (n = 5) mice. The HFHC diet contained 0.15% cholesterol, 6% fat, and 0.8% sodium chloride (CRF-1, Research Diets, Inc., USA). To induce the atherogenic effects of the HFHC diet, the 8-week-old mice were fed a HFHC diet for 12 weeks. During the 12 weeks, animals were randomized to receive intraperitoneal injections (100 mg/kg) twice weekly with pLTA (n = 5) or an equal volume of placebo (PBS, n = 5). In all experiments, body weights were monitored throughout the treatment period. Twelve weeks after treatment with pLTA or the placebo, the mice were sacrificed by CO2 inhalation for further studies.
Histological examinations
The cross-sectional areas of atherosclerotic lesions were quantified by evaluating the lesion size in the aortic sinus. Briefly, heart and aorta were perfused with PBS for 10 min and 4% paraformaldehyde for 5 min and then fixed for at least 24 h. The isolated hearts and aortas were embedded in Tissue-Tek OCT compound and frozen at −70°C. All samples were sectioned using a cryostat at −20°C, and six consecutive 5 μm thick sections were cut from the aorta where the valve cusps were visible. Plaques were stained with Oil red O and counterstained with hematoxylin. The lesion area (micrometer squared) of three sections was then quantified by computer-assisted morphometry (Image-Pro Plus, USA), and the average lesion area was calculated in each animal. Immunohistochemical (IHC) studies were conducted using a commercially available kit (DAKO, USA) with anti-mouse macrophage/monocyte antibody (MOMA-2, Serotec, UK). Cross-sections of frozen samples were stained with MOMA-2 and HRP-conjugated goat anti-rabbit secondary antibody. After the IHC reactions, a positive signal was calculated by quantifying the lesion area (micrometer squared) of three sections as described above. Matrix metalloproteinase-9 (MMP-9) and CD31 antigen expression in the atherosclerotic lesion were analyzed by confocal microscopy. To visualize the target proteins in the lesion, fluorescence-labeled antibodies were used and observed at an excitation of 488 nm.
Gelatin zymography
THP-1 cells were incubated in the presence or absence of pLTA (10 μg/ml) for 12 h. After washing with PBS, cells were incubated with FBS-free RPMI 1640 media containing 0.1 μg/ml LPS for 24 h. Culture supernatants were electrophoresed on a 10% SDS-PAGE gel (0.125 M Tris-HCL, pH 6.8, 4% SDS, 20% glycerol, 0.01% bromophenol blue) copolymerized with 1% gelatin as the substrate. After electrophoresis was terminated, the gel was incubated for 1 h at 37°C in a 2.5% Triton X-100 solution, washed in 50 mM Tris-HCl buffer (pH 7.5) for 30 min, and incubated for 16 h in incubation buffer containing 200 mM NaCl, 10 mM CaCl2, 20 nM ZnCl2, and 0.002% NaN3. The gel was stained with 0.1% Coomassie Brilliant Blue G-250, 2% acetic acid, and 40% methanol, and then destained with a solution containing 30% methanol and 10% acetic acid.
Statistical analysis
Results were expressed as mean ± standard deviation (SD), and statistical analysis was performed using a two-tailed unpaired student t-test. A p value of < 0.05 was considered statistically significant.
RESULTS
pLTA inhibits LPS-induced inflammatory cytokine production in THP-1 cells
Chronic expression of cytokines induces the accumulation of macrophages to arterial intima, causing the formation of atherosclerosis (Johnson and Newby, 2009; Kim et al., 2007). In this study, we examined the effects of pLTA (0 to 10 μg/ml) and LPS (0 to 10 ng/ml) on the production of cytokines. A dose-dependent increase of TNF-α (Fig. 1A) and IL-8 (Fig. 1B) mRNA level was displayed in pLTA untreated cells, while pretreatment of pLTA reduced LPS-mediated mRNA levels of TNF-α and IL-8 in a pLTA a dose-dependent manner. Similar to mRNA levels, TNF-α and IL-8 protein expression were increased by LPS treatment, and pretreatment with pLTA significantly reduced LPS-mediated expression of TNF-α (Fig. 1C) and IL-8 (Fig. 1D). Down-regulation of TNF-α and IL-8 was not concomitant with cell death which was confirmed by a cell viability assay (data not shown).
Fig. 1.

pLTA inhibits TNF-α and IL-8 expression in LPS-stimulated monocytes. THP-1 cells were stimulated with LPS in the presence or absence of various concentrations of pLTA. (A, B) mRNA levels of TNF-α and IL-8 were determined by RT-PCR. THP-1 cells were pretreated with 10 μg/ml of pLTA for 12 h followed by treatment with 0.1 μg/ml LPS for 6 h. β-actin was used as a normalization control. (C, D) Levels of TNF-α and IL-8 were measured by ELISA (*p < 0.05; **p < 0.01).
pLTA inhibits the production of inflammatory factors in LPS-treated RAW264.7 cells
To examine the effects of pLTA in a mouse macrophage cell line, RAW264.7 cells were treated with pLTA followed by LPS. Pretreatment with pLTA showed about 50% reduction of LPS-mediated TNF-α production (Fig. 2A). Since nitric oxide (NO), which is synthesized by nitric oxide synthase (NOS), plays an important role in signaling pathways as well as inflammatory systems, we examined the levels of NO production. The production of NO in LPS-stimulated cells was increased up to 2-fold, while pLTA pretreatment showed about 30% reduction of LPS-induced NO production (Fig. 2B). In addition, pLTA pretreatment inhibited LPS-induced iNOS production (Fig. 2C). pLTA-mediated down-regulation of inflammatory factors was not concurrent with cell death (data not shown), indicating that pLTA has an anti-inflammatory effect against LPS in mouse macrophages.
Fig. 2.
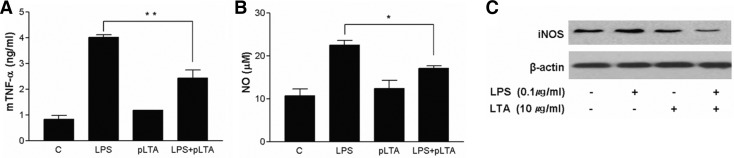
pLTA reduces production of TNF-α and NO as well as expression of iNOS in macrophages. RAW264.7 cells were stimulated with 0.1 μg/ml LPS in the presence or absence of pLTA. (A) Levels of TNF-α were measured in culture supernatants by ELISA 12 h after treatment. (B) Production of nitrite in culture media was measured by Griess reagent 12 h after treatment. (C) Expression levels of iNOS were measured in cell lysates by Western blotting 12 h after treatment.
pLTA down-regulates LPS-mediated signal transduction
LPS activates IκB-α and MAPKs through the TLR4 signaling pathway, which results in the translocation of transcription factors into the nucleus and the production of various cytokines and chemokines. In THP-1 cells stimulated with 0.1 μg/ml LPS, phosphorylation of ERK, p38, or JNK was increased compared to untreated control cells, while LPS-induced phosphorylation was decreased in pLTA pretreated cells (Fig. 3A). pLTA pretreatment attenuated LPS-mediated IκB-α reduction (Fig. 3B), which inhibited the translocation of activated NF-κB into nucleus (Fig. 3C). In all experiments, pLTA alone did not affect the activation of MAPKs and NF-κB signaling pathways.
Fig. 3.

pLTA pretreatment inhibits LPS-induced activation of signaling molecules. THP-1 cells were pretreated with either media or 10 μg/ml pLTA for 12 h followed by treatment with LPS for 30 min. (A) Cell lysates were blotted with the indicated antibodies. Total ERK, p38, and JNK1/2 were used as loading controls. (B) Cytosolic extracts of cell lysates were blotted with antibodies against IκB-α. β-actin was used as a loading control. (C) THP-1 cells were treated with LPS in the presence or absence of pLTA for 1 h. Nuclear translocation of NF-κB was analyzed by confocal microscopy using NF-κB p65 specific antibodies (green). Nuclei are stained blue with DAPI.
pLTA inhibits adhesion of THP-1 cells to HUVEC
Monocytes and macrophages are adherent to arterial intima and disturb blood flow (Young et al., 2002). We examined whether pLTA inhibits THP-1 cell adhesion to HUVEC. After LPS treatment, adhesion of THP-1 cells to HUVEC was increased up to 3-fold compared to untreated controls (Fig. 4A), while adhesion of pLTA-pretreated THP-1 cells to HUVEC displayed ∼60% reduction compared to adhesion of LPS-treated THP-1 cells (Fig. 4B). Intracellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1), as well as some of the integrins, induce firm adhesion of inflammatory cells to vascular surfaces. Expression of VCAM-1, ICAM-1, and E-selectin has been consistently observed in atherosclerotic plaques (Blankenberg et al., 2003). Expression of ICAM-1, VCAM-1, and E-selectin in TNF-α stimulated HUVEC was inhibited by pLTA pretreatment in a dose-dependent manner (Fig. 4C). Similarly, a high dose of pLTA decreased TNF-α-induced mRNA levels of ICAM-1, VCAM-1, and E-selectin (Fig. 4D).
Fig. 4.

pLTA inhibits adhesion of LPS-stimulated THP-1 cells to HUVEC. [C14]-acetate-labeled THP-1 cells were preincubated for 12 h with 10 μg/ml pLTA. Cells were washed twice with PBS and incubated with 0.1 μg/ml LPS for 12 h. THP-1 cells were added to the HUVEC monolayer and allowed to adhere for 1 h. (A) THP-1 cells attached to HUVEC were observed under a microscope at 400 X magnification. (B) Adhesion was measured as described in the “Materials and Methods”. (C) Effects of pLTA on TNF-α-induced mRNA expression of ICAM-1, VCAM-1, and E-selectin. HUVEC were pre-incubated with 10 μg/ml pLTA for 12 h and then stimulated with 2 ng/ml TNF-α for 6 or 12 h. Levels of mRNA for adhesion molecules were determined by RT-PCR. The results shown are from a single experiment and are representative of three separate experiments.
pLTA reduces monocyte and macrophage infiltration in ApoE−/− mice aortic sinus
We performed in vivo experiments to determine whether pLTA would have the same efficiency in a mouse model. Following the development of atherosclerosis in ApoE knockout mice (n = 5 for each group) using a HFHC diet, pLTA or a vehicle were injected twice a week for 12 weeks. MOMA-2 staining showed that pLTA inhibited the infiltration of monocytes and macrophages into arterial intima compared to vehicle injection (Fig. 5A, upper panel). Quantification of those areas that stained positively for MOMA-2 antibody showed that monocyte/macrophage infiltration was reduced in mice aorta injected with pLTA (Fig. 5A, lower panel). The upper panel of Fig. 5B shows the same cross-sections as (A), which are stained with Oil Red O. Areas of aortic sinus lesions stained with Oil Red O were quantified by computer assisted morphometry, indicating that monocyte/macrophage infiltration in the experiment using ApoE−/− mice was not occurred (Fig. 5B, lower panel). Thus, Fig. 5B suggests that pLTA itself did not induce lipid accumulation and pLTA has no risk for atherosclerosis, so it could be a reagent for atherosclerosis prevention.
Fig. 5.

pLTA reduces the accumulation of macrophages and monocytes in hyperlipidemic ApoE−/− mice. (A) Upper panel shows cross-sections of mouse macrophages in the aortic sinus of ApoE−/− mice which stained positively for the monoclonal antibody MOMA-2. The mice were fed a HFHC diet and injected with PBS or pLTA. Lower panel shows % of monocytes/macrophage positive area in total lesion area. Three sections from each animal were stained to determine the mean. Data is represented as the percentage of the total lesion area. (B) Upper panel shows the same crosssections as (A) stained with Oil Red O. Areas of aortic sinus lesions staining positively for Oil Red O were quantified by computer assisted morphometry (Lower panel) (*p < 0.05).
pLTA reduces MMP-9 expression and endothelial cell destruction in atherosclerotic aortic sinus
Macrophages, foam cells, and mast cells express matrix metalloproteinases (MMPs) in atherosclerotic plaques. The degradation of intimal collagens by MMPs increases plaque instability (Ross, 1999). Here, we examined the expression of CD31, vascular endothelial cell markers, and MMP-9 in the aortic sinus of ApoE−/− mice. pLTA-injected mice reduced the expression of MMP-9 in HFHC-treated mice compared to PBS-injected mice, suggesting that disruption of arterial endothelial cells can be inhibited by pLTA injection (Fig. 6A). Preservation of CD31 was observed in the atherosclerotic aortic sinus of pLTA-treated ApoE−/− mice (Fig. 6B). When we quantified CD31 staining area by computerized imaging analysis, it was increased by pLTA as compared to PBS treated control (C). Because zymography reflects both the activity of the renatured enzyme and quantity of protein expressed, the gelatin zymography assay was performed to determine whether pLTA specifically affects the activity and protein expression of MMP-9. Without stimulation, THP-1 cells were sufficient to stimulate MMP-9 protein production. Addition of LPS led to an expected increase of a single protein band at 92 kDa, consistent with the expression of pro-MMP-9. Importantly, in the presence of pLTA, MMP-9 activity was decreased below basal level (Fig. 6D). Atherosclerotic plaque progression involves smooth muscle cell recruitment to the intima of the vessel wall, although controversy currently exists on the exact origin of these recruited cells (Caplice et al., 2003). Thus, we observed smooth muscle state in the atherosclerotic lesion of ApoE−/− mice after treatment of HFHC diet following pLTA or PBS injection. As shown in Fig. 6E, smooth muscle in lesion area was preserved by pLTA as compared to pLTA untreated KO mice, but it was not significant. This result indicates that pLTA plays a role in stabilizing the atherosclerotic plaque (Fig. 6E, lower panel).
Fig. 6.

pLTA treatment reduces MMP-9 expression and endothelial cell destruction. Immunofluorescence images show changes in the expression of (A) MMP-9 or (B) CD31 in the aortic sinus of ApoE−/− mice. For all color images, DAPI is blue and target proteins (MMP-9, CD31) are green (L, lumen; M, media). (C) Quantification of CD31 green fluorescence in aortic sinus is shown. (D) THP-1 cells were incubated in the presence or absence of pLTA (10 μg/ml) for 12 h. After incubation, cells were washed and then further incubated for 24 h in FBS-free media containing LPS (0.1 μg/ml). Culture supernatants were electrophoresed on a 10% SDS-PAGE gel copolymerized with 1% gelatin. Gelatinolytic activities were detected as unstained bands against the background of Coomassie blue stained gelatin. LPS-induced MMP-9 activation was inhibited by pLTA pretreatment. (E) Area of smooth muscle cells in aortic sinus lesion is stained by antismooth muscle actin.
pLTA down-regulates atherosclerotic markers
COX-2 is required for the synthesis of prostanoids, which play an important role in inflammation and mitogenesis. In particular, COX-2 expressed by monocytes and macrophages increases atherosclerosis in murine models (Burleigh et al., 2002) and affects plaque instability through the induction of metalloproteinase (Cipollone et al., 2001). It is known that the COX-2 inhibitor nimesulide reduces atherosclerosis up to 30%, and the administration of rofecoxib reduces atherosclerosis in LDLR-deficient mice (Belton and Fitzgerald, 2003). In this study, we observed that LPS induced COX-2 expression in THP-1 cells, whereas LPS-mediated COX-2 induction was significantly diminished in pLTA pretreated cells (Fig. 7A). COX-2 expression from RAW264.7 cells pretreated with pLTA was also down-regulated (data not shown) and not detected in untreated or pLTA only treated cells.
Fig. 7.

Treatment of THP-1 cells with pLTA inhibits expression of atherosclerotic biomarkers. THP-1 cells were treated with or without 10 μg/ml pLTA for 12 h, and then further treated with 0.1 μg/ml LPS for 24 h. (A) COX-2, (B) Bax, and (C) HSP27 expression were detected by Western blot analysis using specific antibodies.
The pro-apoptotic protein Bax belongs to the Bcl-2 family. Bax controls apoptosis in advanced atherosclerotic lesions and is increased in atherosclerotic plaques (Liu et al., 2005). Like COX-2, Bax was significantly increased in LPS-treated cells but down-regulated in pLTA pretreated cells (Fig. 7B). In addition to COX-2 and Bax, heat shock protein 27 (HSP27) is increased in atherosclerotic plaques (Park et al., 2006). We observed that phosphorylation of HSP27 was significantly increased in LPS-treated cells, and pLTA pretreated cells inhibited LPS-induced phosphorylation of HSP27 (Fig. 7C). These findings indicate that pLTA may be involved in the alleviation of pathogenmediated atherosclerosis.
pLTA inhibits the expression of cell surface receptors, which involved in LPS-mediated inflammation
LPS binds to TLR4 to initiate a cascade of signaling events. Therefore, induction of TLR4 after association with LPS triggers a strong signaling cascade. Indeed, LPS significantly increased TLR4 mRNA expression. However, LPS-induced TLR4 expression was suppressed in THP-1 cells pretreated with pLTA. The expression of TLR2, however, was not affected by LPS and pLTA treatment (Fig. 8A). Recruitment of inflammatory cells to the intima is an essential step in the development and progression of atherosclerosis. This process is triggered by local production of chemokines and chemokine receptors from inflammatory cells and activated endothelial cells, respectively (Charo and Peters, 2003; Combadière et al., 2003; Luchtefeld et al., 2010). In this study, the expression of chemokine receptor CCR7 was induced by LPS, while its expression was inhibited by pLTA pretreatment. However, LPS and pLTA did not affect the expression of CCR2 and CX3CR1 mRNA (Fig. 8B). Interestingly, pLTA only did not induce expression of TLR4 and CCR7. This finding indicates that pLTA has no inducible effects but helps to inhibit LPS-induced TLR or chemokine receptor expression, which suppresses the recruitment of inflammatory cells to aortic sinus lesions.
Fig. 8.

Down-regulation of TLRs and chemokine receptors. THP-1 cells were treated with or without 10 μg/ml pLTA for 12 h, and then further treated with 0.1 μg/ml LPS for 24 h. mRNA expression of (A) TLR2 and TLR4 and (B) CCR2, CCR7, and CX3CR1 was examined by RT-PCR and visualized on a 0.8% agarose gel. The β-actin was used as an endogenous positive control.
DISCUSSION
Atherosclerosis is formed by many cell types such as endothelial cells, smooth muscle cells of blood vessels, monocytes/macrophages, foam cells, and T cells (Neri Serneri et al., 1997). Fatty streaks formed by serial differentiation of monocytes are an early symptom of atherosclerosis. Moreover, many reports suggest that monocytes play an important role in the formation of atherosclerosis (Han et al., 2010; Johnson and Newby, 2009; Stöger et al., 2010). Therefore, in this study, we examined human monocyte-like cells, THP-1 and the mouse macrophage cell line RAW264.7, in the development of inflammation and atherosclerosis. Interestingly, pLTA inhibited adhesion of THP-1 cells to HUVEC in vitro, and infiltration of monocytes and macrophages was also reduced in pLTA-injected APOE−/− mice. Infiltration of monocytes into subendothelium requires expression of adhesion molecules such as VCAM and ICAM. Once monocytes attach to endothelial cells of blood vessels using adhesion molecules, they infiltrate into the subendothelium through the gap between endothelial cells. In subendothelium, monocytes differentiate into macrophages, which uptake oxidized LDL and then become foam cells. These cells express growth factors, inflammatory cytokines, and chemotactic factors, which induce multiplication of smooth muscle cells in the arterial intima-media. The smooth muscle cells then move to intima, resulting in thick and elevated intima. In this process, smooth muscle cells can be switched from contractile form to synthetic form. Macrophages and foam cells become apoptotic cells, and the accumulation of cell debris and lipids creates the core of vulnerable plaques (Young et al., 2002). pLTA-mediated inhibition of monocytes/macrophages infiltration likely occurs via inhibition of cell adhesion. In our study, we showed that pLTA inhibited expression of adhesion molecules, which may affect adhesion of monocytes to the subendothelium. pLTA concentration used in the in vitro or in vivo experiment was decided based on the previous results (Kim et al., 2008). pLTA has shown inhibitory effect against 0.5 μg/ml LPS when 10 μg/ml or more pLTA was added in THP-1 cells, and inhibitory effect was reached highest level at 100 μg/ml pLTA. Similarly, we injected mice intraperitoneally (I.P.) with various dose of pLTA and observed mouse mortality against LPS injection. Mice injected with 50 mg/kg or more pLTA did not show mortality. Mice preinjected i.p. with over 100 mg/kg pLTA recovered their health within 2 days, whereas mice preinjected with 50 mg/kg pLTA recovered painfully after 4 days. Thus, to examine the inhibitory effect of pLTA against LPS-mediated atherosclerosis formation, monocyte or macrophage cell lines were treated with 10 μg/ml pLTA in the in vitro experiment, while ApoE−/− mice were injected with 100 mg/kg pLTA in the in vivo experiment.
In the process of atherosclerosis development, apoptosis occurs in the core, and the lipid content of apoptotic cells increases. Healthy smooth muscle cells are replaced by macrophages, foam cells, and mast cells, which cells express MMPs. The degradation of collagen by MMPs causes weak plaques (Libby, 2003; Ross, 1999). ApoE knockout mice injected with pLTA showed reduced MMP-9 in the arterial intima, and MMP-9 mRNA levels as well as a few cytokines and chemokines from cell lines were also reduced. Down-regulation of inflammatory responses was mediated by inhibition of signaling molecules such as IκBα and MAPKs. Recently, development of atherosclerosis is considered to be an inflammatory disease because alteration in the atherosclerotic process is mainly caused by inflammation and this is very similar to the inflammation observed in the synovium of rheumatoid patients. As an autoimmune disease, atherosclerosis interacts with antigens such as heat shock proteins. The expression of HSP27 is significantly increased in atherosclerotic lesions (Park et al., 2006); therefore, HSP27 could be a biomarker for the development of atherosclerosis. In this study, phosphorylation of HSP27 was increased by LPS treatment; however, we observed that pLTA reduced LPS-induced HSP27 phosphorylation. In addition to MMP-9 and HSP27, we observed an increase of cyclooxygenase-2 (COX-2) in LPS treated cells, which was reduced by pLTA treatment. COX-2 promotes early atherosclerotic lesion formation in ApoE deficient mice (Burleigh et al., 2005). Activation of MMP-9, HSP27, and COX-2 may play an important role in atherosclerosis and therefore these molecules should be considered as potential biomarkers in pathogen-mediated atherosclerosis. Thus, pLTA-mediated down-regulation of these atherosclerotic markers could be an indicator of atherosclerotic alleviation.
It is well known that TLRs recognize various pathogen-associated molecular patterns (PAMPs), and this recognition initiates inflammatory responses through the MyD88-dependent or -independent pathway. The MyD88-dependent pathway activates NF-κB and the MyD88-independent pathway activates interferon regulatory factor (IRF), which results in the induction of inflammatory cytokines and IFN-γ, respectively (Loiarro et al., 2010). Because TLR-mediated inflammation can switch monocytes to foam cells, the regulation of TLR is important for the treatment of atherosclerosis. In this study, we showed that expression of TLR4 was significantly reduced by pLTA and this was concomitant with inactivation of NF-κB and MAP Kinases. The reduced expression of TLR4 by pLTA likely led to down-regulation of adhesion molecules such as ICAM-1, VCAM-1, and E-selectin. These results suggest that pLTA inhibited the overall pathway of inflammation; therefore, monocytes could not infiltrate atherosclerotic lesions or be activated within the lesions. TLR2 and TLR4 are involved in the formation of atherosclerosis through the induction of signaling pathways by endogenous and exogenous ligands (Hodgkinson et al., 2008; Tobias and Curtiss, 2007). TLR2, in particular, inhibited diet- and pathogen-associated atherosclerosis in ApoE knockout mice (Madan and Amar, 2008). In our previous study, we showed that pLTA induces “tolerance” against LPS via TLR2 signaling (Kim et al., 2008; 2011). pLTA-mediated tolerance alleviates LPS-induced inflammation, and TLR2-TLR4 crosstalk plays an important role in this process. Our data also suggest that the anti-inflammatory effects of pLTA against LPS help to prevent monocyte infiltration into the aorta. Therefore, our studies suggest that pLTA may be used for the treatment of inflammatory diseases such as sepsis, rheumatoid arthritis, atopy, autoimmune disease, angiogenesis, metastasis, and atherosclerosis.
In conclusion, atherosclerotic formation is initiated by infiltration of monocytes into the aorta. In this study, we showed that pLTA inhibited infiltration of monocytes in ApoE−/− mice. pLTA also inhibited LPS-induced inflammation in monocytes, which may help to alleviate the formation of inflammatory atherosclerosis. Although more studies are needed to determine the role of pLTA in the alleviation of atherosclerosis, our data provides the first evidence that the probiotic LTA can be used for the development of new therapeutic reagents to treat patients with atherosclerosis.
Acknowledgments
This research was supported by the Basic Science Research Program through a National Research Foundation of Korea grant funded by the Korean government (Ministry of Education, Science and Technology) (KRF-2008-313-F00132) and (No. 2010-0012091).
REFERENCES
- Barlic J., Zhu W., Murphy P.M. Atherogenic lipids induce high-density lipoprotein uptake and cholesterol efflux in human macrophages by up-regulating transmembrane chemokine CXCL16 without engaging CXCL16-dependent cell adhesion. J. Immunol. 2009;182:7928–7936. doi: 10.4049/jimmunol.0804112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belton O., Fitzgerald D. Cyclooxygenase-2 inhibitors and atherosclerosis. J. Am. Coll. Cardiol. 2003;41:1820–1822. doi: 10.1016/s0735-1097(03)00305-x. [DOI] [PubMed] [Google Scholar]
- Blankenberg S., Barbaux S., Tiret L. Adhesion molecules and atherosclerosis. Atherosclerosis. 2003;170:191–203. doi: 10.1016/s0021-9150(03)00097-2. [DOI] [PubMed] [Google Scholar]
- Burleigh M.E., Babaev V.R., Oates J.A., Harris R.C., Gautam S., Riendeau D., Marnett L.J., Morrow J.D., Fazio S., Linton M.F. Cyclooxygenase-2 promotes early atherosclerotic lesion formation in LDL receptor-deficient mice. Circulation. 2002;105:1816–1823. doi: 10.1161/01.cir.0000014927.74465.7f. [DOI] [PubMed] [Google Scholar]
- Burleigh M.E., Babaev V.R., Yancey P.G., Major A.S., McCaleb J.L., Oates J.A., Morrow J.D., Fazio S., Linton M.F. Cyclooxygenase-2 promotes early atherosclerotic lesion formation in ApoE-deficient and C57BL/6 mice. J. Mol. Cell. Cardiol. 2005;39:443–452. doi: 10.1016/j.yjmcc.2005.06.011. [DOI] [PubMed] [Google Scholar]
- Caplice N.M., Bunch T.J., Stalboerger P.G., Wang S., Simper D., Miller D.V., Russell S.J., Litzow M.R., Edwards W.D. Smooth muscle cells in human coronary atherosclerosis can originate from cells administered at marrow transplantation. Proc. Natl. Acad. Sci. USA. 2003;100:4754–4759. doi: 10.1073/pnas.0730743100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charo I.F., Peters W. Chemokine receptor 2 (CCR2) in atherosclerosis, infectious diseases, and regulation of T-cell polarization. Microcirculation. 2003;10:259–264. doi: 10.1038/sj.mn.7800191. [DOI] [PubMed] [Google Scholar]
- Chen F.L., Yang Z.H., Wang X.C., Liu Y., Yang Y.H., Li L.X., Liang W.C., Zhou W.B., Hu R.M. Adipophilin affects the expression of TNF-alpha, MCP-1, and IL-6 in THP-1 macrophages. Mol. Cell. Biochem. 2010;337:193–199. doi: 10.1007/s11010-009-0299-7. [DOI] [PubMed] [Google Scholar]
- Cipollone F., Prontera C., Pini B., Marini M., Fazia M., De Cesare D., Iezzi A., Ucchino S., Boccoli G., Saba V., et al. Overexpression of functionally coupled cyclooxygenase-2 and prostaglandin E synthase in symptomatic atherosclerotic plaques as a basis of prostaglandin E (2)-dependent plaque instability. Circulation. 2001;104:921–927. doi: 10.1161/hc3401.093152. [DOI] [PubMed] [Google Scholar]
- Combadière C., Potteaux S., Gao J.L., Esposito B., Casanova S., Lee E.J., Debré P., Tedgui A., Murphy P.M., Mallat Z. Decreased atherosclerotic lesion formation in CX3CR1/apolipoprotein E double knockout mice. Circulation. 2003;107:1009–1016. doi: 10.1161/01.cir.0000057548.68243.42. [DOI] [PubMed] [Google Scholar]
- Erridge C. The roles of Toll-like receptors in atherosclerosis. J. Innate Immun. 2009;1:340–349. doi: 10.1159/000191413. [DOI] [PubMed] [Google Scholar]
- Glass C.K., Witztum J.L. Atherosclerosis. Cell. 2001;104:503–516. doi: 10.1016/s0092-8674(01)00238-0. [DOI] [PubMed] [Google Scholar]
- Gupta V., Garg R. Probiotics. Indian J. Med. Microbiol. 2009;27:202–209. doi: 10.4103/0255-0857.53201. [DOI] [PubMed] [Google Scholar]
- Han X., Kitamoto S., Wang H., Boisvert W.A. Interleukin-10 overexpression in macrophages suppresses atherosclerosis in hyperlipidemic mice. FASEB J. 2010;24:2869–2880. doi: 10.1096/fj.09-148155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- Hodgkinson C.P., Laxton R.C., Patel K., Ye S. Advanced glycation end-product of low density lipoprotein activates the toll-like 4 receptor pathway implications for diabetic atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2008;28:2275–2281. doi: 10.1161/ATVBAHA.108.175992. [DOI] [PubMed] [Google Scholar]
- Johnson J.L., Newby A.C. Macrophage heterogeneity in atherosclerotic plaques. Curr. Opin. Lipidol. 2009;20:370–378. doi: 10.1097/MOL.0b013e3283309848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K.M., Choi J.Y., Yoo S.E., Park M.Y., Lee B.S., Ko Y.H., Sung S.H., Shin H. M., Park J.E. HMCO5, herbal extract, inhibits NF-kappaB expression in lipopolysaccharide treated macrophages and reduces atherosclerotic lesions in cholesterol fed mice. J. Ethnopharmacol. 2007;144:316–324. doi: 10.1016/j.jep.2007.08.029. [DOI] [PubMed] [Google Scholar]
- Kim H.G., Kim N.R., Gim M.G., Lee J.M., Lee S.Y., Ko M.Y., Kim J.Y., Han S.H., Chung D.K. Lipoteichoic acid isolated from Lactobacillus plantarum inhibits lipopolysaccharide-induced TNF-alpha production in THP-1 cells and endotoxin shock in mice. J. Immunol. 2008;180:2553–2561. doi: 10.4049/jimmunol.180.4.2553. [DOI] [PubMed] [Google Scholar]
- Kim H.G., Lee S.Y., Kim N.R., Lee H.Y., Ko M.Y., Jung B.J., Kim C.M., Lee J.M., Park J.H., Han S.H., et al. Lactobacillus plantarum lipoteichoic acid down-regulated Shigella flexneri peptidoglycan-induced inflammation. Mol. Immunol. 2011;48:382–391. doi: 10.1016/j.molimm.2010.07.011. [DOI] [PubMed] [Google Scholar]
- Libby P. Vascular biology of atherosclerosis: overview and state of the art. Am. J. Cardiol. 2003;91:3A–6A. doi: 10.1016/s0002-9149(02)03143-0. [DOI] [PubMed] [Google Scholar]
- Libby P., Ridker P.M., Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–1143. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- Liu J., Thewke D.P., Su Y.R., Linton M.F., Fazio S., Sinensky M.S. Reduced macrophage apoptosis is associated with accelerated atherosclerosis in low-density lipoprotein receptor-null mice. Arterioscler. Thromb. Vasc. Biol. 2005;25:174–179. doi: 10.1161/01.ATV.0000148548.47755.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loiarro M., Ruggiero V., Sette C. Targeting TLR/IL-1R signalling in human diseases. Mediators Inflamm. 2010;2010:674363. doi: 10.1155/2010/674363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchtefeld M., Grothusen C., Gagalick A., Jagavelu K., Schuett H., Tietge U.J.F., Pabst O., Grote K., Drexler H., Förster R., et al. Chemokine receptor 7 knockout attenuates atherosclerotic plaque development. Circulation. 2010;122:1621–1628. doi: 10.1161/CIRCULATIONAHA.110.956730. [DOI] [PubMed] [Google Scholar]
- Lusis A.J. Atherosclerosis. Nature. 2000;407:233–241. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan M., Amar S. Toll-like receptor-2 mediates diet and/or pathogen associated atherosclerosis: proteomic findings. PLoS One. 2008;3:e3204. doi: 10.1371/journal.pone.0003204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neri Serneri G.G., Prisco D., Martini F., Gori A.M., Brunelli T., Poggesi L., Rostagno C., Gensini G.F., Abbate R. Acute T-cell activation is detectable in unstable angina. Circulation. 1997;95:1806–1812. doi: 10.1161/01.cir.95.7.1806. [DOI] [PubMed] [Google Scholar]
- O’Neill L.A., Bryant C.E., Doyle S.L. Therapeutic targeting of Toll-like receptors for infectious and inflammatory diseases and cancer. Pharmacol. Rev. 2009;61:177–197. doi: 10.1124/pr.109.001073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H.K., Park E.C., Bae S.W., Park M.Y., Kim S.W., Yoo H.S., Tudev M., Ko Y.H., Choi Y.H., Kim S., et al. Expression of heat shock protein 27 in human atherosclerotic plaques and increased plasma level of heat shock protein 27 in patients with acute coronary syndrome. Circulation. 2006;114:886–893. doi: 10.1161/CIRCULATIONAHA.105.541219. [DOI] [PubMed] [Google Scholar]
- Patel S., Drew B.G., Nakhla S., Duffy S.J., Murphy A.J., Barter P.J., Rye K.A., Chin-Dusting J., Hoang A., Sviridov D., et al. Reconstituted high-density lipoprotein increases plasma high-density lipoprotein anti-inflammatory properties and cholesterol efflux capacity in patients with type 2 diabetes. J. Am. Coll. Cardiol. 2009;53:962–971. doi: 10.1016/j.jacc.2008.12.008. [DOI] [PubMed] [Google Scholar]
- Prescott S.M., McIntyre T.M., Zimmerman G.A., Stafforini D.M. Sol Sherry lecture in thrombosis: molecular events in acute inflammation. Arterioscler. Thromb. Vasc. Biol. 2002;22:727–733. doi: 10.1161/01.atv.0000016153.47693.b2. [DOI] [PubMed] [Google Scholar]
- Ross R. Atherosclerosis is an inflammatory disease. Am. Heart J. 1999;138:S419–S420. doi: 10.1016/s0002-8703(99)70266-8. [DOI] [PubMed] [Google Scholar]
- Stöger J.L., Goossens P., de Winther M.P. Macrophage heterogeneity: relevance and functional implications in atherosclerosis. Curr. Vasc. Pharmacol. 2010;8:233–248. doi: 10.2174/157016110790886983. [DOI] [PubMed] [Google Scholar]
- Tobias P.S., Curtiss L.K. Toll-like receptors in atherosclerosis. Biochem. Soc. Trans. 2007;35:1453–1455. doi: 10.1042/BST0351453. [DOI] [PubMed] [Google Scholar]
- Westerterp M., Berbee J.F., Pires N.M., van Mierlo G.J., Kleemann R., Romijn J.A., Havekes L.M., Rensen P.C. Apolipoprotein C-I is crucially involved in lipopolysaccharide-induced atherosclerosis development in apolipoprotein E-knockout mice. Circulation. 2007;116:2173–2181. doi: 10.1161/CIRCULATIONAHA.107.693382. [DOI] [PubMed] [Google Scholar]
- Young J.L., Libby P., Schonbeck U. Cytokines in the pathogenesis of atherosclerosis. Thromb. Haemost. 2002;88:554–567. [PubMed] [Google Scholar]