

Abstract
Focal adhesion kinase (FAK) is a protein tyrosine kinase (PTK) crucial in regulation of cell migration and proliferation. In addition to its canonical roles as a cytoplasmic kinase downstream of integrin and growth factor receptor signaling, recent studies revealed new aspects of FAK action in the nucleus. Nuclear FAK promotes p53 and GATA4 degradation via ubiquitination, resulting in enhanced cell proliferation and reduced inflammatory responses. FAK can also serve as a co-transcriptional regulator that alters a gene transcriptional activity. These findings established a new paradigm of FAK signaling from cellular adhesions to the nucleus. Although physiological stimuli for controlling FAK nuclear localization have not been completely characterized, FAK shuttles from focal adhesions to the nucleus to directly convey extracellular signals. Interestingly, nuclear translocation of FAK becomes prominent in kinase-inhibited conditions such as in de-adhesion and pharmacological FAK inhibition, while a small fraction of nuclear FAK is observed a normal growth condition. In this review, roles of nuclear FAK in regulating transcription factors will be discussed. Furthermore, a potential use of a pharmacological FAK inhibitor to target nuclear FAK function in diseases such as inflammation will be emphasized.
Keywords: cell proliferation, FAK, FERM, GATA4, inflammation, p53
INTRODUCTION
Extracellular signals through integrin and growth factor receptor modulate focal adhesion kinase (FAK) function in development, cell migration, cell proliferation, tumor metastasis and angiogenesis (McLean et al., 2005; Mitra et al., 2005; Schaller, 2010). Increasing attention to the importance of FAK activity in triggering cancer progression has elicited, and small molecule FAK inhibitors have been extensively tested to develop anti-cancer therapeutics (Roberts et al., 2008; Stokes et al., 2011; Walsh et al., 2010).
In an effort to elucidate FAK signaling, series of genetic FAK mouse analyses have been performed. Conventional FAK knockouts become early embryonic lethal at embryonic day 8.5 (E8.5) with gastrulation defects (Furuta et al., 1995; Ilić et al., 1995). One important observation was that FAK-/- mouse embryonic fibroblasts (MEFs) did not proliferate, but the addi-tional deletion of tumor suppressor p53 rescued FAK-/- cells from a proliferation block (Ilic et al., 1995; Lim et al., 2008a). However, FAK-/-p53-/- double knockout MEFs exhibited a strong cell migration defect, and this cell line became an established FAK-/-cells. Endothelial cell (EC) specific FAK knockouts also become early embryonic lethal due to vascular defects (Braren et al., 2006; Shen et al., 2005).
Recently, revisiting FAK-/- mouse phenotypes revealed that FAK knockout causes p53 up-regulation and subsequent p21 (a cyclin dependent kinase inhibitor)-mediated cell proliferation block resulting in FAK-/- embryo lethality (Lim et al., 2008a). However, in the presence of FAK, p53 levels are regulated via ubiquitin E3 ligase mdm-2-mediated turnover by FAK N-terminal FERM (band 4.1, ezrin, radixin, moesin homology) domain (Fig. 1), but not by the kinase activity. Moreover, this study documented an important finding that FAK localizes to the nucleus via the FERM to suppress p53 levels. Therefore, FAK possesses dual functions; a kinase-dependent function which is responsible for substrate phosphorylation, and a kinase-independent function as a scaffold recruiting diverse proteins (Lim et al., 2008b). However, FAK-/- mouse model failed to fully address whether the defective phenotypes were caused by the loss of either a kinase or a scaffold role of FAK.
Fig. 1.
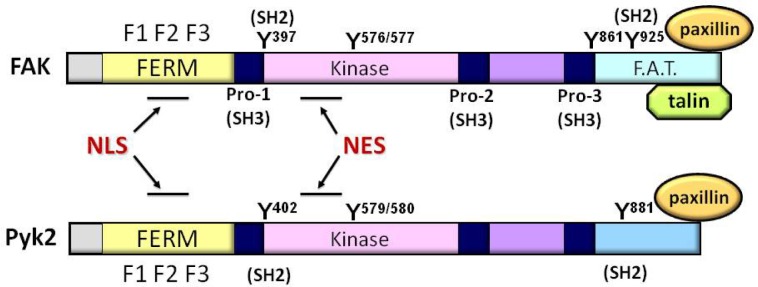
Schematics of FAK and Pyk2. FAK and Pyk2 have a conserved central kinase domain (amino acid homology 60%). Both proteins contain a unique N-terminal FERM (band 4.1, ezrin, radixin, and moesin homology) domain which consists of three subdomains, F1, F2, and F3. FAK C-terminal domain, also called as focal adhesion targeting (FAT) domain, is indirectly linked to integrin adhesomes by interacting with major focal adhesion components such as talin and paxillin, while the Pyk2 C-terminus does not have a binding motif for talin and is shorter than the FAK C-terminus by ∼50 amino acids. In both FAK and Pyk2, the nuclear localization sequence (NLS) is located within F2 domain of FERM, and the nuclear export signal (NES) is also well conserved in the kinase domains. There are three proline-rich domains providing the binding sites for Src homology 3 (SH3) domain-containing proteins such as p130Cas. FAK has five major tyrosine (Y) phosphorylation sites (Y397, Y576, Y577, Y861, and Y925). Y576/Y577 serves as the activation loop and tyrosine phosphorylation of Y397 autophosphorylation or Y925 sites can provided the binding site for Src homology 2 (SH2) domain-containing proteins such as Src tyrosine kinase or Grb2. Pyk2 has the equivalent autophosphorylation site at Y402 for providing Src binding motif, the activation loop Y579/Y580 and the C-terminally located SH2 containing-protein binding site at Y881.
To gain further insights of FAK kinase-dependent function during development, a FAK kinase-dead (KD) knock-in mouse (a point mutation in lysine 454 to arginine prevents ATP binding) was introduced to mouse (Lim et al., 2010b; Zhao et al., 2010). The key idea on this genetic mouse is that the mouse will express a full-length FAK protein but lacks its kinase activity. The KD FAK mouse is early embryonic lethal at E9.5 due to a number of defects, demonstrating that the kinase activity is essential in early embryo developmental processes including vessel formation and chorio-allantois fusion (Inman and Downs, 2007; Lim et al., 2010b; 2012).
Interesting, unlike FAK-/- MEFs, KD FAK cells did not inhibit cell proliferation, supporting that the intact FAK FERM in KD FAK cells are still able to enhance p53 degradation in the nucleus to promote cell survival via a kinase-independent pathway (Lim et al., 2010b). Another remarkable finding from this study is that FAK activity regulates developmental vascular cell adhesion molecule-1 (VCAM-1) expression which is critical in chorio-allantois fusion process (Lim et al., 2012). VCAM-1 receptor on the allantois serves as a counterpart ligand for α4 integrin on the chorion side (Gurtner et al., 1995; Kwee et al., 1995). This study further shed light on a new role of FAK in inflammatory VCAM-1 expression which is critical in inflammation process (Libby, 2002). FAK inhibition in endothelial cells block tumor necrosis factor-α (TNF-α) stimulated VCAM-1 expression by enhancing the degradation of transcription factor GATA4 through nuclear FAK scaffold action (Lim et al., 2012).
This review will focus on mechanistic aspects of FAK shuttling from the cytosol to the nucleus, new roles of nuclear FAK, and its implication in treatment of inflammatory diseases by targeting nuclear FAK.
Structure and general features of FAK family kinases
FAK is a protein tyrosine kinase (PTK) which is expressed in most tissues except non-adherent blood cells. Pyk2, another member of FAK family kinase, is highly expressed in hemato-poietic, endothelial, and neuronal cells (Dikic and Schlessinger, 1998; Lev et al., 1995). Although both kinases exhibit structural similarities, their subcellular localization and functions are distinct (Klingbeil et al., 2001).
FAK and Pyk2 protein have three major domains, a N-terminal FERM (band 4.1, ezrin, radixin, moesin homology) domain, a central kinase domain, and a C-terminal domain (Lim et al., 2008b) (Fig. 1). FAK C-terminal domain is called as focal adhesion targeting (FAT) domain which contains binding sites for both paxillin and talin, thereby connects to cellular adhesion sites called focal adhesions (FAs) while the C-terminal domain of Pyk2 does not have talin binding motif and do not normally localize to the FAs (Klingbeil et al., 2001; Lawson et al., 2012). This difference makes Pyk2 subcellular localization more favorable to perinuclear region than FAs.
FAK FERM domain consists of three subdomains, F1, F2, and F3 (Ceccarelli et al., 2006). As a positively charged basic patch within F2 domain can bind to PIP2 (phosphatidyl 4,5 inositol bisphosphate), FAK FERM can localize to the membrane (Cai et al., 2008).
These unique structural features of FAK allow FAK to function as a kinase and scaffold. First of all, FAK can phosphorylate a number of structural and signaling proteins such as paxillin, p130Cas, and α-actinin. Secondly, multiple proteins interact with FAK through the structural domains (e.g., FERM, proline-rich, FAT domain), and with Src homology 2 (SH2) containing signaling proteins which bind to phosphorylated tyrosine (pY) residues within FAK. Upon activation via integrin and cell surface receptor signaling, FAK is phosphorylated at tyrosine 397 autophosphorylation site (pY397), creating a major binding site for SH2 domain containing proteins such as Src tyrosine kinase (Schlaepfer et al., 2004). Then, Src-FAK complex promotes phosphorylation of FAK Y576/Y577 activation loop and further phosphorylates Y861 and Y925, providing a binding site for other SH2-containing proteins such as Grb2 (Schlaepfer and Hunter, 1996).
Importantly, from KD FAK mouse study, the loss of the kinase activity did not inhibit KD MEF cell growth. Therefore, it seems that the kinase activity is not essential for cell proliferation and FAK regulation of p53 and p21 in cell proliferation is achieved by a kinase-independent mechanism (Lim et al., 2010b). This study demonstrates the first clue that FAK plays a role in the nucleus to regulate a nuclear factor (Lim et al., 2008a).
FAK shuttles between focal adhesions and the nucleus
The evidence of FAK nuclear localization was shown by the treatment of a nuclear export inhibitor, Leptomycin B, which promotes nuclear accumulation of green fluorescent protein tagged FAK (GFP-FAK) wild-type in FAK-/- MEF cells (Lim et al., 2008a). Further analyses on nuclear localization signal (NLS) and nuclear export sequence (NES) revealed that the FAK NLS is within FAK FERM domain F2 lobe with a surface-exposed basic residue clustering of K190, K191, K216, K218, R221 and K222 (Lim et al., 2008a). FAK NES signal consists of a patch of leucine-rich amino acid sequences in the kinase domain (L518, L520, L523, and L525) (Ossovskaya et al., 2008). These nuclear entry and export sequences are also well conserved in Pyk2 (Fig. 1). FAK FERM domain alone predominantly localizes to the nucleus since the FERM does not possess NES.
FAK deletion causes upregulated p53 levels, and under this condition, a compensatory Pyk2 expression is often observed (Lim et al., 2008c; Weis et al., 2008). Pyk2 also localizes to the nucleus and promotes p53 turnover to enhance cell proliferation upon FAK deletion in FAK-/-p21-/- MEFs. Interestingly, Pyk2 knockdown in murine ID8 ovarian carcinoma also increased p53 levels and caused p21-mediated cell cycle arrest at G1 phase. Pyk2 FERM expression in Pyk2 knockdown ID8 cells can reduce p53 levels via ubiquitin E3 ligase mdm-2-mediated ubiquitination, and rescue the cell cycle block (Lim et al., 2010a). These results support that a kinase-independent function of Pyk2 FERM is similarly achieved in the nucleus to reduce p53 levels.
A number of FERM containing proteins such as kindlins (−1, −2, and −3) localize to the nucleus (Frame et al., 2010). Although not all of them have specific roles identified in the nucleus, Merlin encoded by the neurofibromatosis type 2 (NF2) gene suppresses ubiquitin E3 ligase CRL4DCAF1 in the nucleus inhibits cell proliferation (Cooper et al., 2011). Prototype FERM proteins such as ezrin, radixin, and moesin (ERMs) are also found in the nucleus, however, the role of these proteins in the nucleus remains unclear. Among nuclear localizing FERM containing proteins, FAK is a best characterized model of cytoplasm-nucleus shuttling.
FAK is not the only FA protein that travels to the nucleus, and other FA proteins such as zyxin and paxillin family which belong to LIM domain proteins also translocate to the nucleus (Dong et al., 2009). As both proteins do not bind to DNA directly, they may function as co-receptors for certain transcriptional factors in the nucleus (Wang and Gilmore, 2003).
Although zyxin and paxillin family proteins have an NES at the N-terminal domain, no obvious NLS containing sequence has been identified. It is possible that FAK interaction with these proteins may co-regulates nuclear translocation from integrin adhesome structures.
Signals stimulating FAK nuclear localization
Although FAK shuttles from the cytoplasm to the nucleus, a few physiological stimuli have been documented (Fig. 2). First, stress signals induce FAK mobilization from the cytosol to the nucleus. Treatment of human ECs with staurosporine, an apoptotic inducer, promotes FAK nuclear accumulation after the concomitant loss of FAK from focal adhesions (Lim et al., 2008a). Oxidative stress by H2O2 treatment in muscle cells and myotubes also promotes FAK nuclear localization, resulting in muscle cell differentiation (Luo et al., 2009).
Fig. 2.

A working model for FAK shuttling between the cytosol and the nucleus. In a normal growth condition, FAK’s localization is prominent in focal adhesions and the cytosol. However, cell de-adhesion from the substratum or stress signals such as a chemical stress or an oxidative stress promote FAK mobilization from integrin adhesion sites to the nucleus via the FERM NLS (left). In the nucleus, FAK interacts with tumor suppressor p53 and recruits ubiquitin E3 ligase mdm-2, resulting in p53 turnover to enhance cell survival. FAK-p53 regulation occurs in a kinase-independent manner. Nuclear FAK interacts with methyl CpG-binding protein 2 (MBD2) to disrupt the function of MBD2-HDAC1 (histone deacetylase 1) repressor complex, resulting in activating gene expression such as myogenin which promotes muscle differentiation. In pro-inflammatory signaling (right), TNF-α activates FAK and in turn, FAK activates mitogen activated protein kinases (MAPKs) to induce inflammatory vascular endothelial cell adhesion molecule-1 (VCAM-1) expression. FAK inhibition blocks MAPK activation, but additional active MAPK expression did not rescue inflammatory VCAM-1 expression. Surprisingly, FAK inhibition also promotes FAK accumulation in the nucleus. Nuclear FAK FERM binds to GATA4 essential for inflammatory VCAM-1 expression, and promotes GATA4 turnover by recruiting ubiquitin E3 ligase C-terminus Hsp70 interacting protein (CHIP), consequently preventing inflammatory VCAM-1 expression. FAK-GATA4 regulation is mechanistically similar to FAK-p53 turnover process as FAK FERM domain in the nucleus functions as a scaffold for protein degradation in a kinase-independent manner. One potential mechanism that kinase-inhibited FAK is preferentially accumulated in the nucleus would be masking of the NES of FAK by the FERM domain, thereby nuclear FAK export is inhibited. As cell de-adhesion prevents inflammatory VCAM-1 expression, a potential cross-talk between integrin and TNF-α receptor signaling has been proposed (Lim et al., 2012).
Second, cell de-adhesion from the substratum causes FAK nuclear localization, potentially increasing a pool of “free FAK” available in the cytosol by disengagement from focal adhesion targeted FAK (Lim et al., 2008a). A recent report showed that X-chromosome linked inhibitor of apoptosis protein (XIAP) recruits FAK to FAs and activates FAK under a shear stress condition. XIAP knockdown reduces shear stress-enhanced FAK phosphorylation at pY576 and promotes shear stress-triggered translocation of FAK to the nucleus (Ahn and Park, 2010). Possibly, membrane anchors such as FA proteins may hold FAK at the adhesion sites in a normal condition to keep FAK in the cytosol. In a similar manner, a Pyk2 mutation in proline-rich domain (proline 859 to alanine mutation prevents binding of SH3-containing proteins such as p130Cas) promotes Pyk2 nuclear localization (Aoto et al., 2002).
Third, FAK inhibition promotes FAK nuclear localization. Interestingly, from genetic KD FAK studies, KD FAK cells exhibits a stronger nuclear localization of FAK compared to wild-type (WT) FAK (Lim et al., 2010b). A consistent result was demonstrated from a pharmacological FAK inhibitor (PF-562,271, PF-271, Pfizer) study as the inhibitor strongly increased FAK nuclear localization (Lim et al., 2012). A crystal structural study has revealed a potential mechanism in FAK kinase inhibition and activation (Lietha et al., 2007). The inactive state of FAK is maintained as FAK FERM domain directly binds the kinase domain to block the access to the catalytic site. It is possible that when FAK FERM-kinase interaction is stabilized, the NES may be masked by the FERM, but the NLS is always exposed. Therefore, the kinase-inhibited FAK is retained in the nucleus (Fig. 2). It is possible that some other factors may bind to the kinase-inhibited FAK conformation and hold FAK in the nucleus. These speculations suggest that the active form of FAK might preferentially localize at the cytoplasm or adhesions rather in the nucleus.
Potential roles of nuclear FAK
One of the important functions of nuclear FAK is that FAK FERM provides a scaffold to bring p53 and ubiquitin E-3 ligase mdm-2 and promote p53 ubiquitination and degradation via 26S proteasomal pathway (Table 1). Under conditions of a genotoxic or a chemical stress, the level of tumor suppressor p53 increases in the nucleus, which determines cell fate to cell cycle arrest or apoptosis. FAK leaves from FA sites to the nucleus via FERM NLS and forms a p53 degradation complex by recruiting both p53 and E3 ligase mdm-2 (Lim et al., 2008a). Therefore, nuclear FAK plays a key role in reducing p53-mediated cell cycle arrest to enhance cell survival under stress conditions in a kinase-independent fashion (Lim et al., 2008b).
Table 1.
Nuclear FAK interacting proteins
| Protein | Outcome of FAK interaction | DNA binding | Reference |
|---|---|---|---|
| p53* | Degradation | + | Lim et al. (2008a) |
| mdm-2* | p53 ubiquitination | − | Lim et al. (2008a) |
| GATA4 | Degradation | + | Lim et al. (2012) |
| CHIP | GATA4 ubiquitiantion | − | Lim et al. (2012) |
| MBD2 | Dissociation from HDAC1 | + | Luo et al. (2009) |
| Enhance myogenin transcription |
Pyk2 also interacts with p53 and mdm-2 (Lim et al., 2010a).
In inflammation signaling, a pro-inflammatory cytokine tumor necrosis factor-α (TNF-α) or interlukin-1β (IL-1β) induce inflammatory gene expression via FAK (Lim et al., 2012). FAK inhibition prevents inflammatory VCAM-1 expression which plays a critical role in recruiting leukocytes via its α4 integrin to the inflamed sites to enhance inflammation responses (Carter and Wicks, 2001; Cybulsky et al., 2001; Libby et al., 2002). Interestingly, FAK inhibition in MEFs and ECs also promotes FAK accumulation in the nucleus and facilitates the FERM domain-mediated turnover of transcription factor GATA4 which is essential for VCAM-1 expression by recruiting ubiquitin E3 ligase CHIP (C-terminus Hsp70 interacting protein) (Ahmad et al., 1998; Dai et al., 2003; Kobayashi et al., 2007; Minami and Aird, 2001; Molkentin, 2000). FAK-GATA4 regulation is similar to that of FAK-p53, suggesting that nuclear FAK FERM is indeed a scaffold facilitating the turnover of transcription factors.
Oxidative stress caused by H2O2 facilitates FAK nuclear localization in muscle cells and myotubes (Luo et al., 2009). Nuclear FAK binding to methyl CpG-binding protein 2 (MBD2) induces myogenin transcription to promote differentiation of muscles. In this process, nuclear-localized FAK binding to MBD2 promotes dissociation of histone deacetylase 1 (HDAC1) from a MBD2-HDAC1 complex which inhibits myogenin transcription. This study suggested that FAK-MBD2 interaction may play a role in heterochromatin remodeling to activate a gene expression (Mei and Xiong, 2010).
Overall, nuclear FAK controls various transcription factors, resulting in alteration of gene regulation in a kinase-independent manner. It will be intriguing to investigate whether FAK directly binds to DNA to control a gene transcriptional activity.
New roles of FAK in inflammation signaling
Inflammatory cytokines including TNF-α and IL-1β activate inflammatory gene expression via mitogen activated protein kinases (MAPKs) cascade and nuclear factor kappa B (NF-κB) activation (Karin and Gallagher, 2009; Pober, 2002). Inhibition of MAPK or NF-κB pathway significantly reduces inflammatory gene expression. Recent findings suggested that TNF-α activates MAPKs via FAK, and genetic and pharmacological FAK inhibition blocks inflammatory VCAM-1 expression (Lim et al., 2012). Although NF-κB is a critical player in inflammation signaling network, FAK inhibition does not block NF-κB activation but still does block VCAM-1 expression. Additionally, active MAPK expression in KD FAK cells did not rescue VCAM-1 expression. It turns out that FAK inhibition via a small molecule FAK inhibitor (PF-271) or genetic KD FAK promotes turnover of GATA4 transcription factor required for VCAM-1 expression. This is mediated by nuclear FAK scaffold function through interaction with GATA4 and ubiquitin E3 ligase CHIP (Lim et al., 2012). The finding suggests that FAK inhibition can provide anti-inflammatory effects via nuclear-localized FAK, but importantly, the inflammatory signaling pathway through FAK is not dependent of NF-κB activation. This study opened a new role of FAK in inflammatory signaling.
CONCLUSION
The observation that FAK shuttles between focal adhesions and the nucleus extents FAK function to gene expression beyond its cytoplasmic kinase signaling. Under a normal condition, only a small amount of nuclear FAK can be observed. Various stimuli such as stress signals, de-adhesion condition, and the kinase inhibition can trigger nuclear localization of FAK to enable this protein to start whole new jobs in the nucleus. In addition to MAPK activation, the new roles of FAK in inflammation signaling by regulating GATA4 transcription factor, raised a great possibility of using pharmacological FAK inhibitors as anti-inflammatory drug by targeting FAK to the nucleus to block the specific inflammation signaling pathway. These small molecule FAK inhibitors have been tested in preclinical and clinical trials as an anti-cancer drugs. To date, phase I human clinical trial of a FAK inhibitor, PF-271 (Pfizer), has been completed and implicated FAK as a promising anti-cancer target (Bagi et al., 2009). FAK inhibitor may act as a dual role in cancers: 1) kinase inhibition to directly prevent cell migration and tumor metastasis, 2) unidentified nuclear FAK regulation of gene expression in various cancers to prevent a malignancy. It will be intriguing to investigate a role of nuclear FAK in cancer progression by the nuclear FAK promoting effect of a FAK pharmacological FAK inhibitor. There will be more exciting stories to come from future nuclear FAK studies which may unveil the secret of nuclear FAK to address whether FAK directly regulates gene regulation through DNA binding and/or post transcriptional regulation such as splicing and miRNA regulation.
Acknowledgments
This work is supported by American heart association national scientist development grant 12SDG10970000 (to S. L.) and the 2012 Mitchell Cancer Fund (to S. L.).
REFERENCES
- Ahmad M, Theofanidis P, Medford RM. Role of activating protein-1 in the regulation of the vascular cell adhesion molecule-1 gene expression by tumor necrosis factor-alpha. J Biol Chem. 1998;273:4616–4621. doi: 10.1074/jbc.273.8.4616. [DOI] [PubMed] [Google Scholar]
- Ahn S, Park H. XIAP is essential for shear stress-enhanced Tyr-576 phosphorylation of FAK. Biochem Biophys Res Commun. 2010;399:256–261. doi: 10.1016/j.bbrc.2010.07.064. [DOI] [PubMed] [Google Scholar]
- Aoto H, Sasaki H, Ishino M, Sasaki T. Nuclear translocation of cell adhesion kinase beta/proline-rich tyrosine kinase 2. Cell Struct Funct. 2002;27:47–61. doi: 10.1247/csf.27.47. [DOI] [PubMed] [Google Scholar]
- Bagi CM, Christensen J, Cohen DP, Roberts WG, Wilkie D, Swanson T, Tuthill T, Andresen CJ. Sunitinib and PF-562,271 (FAK/Pyk2 inhibitor) effectively block growth and recovery of human hepatocellular carcinoma in a rat xenograft model. Cancer Biol Ther. 2009;8:856–865. doi: 10.4161/cbt.8.9.8246. [DOI] [PubMed] [Google Scholar]
- Braren R, Hu H, Kim YH, Beggs HE, Reichardt LF, Wang R. Endothelial FAK is essential for vascular network stability, cell survival, and lamellipodial formation. J Cell Biol. 2006;172:151–162. doi: 10.1083/jcb.200506184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Lietha D, Ceccarelli DF, Karginov AV, Rajfur Z, Jacobson K, Hahn KM, Eck MJ, Schaller MD. Spatial and temporal regulation of focal adhesion kinase activity in living cells. Mol Cell Biol. 2008;28:201–214. doi: 10.1128/MCB.01324-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter RA, Wicks IP. Vascular cell adhesion molecule 1 (CD106): a multifaceted regulator of joint inflammation. Arthritis Rheum. 2001;44:985–994. doi: 10.1002/1529-0131(200105)44:5<985::AID-ANR176>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Ceccarelli DF, Song HK, Poy F, Schaller MD, Eck MJ. Crystal structure of the FERM domain of focal adhesion kinase. J Biol Chem. 2006;281:252–259. doi: 10.1074/jbc.M509188200. [DOI] [PubMed] [Google Scholar]
- Cooper J, Li W, You L, Schiavon G, Pepe-Caprio A, Zhou L, Ishii R, Giovannini M, Hanemann CO, Long SB, et al. Merlin/NF2 functions upstream of the nuclear E3 ubiquitin ligase CRL4DCAF1 to suppress oncogenic gene expression. Sci Signal. 2011;4:pt6. doi: 10.1126/scisignal.2002314. [DOI] [PubMed] [Google Scholar]
- Cybulsky MI, Iiyama K, Li H, Zhu S, Chen M, Iiyama M, Davis V, Gutierrez-Ramos JC, Connelly PW, Milstone DS. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J Clin Invest. 2001;107:1255–1262. doi: 10.1172/JCI11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Q, Zhang C, Wu Y, McDonough H, Whaley RA, Godfrey V, Li HH, Madamanchi N, Xu W, Neckers L, et al. CHIP activates HSF1 and confers protection against apoptosis and cellular stress. EMBO J. 2003;22:5446–5458. doi: 10.1093/emboj/cdg529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikic I, Schlessinger J. Identification of a new Pyk2 isoform implicated in chemokine and antigen receptor signaling. J Biol Chem. 1998;273:14301–14308. doi: 10.1074/jbc.273.23.14301. [DOI] [PubMed] [Google Scholar]
- Dong JM, Lau LS, Ng YW, Lim L, Manser E. Paxillin nuclear-cytoplasmic localization is regulated by phosphorylation of the LD4 motif: evidence that nuclear paxillin promotes cell proliferation. Biochem J. 2009;418:173–184. doi: 10.1042/BJ20080170. [DOI] [PubMed] [Google Scholar]
- Frame MC, Patel H, Serrels B, Lietha D, Eck MJ. The FERM domain: organizing the structure and function of FAK. Nat Rev Mol Cell Biol. 2010;11:802–814. doi: 10.1038/nrm2996. [DOI] [PubMed] [Google Scholar]
- Furuta Y, Ilic D, Kanazawa S, Takeda N, Yamamoto T, Aizawa S. Mesodermal defect in late phase of gastrulation by a targeted mutation of focal adhesion kinase, FAK. Oncogene. 1995;11:1989–1995. [PubMed] [Google Scholar]
- Gurtner GC, Davis V, Li H, McCoy MJ, Sharpe A, Cybulsky MI. Targeted disruption of the murine VCAM1 gene, essential role of VCAM-1 in chorioallantoic fusion and placentation. Genes Dev. 1995;9:1–14. doi: 10.1101/gad.9.1.1. [DOI] [PubMed] [Google Scholar]
- Ilić D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, Nomura S, Fujimoto J, Okada M, Yamamoto T. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 1995;377:539–544. doi: 10.1038/377539a0. [DOI] [PubMed] [Google Scholar]
- Inman KE, Downs KM. The murine allantois, emerging paradigms in development of the mammalian umbilical cord and its relation to the fetus. Genesis. 2007;45:237–258. doi: 10.1002/dvg.20281. [DOI] [PubMed] [Google Scholar]
- Karin M, Gallagher E. TNFR signaling, ubiquitin-conjugated TRAFfic signals control stop-and-go for MAPK signaling complexes. Immunol Rev. 2009;228:225–240. doi: 10.1111/j.1600-065X.2008.00755.x. [DOI] [PubMed] [Google Scholar]
- Klingbeil CK, Hauck CR, Hsia DA, Jones KC, Reider SR, Schlaepfer DD. Targeting Pyk2 to beta 1-integrin-containing focal contacts rescues fibronectin-stimulated signaling and haptotactic motility defects of focal adhesion kinase-null cells. J Cell Biol. 2001;152:97–110. doi: 10.1083/jcb.152.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S, Mao K, Zheng H, Wang X, Patterson C, O’Connell TD, Liang Q. Diminished GATA4 protein levels contribute to hyperglycemia-induced cardiomyocyte injury. J Biol Chem. 2007;282:21945–21952. doi: 10.1074/jbc.M703048200. [DOI] [PubMed] [Google Scholar]
- Kwee L, Baldwin HS, Shen HM, Stewart CL, Buck C, Buck CA, Labow MA. Defective development of the embryonic and extraembryonic circulatory systems in vascular cell adhesion molecule (VCAM-1) deficient mice. Development. 1995;121:489–503. doi: 10.1242/dev.121.2.489. [DOI] [PubMed] [Google Scholar]
- Lawson C, Lim ST, Uryu S, Chen XL, Calderwood DA, Schlaepfer DD. FAK promotes recruitment of talin to nascent adhesions to control cell motility. J Cell Biol. 2012;196:223–232. doi: 10.1083/jcb.201108078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lev S, Moreno H, Martinez R, Canoll P, Peles E, Musacchio JM, Plowman GD, Rudy B, Schlessinger J. Protein tyrosine kinase PYK2 involved in calcium-induced regulation of ion channel and MAP kinase functions. Nature. 1995;376:737–745. doi: 10.1038/376737a0. [DOI] [PubMed] [Google Scholar]
- Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–1143. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- Lietha D, Cai X, Ceccarelli DF, Li Y, Schaller MD, Eck MJ. Structural basis for the autoinhibition of focal adhesion kinase. Cell. 2007;129:1177–1187. doi: 10.1016/j.cell.2007.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim ST, Chen XL, Lim Y, Hanson DA, Vo TT, Howerton K, Larocque N, Fisher SJ, Schlaepfer DD, Ilic D. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol Cell. 2008a;29:9–22. doi: 10.1016/j.molcel.2007.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S-T, Mikolon D, Stupack DG, Schlaepfer DD. FERM control of FAK function, Implications for cancer therapy. Cell Cycle. 2008b;7:2306–2314. doi: 10.4161/cc.6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim Y, Lim ST, Tomar A, Gardel M, Bernard-Trifilo JA, Chen XL, Uryu SA, Canete-Soler R, Zhai J, Lin H, et al. PyK2 and FAK connections to p190Rho guanine nucleotide exchange factor regulate RhoA activity, focal adhesion formation, and cell motility. J Cell Biol. 2008c;180:187–203. doi: 10.1083/jcb.200708194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim ST, Miller NL, Nam JO, Chen XL, Lim Y, Schlaepfer DD. PYK2 inhibition of p53 as an adaptive and intrinsic mechanism facilitating cell proliferation and survival. J Biol Chem. 2010a;285:1743–1753. doi: 10.1074/jbc.M109.064212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim ST, Chen XL, Tomar A, Miller NL, Yoo J, Schlaepfer DD. Knock-in mutation reveals an essential role for focal adhesion kinase activity in blood vessel morphogenesis and cell motility-polarity but not cell proliferation. J Biol Chem. 2010b;285:21526–21536. doi: 10.1074/jbc.M110.129999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim ST, Miller NL, Chen XL, Tancioni I, Walsh CT, Lawson C, Uryu S, Weis SM, Cheresh DA, Schlaepfer DD. Nuclear-localized focal adhesion kinase regulates inflammatory VCAM-1 expression. J Cell Biol. 2012;197:907–919. doi: 10.1083/jcb.201109067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo SW, Zhang C, Zhang B, Kim CH, Qiu YZ, Du QS, Mei L, Xiong WC. Regulation of heterochromatin remodelling and myogenin expression during muscle differentiation by FAK interaction with MBD2. EMBO J. 2009;28:2568–2582. doi: 10.1038/emboj.2009.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean GW, Carragher NO, Avizienyte E, Evans J, Brunton VG, Frame MC. The role of focal-adhesion kinase in cancer - a new therapeutic opportunity. Nat. Rev Cancer. 2005;5:505–515. doi: 10.1038/nrc1647. [DOI] [PubMed] [Google Scholar]
- Mei L, Xiong WC. FAK interaction with MBD2, A link from cell adhesion to nuclear chromatin remodeling? Cell Adh Migr. 2010;4:77–80. doi: 10.4161/cam.4.1.10343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minami T, Aird WC. Thrombin stimulation of the vascular cell adhesion molecule-1 promoter in endothelial cells is mediated by tandem nuclear factor-kappa B and GATA motifs. J Biol Chem. 2001;276:47632–47641. doi: 10.1074/jbc.M108363200. [DOI] [PubMed] [Google Scholar]
- Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase, in command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6:56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- Molkentin JD. The zinc finger-containing transcription factors GATA-4, -5, and -6. Ubiquitously expressed regulators of tissue-specific gene expression. J Biol Chem. 2000;275:38949–38952. doi: 10.1074/jbc.R000029200. [DOI] [PubMed] [Google Scholar]
- Ossovskaya V, Lim ST, Ota N, Schlaepfer DD, Ilic D. FAK nuclear export signal sequences. FEBS Lett. 2008;582:2402–2406. doi: 10.1016/j.febslet.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pober JS. Endothelial activation, intracellular signaling pathways. Arthritis Res. 2002;4(Suppl 3):S109–116. doi: 10.1186/ar576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts WG, Ung E, Whalen P, Cooper B, Hulford C, Autry C, Richter D, Emerson E, Lin J, Kath J, et al. Antitumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF-562,271. Cancer Res. 2008;68:1935–1944. doi: 10.1158/0008-5472.CAN-07-5155. [DOI] [PubMed] [Google Scholar]
- Schaller MD. Cellular functions of FAK kinases, insight into molecular mechanisms and novel functions. J Cell Sci. 2010;123:1007–1013. doi: 10.1242/jcs.045112. [DOI] [PubMed] [Google Scholar]
- Schlaepfer DD, Hunter T. Evidence for in vivo phosphorylation of the Grb2 SH2-domain binding site on focal adhesion kinase by Src-family protein-tyrosine kinases. Mol Cell Biol. 1996;16:5623–5633. doi: 10.1128/mcb.16.10.5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer DD, Mitra SK, Ilic D. Control of motile and invasive cell phenotypes by focal adhesion kinase. Biochim. Biophys Acta. 2004;1692:77–102. doi: 10.1016/j.bbamcr.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Shen TL, Park AYJ, Alcaraz A, Peng X, Jang I, Koni P, Flavell RA, Gu H, Guan JL. Conditional knockout of focal adhesion kinase in endothelial cells reveals its role in angiogenesis and vascular development in late embryogenesis. J Cell Biol. 2005;169:941–952. doi: 10.1083/jcb.200411155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes JB, Adair SJ, Slack-Davis JK, Walters DM, Tilghman RW, Hershey ED, Lowrey B, Thomas KS, Bouton AH, Hwang RF, et al. Inhibition of focal adhesion kinase by PF-562,271 inhibits the growth and metastasis of pancreatic cancer concomitant with altering the tumor microenvironment. Mol Cancer Ther. 2011;10:2135–2145. doi: 10.1158/1535-7163.MCT-11-0261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh C, Tanjoni I, Uryu S, Tomar A, Nam JO, Luo H, Phillips A, Patel N, Kwok C, McMahon G, et al. Oral delivery of PND-1186 FAK inhibitor decreases spontaneous breast to lung metastasis in pre-clinical tumor models. Cancer Biol Ther. 2010;9:776–788. doi: 10.4161/cbt.9.10.11433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Gilmore TD. Zyxin and paxillin proteins, focal adhesion plaque LIM domain proteins go nuclear. Biochim. Biophys Acta. 2003;1593:115–120. doi: 10.1016/s0167-4889(02)00349-x. [DOI] [PubMed] [Google Scholar]
- Weis SM, Lim ST, Lutu-Fuga KM, Barnes LA, Chen XL, Göthert JR, Shen TL, Guan JL, Schlaepfer DD, Cheresh DA. Compensatory role for Pyk2 during angiogenesis in adult mice lacking endothelial cell FAK. J Cell Biol. 2008;181:43–50. doi: 10.1083/jcb.200710038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Peng X, Sun S, Park AY, Guan JL. Role of kinase-independent and -dependent functions of FAK in endothelial cell survival and barrier function during embryonic development. J Cell Biol. 2010;189:955–965. doi: 10.1083/jcb.200912094. [DOI] [PMC free article] [PubMed] [Google Scholar]