

Abstract
Fanconi anemia is an inherited bone marrow failure syndrome, characterised by failing DNA repair. Hematopoetic stem cell transplantation, known to be curative for the bone marrow failure, does neither prevent or cure other manifestations such as the development of malignancies. We describe a 26-year-old male patient with known Fanconi anemia and Marfan syndrome who in 1994 underwent a successful bone marrow transplantation of stem cells from his HLA-identical sister. In 2006, three hepatocellular carcinoma (HCC) lesions in the liver were detected and promptly resected. The resection specimen contained 3 lesions, all showing activation of the beta-catenin pathway: a well differentiated steatotic HCC with remnants of the underlying adenoma from which it arose, an adenoma with small foci of well differentiated HCC and a cholestatic adenoma. Known risk factors for developing HCC include Fanconi anemia itself and the use of androgens (oxymetholone) for a period of 3 years preceeding transplantation. Because of the increased risk of developing additional HCC’s, liver transplantation was proposed, taking into account that immunosuppression increases the risk of other malignancies. By using part of the liver of the HLA-identical sister, already acting as bone marrow donor 13 years before, immunosuppression could be avoided.
A left lobe liver transplantation was performed without immediate complications for donor and acceptor on July 2, 2007. Nine months after liver transplantation the recipient developed an anastomotic biliary stricture that had to be dilated by percutaneous transhepatic cholangiography. Two months later however, the stenosis recurred, necessitating a surgical reanastomosis (hepaticojejunostomy). Five years after liver transplantation the patient is still doing well.
This case report is twofold special being the first case reporting Fanconi anemia linked to Marfan syndrome and being the first reported case of Fanconi anemia who was treated for hepatocellular carcinoma by liver transplantation from a living related HLA-identical donor without the use of immunosuppression.
Keywords: Fanconi anemia, bone marrow transplantation, liver transplantation, hepatocellular adenoma/carcinoma
Background
Fanconi anemia, the most common inherited aplastic anemia, is a multigenic autosomal recessive or X-linked disorder with at least 13 complementation groups (FA-A to FA-N) being involved (1). The proteins encoded by the Fanconi genes contribute to controlling apoptosis, oxygen sensitivity, response to cytokines or maintaining genomic stability (1). Over 90 % of patients develop pancytopenia. About 25% of Fanconi anemia patients eventually develop malignancies, especially haematological tumors (60%) such as myelodysplastic syndrome, acute myelocytic lymphoma and head and neck squamous cell carcinomas (500 times higher risk than that in the normal population) (2). Clonal chromosomal changes may be the predisposing factor for malignancy.
Hematopoetic stem cell transplantation is only curative for the bone marrow failure. Outside the bone marrow DNA repair remains deficient. Androgen treatment improves hypocellularity in Fanconi anemia but is not curative and has adverse side effects including virilisation and rarely peliosis hepatis, hepatic adenoma, hepatocellular carcinoma (HCC) and hepatic cystic diseases (3–5).
We describe the presence of Fanconi anemia in a 26-year-old man who also has Marfan syndrome. He underwent a bone marrow transplantation from his HLA-identical sister at the age of 12 and developed hepatocellular carcinoma which was treated by a living related liver transplantation with a liver segment from the same donor, several years after the bone marrow transplantation.
Case presentation
A 26-year-old male patient was diagnosed in 1990 (at the age of 9 year) with Fanconi anemia by demonstration of mitomycine C induced increased chromosomal breakage in the lymphocyte culture. The patient presented with pancytopenia and had the typical physical features of Fanconi anemia, including short statue (169cm, 52 kg) and delicate posture, microcephaly and bird-like face, Sprengel deformity and a horseshoe kidney. Cell analytic studies and retroviral complementation analysis indicates that the patient belongs to the FA-G complementation group. A homozygous pathogenic mutation in exon 5 has been identified: C620delT, which is a frameshift mutation predicting premature termination of translation. He initially responded well on a treatment with androgens (oxymetholone 1mg/kg/d orally) during 3 years (January 1991 until December 1993). In 1994, the patient underwent a bone marrow transplantation from his HLA-identical (A2-A10 (26)-B5-B27-DR1-DR7-DQW1) two year younger sister. The conditioning regimen consisted of ATG at a total dose of 20 mg/kg in 4 days (day–6, -5, -4, -3), cyclophosphamide at a total dose of 20 mg/kg over 4 days (day -5, -4, -3, -2 ) and total nodular irradiation 5 Gy in 2 sessions (D–1).
Besides his Fanconi anemia he was also diagnosed with Marfan disease associated with the fibrillin-1 (FBN1) mutation (insertion of 6 nucleotides in FBN1 exon 36) and clinically characterised by lens luxation, mild proximal aortic dilatation and scoliosis, the latter being treated surgically in 1996.
In December 2006, he developed a hepatocellular carcinoma (HCC) of 3 cm in segment 7 diagnosed on CT scan (Figure 1a) and Magnetic Resonance Imaging (Figure 1b, 1c) for which a laparoscopic resection was performed. At the time of resection two other lesions of 0.8 cm and 0.9 cm both in segment 5 were found and laparoscopically resected. Standard histopathological examination and histochemical stains were performed as described previously (6). Immunohistochemistry for beta-catenin, glutamine synthetase and glypican-3 were performed according to recently described protocols (7;8).
Figure 1.
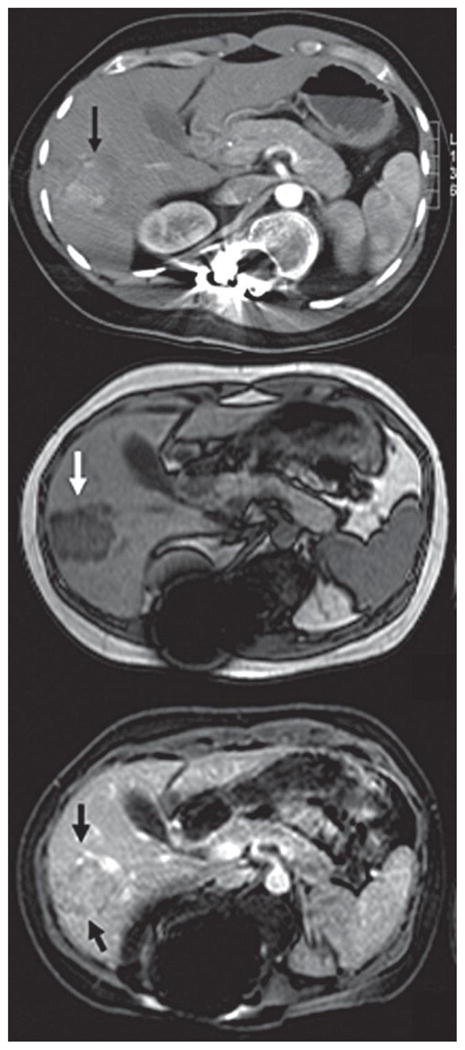
Figure 1a. CT scan in arterial phase showing a hepatocellular carcinoma in segment 7
Figure 1b. MRI in T1 weighed opposed phase shows a hypointense lesion of 3 cm in segment 7 (white arrow)
Figure 1c. MRI in T1 weighed gadolinium enhanced phase, shows an iso-to hyperintense lesion of 3 cm in segment 7 (black arrows)
The lesion of 0.8 cm (Figure 2) consisted of hepatocytes that formed trabeculae that were 2 and occasionally 3 cells thick. Reticulin was preserved throughout the lesion and the nuclei of the hepatocytes showed no obvious atypia. Portal tracts, ductular reaction, pseudoglandular structures, inflammation and sinusoidal dilatation were all absent. There was limited steatosis and some hepatocytes showed eosinophilic inclusions, possibly corresponding to fibrinogen. Numerous bilirubin droplets were present in the lesion, both within hepatocytes and canaliculi (Figure 2a). A considerable proportion of the hepatocytes showed feathery degeneration of the cytoplasm and these cells were positive on the CK7-staining (Figure 2b), indicating the presence of severe cholate stasis. On the staining for beta-catenin only 5% of hepatocytes had a positive nucleus, but the lesion was diffusely and intensely positive on the glutamine synthetase staining (Figure 2c). The lesion was negative on the staining for glypican-3. Based on these findings, the lesion was diagnosed as a cholestatic adenoma (9) with activation of the beta-catenin pathway, but without morphological or immunohistochemical evidence of malignant transformation.
Figure 2.
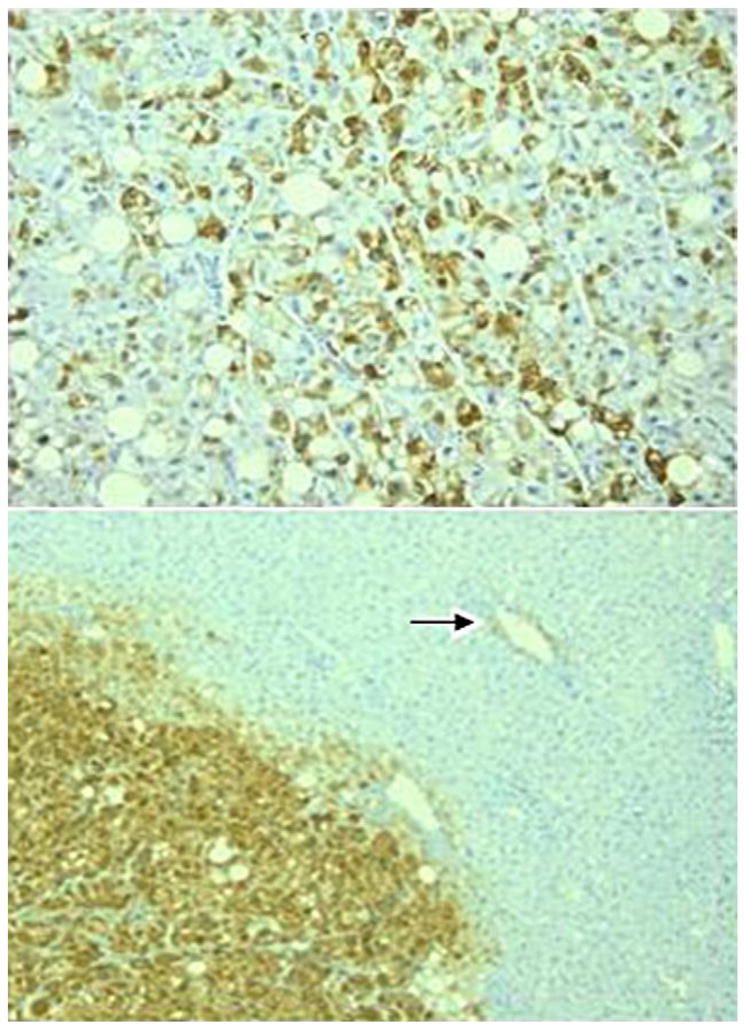
Figure 2a. The cholestatic adenoma contains numerous bilirubin droplets (short arrows) and a considerable number of hepatocytes show feathery degeneration (long arrows). Original magnification ×400.
Figure 2b. The staining for cytokeratin 7 is positive in the hepatocytes showing feathery degeneration, confirming the presence of cholate stasis. Original magnification ×200.
Figure 2c. The cholestatic adenoma diffusely and strongly expresses glutamine synthetase, while the surrounding parenchyma shows only weak staining in the centrolobular area. Original magnification ×100.
The lesion of 0.7 cm (Figure 3) consisted of trabeculae of hepatocytes with a thickness of 2 and occasionally 3 cells. In some areas the reticulin pattern was lost. The hepatocytes showed mild to moderate atypia and several pseudoglandular structures were present (Figure 3a and 3b). Portal tracts, ductular reaction, steatosis, inflammation and sinusoidal dilatation were all absent. The staining for beta-catenin (Figure 3c) and glutamine synthetase showed diffuse and strong nuclear and cytoplasmic positivity, respectively. The lesion was multifocally positive on the staining for glypican-3. Based on these findings, the lesion was diagnosed as a beta-catenin-activated adenoma with foci of well-differentiated HCC.
Figure 3.

Figure 3a. The beta-catenin-activated adenoma shows pseudoglandular structures (arrow) and hepatocytes with nuclei that contain coarsly granular chromatin. Original magnification ×400.
Figure 3b. Within the adenoma, there are some areas (upper part of figure) that show loss of the reticulin pattern, indicating malignant transformation into a well-differentiated HCC. Again, several pseudoglandular structures are recognizable (arrow). Original magnification ×200.
Figure 3c. The adenoma shows diffuse and strong nuclear expression of beta-catenin, while the surrounding parenchyma only shows membranous staining of hepatocytes. Original magnification ×200.
The lesion of 3 cm (Figure 4) showed diffuse steatosis and consisted of hepatocytes with atypical nuclei, organised in trabeculae with loss of reticulin in most areas Figure 4a, 4b and 4c). Portal tracts, ductular reaction, pseudoglandular structures, inflammation and sinusoidal dilatation were all absent. The staining for beta-catenin (Figure 4d) and glutamine synthetase showed diffuse and strong nuclear and cytoplasmic positivity, respectively. The lesion was diffusely positive on the staining for glypican-3. Based on these findings, the lesion was diagnosed as a well-differentiated HCC originating from a steatotic adenoma with activation of the beta-catenin pathway. The surrounding liver parenchyma a well as the explant liver showed no abnormalities.
Figure 4.
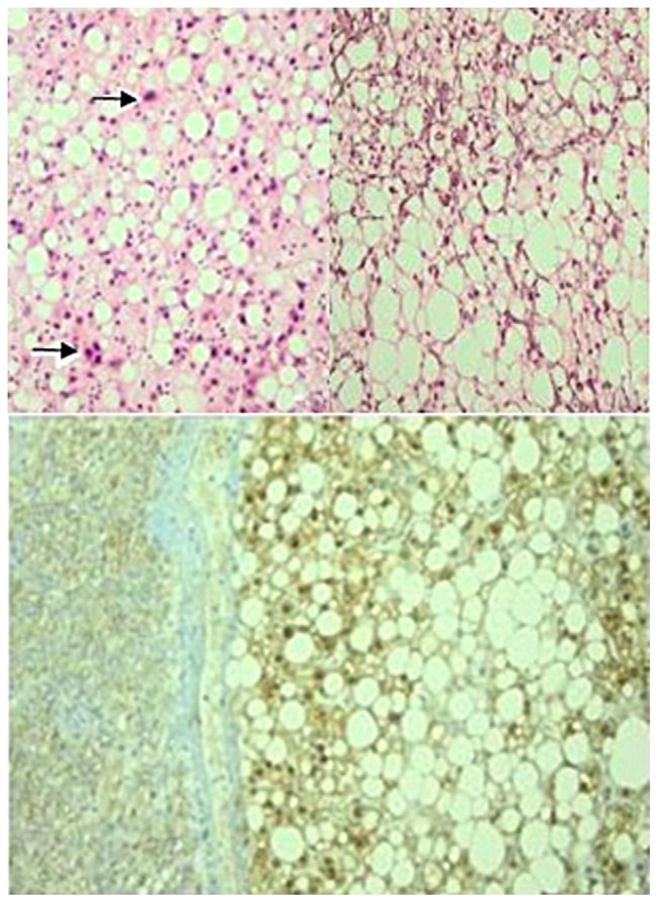
Figure 4a and 4b. The well-differentiated hepatocellular carcinoma shows diffuse steatosis and hyperchromatic, irregular nuclei (arrows). Original magnifications ×400 and ×200.
Figure 4c. Although the reticulin pattern was lost in most areas of the lesion, there are some foci in which reticulin was at least partly retained, delineating 2 to 3 cell-thick trabecules (upper part). Original magnification ×200.
Figure 4d. The adenoma shows diffuse and strong nuclear expression of beta-catenin, while the surrounding parenchyma only shows membranous staining of hepatocytes. Original magnification ×200.
With the FISH test, 46% of the examined cells had an X and Y chromosome, pointing to a male origin. The alpha fetoprotein level was normal, viral tests, ferritin, iron saturation, alpha 1-antitrypsin, ceruloplasmin and autoimmune tests for chronic liver disease were all normal. The patient recovered easily from this surgical intervention. We presumed that the inherited DNA repair disorder, characteristic for Fanconi anemia, and the 3 year exposure to androgens would give rise to other HCC lesions in this high risk patient. A liver transplantation was proposed, taking the increased risk of other malignancies during immune suppression into account. However, immunosuppression would be of no need if the same donor is providing both liver and hematopoietic cells. His 2 year younger HLA-identical sister who figured as a bone marrow donor in 1994 and who in the meantime has become a healthy adult woman, was willing to give a part of her liver. Following the ethical committee rules of our hospital, care was taken not to put any moral pressure on the sister. Brother and sister were treated by different doctors and the sister was also seen by the donor’s advocate.
The patient’s pretransplantation workout did not reveal any other malignancy, especially in the head and neck region. There were neither other contraindications for liver transplantation.
There were also no contraindications for safe liver donation in the sister. Her liver volume was estimated to be 1600 cc, the right lobe representing 1050 cc and left lobe 550 cc. Both donor and acceptor signed the informed consent form, as approved by the ethical committee of the Ghent University Hospital.
A left liver lobe transplantation was performed on July 2, 2007. The actual graft weight was 374 grams at the time of surgery, with a graft weight/recipient weight ratio of 0.73%. Due to the absence of portal hypertension and a portal vein flow of 120 mL/min/100 g with a hepatic artery flow of 150 mL, no attempt was done to inflow modification according to our technique (10;11). The donor was discharged in good health 7 days later, the patient 17 days later. The explanted liver weighted 820 grams and showed no evidence of hepatocellular adenoma, HCC or other abnormalities. Corticosteroids were given at time of transplantation at a dose of 1000 mg intravenously, followed by tapering to zero in 1 week. No other immunosuppression was given.
Nine months posttransplantation, the recipient developed an anastomotic biliary stricture for which a percutaneous transhepatic cholangiography with dilatation was performed. A liver biopsy performed at the same time did not show any evidence of rejection. Two months later, however, restenosis occurred with a need for surgical repair (hepaticojejunostomy). The patient recovered quickly and was discharged 14 days later. His sister who donated her left liver lobe is doing well without any complication now 5 years after resection.
Conclusion
This is the first case report of a Fanconi anemia in association with Marfan syndrome, both very rare diseases. Besides, this is the first case report of a liver transplantation in a Fanconi anemia patient for hepatocellular carcinoma. In childhood, this patient underwent a successful bone marrow transplantation using stem cells from his HLA-identical sister, with full donor chimerism after hematopoietic stem cell transplant. A living related liver transplantation was performed, using the same donor to avoid the use of immunosuppression.
Fanconi anemia is a rare autosomal recessive syndrome characterised by progressive lethal pancytopenia, skeletal abnormalities, hyperpigmentation, increased chromosomal abnormalities/instability and defective DNA repair a with high incidence of leukaemia, hepatocellular and squamous cell carcinoma (1;12;13). In recent years, the Fanconi anemia treatment has been significantly improved, resulting in a better survival into adulthood (1). Our patient was 26 years old when 3 hepatocellular carcinoma (HCC) lesions were detected and resected.
Patients with Fanconi anemia are at risk for developing adenomas and hepatocellular carcinomas (14). In our patient, the risk factors for the development of an adenoma and a hepatocellular carcinoma are dual: the Fanconi anemia by itself, due to the increased chromosomal instability and a defective DNA repair system, and the exposure for 3 years to androgens, oxymetholone in particular. Iron overload, the third risk factor for HCC, could not be withheld in our patient (8). Assuming that this patient is at high risk for developing other HCC lesions, taking these 2 above mentioned risk factors into account, a liver transplantation was proposed. Immunosuppression, a probable risk factor for the development of other malignancies in this kind of patient, could be avoided by using the same donor, his younger sister who already figured as a bone marrow donor in 1994, and who was willing to renounce a part of her liver. The immune competent cells in the recipient were from the same HLA-identical donor origin and would not react against the liver cells which were recognised as ‘self’.
Velazquez et al. reviewed the literature about the relationship between androgens and liver tumors in Fanconi anemia and non-Fanconi anemia patients (14). All patients on anabolic steroids are at risk to develop liver tumours, however, Fanconi anemia patients are more prone to develop HCC, as they develop liver tumours at younger age, after smaller amounts of androgens and after shorter androgen exposure (14–19). Oxymetholone, a 17-α-alkylated androgen, (used by our patient) and methyltestosterone are most often associated with the development of HCC, while danazol is more associated with adenomas (12;14;20;21). As oxymetholone and methyltestosteroneare are more often used in Fanconi anemia patients and danazol in non-aplastic anemia patients, it is quite impossible to determine if the development of HCC is also related to the type of androgens.
The partial liver resection specimen contained 3 adenomas of which 2 showed incipient and advanced malignant transformation into a well-differentiated HCC. Remarkably, only one lesion, the beta-catenin-activated adenoma with foci of well-differentiated HCC, fitted into the new Bordeaux classification of hepatocellular adenoma (22). The smallest lesion corresponded to a cholestatic adenoma, a rarely reported subtype of hepatocellular adenoma (9). Chandra et al. (8) reported that cholestasis in hepatocellular adenomas in children only occur in those who received androgen therapy and Khoo et al. (9) reported a cholestatic adenoma in a child with hirsutism, a feature which can be caused by elevated androgen levels. As a consequence, the current case adds evidence to the hypothesis that androgens play an etiological role in the development of cholestatic adenoma. Due to the rarity of this type of adenoma, it has not been assigned to a specific subclass in the recent Bordeaux classification (22). The current case appears to belong to the beta-catenin-activated subclass at the immunohistochemical level, but not at the morphological level. Further investigations on a larger number of cases is clearly needed. The largest lesion was a beta-catenin-activated adenoma of which a large part was transformed into a well-differentiated HCC. However, the diffuse steatosis prevails its inclusion into the beta-catenin-activated subtype of adenoma. Indeed, diffuse steatosis is not compatible with beta-catenin activation and malignant transformation in the Bordeaux classification (22).
Despite most hepatic adenoma disappear after stopping androgen treatment, some adenomas and HCC’s can develop years after the androgen use (between 1 and 24 years) (14;23). In our patient the diagnosis of HCC was made 13 years after the last intake of androgens. Perhaps, in this high risk group of patients a yearly ultrasound should be advised.
In conclusion, this is the first case which reports Fanconi anemia together with Marfan disease in literature and where the patient underwent a living related liver transplantation for hepatocellular carcinoma without the use of immunosuppression as the liver was derived from the HLA-identical sister who already donated bone marrow, for the treatment of the pancytopenia with hematopoietc stem cell transplantation 13 years earlier.
Acknowledgments
Thanks to Mr Ralf Dietrich from the Fanconi patient’s organisation who was so kind to pick up the different tissue samples and bring them to different universities in Germany.
Footnotes
Consent
Written informed consent was obtained from the patient for publication of this Case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All authors listed have contributed sufficiently to the project to be included as authors, and have read and approved the final manuscript.
Contributor Information
Isabelle Colle, Email: isabelle.colle@ugent.be.
Geneviève Laureys, Email: genevieve.laureys@ugent.be.
Sarah Raevens, Email: sarah.raevens@ugent.be.
Louis Libbrecht, Email: louisj.libbrecht@ugent.be.
Koen Reyntjens, Email: k.reyntjens@ugent.be.
Anja Geerts, Email: anja.geerts@ugent.be.
Xavier Rogiers, Email: xavier.rogiers@uzgent.be.
Roberto Troisi, Email: roberto.troisi@ugent.be.
Holger Hoehn, Email: hoehn@biozentrum.uni-wuerzburg.de.
Detlev Schindler, Email: schindler@biozentrum.uni-wuerzburg.de.
Helmut Hanenberg, Email: HHanenbe@iupui.edu.
Vincent De Wilde, Email: vincent.dewilde@azbrugge.be.
Hans Van Vlierberghe, Email: hans.vanvlierberghe@ugent.be.
Reference List
- 1.Green AM, Kupfer GM. Fanconi anemia. Hematol Oncol Clin North Am. 2009 Apr;23(2):193–214. doi: 10.1016/j.hoc.2009.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alter BP. Cancer in Fanconi anemia, 1927–2001. Cancer. 2003 Jan 15;97(2):425–40. doi: 10.1002/cncr.11046. [DOI] [PubMed] [Google Scholar]
- 3.Shapiro P, Ikeda RM, Ruebner BH, Connors MH, Halsted CC, Abildgaard CF. Multiple hepatic tumors and peliosis hepatis in Fanconi’s anemia treated with androgens. Am J Dis Child. 1977 Oct;131(10):1104–6. doi: 10.1001/archpedi.1977.02120230050009. [DOI] [PubMed] [Google Scholar]
- 4.Soe KL, Soe M, Gluud C. Liver pathology associated with the use of anabolic-androgenic steroids. Liver. 1992 Apr;12(2):73–9. doi: 10.1111/j.1600-0676.1992.tb00560.x. [DOI] [PubMed] [Google Scholar]
- 5.Touraine RL, Bertrand Y, Foray P, Gilly J, Philippe N. Hepatic tumours during androgen therapy in Fanconi anaemia. Eur J Pediatr. 1993 Aug;152(8):691–3. doi: 10.1007/BF01955250. [DOI] [PubMed] [Google Scholar]
- 6.Meuleman P, Libbrecht L, De VR, de HB, Gevaert K, Vandekerckhove J, Roskams T, Leroux-Roels G. Morphological and biochemical characterization of a human liver in a uPA-SCID mouse chimera. Hepatology. 2005 Apr;41(4):847–56. doi: 10.1002/hep.20657. [DOI] [PubMed] [Google Scholar]
- 7.Bioulac-Sage P, Rebouissou S, Thomas C, Blanc JF, Saric J, Sa CA, Rullier A, Cubel G, Couchy G, Imbeaud S, Balabaud C, Zucman-Rossi J. Hepatocellular adenoma subtype classification using molecular markers and immunohistochemistry. Hepatology. 2007 Sep;46(3):740–8. doi: 10.1002/hep.21743. [DOI] [PubMed] [Google Scholar]
- 8.Chandra RS, Kapur SP, Kelleher J, Jr, Luban N, Patterson K. Benign hepatocellular tumors in the young. A clinicopathologic spectrum. Arch Pathol Lab Med. 1984 Feb;108(2):168–71. [PubMed] [Google Scholar]
- 9.Khoo US, Nicholls JM, Lee JS, Saing H, Ng IO. Cholestatic liver cell adenoma in a child with hirsutism and elevated serum levels of cortisol and ACTH. Histopathology. 1994 Dec;25(6):586–8. doi: 10.1111/j.1365-2559.1994.tb01379.x. [DOI] [PubMed] [Google Scholar]
- 10.Troisi R, Cammu G, Militerno G, De BL, Decruyenaere J, Hoste E, Smeets P, Colle I, Van VH, Petrovic M, Voet D, Mortier E, Hesse UJ, de HB. Modulation of portal graft inflow: a necessity in adult living-donor liver transplantation? Ann Surg. 2003 Mar;237(3):429–36. doi: 10.1097/01.SLA.0000055277.78876.B7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Troisi R, Ricciardi S, Smeets P, Petrovic M, Van MG, Colle I, Van VH, de HB. Effects of hemi-portocaval shunts for inflow modulation on the outcome of small-for-size grafts in living donor liver transplantation. Am J Transplant. 2005 Jun;5(6):1397–404. doi: 10.1111/j.1600-6143.2005.00850.x. [DOI] [PubMed] [Google Scholar]
- 12.Linares M, Pastor E, Gomez A, Grau E. Hepatocellular carcinoma and squamous cell carcinoma in a patient with Fanconi’s anemia. Ann Hematol. 1991 Jul;63(1):54–5. doi: 10.1007/BF01714963. [DOI] [PubMed] [Google Scholar]
- 13.Lustig JP, Lugassy G, Neder A, Sigler E. Head and neck carcinoma in Fanconi’s anaemia--report of a case and review of the literature. Eur J Cancer B Oral Oncol. 1995 Jan;31B(1):68–72. doi: 10.1016/0964-1955(94)00044-5. [DOI] [PubMed] [Google Scholar]
- 14.Velazquez I, Alter BP. Androgens and liver tumors: Fanconi’s anemia and non-Fanconi’s conditions. Am J Hematol. 2004 Nov;77(3):257–67. doi: 10.1002/ajh.20183. [DOI] [PubMed] [Google Scholar]
- 15.Abbondanzo SL, Manz HJ, Klappenbach RS, Gootenberg JE. Hepatocellular carcinoma in an 11-year-old girl with Fanconi’s anemia. Report of a case and review of the literature. Am J Pediatr Hematol Oncol. 1986;8(4):334–7. doi: 10.1097/00043426-198624000-00012. [DOI] [PubMed] [Google Scholar]
- 16.Holder LE, Gnarra DJ, Lampkin BC, Nishiyama H, Perkins P. Hepatoma associated with anabolic steroid therapy. Am J Roentgenol Radium Ther Nucl Med. 1975 Aug;124(4):638–42. doi: 10.2214/ajr.124.4.638. [DOI] [PubMed] [Google Scholar]
- 17.Johnson FL, Lerner KG, Siegel M, Feagler JR, Majerus PW, Hartmann JR, Thomas ED. Association of androgenic-anabolic steroid therapy with development of hepatocellular carcinoma. Lancet. 1972 Dec 16;2(7790):1273–6. doi: 10.1016/s0140-6736(72)92649-9. [DOI] [PubMed] [Google Scholar]
- 18.Kew MC, Van CB, Prowse CM, Skikne B, Wolfsdorf JI, Isdale J, Krawitz S, Altman H, Levin SE, Bothwell TH. Occurrence of primary hepatocellular cancer and peliosis hepatis after treatment with androgenic steroids. S Afr Med J. 1976 Jul 24;50(32):1233–7. [PubMed] [Google Scholar]
- 19.Sarna G, Tomasulo P, Lotz MJ, Bubinak JF, Shulman NR. Multiple neoplasms in two siblings with a variant form of Fanconi’s anemia. Cancer. 1975 Sep;36(3):1029–33. doi: 10.1002/1097-0142(197509)36:3<1029::aid-cncr2820360328>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 20.Farrell GC. Fanconi’s familial hypoplastic anaemia with some unusual features. Med J Aust. 1976 Jan 31;1(5):116–8. doi: 10.5694/j.1326-5377.1976.tb109278.x. [DOI] [PubMed] [Google Scholar]
- 21.Mulvihill JJ, Ridolfi RL, Schultz FR, Borzy MS, Haughton PB. Hepatic adenoma in Fanconi anemia treated with oxymetholone. J Pediatr. 1975 Jul;87(1):122–4. doi: 10.1016/s0022-3476(75)80087-4. [DOI] [PubMed] [Google Scholar]
- 22.Bioulac-Sage P, Laumonier H, Couchy G, Le BB, Sa CA, Rullier A, Laurent C, Blanc JF, Cubel G, Trillaud H, Zucman-Rossi J, Balabaud C, Saric J. Hepatocellular adenoma management and phenotypic classification: the Bordeaux experience. Hepatology. 2009 Aug;50(2):481–9. doi: 10.1002/hep.22995. [DOI] [PubMed] [Google Scholar]
- 23.Guy JT, Auslander MO. Androgenic steroids and hepatocellular carcinoma. Lancet. 1973 Jan 20;1(7795):148. doi: 10.1016/s0140-6736(73)90212-2. [DOI] [PubMed] [Google Scholar]