

Abstract
Proteins have evolved to compensate for detrimental mutations. However, compensatory mechanisms for protein defects are not well understood. Using ketosteroid isomerase (KSI), we investigated how second-site mutations could recover defective mutant function and stability. Previous results revealed that the Y30F mutation rescued the Y14F, Y55F and Y14F/Y55F mutants by increasing the catalytic activity by 23-, 3- and 1.3-fold, respectively, and the Y55F mutant by increasing the stability by 3.3 kcal/mol. To better understand these observations, we systematically investigated detailed structural and thermodynamic effects of the Y30F mutation on these mutants. Crystal structures of the Y14F/Y30F and Y14F/Y55F mutants were solved at 2.0 and 1.8 Å resolution, respectively, and compared with previoulsy solved structures of wild-type and other mutant KSIs. Structural analyses revealed that the Y30F mutation partially restored the active-site cleft of these mutant KSIs. The Y30F mutation also increased Y14F and Y14F/Y55F mutant stability by 3.2 and 4.3 kcal/mol, respectively, and the melting temperatures of the Y14F, Y55F and Y14F/Y55F mutants by 6.4°C, 5.1°C and 10.0°C, respectively. Compensatory effects of the Y30F mutation on stability might be due to improved hydrophobic interactions because removal of a hydroxyl group from Tyr30 induced local compaction by neighboring residue movement and enhanced interactions with surrounding hydrophobic residues in the active site. Taken together, our results suggest that perturbed active-site geometry recovery and favorable hydrophobic interactions mediate the role of Y30F as a second-site suppressor.
Keywords: active-site recovery, ketosteroid isomerase, more hydrophobic interactions, rescue mechanism, second-site suppressor
INTRODUCTION
Most mutations that occur in nature are deleterious to protein function or stability. Defects caused by deleterious substitutions can be rescued by second-site suppressor mutations. Recovery of mutational effects on catalysis or stability by second-site mutations has been observed in many proteins such as p53 and HIV capsid protein (Brachmann et al., 1998; Noviello et al., 2011). However, the mechanisms by which defective protein stabilities or activities are recovered by second-site mutations are not known, although few structural analyses were performed to understand how second-site suppressor mutations rescue the effects of deleterious mutations (Merabet et al., 2010; Poteete et al., 1997; Suad et al., 2009; Wilson et al., 1992). Therefore, more systematic approaches are needed to understand the mechanisms underlying mutation restoration because this mechanism can provide insight into how to overcome the original defect (Joerger et al., 2006).
Δ5-3-Ketosteroid isomerase (KSI) catalyzes the allylic isomerization of the 5,6 double bond of Δ5-3-ketosteroids to the 4,5 position (Scheme 1) (Ha et al., 2001; Pollack, 2004). Two KSIs from Pseudomonas putida and Comamonas testosteroni bacteria have been most intensively studied in terms of structure-function relationships (Kim et al., 1997; 2000; Kraut et al., 2010; Lee et al., 2008; Sigala et al., 2009; Wu et al., 1997). Although the two KSIs share only 34% sequence identity, three-dimensional structures of the two KSIs are remarkably similar to each other (Kim et al., 1997; Wu et al., 1997). The tyrosine triad (Tyr14, Tyr55 and Tyr30; P. putida KSI residues are numbered according to those of C. testosteroni KSI throughout the text) forms a hydrogen bond network together with Asp99 and a water molecule at the bottom of the deep hydrophobic active-site cavity in P. putida KSI (Fig. 1) (Kim et al., 1997). Previously, our studies indicated that Y30F mutation introduction increased the Y14F, Y55F and Y14F/Y55F mutant kcat values by 23-, 3- and 1.3-fold, respectively (Choi et al., 2001; Kim et al., 2000). Moreover, Y30F/Y55F mutant stability was increased by 3.3 kcal/mol compared with the Y55F mutant (Kim et al., 2000). These results suggested that the Y30F mutation could function as a second-site suppressor.
Scheme 1..
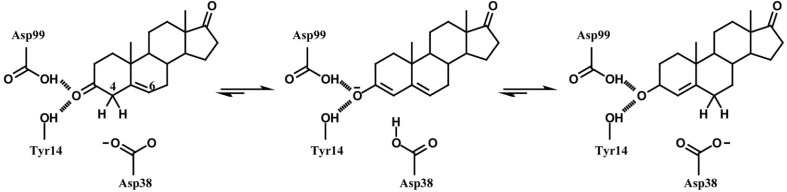
Catalytic mechanism of KSI. KSI catalyzes the isomerization reaction; C4 proton of the substrate 5-androstene-3,17-dione is transferred to the enzyme catalytic base Asp38 to generate the dienolate intermediate, and the same proton is then transferred to C6 to generate 4-androstene-3,17-dione. Both Tyr14 and Asp99 stabilize the intermediate by forming a hydrogen bond with the C3-O of the steroid.
Fig. 1.

The KSI hydrogen bond network involving Tyr14, Tyr30, Tyr55, Asp99 and Water504. The residues involved in the hydrogen bond network are shown as yellow stick models. The PyMOL program was used to draw the figure (www.pymol.org). A potential hydrogen bond is represented by the dashed line.
In the present study, we systematically performed thermodynamic studies, solved Y14F/Y30F and Y14F/Y55F mutant structures and analyzed compensatory effects of the Y30F mutation on mutant KSI catalysis, stability and structure to better understand how second-site compensatory mutations rescue mutant protein stability and activity. As shown in catalytic activity, the additional replacement of Tyr30 with phenylalanine rescued Y14F, Y55F and Y14F/Y55F mutants by increasing the stability. The compensatory effect of the Y30F mutation on stability might be because of increased hydrophobic interactions between the phenyl ring of Tyr30 and surrounding hydrophobic residues. Moreover, structural analyses of Y14F/Y30F and Y14F/Y55F mutants together with previously solved structures of other mutants indicated that the distorted geometry around the active site could be partially recovered to that of wild-type (WT) by additionally mutating Tyr30 to phenylalanine. Our results demonstrate that recovering the active-site geometry and increasing favorable hydrophobic interactions within the active site are important for the role of Y30F as a second-site suppressor.
MATERIALS AND METHODS
Materials
Ultrapure urea and other chemicals used for buffer solutions were purchased from Sigma (USA). Oligonucleotide primers were from Bionics (Korea). DNA pfu polymerase was obtained from SolGent (Korea). dpn1 was purchased from Roche Applied Science (USA). The superose 12 gel filtration and Mono Q anionic exchange columns were obtained from Amersham Bioscience (USA).
Site-directed mutagenesis, expression and purification
Each mutant KSI was constructed using the QuickChange Site-Directed Mutagenesis Kit (Stratagene) using the pKK 223-3 vector (carrying the WT KSI) as a template and a two-primer set for each mutation: 5′-GGCCCGTTTCATCGAGCTGG-3′ and 5′-CCAGCTCGATGAAACGGGCC-3′ (Y14F), 5′-GCAGATGTTCGCCGATGACG-3′ and 5′-CGTCATCGGCGAACATCTGC-3′ (Y30F), 5′-CCGCGTTCTTTCGCCAGGG-3′ and 5′-CCCTGGCGAAAGAACGCGG-3′ (Y55F). For each primer, the mutated bases are underlined. All the mutants were sequenced to confirm that the desired mutation was present. WT and mutant KSIs were overexpressed in E. coli BL21 (DE3) that harbored an expression vector plasmid containing the respective KSI gene. All the enzymes were purified by deoxycholate affinity chromatography, Superose 12 gel filtration chromatography and Mono Q anionic exchange chromatography. Purified proteins were homogeneous as assessed by SDS-PAGE analysis.
Urea-induced denaturation studies
Urea-mediated equilibrium KSI unfolding was performed as described previously (Kim et al., 2000). Changes in the KSI protein optical properties were compared by normalizing each transition curve with the apparent fraction of the unfolded form, FU:
| (1) |
where FU is the fraction of unfolded forms, Y is the observed molar ellipticity at a given urea concentration, and YN and YU are the observed values for the native and unfolded states, respectively, at the same urea concentration. The denaturation equilibrium constant (KU) and free energy change (ΔGU) were determined according to a two-state denaturation model by utilizing the following equations:
| (2) |
| (3) |
where PT is the total protein concentration, ΔGUH2O is the free energy change in the absence of urea and m is a measure of dependency of ΔGU on urea concentration. ΔGUH2O and m values were obtained by fitting the urea denaturation curve data to Eqn. (4) using a software program (Synergy Software, Kaleidagraph version 3.6):
| (4) |
The difference in the free-energy change for unfolding, ΔΔGU, between WT and each mutant KSI was obtained Eqn. (5):
| (5) |
where ΔGU and ΔGUm are the free-energy changes for the unfolding of WT and each mutant KSI, respectively.
Thermal-induced denaturation studies
Thermal unfolding was performed in triplicate by monitoring the change in ellipticity at 222 nm. The temperature was increased from 25 to 105°C at rate of 1°C/min using a Jasco PTC-348W peltier temperature control system. Circular dichroism (CD) signals at 222 nm were collected at every 1°C. The data obtained from CD measurements were analyzed assuming a two-state process. The unfolded protein fraction (Fu) at each temperature was calculated using Eqn. (1). The protein unfolding equilibrium constant is KU = FU/(1 - FU). The midpoint melting temperature (Tm) was obtained at KU = 1.
Crystallization and structure determination
Crystals of the Y14F/Y55F and Y14F/Y30F mutants were grown by the hanging-drop vapor diffusion method as described previously (Jang et al., 2004). For data collection, crystals were briefly immersed in the same precipitant solution containing 10–15% (v/v) glycerol before they were frozen at 100 K utilizing a cryostream cooler (Oxford Cryosystems). Diffraction data were obtained from the Beamline 6B at the Pohang Accelerator Laboratory. The diffraction data for each mutant KSI were indexed, integrated and scaled using the HKL2000 software package (Otwinowski and Minor, 1997). The Y14F/Y55F and Y14F/Y30F mutant structures were determined by molecular replacement utilizing the WT KSI coordinates (PDB code 1OPY), and further refinement was performed using the CNS program (Brunger et al., 1998). The atomic coordinates for the Y14F/Y30F (4K1U) and Y14F/Y55F (4K1V) mutants were deposited in the Protein Data Bank.
Solvent-accessible surface area
The solvent-accessible surface area (SAS) was calculated from the atomic coordinates obtained by X-ray crystallography with the Molecular Surface software program, which was supplied with Quanta software (Accelrys) (Richards, 1977). The probe radius for the calculation was 1.4 Å.
RESULTS AND DISCUSSION
It was previously suggested that the Y30F mutation could be a second-site suppressor because it increased the Y14F, Y55F and Y14F/Y55F mutant catalytic activities by 23-, 3- and 1.3-fold, respectively, and the Y55F mutant stability by 3.3 kcal/mol (Choi et al., 2001; Kim et al., 2000). In this study, we systematically performed structural and thermodynamic analyses to further investigate how the Y30F mutation rescued the mutant catalytic activities and stabilities at the molecular level.
The Y30F mutation rescued the mutant KSIs by restoring active-site geometry
To understand how the mutations rescued KSI catalytic activity, Y14F/Y30F and Y14F/Y55F crystal structures were determined at 2.0 and 1.8 Å resolution, respectively, and compared with previously reported WT and other mutant KSI structures. The crystallographic data are presented in Table 1. Although the overall structures of these mutant proteins were very similar, noticeable differences were found in the active-site geometry between the WT and mutant KSIs. Superposing the WT structure with the Y14F or Y14F/Y30F mutants revealed that the additional Y30F mutation partially restored the structural perturbation of the active site by the Y14F mutation. Previous studies on the Y14F crystal structure revealed that the Y14F mutation caused the phenyl ring of residue 14 to move toward the hydrophobic core (Choi et al., 2001). Moreover, the Y14F mutation displaced a water molecule (Wat504) away from its original position, causing an unfavorable interaction between Asp99 and Wat504 (Fig. 2A). However, the Y30F mutation induced structural rearrangement of the surrounding residues when it was introduced into the Y14F backbone (Supplementary Fig. 1). Ile26 moved toward residue 30, while the phenyl ring of residue 14 was partially recovered to its original location, and the Wat504 molecule returned to its original position. Therefore, the additional Y30F mutation restored the Asp99-Wat504 interaction to the WT by shortening the hydrogen bond between Asp99 and Wat504 from 3.3 to 2.6 Å (Fig. 2B). Restoration of the hydrogen bond between the carboxyl group of Asp99 and Wat504 could explain the 23-fold increase in the Y14F/Y30F kcat compared with that of Y14F, because Asp99 stabilized the dienolate intermediate by forming a hydrogen bond with the bound steroid C3-O and Tyr14 (Scheme 1) (Cho et al., 1999; Choi et al., 2000). A similar positional restoration of the active site was reported in the Y30F/Y55F mutant (Figs. 2C and 2D) (Kim et al., 2000). The additional Y30F mutation compensated for the Y55F mutation-induced decrease in catalytic activity, because the mutation moved Tyr14 into the original WT position. When the second-site mutation was introduced, restoration of the WT geometry was also observed in other proteins such as T4 lysozyme and tryptophan synthase (Nagata et al., 1989; Poteete et al., 1991). Introduction of Y18H or Y18D second-site mutations in T4 lysozyme compensated for the T26Q mutation-induced defects (Poteete et al., 1991). The second-site mutations in T4 lysozyme reoriented Gln26 into the position that was originally occupied in WT T4 lysozyme and increased the activity of the Y18F/T26Q or Y18D/T26Q mutants. Our results suggest that restoring active-site geometry could compensate for deleterious mutations compared with previous studies on T4 lysozyme and tryptophan synthase.
Table 1.
Data collection and structure refinement statistics
| Data collection | Y14F/Y30F (PDB code 4K1U) | Y14F/Y55F (PDB code 4K1V) |
|---|---|---|
| Space group | P212121 | C2221 |
| Unit cell dimensions | ||
| a, b, c (Å) | 35.28, 73.09, 95.08 | 35.35, 94.72, 73.89 |
| Wavelength (Å) | 1.1271 | 1.1271 |
| Resolution (Å) | 50.0-2.0 | 50.0-1.8 |
| Rsyma | 6.6 (18.4)b | 5.7 (12.8) |
| I/σ(I) | 31.9 (5.5) | 50.2 (12.4) |
| Completeness (%) | 95.5 (81.0) | 92.4 (82.0) |
| Redundancy | 2.8 | 2.5 |
|
| ||
| Refinement | ||
|
| ||
| Resolution (Å) | 20.0-2.0 | 20.0-1.8 |
| No. of reflections | 16,120 | 10,875 |
| Rworkc / Rfree | 22.5 / 26.2 | 20.0 / 20.7 |
| No. atoms | ||
| Protein | 1,892 | 959 |
| Water | 94 | 68 |
| R.m.s deviations | ||
| Bond lengths (Å) | 0.0095 | 0.0053 |
| Bond angles (°) | 1.4221 | 1.2471 |
| Ramachandran plot (%) | ||
| Most favored region | 89.6 | 90.2 |
| Additionally allowed | 10.4 | 9.8 |
| Average B-values (Å2) | ||
| Protein | 27.0 | 17.3 |
| Water | 30.0 | 27.8 |
Rsym = ∑|Iobs - Iavg| / Iobs, where Iobs is the observed intensity of individual reflection and Iavg is average over symmetry equivalents.
The numbers in parentheses are statistics from the highest resolution shell.
Rwork = ∑‖Fo| - |Fc ‖ / ∑ |Fo|, where |Fo| and |Fc| are the observed and calculated structure factor amplitudes, respectively. Rfree was calculated with 5% of the data.
Fig. 2.

Superposition of the WT and mutant KSI active-site residues. (A) WT KSI [light gray; PDB code 1OPY (Kim et al., 1997)] and Y14F [dark gray; PDB code 1EA2 (Choi et al., 2001)]. (B) WT KSI (light gray; PDB code 1OPY) and Y14F/Y30F (dark gray; PDB code 4K1U). (C) WT KSI (light gray; PDB code 1OPY) and Y55F [dark gray; PDB code 1DMM (Kim et al., 2000)]. (D) WT KSI (light gray; PDB code 1OPY) and Y30F/Y55F [dark gray; PDB code 1DMN (Kim et al., 2000)]. (E) WT KSI (light gray; PDB code 1OPY) and Y14F/Y55F (dark gray; PDB code 4K1V). (F) WT KSI (light gray; PDB code 1OPY) and Y14F/Y30F/Y55F [dark gray; PDB code 1E97 (Choi et al., 2001)]. Quanta software (Accelrys) was used for the superposition. The figure was drawn with the PyMOL program.
Interestingly, the active-site geometries in the Y14F/Y55F and Y14F/Y30F/Y55F mutants were similar to WT, and the active site was marginally restored by introducing the Y30F mutation (Figs. 2E and 2F). Structural comparison between the Y14F and Y14F/Y55F mutants revealed that the compensatory effect of Y55F mutation on the Y14F mutant in P. putida KSI could originate from active-site geometry recovery (Figs. 2A and 2E). In the Y14F/Y55F mutants, the phenyl ring in residue 14 was reoriented into its original position, which formed more favorable aromatic and hydrophobic interactions with the phenyl ring in residue 55 (Fig. 2E). Because the Y14F/Y55F mutant active-site geometry was restored by the additional Y55F mutation, the Y30F mutation-mediated compensation in Y14F/Y30F/Y55F mutant could not be significant. Previous kinetic analyses of mutant KSIs indicated that the Y14F/Y55F mutant catalytic activity in P. putida KSI was increased 27-fold relative to the Y14F mutant (Choi et al., 2001). A similar restoration of catalytic activity (30-fold) was also observed in C. testosteroni KSI (Li et al., 1993). A slight increase in activity (1.3-fold) was observed in the Y14F/Y30F/Y55F mutant compared with the Y14F/Y30F mutant, because the effect of the additional Y30F mutation on the structural change was marginal. Structural and kinetic analyses consistently indicated that structural restoration of the active site by second-site mutation was important for rescuing defective mutant KSI function.
The Y30F mutation rescued the mutant KSI by increasing stability
To investigate whether the Y30F mutation also compensated for the mutant KSI decreased stabilities, the unfolding free-energy changes, ΔGU, were determined (Fig. 3). ΔGUH2O, m and [urea]50% values for WT and mutant KSIs were obtained (Supplementary Table 1). The ΔGUH2O values for the WT, and the Y14F, Y30F, Y55F and Y30F/Y55F mutants were consistent with previous data (Kim et al., 2000). When the Y30F mutation was introduced, Y14F/Y30F mutant stability was increased by 3.2 kcal/mol compared with the Y14F mutant. Similarly, Y14F/Y30F/Y55F mutant stability was increased by 4.3 kcal/mol compared with the Y14F/Y55F mutant. To confirm the stability increase by the additional Y30F mutation, thermal denaturation experiments were also performed. The thermal unfolding curves in the fraction of unfolded forms versus temperature plot are demonstrated in Fig. 4. The Tm values were obtained when the equilibrium constant (KU) =1 (Supplementary Table 2). The Y14F, Y55F and Y14F/Y55F Tm values were 6.2°C, 3.9°C and 7.1°C lower than WT, respectively. Interestingly, as demonstrated in ΔGUH2O, the additional Y30F mutation also increased the Tm values of Y14F, Y55F and Y14F/Y55F by 6.4°C, 5.1°C and 10.0°C, respectively, compared with each corresponding mutant. Those results indicated that the additional Y30F mutation also rescued the Y14F, Y55F and Y14F/Y55F mutants by significantly increasing KSI stability.
Fig. 3.

Urea-induced unfolding equilibrium transition curves of WT and mutant KSIs. The data were obtained with 15 μM protein in buffer containing 20 mM potassium phosphate, pH 7.0, and 1 mM DTT. (A) Fractional unfolding of each protein was calculated from the change at 222 nm after correction for pre- and post-transition baselines. The data points were fitted to Eqn. (4) to obtain the transition curve, which gave us the ΔGUH2O, [urea]50% and m values listed in Supplementary Table 1. (B) The bar graphs demonstrate the ΔGUH2O values for WT and mutant KSIs.
Fig. 4.

Thermal unfolding transitions of WT and mutant KSIs. Thermal unfolding curves were obtained by monitoring CD spectra at 222 nm. Protein melting temperature was determined in 20 mM potassium phosphate, pH 7.0, and 1 mM DTT. (A) The unfolded protein fraction was calculated from the molar ellipticity at 222 nm after correction for the pre- and post-transition baselines. (B) The bar graphs demonstrate the WT and mutant KSI Tm values.
To understand how the Y30F mutation improved mutant KSI stability, we compared WT and mutant KSI structures. Crystallographic analysis of WT KSI demonstrated that the Tyr30 side chain was completely buried (SAS = 0) and pointed towards the densely packed hydrophobic core where it could interact with a number of neighboring hydrophobic residues such as Ile26, Val27, Met29, Ala34, Val36, Ile51 and Met112 (Fig. 5) (Kim et al., 1997). Removing the hydroxyl group from Tyr30 induced local compaction by moving surrounding residues such as Ile26 and Met112 and increased the contact numbers between the aromatic moiety of residue 30 and its surrounding hydrophobic residues in KSI (Supplementary Table 3). Thus, the increased stability of the Y14F/Y30F, Y30F/Y55F and Y14F/Y30F/Y55F mutants compared with the corresponding mutant lacking Y30F might be because of Y30F-induced improvements in hydrophobic interactions within the active site. Consistent with our results, Golovanov et al. demonstrated that the increased inter-residue hydrophobic contact could contribute to protein stability in T4 lysozyme and barnase (Golovanov et al., 2000). Interestingly, Tyr30 of P. putida KSI was homologously substituted with the phenylalanine in C. testosteroni KSI. The C. testosteroni KSI F30Y mutation decreased its stability by 1.2 kcal/mol, suggesting that phenylalanine at position 30 within the hydrophobic core might be more favorable than tyrosine. Improved hydrophobic interactions by second-site mutations were also reported previously in E. coli DNA gyrase and phospholipase A2 (PLA2) (Blance et al., 2000; Sekharudu et al., 1992). The second-site substitution of Thr57 with isoleucine in E. coli DNA gyrase could compensate for the G164V mutation-induced loss of stability by making new hydrophobic interactions with Val156 and Ile140 (Blance et al., 2000). The Y52F/Y73F PLA2 double mutant structural studies demonstrated that the increased phenyl group hydrophobic interactions compensated for the loss of tyrosine residue hydrogen bonds (Sekharudu et al., 1992). As demonstrated in DNA gyrase and PLA2, the improved hydrophobic interactions within the core might contribute to the Y30F mutation-mediated stability restoration.
Fig. 5.

The local environment of Tyr30 in KSI. (A) Tyr30 is fully buried in the active site as determined by solvent accessible surface area calculations (see “Materials and Methods”), and it is shown as a stick model (yellow). (B) The tyrosine residue at position 30 is surrounded by a number of hydrophobic groups including Ile26, Met29, Ala34, Val36, Ile51, Met101, Met109 and the aromatic moieties of both Tyr14 and Tyr55. The figure was generated by PyMOL.
Compensatory effect against the disrupted hydrogen bond network
Many lines of evidence have suggested that hydrogen bond networks are important for catalysis and stability (Polander and Barry, 2012; Ramilo et al., 1999). Therefore, deleterious mutations occurring at the residues that constitute a hydrogen bond network must be compensated for by other second-site mutations near or far away from the primary mutation site. Interestingly, among the residues constituting the hydrogen bond network in KSI, tyrosine to phenylalanine residue mutations compensated for each other. The Y14F mutation-induced defect was rescued by the nearby Y55F mutation. Another neighboring Y30F mutation compensated for both Y14F and Y55F mutants. However, neither the Y30F nor the Y55F mutation rescued the Asp99 to leucine mutation, which is another residue that is involved in the hydrogen bond network (Jang et al., 2004). These tyrosine residues are spatially close to each other as demonstrated in Fig. 1. Spatially close compensatory mutations were also observed in the foot-and-mouth disease virus capsid and in the DNA bacteriophage ΦX174 (Mateo and Mateu, 2007; Poon and Chao, 2005). Analysis of compensatory mutation frequency in DNA bacteriophage ΦX174 revealed that many compensatory mutations occur at spatially close residues (Poon and Chao, 2005). Consequently, our results suggest that KSI might escape from the deleterious effects by disrupting the hydrogen bond network through compensatory effects of tyrosine-to-phenylalanine mutations that constitute the network.
In conclusion, we found that the Y30F mutation could rescue the Y14F, Y55F and Y14F/Y55F mutant catalytic activities and stabilities. The catalytic rescues of these mutants by the additional Y30F mutation originated from active-site geometry restoration as demonstrated by X-ray crystallography. More favorable hydrophobic interactions between the Tyr30 phenyl ring and hydrophobic residues surrounding the active site may contribute to the stabilization of these mutants. Our studies could provide insight into the rescue mechanisms of protein activity and stability at the atomic level.
Supplementary Material
Acknowledgments
This research was supported by a grant from the Next-Generation BioGreen 21 Program (No. PJ009503), Rural Development Administration, Korea and by a grant from the National R & D Program for Cancer Control, Ministry for Health and Welfare, Republic of Korea (1320240). We thank the staff at beam-lines 6B and 6C, Pohang Accelerator Laboratory, Korea, for help with data collection, and Mr. Han Seop Shin for technical assistance. We also thank Dr. Ki Joon Cho (Korea University, Korea) for helpful discussion.
Note:
Supplementary information is available on the Molecules and Cells website (www.molcells.org).
REFERENCES
- Blance SJ, Williams NL, Preston ZA, Bishara J, Smyth MS, Maxwell A. Temperature-sensitive suppressor mutations of the Escherichia coli DNA gyrase B protein. Protein Sci. 2000;9:1035–1037. doi: 10.1110/ps.9.5.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann RK, Yu K, Eby Y, Pavletich NP, Boeke JD. Genetic selection of intragenic suppressor mutations that reverse the effect of common p53 cancer mutations. EMBO J. 1998;17:1847–1859. doi: 10.1093/emboj/17.7.1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Cho HS, Ha NC, Choi G, Kim HJ, Lee D, Oh KS, Kim KS, Lee W, Choi KY, Oh BH. Crystal structure of delta(5)-3-ketosteroid isomerase from Pseudomonas testosteroni in complex with equilenin settles the correct hydrogen bonding scheme for transition state stabilization. J Biol Chem. 1999;274:32863–32868. doi: 10.1074/jbc.274.46.32863. [DOI] [PubMed] [Google Scholar]
- Choi G, Ha NC, Kim SW, Kim DH, Park S, Oh BH, Choi KY. Asp-99 donates a hydrogen bond not to Tyr-14 but to the steroid directly in the catalytic mechanism of Delta 5-3-ketosteroid isomerase from Pseudomonas putida biotype B. Biochemistry. 2000;39:903–909. doi: 10.1021/bi991579k. [DOI] [PubMed] [Google Scholar]
- Choi G, Ha NC, Kim MS, Hong BH, Oh BH, Choi KY. Pseudoreversion of the catalytic activity of Y14F by the additional substitution(s) of tyrosine with phenylalanine in the hydrogen bond network of delta 5-3-ketosteroid isomerase from Pseudomonas putida biotype B. Biochemistry. 2001;40:6828–6835. doi: 10.1021/bi002767+. [DOI] [PubMed] [Google Scholar]
- Golovanov AP, Vergoten G, Arseniev AS. Stabilization of proteins by enhancement of inter-residue hydrophobic contacts: lessons of T4 lysozyme and barnase. J Biomol Struct Dyn. 2000;18:477–491. doi: 10.1080/07391102.2000.10506682. [DOI] [PubMed] [Google Scholar]
- Ha NC, Choi G, Choi KY, Oh BH. Structure and enzymology of Delta5-3-ketosteroid isomerase. Curr Opin Struct Biol. 2001;11:674–678. doi: 10.1016/s0959-440x(01)00268-8. [DOI] [PubMed] [Google Scholar]
- Jang DS, Cha HJ, Cha SS, Hong BH, Ha NC, Lee JY, Oh BH, Lee HS, Choi KY. Structural double-mutant cycle analysis of a hydrogen bond network in ketosteroid isomerase from Pseudomonas putida biotype B. Biochem J. 2004;382:967–973. doi: 10.1042/BJ20031871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joerger AC, Ang HC, Fersht AR. Structural basis for understanding oncogenic p53 mutations and designing rescue drugs. Proc. Natl. Acad. Sci USA. 2006;103:15056–15061. doi: 10.1073/pnas.0607286103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SW, Cha SS, Cho HS, Kim JS, Ha NC, Cho MJ, Joo S, Kim KK, Choi KY, Oh BH. High-resolution crystal structures of delta5-3-ketosteroid isomerase with and without a reaction intermediate analogue. Biochemistry. 1997;36:14030–14036. doi: 10.1021/bi971546+. [DOI] [PubMed] [Google Scholar]
- Kim DH, Jang DS, Nam GH, Choi G, Kim JS, Ha NC, Kim MS, Oh BH, Choi KY. Contribution of the hydrogen-bond network involving a tyrosine triad in the active site to the structure and function of a highly proficient ketosteroid isomerase from Pseudomonas putida biotype B. Biochemistry. 2000;39:4581–4589. doi: 10.1021/bi992119u. [DOI] [PubMed] [Google Scholar]
- Kraut DA, Sigala PA, Fenn TD, Herschlag D. Dissecting the paradoxical effects of hydrogen bond mutations in the ketosteroid isomerase oxyanion hole. Proc. Natl. Acad. Sci USA. 2010;107:1960–1965. doi: 10.1073/pnas.0911168107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Yoon YJ, Jang do S, Kim C, Cha HJ, Hong BH, Choi KY, Lee HC. 15N NMR relaxation studies of Y14F mutant of ketosteroid isomerase: the influence of mutation on backbone mobility. J Biochem. 2008;144:159–166. doi: 10.1093/jb/mvn053. [DOI] [PubMed] [Google Scholar]
- Li YK, Kuliopulos A, Mildvan AS, Talalay P. Environments and mechanistic roles of the tyrosine residues of delta 5-3-ketosteroid isomerase. Biochemistry. 1993;32:1816–1824. doi: 10.1021/bi00058a016. [DOI] [PubMed] [Google Scholar]
- Mateo R, Mateu MG. Deterministic, compensatory mutational events in the capsid of foot-and-mouth disease virus in response to the introduction of mutations found in viruses from persistent infections. J Virol. 2007;81:1879–1887. doi: 10.1128/JVI.01899-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merabet A, Houlleberghs H, Maclagan K, Akanho E, Bui TT, Pagano B, Drake AF, Fraternali F, Nikolova PV. Mutants of the tumour suppressor p53 L1 loop as second-site suppressors for restoring DNA binding to oncogenic p53 mutations: structural and biochemical insights. Biochem J. 2010;427:225–236. doi: 10.1042/BJ20091888. [DOI] [PubMed] [Google Scholar]
- Nagata S, Hyde CC, Miles EW. The alpha subunit of tryptophan synthase. Evidence that aspartic acid 60 is a catalytic residue and that the double alteration of residues 175 and 211 in a second-site revertant restores the proper geometry of the substrate binding site. J Biol Chem. 1989;264:6288–6296. [PubMed] [Google Scholar]
- Noviello CM, Lopez CS, Kukull B, McNett H, Still A, Eccles J, Sloan R, Barklis E. Second-site compensatory mutations of HIV-1 capsid mutations. J Virol. 2011;85:4730–4738. doi: 10.1128/JVI.00099-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. In: Carter CW Jr, Sweet RM, editors. Methods in Enzymology Vo 276, Macromolecular Crystallography, part A. Academic Press; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- Polander BC, Barry BA. A hydrogen-bonding network plays a catalytic role in photosynthetic oxygen evolution. Proc. Natl. Acad. Sci USA. 2012;109:6112–6117. doi: 10.1073/pnas.1200093109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollack RM. Enzymatic mechanisms for catalysis of enolization: ketosteroid isomerase. Bioorg Chem. 2004;32:341–353. doi: 10.1016/j.bioorg.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Poon A, Chao L. The rate of compensatory mutation in the DNA bacteriophage phiX174. Genetics. 2005;170:989–999. doi: 10.1534/genetics.104.039438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poteete AR, Sun DP, Nicholson H, Matthews BW. Second-site revertants of an inactive T4 lysozyme mutant restore activity by restructuring the active site cleft. Biochemistry. 1991;30:1425–1432. doi: 10.1021/bi00219a037. [DOI] [PubMed] [Google Scholar]
- Poteete AR, Rennell D, Bouvier SE, Hardy LW. Alteration of T4 lysozyme structure by second-site reversion of deleterious mutations. Protein Sci. 1997;6:2418–2425. doi: 10.1002/pro.5560061115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramilo CA, Leveque V, Guan Y, Lepock JR, Tainer JA, Nick HS, Silverman DN. Interrupting the hydrogen bond network at the active site of human manganese super-oxide dis-mutase. J Biol Chem. 1999;274:27711–27716. doi: 10.1074/jbc.274.39.27711. [DOI] [PubMed] [Google Scholar]
- Richards FM. Areas, volumes, packing and protein structure. Annu Rev Biophys Bioeng. 1977;6:151–176. doi: 10.1146/annurev.bb.06.060177.001055. [DOI] [PubMed] [Google Scholar]
- Sekharudu C, Ramakrishnan B, Huang B, Jiang RT, Dupureur CM, Tsai MD, Sundaralingam M. Crystal structure of the Y52F/Y73F double mutant of phospholipase A2: increased hydrophobic interactions of the phenyl groups compensate for the disrupted hydrogen bonds of the tyrosines. Protein Sci. 1992;1:1585–1594. doi: 10.1002/pro.5560011206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigala PA, Caaveiro JM, Ringe D, Petsko GA, Herschlag D. Hydrogen bond coupling in the ketosteroid isomerase active site. Biochemistry. 2009;48:6932–6939. doi: 10.1021/bi900713j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suad O, Rozenberg H, Brosh R, Diskin-Posner Y, Kessler N, Shimon LJ, Frolow F, Liran A, Rotter V, Shakked Z. Structural basis of restoring sequence-specific DNA binding and transactivation to mutant p53 by suppressor mutations. J Mol Biol. 2009;385:249–265. doi: 10.1016/j.jmb.2008.10.063. [DOI] [PubMed] [Google Scholar]
- Wilson KP, Malcolm BA, Matthews BW. Structural and thermodynamic analysis of compensating mutations within the core of chicken egg white lysozyme. J Biol Chem. 1992;267:10842–10849. [PubMed] [Google Scholar]
- Wu ZR, Ebrahimian S, Zawrotny ME, Thornburg LD, Perez-Alvarado GC, Brothers P, Pollack RM, Summers MF. Solution structure of 3-oxo-delta5-steroid isomerase. Science. 1997;276:415–418. doi: 10.1126/science.276.5311.415. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.