

Abstract
Insights gained from characterizing MHC-peptide-TCR interactions have held the promise that directed structural modifications can have predictable functional consequences. The ability to manipulate T cell reactivity synthetically or through genetic engineering might thus be translated into new therapies for common diseases such as cancer and autoimmune disorders. In the current study, we determined the crystal structure of HLA-DR4 in complex with the non-mutated dominant gp100 epitope, gp10044–59, associated with many melanomas. Altered peptide ligands (APLs) were designed to enhance MHC binding and hence T cell recognition of gp100 in HLA-DR4+ melanoma patients. Increased MHC binding of several APLs was observed, validating this approach biochemically. Nevertheless, heterogeneous preferences of CD4+ T cells from several HLA-DR4+ melanoma patients for different gp100 APLs suggested highly variable TCR usage, even among six patients who had been vaccinated against the wild type gp100 peptide. This heterogeneity prevented the selection of an APL candidate for developing an improved generic gp100 vaccine in melanoma. Our results are consistent with the idea that even conservative changes in MHC anchor residues may result in subtle, yet crucial, effects on peptide contacts with the TCR or on peptide dynamics, such that alterations intended to enhance immunogenicity may be unpredictable or counterproductive. They also underscore a critical knowledge gap that needs to be filled, before structural and in vitro observations can be used reliably to devise new immunotherapies for cancer and other disorders.
INTRODUCTION
Melanoma is an aggressive form of skin cancer which is curable in its early stages but carries a poor prognosis following distant organ metastasis (1). It is also highly immunogenic, as evidenced by endogenous anti-melanoma T and B cell responses and the susceptibility of melanoma to drugs with a purely immunological mode of action, such as interleukin-2 (2), anti-CTLA-4 (3), anti-PD-1 (4) and anti-PD-L1 (5). Efficient vaccination with tumor specific antigens can re-direct the anti-tumor immune response and provide synergistic treatment effects when combined with systemic immune-enhancing agents (6–8). Thus, there is a need to develop optimal cancer vaccines and tumor antigen-specific detection methods for monitoring treatment outcomes in vitro. Rational chemical modification of tumor specific peptide antigens to increase their immunogenicity, based on structural models, may facilitate this approach.
Gp100, a melanocyte lineage-specific transmembrane glycoprotein, is expressed in most melanomas and is involved in a multiple-step process of pigment production (9). Gp100 has been a widely-used target for melanoma immunotherapy since the demonstration that tumor infiltrating lymphocytes and circulating T cells from melanoma patients commonly recognize this antigen (10, 11). Despite the fact that the most gp100-directed melanoma therapies have focused on stimulating CD8+ T cell responses, CD4+ T cells play a central role in inducing and maintaining tumor specific CD8+ T cells (12). Devising immunotherapies which can efficiently raise specific CD4+ T cell responses is therefore an important goal.
A gp100 MHC II restricted peptide, gp10044–59, was identified from HLA-DRB1*0401 (hereafter HLA-DR4) positive melanoma cell lines (13) and was subsequently validated as a dominant epitope in a transgenic animal model (14). This peptide can generate melanoma-specific CD4+ T cells from the peripheral blood of melanoma patients following repetitive in vitro stimulation (14, 15). Nevertheless, in a clinical trial using gp10044–59 as a vaccine, no enhancement of gp100-specific reactivity was detected in the peripheral blood of patients following vaccination, dampening enthusiasm for its therapeutic potential (16).
Because gp10044–59 is a non-mutated self antigen with intermediate binding affinity for HLA-DR4 (15), we hypothesized that altered peptide ligands (APLs) with single amino acid substitutions could be designed to confer higher MHC binding affinity and hence improved immunogenicity. Such APLs derived from gp100 MHC I-restricted epitopes have been employed as melanoma vaccines (17–20). Whereas unmodified HLA-A2-restricted gp100209–217 and gp100280–288 peptides induced melanoma-reactive CTLs from limited numbers of melanoma patients in vitro, and numerous re-stimulations were required, the APLs gp100209–217(210M) and gp100280–288(288V) with enhanced MHC affinity showed superior immunogenicity in vitro and in vivo (17). Similarly, in mice, a variant of gp100 that bound H-2Db with increased affinity induced high frequencies of melanoma-specific CTLs in the endogenous CD8+ repertoire (21).
APLs based on MHC II-restricted epitopes have rarely been explored, since these peptides are heterogeneous in length and more degenerate in MHC binding specificity than class I-restricted peptides (22), making it difficult to precisely define MHC II-specific peptide binding motifs. However, combined information from MHC II-peptide crystal structures, ligand sequencing and binding affinity determinations has enriched our knowledge of the general chemical properties permitting optimal peptide binding to HLA-DR4. A dominant large hydrophobic or aromatic residue in the P1 binding position, a hydroxylated residue at P6, and a hydrophobic or polar residue at P9 appear to be favored (22–25).
In order to obtain precise information about the binding characteristics of gp10044–59 to HLA-DR4, as a basis for designing optimal melanoma vaccines and immunomonitoring tools, we determined the crystal structure of this MHC-peptide complex. APLs based on structural data were compared to the wild type peptide for their ability to detect gp100-specific reactivity in melanoma patients vaccinated against the wild type peptide, or to raise melanoma-specific T cells from pre-vaccination peripheral blood mononuclear cells (PBMCs).
MATERIALS AND METHODS
Isolation and sequencing of native gp100 peptides complexed to HLA-DR4
Peptide-HLA-DR complexes were isolated from cultured 1102-mel melanoma cells on an anti-HLA-DR affinity column and peptides were eluted as described (26). Peptides were then analyzed by nanoflow HPLC-microelectrospray ionization coupled to either a hybrid linear quadrupole ion trap-Fourier-transform ion cyclotron resonance (LTQ-FT) mass spectrometer or an LTQ-Orbitrap mass spectrometer (Thermo Fisher Scientific) modified to perform electron transfer dissociation (ETD). Data were acquired as previously described (27). In brief, a precolumn loaded with 1e7 cell equivalents of MHC peptides was connected with polytetrafluoroethylene tubing (0.06-inch o.d. and 0.012-inch i.d.; Zeus Industrial Products) to the end of an analytical HPLC column (360-μm o.d. and 50 μm i.d.) containing 7 cm of C18 reverse-phase packing material (5-μm particles; YMC). Peptides were eluted into the mass spectrometer at a flow rate of 60 nL/min with a gradient: A = 0.1 M acetic acid (Sigma-Aldrich) in H2O; B = 70% acetonitrile (Malinckrodt) and 0.1M acetic acid in H2O; 0–60% B in 40 min, 60–100% B in 5 min. Parameters used to acquire ETD/MS/MS spectra in a data-dependent mode on the modified LTQ instrument have been described previously (28).
Sequence analysis was performed by searching MS/MS spectral data against a database consisting only of the gp100 protein using the Open Mass Spectrometry Search Algorithm (OMSSA) software to generate a list of candidate spectra (29). A precursor mass tolerance of ± .01 Da was used, MS/MS spectra were searched using a monoisotopic fragment ion mass tolerance of ± 0.5 Da. Data were searched allowing variable modifications for phosphorylation of serine, threonine and tyrosine residues and oxidation of methionine, with a total of 1024 variable modifications per peptide being allowed. Other parameters utilized were; peptide charge range from +2 to +5, +2 charge state products allowed for peptides of charge +3 and above, peptide size range 4–25 a.a. with no enzyme restriction. Peptide sequence assignments were validated by manual interpretation of the corresponding ETD and/or CAD MS/MS spectra. Approximate copy numbers per melanoma cell for each peptide were determined by comparing peak areas of the observed parent ions to that of angiotensin I and vasoactive intestinal peptide (DRVYIHPFHL HSDAVFTDNYTR, 100 fmol; Sigma-Aldrich) spiked into the sample mixture.
Synthesis of HLA-DR4-restricted peptides
Synthetic peptides used in this study were made with Fmoc chemistry, isolated by HPLC to ≥90% purity, and validated with mass spectrometry (Global Peptides or Pi Proteomics). Peptides included: gp100 overlapping peptides (Table I); wild type (WT) gp10044–59 (WTWNRQLYPEWTEAQRLD); gp10044–59 APLs (Table II); influenza hemagglutinin (HA)307–319 (PKYVKQNTLKLAT); and CDC27758–772 (MNFSWAMDLDPKGAN) (30).
Table I.
Naturally processed HLA-DR4-restricted overlapping gp100 peptides.
| Residues | Copy number/cell | Amino acid sequence |
|---|---|---|
| 44–59 | 4500 | WNRQLYPEWTEAQRLD |
| 44–57 | 2000 | W------------R |
| 45–57 | 900 | N-----------R |
| 45–59 | 500 | N-------------D |
| 46–57 | 300 | R----------R |
| 46–59 | 800 | R------------D |
| 40–57 | 100 | RTKAW------------R |
All peptides have the same core binding region (in bold) as wild type gp10044–59.
Table II.
gp10044–59 APLs and their affinity for HLA-DR4
| gp10044–59 and APLs | Amino acid sequence | Substituted position | IC50 (nM) |
|---|---|---|---|
| WT | WNRQLYPEWTEAQRLD | NA | 627 |
| L48F | ----F----------- | P1* | 280 |
| Y49M | -----M---------- | P2 | 521 |
| P50A | ------A--------- | P3 | 189 |
| E51A | -------A-------- | P4* | 199 |
| E51Q | -------Q-------- | P4* | 200 |
| W52A | --------A------- | P5 | 372 |
| T53V | ---------V------ | P6* | 489 |
| E54L | ----------L----- | P7* | 65 |
| E54T | ----------T----- | P7* | 104 |
| A55G | -----------G---- | P8 | 553 |
| Q56A | ------------A--- | P9* | 276 |
| Q56I | ------------I--- | P9* | 926 |
Indicates MHC anchor residues according to crystal structure. IC50, 50% inhibitory concentration. WT, wild type gp100 peptide sequence. NA, not applicable.
MHC-peptide binding affinity assay
Competition assays to quantitatively measure the binding affinities of native and altered gp100 peptides for purified HLA-DR4 were performed essentially as detailed elsewhere (31–33). Briefly, purified HLA-DR4 molecules (5–500 nM) were incubated with various concentrations of unlabeled peptide inhibitors and 0.1–1 nM 125I-radiolabeled probe peptide (human MBP85–101, sequence PVVHFFKNIVTPRTPPY) for 48h in PBS containing 0.05–0.15% Nonidet P-40 (NP40) in the presence of a protease inhibitor cocktail. MHC binding of the radiolabeled peptide was determined by capturing MHC/peptide complexes on L243 (anti-HLA-DRA) antibody-coated Lumitrac 600 plates (Greiner Bio-one, Frickenhausen, Germany), and measuring bound cpm using the TopCount (Packard Instrument Co., Meriden, CT) microscintillation counter. Peptides were typically tested at six different concentrations covering a 100,000-fold dose range, in three or more independent assays. Under the conditions utilized, where [label]<[MHC] and IC50 ≥ [MHC], the measured IC50 values are reasonable approximations of the KD values (34, 35).
MHC-peptide protein preparation, crystallization, data collection, and structure determination
The gp10044–59–HLA-DR4 complex was assembled in two steps in order to maximize the yield of recombinant protein. First, HLA-DR4 bearing CLIP87–101 peptide (PVSKMRMATPLIMQA) was prepared by in vitro folding from bacterial inclusion bodies. Second, gp10044–59 was loaded into CLIP–HLA-DR4 using the peptide-exchange catalyst HLA-DM. Briefly, the extracellular portions of the HLA-DR4 α and β chains (residues 1–181 and 1–192, respectively) were expressed separately as inclusion bodies in Escherichia coli BL21 (DE3) cells (Novagen). Inclusion bodies were dissolved in 8 M urea, 50 mM Tris-HCl (pH 8.0), and 10 mM DTT, followed by purification on a Poros HQ20 anion exchange column (PerSeptive Biosystems) in 50 mM Tris-HCl, 8 M urea, and 1 mM DTT at pH 8.0 (DRα) or pH 8.5 (DRβ), using a linear NaCl gradient (36). For in vitro folding, the purified subunits were diluted to a final concentration of 40 mg/L each in a folding solution containing 50 mM Tris-HCl, 30% (w/v) glycerol, 0.5 mM EDTA, 3 mM reduced glutathione, and 0.9 mM oxidized glutathione (pH 8.0). CLIP peptide (GenScript) was added to a final concentration of 5 μM, and the folding mixture was kept for two weeks at 4 °C. The final folding solution was concentrated and dialyzed against 50 mM Mes (pH 6.0). Purification was carried out with sequential Superdex S-200 and Mono Q FPLC columns (GE Healthcare). The CLIP–HLA-DR4 complex was concentrated to 0.8 mg/ml and loaded with gp10044–59 by overnight incubation at 37 °C in 100 mM sodium citrate-HCl (pH 5.8) containing 200 μM gp10044–59 and 0.2 mg/ml soluble HLA-DM.
The gp10044–59–HLA-DR4 complex was crystallized at room temperature in hanging drops by mixing equal volumes of the protein solution at 5 mg/ml and a reservoir solution of 14% (w/v) polyethylene glycol (PEG) 8000, 0.2 M magnesium acetate, and 0.1 M HEPES (pH 7.0). For data collection, crystals were transferred to a cryoprotectant solution [mother liquor containing 30% (w/v) PEG 8000], prior to flash-cooling in a nitrogen stream. X-ray diffraction data to 2.5 Å resolution were recorded at beamline X29 of the Brookhaven National Synchrotron Light Source with an ADSC Quantum-315 CCD detector. All data were indexed, integrated, and scaled with the program HKL 2000 (37). Data collection statistics are shown in Table III.
Table III.
Data collection and refinement statistics
| gp10044–59–HLA-DR4 | |
|---|---|
| Data collection statistics | |
| Space group | P21212 |
| Unit cell (Å, °) | a = 90.3, b = 117.6, c = 41.9 |
| Resolution (Å) | 30.0–2.50 |
| Observations | 144,536 |
| Unique reflections | 14,952 |
| Completeness (%)a | 93.0 (71.2) |
| Mean I/σ(I)a | 37.6 (6.2) |
| Rsym (%)a,b | 6.1 (24.4) |
| Refinement statistics | |
| Resolution range (Å) | 30.0–2.50 |
| Rwork (%)c | 26.9 |
| Rfree (%)c | 29.8 |
| Protein atoms | 3,133 |
| R.m.s. deviations from ideality | |
| Bond lengths (Å) | 0.009 |
| Bond angles (°) | 1.47 |
| Ramachandran plot statistics | |
| Most favored (%) | 86.2 |
| Additionally allowed (%) | 12.6 |
| Generously allowed (%) | 0.9 |
| Disallowed | 0.3 |
Values in parentheses are statistics for the highest resolution shell (2.50–2.54 Å).
Rsym = Σ|Ij – <I>|/ΣIj, where Ij is the intensity of an individual reflection and <I> is the average intensity of that reflection.
Rwork = Σ||Fo| – |Fc||/Σ|Fo|, where Fc is the calculated structure factor. Rfree is as for Rwork but calculated for a randomly selected 10.0% of reflections not included in the refinement.
The structure was solved by molecular replacement with the program Phaser (38) using HLA-DR4 (Protein Data Bank accession code 1D5Z) (39) as a search model. Refinement was carried out using CNS1.2 (40) including iterative cycles of simulated annealing, positional refinement and B factor refinement, interspersed with model rebuilding into δA-weighted Fo– Fc and 2Fo– Fc electron density maps using XtalView (41). Refinement statistics are summarized in Table III. Stereochemical parameters were evaluated with PROCHECK (42). Atomic coordinates and structure factors have been deposited in the Protein Data Bank (accession code 4IS6 http://www.rcsb.org/pdb/home/home.do).
Patients
Patients with unresectable stage IV melanoma who expressed HLA-DRB1*0401 were treated with a synthetic gp100 peptide vaccine. Patients were vaccinated subcutaneously with 5 mg of gp10044–59 peptide emulsified in incomplete Freund’s adjuvant every 3 weeks for a series of 4 inoculations (1 treatment cycle), as described (16). PBMCs were collected by leukapheresis before treatment and 3 weeks after every two vaccinations for in vitro immunologic monitoring studies. Patients underwent radiologic restaging after each treatment cycle. All patients were treated in the Surgery Branch of the National Cancer Institute, National Institutes of Health in Bethesda, Maryland on a clinical trial approved by the Institutional Review Board of the National Cancer Institute, after signing informed consent (16). In addition to DRB1*0401, HLA-DR alleles expressed by the six patients reported here included the following: DRB1*0404, patient #6; DRB1*0701, patients #1, 3 and 5; DRB1*1104, patient #2; DRB1*1501, patient #4; DRB3*02, patient #2; DRB4*01, all patients; DRB5*01, patient #4.
gp100-specific CD4+ T cell clone 100.1.G7
The gp10044–59–specific CD4+ T cell clone 100.1.G7 (G7 clone hereafter) was raised by repetitive in vitro peptide stimulation of peripheral blood lymphocytes (PBLs) from a HLA-DR4+ melanoma patient whose tumor expressed gp100 protein. Briefly, T cell cultures were initiated in 24-well plates in the presence of IL-2. GM-CSF (200 U/ml) and IL-4 (100 U/ml) were added on day 0 to generate dendritic cells as APCs, along with 25 μM peptide. T cells were restimulated every 10–14 days with irradiated peptide-pulsed autologous PBMCs or HLA-DR4+ EBV-transformed B cells and were cloned after 28 days by limiting dilution culture, as previously described (26). Long-term CD4+ T cell clones were cultured in RPMI 1640 medium supplemented with 10% heat inactivated human AB serum, IL-2, −7, and −15, and 20% conditioned medium from lymphokine-activated killer cell cultures.
T cell functional assays: ELISAs
Peptide-specific CD4+ T cells at 1e5–3e5 cells per well were co-cultured overnight in flat-bottom 96-well plates with 1e5 peptide-pulsed HLA-DR4+ allogeneic EBV-B cells or autologous PBMCs. Culture supernatants were harvested for measurement of GM-CSF and IFN-γ secretion by T cells, using ELISA with commercially available kits (R&D Systems).
In vitro stimulation (IVS) and ELISPOT assay
Cryopreserved PBMCs from vaccinated patients were thawed and suspended in complete medium (CM, RPMI 1640 plus 10% heat-inactivated human AB serum, 2 mM glutamine, 10 mM HEPES buffer, and antibiotics) at 1e6 cells/ml in 24-well plates with 20 μM peptide, 200 U/ml GM-CSF and 100 U/ml IL-4. Parallel cultures were grown with IL-7 and IL-15 (25 ng/ml each) instead of GM-CSF and IL-4. Cells were incubated at 37°C. After 3 days, IL-2 was added at a final concentration of 10 IU/ml. At 9–12 days, some cells were harvested for ELISPOT assay and the remaining cells were restimulated with peptide-pulsed irradiated autologous PBMCs and cultured in CM containing 150 IU/ml IL-2. At day 20, cells were harvested again for ELISPOT assay. One day prior to ELISPOT assays, approximately half of the culture volume was replaced with fresh CM (without IL-2).
ELISPOT assays were conducted in MultiScreen-IP Filter Plates coated overnight at 4°C with 50 μl/well of 20 μg/ml mouse anti-human IFN-γ antibody (clone 1D1K, Mabtech). On the following day, the plates were washed and blocked with AB medium for 2 hours at 37 °C. For some assays, fresh cryopreserved PBMCs were thawed into ELISPOT media (EM, RPMI with 10% heat-inactivated human AB serum, 2 mM glutamine, 10 mM HEPES buffer) at 4e6 cells/ml. After 2 hours incubation at 37°C and 5% CO2, 2e5 PBMCs were plated directly to each IFN-γ antibody-coated well. For ELISPOT assays using peptide-stimulated T cell cultures, 1e5 T cells were plated into each IFN-γ antibody-coated well. Then, 1e5 irradiated (5000 rad) autologous PBMCs were added to T cells in each well as APCs. Gp100 peptides were added at 20 μM in 100 μl total volume. An unrelated HLA-DR4-restricted peptide, CDC27758–772, was used as a negative control. PMA-ionomycin stimulation provided a positive control. After overnight incubation at 37°C, cells were discarded and the plates were washed with PBS/0.05% Tween 20 followed by PBS. 100 μl biotinylated mouse anti-human IFN-γ mAb (clone 7-B6-1, Mabtech) diluted at 2 μg/ml in PBS with 0.5% BSA was added to each well and incubated at 37°C for 2 hours. The plates were washed and developed with avidin-peroxidase-complex (Vector Laboratories) and stained with AEC substrate (Sigma). Spots were counted by KS ELISPOT automated reader system (Carl Zeiss). The number of spots was averaged from triplicate wells. Peptide-specific T cells were defined as showing ≥ 20 spots per 1e6 fresh PBMCs or cultured T cells, and ≥ twice the numbers of spots observed in the negative control wells.
RESULTS
T cell recognition of native DR4-restricted gp100 peptides
The DR4-restricted peptide gp10044–59 was originally described by Halder et al. (13) as a dominant peptide displayed on the surface of melanoma cells. The immunogenicity of this peptide was confirmed in DR4-transgenic mice and in human in vitro studies (14, 15), leading to its clinical testing as a melanoma vaccine. Although many MHC II-restricted peptides occur as nested peptide sets, naturally occurring sequences overlapping gp10044–59 have not been previously identified. In order to identify native nested gp100 peptides with potentially enhanced immunogenicity, we searched peptides eluted from DR4 molecules displayed on cultured melanoma cells. A set of seven nested gp100 peptides spanning residues 40–59 was identified. Peptide abundance ranged from 100 to 4500 copies/cell (Table I). We assessed the recognition of these peptides by measuring specific cytokine secretion from the CD4+ gp100-specific DR4-restricted G7 clone (Figure 1). As shown in Figure 2, recognition of gp10044–59 exceeded all other peptides. Truncated peptides lacking the N-terminal residues Trp44 and Asn45, outside the peptide’s MHC core binding sequence, were not well recognized by T cells; this is reminiscent of our findings with another MHC II-restricted melanoma-associated peptide, phospho-MART-1, and highlights the important effects that N-terminal residues outside the binding groove may have on non-mutated (self) epitope recognition (43). Thus, we were unable to identify a naturally processed overlapping peptide that was better recognized by the G7 clone than gp10044–59. Furthermore, this peptide was the most abundantly expressed member of the nested peptide set. Therefore, all subsequent experiments were based on the gp10044–59 native peptide sequence.
Figure 1.
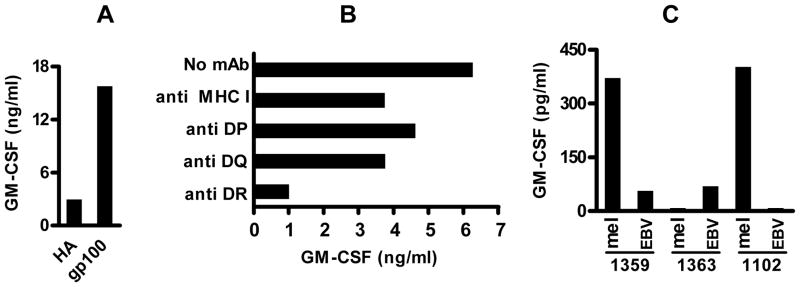
The CD4+ T cell clone 100.1.G7 (“G7”) is gp10044–59-specific and HLA-DR4-restricted. (A) G7 T cells were cultured overnight with allogeneic HLA-DR4+ EBV-B cells pre-pulsed with gp10044–59 or HA307–319 peptides (25 μM). GM-CSF secreted by T cells into culture supernatants was measured by ELISA. Results representative of 4 experiments. (B) Recognition of gp10044–59 peptide-pulsed EBV-B cells by G7 is specifically blocked by an anti-HLA-DR mAb. G7 cells are homozygous for HLA-DR4. (C) Specific recognition of HLA-DR4+ melanoma cells by G7 T cells, which were co-incubated with HLA-DR4+ (1359 and 1102) or HLA-DR4− (1363) EBV-B or melanoma cells overnight. HA, influenza hemagglutinin307–319, an HLA-DR4-restricted peptide that was used as a negative control. EBV, Epstein-Barr Virus. mAb, monoclonal antibody. Mel, melanoma cell lines.
Figure 2.
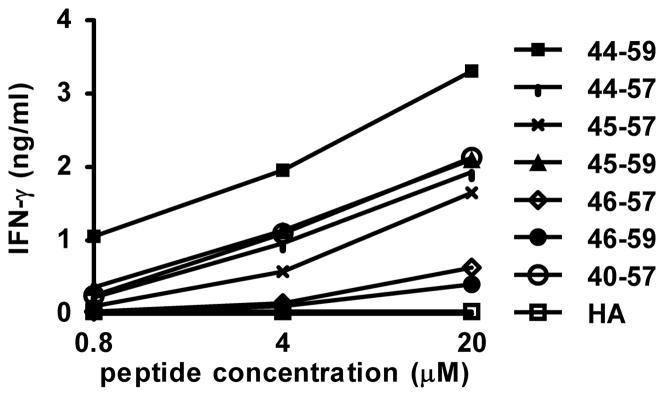
G7 clone recognition of gp10044–59 exceeds recognition of other members of a nested set of naturally processed gp100 peptides. Antigen presenting cells were pulsed with gp10044–59 or nested peptides at the indicated concentrations overnight. G7 cells were added and cultured overnight. Supernatants were harvested and tested for IFN-γ secretion by ELISA. Similar results were obtained for GM-CSF secretion (not shown). HA, influenza hemagglutinin307–319, an HLA-DR4-restricted peptide that was used as a negative control.
Structure of gp10044–59 bound to HLA-DR4
The crystal structure of gp10044–59 complexed to HLA-DR4 was determined to 2.5 Å resolution (Table III). Continuous and unambiguous electron density was observed for gp10044–59 residues Arg46 to Leu58; however, N-terminal residues Trp44 and Asn45 and C-terminal residue Asp59 were not defined in the electron density map (Figure 3A), despite their importance for recognition by clone G7 (Figure 2).
Figure 3.

Structure of the gp10044–59–HLA-DR4 complex. (A) Electron density map for the bound gp10044–59 peptide. The 2Fo – Fc map at 2.5 Å resolution is contoured at 1δ. The peptide is drawn in stick representation with carbon atoms in yellow, oxygen atoms in red, and nitrogen atoms in blue. (B) Top view of the gp10044–59–HLA-DR4 complex, looking down on the peptide-binding groove. The molecular surface of HLA-DR4 is cyan (MHC α-chain) and green (MHC β-chain). (C) Conformation of high- and low-affinity peptides bound to HLA-DR4. The conformation of gp10044–59 (yellow) was compared with those of CII1168–1180 (green), HA307–319 (blue), and MBP111–129 (pink) by superposing the α1β1 domains of HLA-DR4 in the gp10044–59–HLA-DR4, CII1168–1180–HLA-DR4 (Protein Data Bank accession code 2SEB) (47), HA307–319–HLA-DR4 (1J8H) (45), and MBP111–129 –HLA-DR4 (3O6F) (48) complexes. The peptides are viewed from the side of the β1 helix; gp10044–59, CII1168–1180, and HA307–319 bind HLA-DR4 with higher affinity than MBP111–129.
In the structure, the primary anchor residues for gp10044–59 bound to HLA-DR4 are Leu48 (P1) and Thr53 (P6); the secondary anchor residues are Glu51 (P4), Glu54 (P7) and Gln56 (P9). This result is consistent with the sequence motif for HLA-DR4 deduced from phage display and synthetic peptide libraries (24, 33, 44). Leu48 fulfills the requirement for a large nonpolar residue at P1 for efficient binding to HLA-DR4. Thr53 at P6 is a suitable primary anchor as well. By contrast, none of the secondary anchors (P4 Glu51, P7 Glu54 and P9 Gln56) conform to the optimal binding motif for HLA-DR4. Thus, the P4 pocket of HLA-DR4, which is lined by β-chain residues Phe26, Lys71, Ala74 and Tyr78, is hydrophobic and prefers nonpolar residues (24, 33, 44). In the gp10044–59–HLA-DR4 complex, the charged carboxy group of the P4 Glu51 side chain points toward the surface of HLA-DR4 instead of into the P4 pocket (Figure 3B). The P7 pocket of HLA-DR4, formed by β-chain residues Try47, Trp61, Leu67 and Lys71, is likewise hydrophobic, such that the side chain of P7 Glu54 adopts a conformation very similar to that of P4 Glu51. The side chain of P9 Gln56 projects deep into the primarily nonpolar P9 pocket of HLA-DR4 (Figure 4 left panel), which is formed by α-chain residues Asn69, Ile72 and Met73 and β-chain residues Tyr37, Asp57 and Trp61. P9 Gln56 makes extensive van der Waals contacts with these residues, in addition to two side-chain–side-chain hydrogen bonds: P9 Glu56 Oε1–Oη DR4 Tyr37β and P9 Glu56 Nε2–Oδ1 DR4 Asp57β (Figure 4 right panel).
Figure 4.
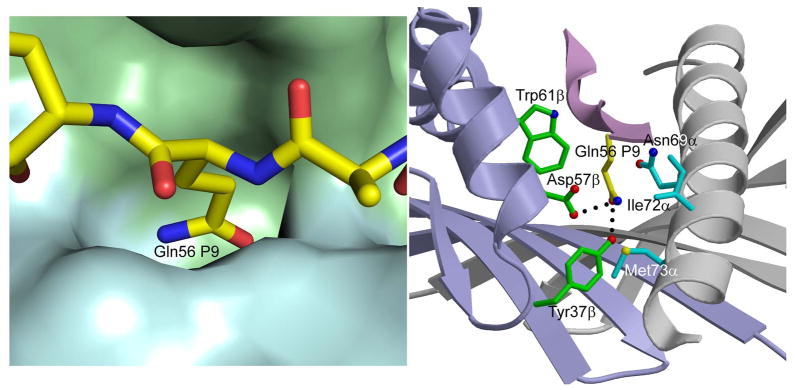
Interaction of P9 Gln56 with HLA-DR4. Left panel, molecular surface of HLA-DR4 (MHC α-chain, cyan; MHC β-chain, green) showing the P9 pocket that accommodates the side chain of P6 Gln56. Right panel, contact residues of HLA-DR4 are drawn and labeled. Hydrogen bonds are indicated by broken black lines.
The conformation of gp10044–59, which binds HLA-DR4 with intermediate affinity (15), was directly compared with those of three other peptides that bind HLA-DR4: 1) a self peptide from collagen II (CII1168–1180) which binds HLA-DR4 with relatively high affinity (24, 44); 2) a foreign peptide from influenza virus hemagglutinin (HA307–319) that also binds HLA-DR4 with high affinity (45); and 3) a self peptide from myelin basic protein (MBP111–129), which binds weakly to HLA-DR4 (46). The main chain of gp10044–59 superposes very closely onto those of CII1168–1180 and HA307–319 in complex with HLA-DR4 (45, 47), from residues P2 to P9 (Figure 3C). The C-terminus of gp10044–59 (residues P10 and P11), which is positioned ~1 Å higher than that of CII1168–1180 or HA307–319, is located outside the peptide-binding groove. However, all three peptides sit deeply in the binding groove. By contrast, the low-affinity MBP111–129 peptide diverges from gp10044–59, CII1168–1180, and HA307–319 at anchor residues P6 and P7 (Figure 3C), due to a longer side chain at P6 (MBP111–129: Gln; gp10044–59: Thr; CII1168–1180: Ala; HA307–319: Thr) (48). The consequent elevation of MBP111–129 results in fewer contacts to HLA-DR4 compared to gp10044–59, CII1168–1180, or HA307–319.
APLs and their affinity for DR4
According to the crystal structure of the DR4-gp10044–59 complex as well as published HLA-DR4 peptide binding motifs (22–24), we designed several altered peptide ligands (APLs) with substituted MHC anchor residues (Table II). The amino acid substitutions L48F (P1), E51Q and E51A (P4), E54L and E54T (P7), and Q56A (P9) are located at MHC anchor residues revealed by crystal structure and were designed to enhance MHC-peptide affinity. Indeed, these anchor-modified APLs were found to have 2–10 fold higher affinities for DR4 than the WT peptide. Y49M (P2), P50A (P3), W52A (P5) and A55G (P8), in which the substituted amino acids are positioned at potential TCR contact residues, had higher or similar MHC-binding affinities compared to WT peptide. As a negative control, Q56I was designed to reduce binding affinity to DR4 due to the large isoleucine residue in P9, incompatible with the corresponding shallow binding groove in HLA-DR4; the MHC binding affinity of this modified peptide was approximately 50% lower than WT (Table II).
Recognition of gp10044–59 APLs by gp10044–59 -specific CD4+ T cell clone G7
T cell recognition of gp10044–59 APLs was first characterized by cytokine secretion from the G7 clone. Peptides modified at putative TCR contact residues -- Y49M (P2), P50A (P3) and W52A (P5) -- were not recognized by the G7 clone, confirming the importance of these residues for T cell specificity (Figure 5). In addition, a replacement of threonine by valine in anchor position 6 also abrogated T cell recognition (not shown), suggesting that P6 has high stringency for a hydroxylated residue. However, G7 secreted 2 to 3 times more IFN-γ and GM-CSF in response to Q56A than to WT peptide (Figure 5), nominating the Q56A APL for further study. Other APLs showed equivalent or lower recognition by G7, compared to WT peptide (Figure 5). The ability of G7 to cross-react with gp10044–59 and Q56A APL is easily understood in structural terms, since these peptides differ by only a single residue, at anchor position P9. The Gln56 side chain of gp10044–59 (and presumably the Ala56 side chain of Q56A APL) is sequestered in the P9 pocket of HLA-DR4 and is therefore not exposed to TCR G7 (Figure 3B).
Figure 5.
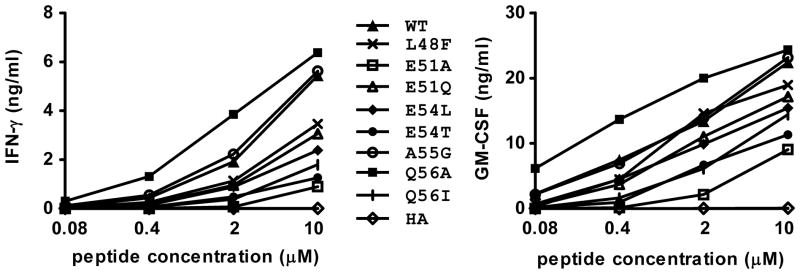
Q56A substitution at the P9 anchor residue of gp10044–59 confers enhanced recognition by the G7 clone. HLA-DR4+ 1102-EBV cells were pulsed with gp100 peptides at the indicated concentrations for 24 hrs. Then G7 cells were added and cultured overnight. Supernatants were harvested and tested for IFN-γ (left) and GM-CSF (right) secretion by ELISA. T cell recognition of the Q56A was enhanced approximately 5-fold compared to WT gp100 peptide. APLs not shown in this figure did not stimulate detectable cytokine secretion, including Y49M (P2 substitution), P50A (P3), W52A (P5) and T53V (P6). Results representative of 3 experiments. HA, influenza hemagglutinin307–319, an HLA-DR4-restricted peptide that was used as a negative control. WT, wild type gp10044–59.
Comparison of gp100 WT peptide versus the Q56A APL in detecting gp100-specific immunity in PBLs from vaccinated patients
In a prior study of melanoma patients receiving the gp100 WT vaccine, no detectable immunity against the vaccinating peptide was detected in post-treatment PBMCs, using a standard IFN-γ ELISPOT assay. Since the Q56A APL had higher affinity for DR4 and provoked greater recognition by the G7 clone, we assessed Q56A for its ability to detect gp100-specific reactivity in pre- and post-treatment PBLs from six patients previously reported in this clinical trial (16) (Figure 6). Uncultured PBLs were assessed for reactivity against WT peptide or the Q56A APL, using an IFN-γ ELISPOT assay. In addition, PBLs were stimulated in vitro (IVS) for 10–20 days with WT or Q56A peptide to amplify gp10044–59 specific T cells, prior to ELISPOT. With IVS, the CD4+ T cell population increased to 60%-90% of cultured T cells (data not shown). We found reproducible evidence that new specific anti-gp100 responses developed after vaccination in 2 of 6 patients, using uncultured or 10–20 day IVS T cells (Patients #1 and 2). In Patient #3, IVS cultures showed greater gp100-specific activity after 4 compared to 2 vaccines; pre-treatment T cells did not proliferate in IVS culture and thus could not be assessed. There was pre-existing immunity against gp10044–59 which persisted after vaccination in uncultured PBMCs and IVS cultures, in Patient #4. Finally, two patients did not manifest evidence of a pre-existing or vaccination-induced anti-gp100 response (data not shown). Among the 4 patients with anti-gp100 immunity, only Patient #3 had improved detection of responses using the Q56A APL, while also recognizing WT peptide. IVS cultures raised against WT or Q56A peptides showed equivalent recognition of the reciprocal peptide in patients #1, 2 and 4 (Figure 6). In summary, IVS-enhanced ELISPOT techniques enabled the detection of gp100-specific immunity in 4 of 6 melanoma patients (in 3 of 4 patients, immunity increased following vaccination). Unlike results with the G7 T cell clone, the Q56A APL was not consistently superior to WT peptide in revealing these responses.
Figure 6.

Detection of gp100 specific immunity in 4 vaccinated melanoma patients. Reactivity against gp10044–59 in uncultured PBMCs or in 10–20 day IVS cultures was determined by IFN-γ ELISPOT assays. 20 μM WT or Q56A peptide was used to stimulate CD4+ T cells in both IVS cultures and ELISPOT assays. Data from uncultured PBMCs from patient #3 are not shown, because all values were < 20 spots/106 cells. CDC27, an unrelated HLA-DR4-restricted control peptide. IVS, in vitro stimulation. Pre, pre-vaccine. P2, P4, P8, PBMCs collected from patients post 2, 4 or 8 vaccinations. SFC, spot forming colonies, mean± SEM. WT, wild type gp10044–59. * P<0.02, one-tailed t test compared to CDC27.
T cells raised against WT peptide prefer a variety of APLs
We proceeded to evaluate a panel of gp100 APLs for their ability to detect gp100-specific CD4+ T cell responses in vitro, by testing their recognition by T cells from vaccinated melanoma patients that were stimulated in vitro with WT peptide. Figure 7A shows results from 3 patients, demonstrating that IVS T cells from each patient had a unique preference for recognizing various APLs. These results suggest heterogeneous TCR usage by gp100 WT-specific T cells from individual patients. Several APLs that were preferred by individual patients (L48F, E54L and E54T) were assessed for their ability to generate gp100-specific CD4+ T cells from the pre-vaccination PBLs of the 6 melanoma patients. None of these APLs was superior to WT or Q56A peptide in stimulating gp100-specific CD4+ T cells, and in some cases APLs provoked APL-specific responses that did not cross-react with WT peptide (not shown). Finally, a peptide pool comprised of WT peptide plus the APLs L48F, E54L, E54T, and Q56A was evaluated for its ability to raise gp100-specific immunity in pre-vaccination T cells or to detect post-vaccination gp100 specific immunity, in melanoma patients #1, 2 and 4, similar to experiments shown in Figure 6. The peptide pool was not superior to the WT peptide in raising or detecting gp100-specific immunity (Figure 7B).
Figure 7.

T cells stimulated in vitro with gp10044–59 WT peptide show selective recognition of a variety of APLs. (A) T cells were derived from melanoma patients receiving a gp100 WT peptide vaccine. Results of IFN-γ ELISAs are shown. Background values for IFN-γ secretion by T cells stimulated with APC + HA307–319 have been subtracted from data shown, as follows: Patient #2, 213 pg/ml (1 μM peptide stimulation) and 208 pg/ml (10 μM peptide stimulation); Patient #4, 187 and 191 pg/ml; Patient #5, 346 and 388 pg/ml. (B) A peptide pool including APLs was not superior to the WT peptide in raising or detecting gp100-specific immunity. Reactivity against gp10044–59 in 10-day IVS cultures was determined by IFN-γ ELISPOT assays. CD4+ T cells were stimulated with 20 μM WT gp100 or peptide pool (WT, L48F, E54L E54T and Q56A) in both IVS cultures and ELISPOT assays. CDC27, an unrelated HLA-DR4-restricted peptide, was used as a negative control. P4, P6, P8, PBMCs collected from patients post 4, 6 or 8 vaccinations. SFC, spot forming colonies, mean± SEM. WT, wild type gp10044–59. * P<0.02, × P<0.05, one-tailed t test compared to HA or CDC27 control peptides.
DISCUSSION
Insights gained from the characterization of MHC-peptide-TCR interactions hold the promise that directed structural modifications can have predictable functional consequences. The ability to manipulate T cell reactivity synthetically or through genetic engineering might thus be translated into new therapies for common diseases such as cancer and autoimmune disorders. In an early example with the HLA-A2-restricted gp100 epitope 209–217, an MHC anchor residue substitution improved MHC-peptide binding affinity, which translated into enhanced CD8 T cell recognition in vitro and increased immunogenicity in the clinic as a melanoma vaccine (8, 17). However, subsequent investigations on the same theme using MHC I-restricted melanoma-associated peptides and melanoma-specific TCRs have yielded variable results, demonstrating that structural modifications of MHC-peptide-TCR interactions may have unpredictable or undesirable functional consequences (49, 50), which may involve collateral damage to normal tissues when tolerance to non-mutated tumor antigens such as gp100 is alleviated. Furthermore, in vitro functional testing may not always be predictive of in vivo effects (51). These difficulties are amplified in the context of MHC II interactions, for which peptide binding motifs are permissive and not rigid, enabling binding of a single peptide to multiple MHC II alleles (52). An APL of an HLA-DR2-restricted myelin basic protein (MBP) epitope containing modified TCR contact residues, designed to antagonize autoreactive T cells in patients with multiple sclerosis, caused disease exacerbation rather than alleviation in some vaccinated patients (53).
In the current study, APLs from a dominant HLA-DR4-restricted gp100 epitope were designed to enhance MHC binding and hence T cell recognition in melanoma patients. APL design was based on precise structural definition of the wild type peptide-MHC II complex, and increased MHC binding was validated by direct affinity measurements. Nevertheless, we observed heterogeneous preferences of CD4+ T cells from several HLA-DR4+ melanoma patients for different gp100 APLs, suggesting highly variable TCR usage even among patients who had been vaccinated against the wild type gp100 peptide. This could reflect heterogeneous responses of TCRs from different patients to APL-DRB1*0401 complexes, but might also reflect immune responses to APLs bound to alternative DR alleles expressed by individual patients. This heterogeneity prevented the selection of an APL candidate for developing an improved generic melanoma vaccine.
Although we did not determine crystal structure of Q56A APL bound to HLA-DR4, it is unlikely that the conformation of this anchor-modified peptide differs significantly from that of wild-type gp10044–59, which could have explained loss of TCR reactivity. Q56A APL and gp10044–59 differ by only a single residue, at anchor position P9. The possible effect on the peptide backbone of replacing P9 Gln by Ala may be assessed by examining two other peptide–HLA-DR4 structures, one involving CII1168–1180, in which P9 is Gly (47), and the other involving HA307–319, in which P9 is Leu (45). As shown in Figure 3C, the main chain of gp10044–59 superposes very closely onto those of CII1168–1180 and HA307–319, even though these three unrelated peptides have different residues at P9 (Gln, Gly, and Leu, respectively). This suggests that the P9 anchor modification in Q56A APL exerts subtle effects on peptide contacts with TCR or on peptide dynamics, such that an alteration intended to enhance immunogenicity may be counterproductive. Indeed, there is growing evidence for an important role of peptide dynamics in modulating TCR recognition (54–56). In one especially relevant study (55), replacing suboptimal anchor residues of the HLA-A2-restricted MART-127–35 melanoma antigen unexpectedly abolished recognition by most MART-1-specific T cell clones, despite a 40-fold improvement in peptide binding to MHC. Moreover, crystal structures of wild-type and anchor-modified MART-127–35 peptides bound to HLA-A2 showed that the anchor modifications did little to alter the conformation of the peptide or the MHC binding groove (49). Instead, NMR and molecular dynamics simulations revealed that the anchor modifications increased the flexibility of both MART-127–35 and HLA-A2 (55). Although the resulting entropic effects improved binding of the peptide to MHC, they also increased the entropic penalty for TCR binding to prohibitive levels, resulting in loss of TCR affinity for peptide–MHC. A similar mechanism may explain the loss of T cell recognition that we observed for Q56A APL. In a related study, TCR recognition of an APL of lymphocytic choriomeningitis virus peptide p33 was found to be entropically driven, whereas recognition of wild-type p33 was enthalpically driven (57). If different peptides can alter peptide-MHC dynamics in ways that affect TCR recognition, the commonly used approach of optimizing anchor residues to enhance peptide binding to MHC may be insufficient to reliably generate improved vaccine candidates, as we found in the present study.
In the gp10044–59–HLA-DR4 structure, N-terminal residues Trp44 and Asn45 lie outside the peptide-binding groove and are not defined in the electron density, suggesting flexibility. Nevertheless, these two N-terminal residues are required for efficient recognition by TCR G7, which is reminiscent of the way some autoreactive TCRs engage self or altered self peptides, including tumor antigens, presented by MHC II molecules (43, 58). For example, X-ray crystallographic studies of tumor-specific TCRs that recognize a somatically mutated human melanoma antigen have revealed substantial alterations in the topology of TCR binding to peptide–MHC compared to anti-foreign TCRs (59, 60). In these anti-tumor TCR–peptide–MHC complexes, the TCR is skewed toward the peptide N-terminus relative to its central position in anti-foreign TCR–peptide–MHC complexes, resulting in low affinity binding (KD > 200 μM) that likely enabled escape from negative thymic selection. In another example, human autoimmune TCR Ob.1A12, which recognizes a self-peptide from myelin basic protein (MBP85–99) bound to HLA-DR2b, was found to only contact the N-terminal half of MBP85–99 (61). TCR Ob.1A12 cross-reacts with an E. coli peptide having limited sequence identity with MBP85–99. Cross-reactivity is due to structural mimicry of a binding hotspot at the N-terminal portion of the bacterial and self-peptide (62).
We attempted to measure the binding of TCR G7 to gp10044–59–HLA-DR4 by surface plasmon resonance (data not shown). However, we could not detect an interaction, even after injecting a high concentration (up to 200 μM) of recombinant TCR G7 over immobilized gp10044–59–HLA-DR4, which precluded further analysis. This result is consistent with previous findings that autoreactive TCRs generally bind self-antigens, including tumor antigens, with very low affinity (58). For example, the human melanoma-specific TCR E8 binds mutTPI–HLA-DR1 with KD > 200 μM (59). The low affinities of G7 and E8 for their self-peptide–MHC ligands likely enabled these autoreactive T cells to escape negative thymic selection (58).
Although an explosion of information in the field of molecular immunology over the past two decades has yielded extraordinarily precise and extensive data on molecular interactions and structure-function relationships in the immune system, there remains a critical gap in knowledge that hinders reliable clinical translation from structural and in vitro observations. As demonstrated in the present study, peptide modifications that improve binding to MHC class II molecules do not necessarily translate to increased antigenicity. Confounding this strategy is the heterogeneity of human CD4+ T cell responses, as well as possible dynamical effects of peptide modifications. Predicting these effects will require the use of molecular dynamics simulations in conjunction with structural information. Such structure-guided computational design may eventually allow successful re-orientation of immune responses against cancer with vaccines and other immunomodulatory therapies.
Abbreviations used in the article
- APL
altered peptide ligand
- HA
influenza hemagglutinin
- IC50
50% inhibitory concentration
- IVS
in vitro stimulation
- MBP
myelin basic protein
- WT
wild type
Footnotes
This work was supported by National Institutes of Health-NIAID Grant AI073654 (to SLT and RAM), NIH-NIAID contract HHSN272200900044C (to AS), and NIH Grant AI033993 (to DFH). We thank H. Robinson (Brookhaven National Synchrotron Light Source) for X-ray data collection. Support for beamline X29 comes from the Offices of Biological and Environmental Research and of Basic Energy Sciences of the Department of Energy, and from the National Center for Research Resources of the National Institutes of Health.
References
- 1.Balch CM, Gershenwald JE, Soong SJ, Thompson JF, Atkins MB, Byrd DR, Buzaid AC, Cochran AJ, Coit DG, Ding S, Eggermont AM, Flaherty KT, Gimotty PA, Kirkwood JM, McMasters KM, Mihm MC, Jr, Morton DL, Ross MI, Sober AJ, Sondak VK. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27:6199–6206. doi: 10.1200/JCO.2009.23.4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heemskerk B, Liu K, Dudley ME, Johnson LA, Kaiser A, Downey S, Zheng Z, Shelton TE, Matsuda K, Robbins PF, Morgan RA, Rosenberg SA. Adoptive cell therapy for patients with melanoma, using tumor-infiltrating lymphocytes genetically engineered to secrete interleukin-2. Hum Gene Ther. 2008;19:496–510. doi: 10.1089/hum.2007.0171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbe C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, Sznol M. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, Martins R, Eaton K, Chen S, Salay TM, Alaparthy S, Grosso JF, Korman AJ, Parker SM, Agrawal S, Goldberg SM, Pardoll DM, Gupta A, Wigginton JM. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li B, VanRoey M, Wang C, Chen TH, Korman A, Jooss K. Anti-programmed death-1 synergizes with granulocyte macrophage colony-stimulating factor--secreting tumor cell immunotherapy providing therapeutic benefit to mice with established tumors. Clin Cancer Res. 2009;15:1623–1634. doi: 10.1158/1078-0432.CCR-08-1825. [DOI] [PubMed] [Google Scholar]
- 7.Hurwitz AA, Yu TF, Leach DR, Allison JP. CTLA-4 blockade synergizes with tumor-derived granulocyte-macrophage colony-stimulating factor for treatment of an experimental mammary carcinoma. Proc Natl Acad Sci U S A. 1998;95:10067–10071. doi: 10.1073/pnas.95.17.10067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwartzentruber DJ, Lawson DH, Richards JM, Conry RM, Miller DM, Treisman J, Gailani F, Riley L, Conlon K, Pockaj B, Kendra KL, White RL, Gonzalez R, Kuzel TM, Curti B, Leming PD, Whitman ED, Balkissoon J, Reintgen DS, Kaufman H, Marincola FM, Merino MJ, Rosenberg SA, Choyke P, Vena D, Hwu P. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N Engl J Med. 2011;364:2119–2127. doi: 10.1056/NEJMoa1012863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kawakami Y, Robbins PF, Rosenberg SA. Human melanoma antigens recognized by T lymphocytes. Keio J Med. 1996;45:100–108. doi: 10.2302/kjm.45.100. [DOI] [PubMed] [Google Scholar]
- 10.Dudley ME, Wunderlich JR, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry RM, Marincola FM, Leitman SF, Seipp CA, Rogers-Freezer L, Morton KE, Nahvi A, Mavroukakis SA, White DE, Rosenberg SA. A phase I study of nonmyeloablative chemotherapy and adoptive transfer of autologous tumor antigen-specific T lymphocytes in patients with metastatic melanoma. J Immunother. 2002;25:243–251. doi: 10.1097/01.CJI.0000016820.36510.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kawakami Y, Eliyahu S, Jennings C, Sakaguchi K, Kang X, Southwood S, Robbins PF, Sette A, Appella E, Rosenberg SA. Recognition of multiple epitopes in the human melanoma antigen gp100 by tumor-infiltrating T lymphocytes associated with in vivo tumor regression. J Immunol. 1995;154:3961–3968. [PubMed] [Google Scholar]
- 12.Pardoll DM, Topalian SL. The role of CD4+ T cell responses in antitumor immunity. Curr Opin Immunol. 1998;10:588–594. doi: 10.1016/s0952-7915(98)80228-8. [DOI] [PubMed] [Google Scholar]
- 13.Halder T, Pawelec G, Kirkin AF, Zeuthen J, Meyer HE, Kun L, Kalbacher H. Isolation of novel HLA-DR restricted potential tumor-associated antigens from the melanoma cell line FM3. Cancer Res. 1997;57:3238–3244. [PubMed] [Google Scholar]
- 14.Touloukian CE, Leitner WW, Topalian SL, Li YF, Robbins PF, Rosenberg SA, Restifo NP. Identification of a MHC class II-restricted human gp100 epitope using DR4-IE transgenic mice. J Immunol. 2000;164:3535–3542. doi: 10.4049/jimmunol.164.7.3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kierstead LS, Ranieri E, Olson W, Brusic V, Sidney J, Sette A, Kasamon YL, Slingluff CL, Jr, Kirkwood JM, Storkus WJ. gp100/pmel17 and tyrosinase encode multiple epitopes recognized by Th1-type CD4+T cells. Br J Cancer. 2001;85:1738–1745. doi: 10.1054/bjoc.2001.2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Phan GQ, Touloukian CE, Yang JC, Restifo NP, Sherry RM, Hwu P, Topalian SL, Schwartzentruber DJ, Seipp CA, Freezer LJ, Morton KE, Mavroukakis SA, White DE, Rosenberg SA. Immunization of patients with metastatic melanoma using both class I- and class II-restricted peptides from melanoma-associated antigens. J Immunother. 2003;26:349–356. doi: 10.1097/00002371-200307000-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parkhurst MR, Salgaller ML, Southwood S, Robbins PF, Sette A, Rosenberg SA, Kawakami Y. Improved induction of melanoma-reactive CTL with peptides from the melanoma antigen gp100 modified at HLA-A*0201-binding residues. J Immunol. 1996;157:2539–2548. [PubMed] [Google Scholar]
- 18.Salgaller ML, Marincola FM, Cormier JN, Rosenberg SA. Immunization against epitopes in the human melanoma antigen gp100 following patient immunization with synthetic peptides. Cancer Res. 1996;56:4749–4757. [PubMed] [Google Scholar]
- 19.Clay TM, Custer MC, McKee MD, Parkhurst M, Robbins PF, Kerstann K, Wunderlich J, Rosenberg SA, Nishimura MI. Changes in the fine specificity of gp100(209–217)-reactive T cells in patients following vaccination with a peptide modified at an HLA-A2.1 anchor residue. J Immunol. 1999;162:1749–1755. [PubMed] [Google Scholar]
- 20.Dudley ME, Nishimura MI, Holt AK, Rosenberg SA. Antitumor immunization with a minimal peptide epitope (G9–209-2M) leads to a functionally heterogeneous CTL response. J Immunother. 1999;22:288–298. doi: 10.1097/00002371-199907000-00002. [DOI] [PubMed] [Google Scholar]
- 21.van Stipdonk MJ, Badia-Martinez D, Sluijter M, Offringa R, van Hall T, Achour A. Design of agonistic altered peptides for the robust induction of CTL directed towards H-2Db in complex with the melanoma-associated epitope gp100. Cancer Res. 2009;69:7784–7792. doi: 10.1158/0008-5472.CAN-09-1724. [DOI] [PubMed] [Google Scholar]
- 22.Rammensee HG, Friede T, Stevanoviic S. MHC ligands and peptide motifs: first listing. Immunogenetics. 1995;41:178–228. doi: 10.1007/BF00172063. [DOI] [PubMed] [Google Scholar]
- 23.Sette A, Sidney J, Oseroff C, del Guercio MF, Southwood S, Arrhenius T, Powell MF, Colon SM, Gaeta FC, Grey HM. HLA DR4w4-binding motifs illustrate the biochemical basis of degeneracy and specificity in peptide-DR interactions. J Immunol. 1993;151:3163–3170. [PubMed] [Google Scholar]
- 24.Hammer J, Valsasnini P, Tolba K, Bolin D, Higelin J, Takacs B, Sinigaglia F. Promiscuous and allele-specific anchors in HLA-DR-binding peptides. Cell. 1993;74:197–203. doi: 10.1016/0092-8674(93)90306-b. [DOI] [PubMed] [Google Scholar]
- 25.Hammer J, Belunis C, Bolin D, Papadopoulos J, Walsky R, Higelin J, Danho W, Sinigaglia F, Nagy ZA. High-affinity binding of short peptides to major histocompatibility complex class II molecules by anchor combinations. Proc Natl Acad Sci U S A. 1994;91:4456–4460. doi: 10.1073/pnas.91.10.4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Depontieu FR, Qian J, Zarling AL, McMiller TL, Salay TM, Norris A, English AM, Shabanowitz J, Engelhard VH, Hunt DF, Topalian SL. Identification of tumor-associated, MHC class II-restricted phosphopeptides as targets for immunotherapy. Proc Natl Acad Sci U S A. 2009;106:12073–12078. doi: 10.1073/pnas.0903852106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Udeshi ND, Compton PD, Shabanowitz J, Hunt DF, Rose KL. Methods for analyzing peptides and proteins on a chromatographic timescale by electron-transfer dissociation mass spectrometry. Nat Protoc. 2008;3:1709–1717. doi: 10.1038/nprot.2008.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chi A, Huttenhower C, Geer LY, Coon JJ, Syka JE, Bai DL, Shabanowitz J, Burke DJ, Troyanskaya OG, Hunt DF. Analysis of phosphorylation sites on proteins from Saccharomyces cerevisiae by electron transfer dissociation (ETD) mass spectrometry. Proc Natl Acad Sci U S A. 2007;104:2193–2198. doi: 10.1073/pnas.0607084104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Geer LY, Markey SP, Kowalak JA, Wagner L, Xu M, Maynard DM, Yang X, Shi W, Bryant SH. Open mass spectrometry search algorithm. J Proteome Res. 2004;3:958–964. doi: 10.1021/pr0499491. [DOI] [PubMed] [Google Scholar]
- 30.Wang RF, Wang X, Atwood AC, Topalian SL, Rosenberg SA. Cloning genes encoding MHC class II-restricted antigens: mutated CDC27 as a tumor antigen. Science. 1999;284:1351–1354. doi: 10.1126/science.284.5418.1351. [DOI] [PubMed] [Google Scholar]
- 31.Sidney J, Southwood S, Oseroff C, del Guercio MF, Sette A, Grey HM. Measurement of MHC/peptide interactions by gel filtration. Curr Protoc Immunol. 2001;Chapter 18(Unit 18):13. doi: 10.1002/0471142735.im1803s31. [DOI] [PubMed] [Google Scholar]
- 32.Greenbaum J, Sidney J, Chung J, Brander C, Peters B, Sette A. Functional classification of class II human leukocyte antigen (HLA) molecules reveals seven different supertypes and a surprising degree of repertoire sharing across supertypes. Immunogenetics. 2011;63:325–335. doi: 10.1007/s00251-011-0513-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Southwood S, Sidney J, Kondo A, del Guercio MF, Appella E, Hoffman S, Kubo RT, Chesnut RW, Grey HM, Sette A. Several common HLA-DR types share largely overlapping peptide binding repertoires. J Immunol. 1998;160:3363–3373. [PubMed] [Google Scholar]
- 34.Gulukota K, Sidney J, Sette A, DeLisi C. Two complementary methods for predicting peptides binding major histocompatibility complex molecules. J Mol Biol. 1997;267:1258–1267. doi: 10.1006/jmbi.1997.0937. [DOI] [PubMed] [Google Scholar]
- 35.Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 36.Frayser M, Sato AK, Xu L, Stern LJ. Empty and peptide-loaded class II major histocompatibility complex proteins produced by expression in Escherichia coli and folding in vitro. Protein Expr Purif. 1999;15:105–114. doi: 10.1006/prep.1998.0987. [DOI] [PubMed] [Google Scholar]
- 37.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 38.Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta Crystallogr D Biol Crystallogr. 2004;60:432–438. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- 39.Bolin DR, Swain AL, Sarabu R, Berthel SJ, Gillespie P, Huby NJ, Makofske R, Orzechowski L, Perrotta A, Toth K, Cooper JP, Jiang N, Falcioni F, Campbell R, Cox D, Gaizband D, Belunis CJ, Vidovic D, Ito K, Crowther R, Kammlott U, Zhang X, Palermo R, Weber D, Guenot J, Nagy Z, Olson GL. Peptide and peptide mimetic inhibitors of antigen presentation by HLA-DR class II MHC molecules. Design, structure-activity relationships, and X-ray crystal structures. J Med Chem. 2000;43:2135–2148. doi: 10.1021/jm000034h. [DOI] [PubMed] [Google Scholar]
- 40.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 41.McRee DE. XtalView/Xfit--A versatile program for manipulating atomic coordinates and electron density. J Struct Biol. 1999;125:156–165. doi: 10.1006/jsbi.1999.4094. [DOI] [PubMed] [Google Scholar]
- 42.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 43.Li Y, Depontieu FR, Sidney J, Salay TM, Engelhard VH, Hunt DF, Sette A, Topalian SL, Mariuzza RA. Structural basis for the presentation of tumor-associated MHC class II-restricted phosphopeptides to CD4+ T cells. J Mol Biol. 2010;399:596–603. doi: 10.1016/j.jmb.2010.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hammer J, Gallazzi F, Bono E, Karr RW, Guenot J, Valsasnini P, Nagy ZA, Sinigaglia F. Peptide binding specificity of HLA-DR4 molecules: correlation with rheumatoid arthritis association. J Exp Med. 1995;181:1847–1855. doi: 10.1084/jem.181.5.1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hennecke J, Wiley DC. Structure of a complex of the human alpha/beta T cell receptor (TCR) HA1.7, influenza hemagglutinin peptide, and major histocompatibility complex class II molecule, HLA-DR4 (DRA*0101 and DRB1*0401): insight into TCR cross-restriction and alloreactivity. J Exp Med. 2002;195:571–581. doi: 10.1084/jem.20011194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Muraro PA, Vergelli M, Kalbus M, Banks DE, Nagle JW, Tranquill LR, Nepom GT, Biddison WE, McFarland HF, Martin R. Immunodominance of a low-affinity major histocompatibility complex-binding myelin basic protein epitope (residues 111–129) in HLA-DR4 (B1*0401) subjects is associated with a restricted T cell receptor repertoire. J Clin Invest. 1997;100:339–349. doi: 10.1172/JCI119539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dessen A, Lawrence CM, Cupo S, Zaller DM, Wiley DC. X-ray crystal structure of HLA-DR4 (DRA*0101, DRB1*0401) complexed with a peptide from human collagen II. Immunity. 1997;7:473–481. doi: 10.1016/s1074-7613(00)80369-6. [DOI] [PubMed] [Google Scholar]
- 48.Yin Y, Li Y, Kerzic MC, Martin R, Mariuzza RA. Structure of a TCR with high affinity for self-antigen reveals basis for escape from negative selection. EMBO J. 2011;30:1137–1148. doi: 10.1038/emboj.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Borbulevych OY, Insaidoo FK, Baxter TK, Powell DJ, Jr, Johnson LA, Restifo NP, Baker BM. Structures of MART-126/27–35 Peptide/HLA-A2 complexes reveal a remarkable disconnect between antigen structural homology and T cell recognition. J Mol Biol. 2007;372:1123–1136. doi: 10.1016/j.jmb.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, Kammula US, Royal RE, Sherry RM, Wunderlich JR, Lee CC, Restifo NP, Schwarz SL, Cogdill AP, Bishop RJ, Kim H, Brewer CC, Rudy SF, VanWaes C, Davis JL, Mathur A, Ripley RT, Nathan DA, Laurencot CM, Rosenberg SA. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114:535–546. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McMahan RH, McWilliams JA, Jordan KR, Dow SW, Wilson DB, Slansky JE. Relating TCR-peptide-MHC affinity to immunogenicity for the design of tumor vaccines. J Clin Invest. 2006;116:2543–2551. doi: 10.1172/JCI26936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vita R, Zarebski L, Greenbaum JA, Emami H, Hoof I, Salimi N, Damle R, Sette A, Peters B. The immune epitope database 2.0. Nucleic Acids Res. 2010;38:D854–862. doi: 10.1093/nar/gkp1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bielekova B, Goodwin B, Richert N, Cortese I, Kondo T, Afshar G, Gran B, Eaton J, Antel J, Frank JA, McFarland HF, Martin R. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83–99) in multiple sclerosis: results of a phase II clinical trial with an altered peptide ligand. Nat Med. 2000;6:1167–1175. doi: 10.1038/80516. [DOI] [PubMed] [Google Scholar]
- 54.Pohlmann T, Bockmann RA, Grubmuller H, Uchanska-Ziegler B, Ziegler A, Alexiev U. Differential peptide dynamics is linked to major histocompatibility complex polymorphism. J Biol Chem. 2004;279:28197–28201. doi: 10.1074/jbc.C400128200. [DOI] [PubMed] [Google Scholar]
- 55.Insaidoo FK, Borbulevych OY, Hossain M, Santhanagopolan SM, Baxter TK, Baker BM. Loss of T cell antigen recognition arising from changes in peptide and major histocompatibility complex protein flexibility: implications for vaccine design. J Biol Chem. 2011;286:40163–40173. doi: 10.1074/jbc.M111.283564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Baker BM, Scott DR, Blevins SJ, Hawse WF. Structural and dynamic control of T-cell receptor specificity, cross-reactivity, and binding mechanism. Immunol Rev. 2012;250:10–31. doi: 10.1111/j.1600-065X.2012.01165.x. [DOI] [PubMed] [Google Scholar]
- 57.Allerbring EB, Duru AD, Uchtenhagen H, Madhurantakam C, Tomek MB, Grimm S, Mazumdar PA, Friemann R, Uhlin M, Sandalova T, Nygren PA, Achour A. Unexpected T-cell recognition of an altered peptide ligand is driven by reversed thermodynamics. Eur J Immunol. 2012;42:2990–3000. doi: 10.1002/eji.201242588. [DOI] [PubMed] [Google Scholar]
- 58.Yin Y, Li Y, Mariuzza RA. Structural basis for self-recognition by autoimmune T-cell receptors. Immunol Rev. 2012;250:32–48. doi: 10.1111/imr.12002. [DOI] [PubMed] [Google Scholar]
- 59.Deng L, Langley RJ, Brown PH, Xu G, Teng L, Wang Q, Gonzales MI, Callender GG, Nishimura MI, Topalian SL, Mariuzza RA. Structural basis for the recognition of mutant self by a tumor-specific, MHC class II-restricted T cell receptor. Nat Immunol. 2007;8:398–408. doi: 10.1038/ni1447. [DOI] [PubMed] [Google Scholar]
- 60.Deng L, Langley RJ, Wang Q, Topalian SL, Mariuzza RA. Structural insights into the editing of germ-line-encoded interactions between T-cell receptor and MHC class II by Valpha CDR3. Proc Natl Acad Sci U S A. 2012;109:14960–14965. doi: 10.1073/pnas.1207186109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hahn M, Nicholson MJ, Pyrdol J, Wucherpfennig KW. Unconventional topology of self peptide-major histocompatibility complex binding by a human autoimmune T cell receptor. Nat Immunol. 2005;6:490–496. doi: 10.1038/ni1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Harkiolaki M, Holmes SL, Svendsen P, Gregersen JW, Jensen LT, McMahon R, Friese MA, van Boxel G, Etzensperger R, Tzartos JS, Kranc K, Sainsbury S, Harlos K, Mellins ED, Palace J, Esiri MM, van der Merwe PA, Jones EY, Fugger L. T cell-mediated autoimmune disease due to low-affinity crossreactivity to common microbial peptides. Immunity. 2009;30:348–357. doi: 10.1016/j.immuni.2009.01.009. [DOI] [PubMed] [Google Scholar]