

Abstract
Background
Despite growing recognition of an etiologic role for inflammation in lung carcinogenesis, few prospective epidemiologic studies have comprehensively investigated the association of circulating inflammation markers with lung cancer.
Methods
We conducted a nested case–control study (n = 526 lung cancer patients and n = 592 control subjects) within the Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial. Control subjects were matched to lung cancer case patients on age, sex, follow-up time (median = 2.9 years), randomization year, and smoking (pack-years and time since quitting). Serum levels of 77 inflammation markers were measured using a Luminex bead-based assay. Conditional logistic regression and weighted Cox models were used to estimate odds ratios (ORs) and cumulative risks, respectively.
Results
Of 68 evaluable markers, 11 were statistically significantly associated with lung cancer risk (P trend across marker categories < .05), including acute-phase proteins (C-reactive protein [CRP], serum amyloid A [SAA]), proinflammatory cytokines (soluble tumor necrosis factor receptor 2 [sTNFRII]), anti-inflammatory cytokines (interleukin 1 receptor antagonist [IL-1RA]), lymphoid differentiation cytokines (interleukin 7 [IL-7]), growth factors (transforming growth factor alpha [TGF-A]), and chemokines (epithelial neutrophil-activating peptide 78 [ENA 78/CXCL5], monokine induced by gamma interferon [MIG/CXCL9], B cell–attracting chemokine 1 [BCA-1/CXCL13], thymus activation regulated chemokine [TARC/CCL17], macrophage-derived chemokine [MDC/CCL22]). Elevated marker levels were associated with increased lung cancer risk, with odds ratios comparing the highest vs the lowest group ranging from 1.47 (IL-7) to 2.27 (CRP). For IL-1RA, elevated levels were associated with decreased lung cancer risk (OR = 0.71; 95% confidence interval = 0.51 to 1.00). Associations did not differ by smoking, lung cancer histology, or latency. A cross-validated inflammation score using four independent markers (CRP, BCA-1/CXCL13, MDC/CCL22, and IL-1RA) provided good separation in 10-year lung cancer cumulative risks among former smokers (quartile [Q] 1 = 1.1% vs Q4 = 3.1%) and current smokers (Q1 = 2.3% vs Q4 = 7.9%) even after adjustment for smoking.
Conclusions
Some circulating inflammation marker levels are associated with prospective lung cancer risk.
Lung cancer is the most common cause of cancer-related death in the United States, with 160340 deaths estimated in 2012 (1). The predominant risk factor for lung cancer is smoking, accounting for approximately 90% of these lung cancer deaths (2). Additionally, lung cancer risk is associated with several indicators of inflammation, including pulmonary fibrosis (3–6), chronic obstructive pulmonary disease (7,8), and chronic pulmonary infections (9–12), even after taking the effects of smoking into consideration. Further, polymorphisms in inflammation genes have been associated with lung cancer risk (13–15). However, few prospective epidemiologic studies have investigated the association of circulating inflammation markers with lung cancer risk.
Most prior studies addressing inflammation markers have been small in size, retrospective in design, or have focused on a few candidate markers. Our group, along with others, has previously studied the association between C-reactive protein (CRP), interleukin 6 (IL-6), and interleukin 8 (IL-8) (16–21) and lung cancer risk, but these markers represent a small subset of the inflammation cascade. The inflammation process is a complex response to stimuli involving the interplay of host cells and signaling molecules, such as proinflammatory and anti-inflammatory cytokines, growth and angiogenesis factors, and chemokines (22–24). No prior study has comprehensively measured a wide range of these inflammation markers in relation to lung cancer risk.
Investigating the role of inflammation in lung carcinogenesis could have relevance for prevention and screening. In addition to providing etiologic insight, the discovery of circulating inflammation markers prospectively associated with lung cancer could aid in identifying individuals at highest lung cancer risk. In this study nested within the Prostate, Lung, Colorectal, and Ovarian (PLCO) Cancer Screening Trial (25,26), we investigated the association of circulating levels of several key components of inflammation, including acute-phase proteins, pro- and anti-inflammatory cytokines, chemokines, growth factors, and angiogenesis factors, with prospective lung cancer risk (27).
Methods
Study Population
We conducted a nested case–control study within the screening arm of the PLCO trial, which recruited approximately 155000 participants aged 50 to 74 years from the general population during the period from 1992 to 2001 (25). Lung cancer screening included a chest x-ray at baseline, followed by three annual screens for smokers or two annual screens for never smokers (26). In addition to demographic, behavioral, and dietary information, blood samples were obtained at baseline and five subsequent annual visits. Lung cancers were ascertained through annual questionnaires and confirmed by medical chart abstraction and death certificate review (25,26). This study was approved by the institutional review boards at each screening center, and all participants gave informed consent.
Of 898 lung cancers that occurred through December 31, 2004, 526 lung cancer case patients were included in this study. We restricted our study to case patients with lung cancers that occurred through 2004 to retain the case–control selection used in our prior studies of inflammation and lung cancer (6,14,16,19,28). Reasons for exclusion of lung cancer case patients were missing/incomplete smoking information, history of cancer, multiple cancers during follow-up, unavailable serum, or missing consent for etiologic studies. Five hundred ninety-two control subjects were matched with lung cancer case patients on age at randomization (55–59, 60–64, 65–69, 70–74 years), sex, randomization year (1993–1995, 1996–1997, 1998–1999, 2000–2001), follow-up time (1-year intervals), smoking status (current, former, never), cumulative smoking at baseline for ever-smokers (0–29, 30–39, 40–49, ≥50 pack-years), and time since quitting for former smokers (<15 and ≥15 years). One control subject was selected for each current or former smoker case patient and three control subjects were selected for each never smoker case patient.
Laboratory Methods
Serum specimens collected at baseline (processed at 2400–3000rpm for 15 minutes, frozen within 2 hours of collection, stored at −80°C) were used to measure circulating levels of 77 markers (Supplementary Table 1, available online). These markers were selected based on a recent methodologic study that evaluated the performance and reproducibility of multiplexed assays for measurement of inflammation markers in serum (27). Markers were measured using a Luminex bead-based assay (Millipore Inc, Billerica, MA). Concentrations were calculated using either a four- or five-parameter standard curve. Serum samples were assayed in duplicate and averaged to calculate concentrations. Case patients and matched control subjects were included on the same analytical batch. Each case–control pair was plated into adjacent wells on each batch. We assayed 34 pairs of blinded duplicates within the same batch and 17 pairs of blinded duplicates across different batches to evaluate assay reproducibility through coefficients of variation and intraclass correlation coefficients calculated on log-transformed values of the markers. Nine markers with more than 90% of values below the lowest limit of detection (LLOD) were excluded from all analyses, resulting in 68 evaluable markers. Log-transformed intraclass correlation coefficients were greater than 0.8 in 90% of these markers (Supplementary Table 1, available online).
Statistical Analyses
Markers were analyzed as both continuous (log-transformed) and categorical variables. Measurements below the LLOD were assigned a value of half the LLOD. Marker levels were categorized into groups based on the proportion of individuals with measurements less than LLOD as follows: markers with less than 25% of individuals below the LLOD (n = 38 markers) were categorized into quartiles (based on the distribution among control subjects); markers with 25% to 50% of individuals below the LLOD (n = 5 markers) were categorized as less than LLOD and tertiles of detectable measurements; markers with 50% to 75% of individuals below the LLOD (n = 7 markers) were categorized as less than LLOD and below and above the median; and markers with 75% to 90% of individuals below the LLOD (n = 18 markers) were categorized as undetectable and detectable.
Conditional logistic regression was used to calculate odds ratios (ORs) and 95% confidence intervals (CIs) for the association of each marker with lung cancer risk. In addition to the matching variables, we adjusted all models for history of chronic bronchitis/emphysema, history of coronary heart disease or heart attack, family history of lung cancer, regular use of aspirin/ibuprofen, body mass index, race, and education. We also stratified analyses by latency (ie, time from serum collection to lung cancer diagnosis, categorized as <2 years and ≥2 years), histology (squamous cell carcinoma, adenocarcinoma, small cell carcinoma, and other histologies), and smoking status (never, former, current). Multiplicative statistical interactions with latency and smoking and heterogeneity across histologies were evaluated using the Wald test.
To calculate an inflammation score for each individual, we built a conditional logistic regression model with simultaneous adjustment for all markers statistically significantly associated with lung cancer (log-transformed) and used backwards step-wise regression to identify markers that retained statistical significance (P < .05). Using these markers, we estimated an inflammation score for each individual through five-fold cross-validation (29). Briefly, we divided the 526 matched case–control pairs into five equal groups, estimated regression coefficients for each marker using data from four groups, and calculated an inflammation score (sum of the product of each regression coefficient with the respective marker level) in the fifth group. In each of the five replicates, we classified individuals into quartiles based on the distribution of the inflammation score among control subjects.
Using the inflammation score, we estimated 10-year cumulative risks of lung cancer stratified by smoking status. These cumulative risks were calculated in weighted Cox regression models and standardized to the age, sex, and smoking (pack-years and time since quit) distributions of the 50862 PLCO screening arm participants eligible for selection into our study (16,30,31). Additional details are presented in the Supplementary Data (available online). The proportional hazards assumption was assessed graphically.
Among control subjects, we conducted ordinal logistic regression analyses to investigate the association of each inflammation marker and the inflammation score with smoking. These models were adjusted for age, sex, history of chronic bronchitis/emphysema, history of coronary heart disease or heart attack, family history of lung cancer, use of aspirin/ibuprofen, body mass index, race, and education. Analyses were not stratified by sex or racial/ethnic group.
We assessed statistical significant at P less than .05. Additionally, we highlight those associations that met Bonferroni significance (P trends < .0007; P = .05/68 markers).
All tests of statistical significance were two-sided. Analyses were carried out in SAS version 9.2 (SAS Inc, Cary, NC) and R version 2.15.2 (R Project for Statistical Computing, Vienna, Austria).
Results
The 526 lung cancer patients and 592 control subjects had similar distributions of the matching factors (Table 1). Case patients were statistically significantly more likely to have lower education, history of bronchitis/emphysema, or a family history of lung cancer. The median time from serum collection to lung cancer diagnosis/control selection was 2.9 years (interquartile range = 1.1–5.1 years).
Table 1.
Characteristics of lung cancer case patients and control subjects
| Characteristic | Control subjects (n = 592) | Lung cancer case patients (n = 526) | P* |
|---|---|---|---|
| Age at randomization, years | —† | ||
| <59 | 104 (17.6) | 96 (18.3) | |
| 60–64 | 167 (28.2) | 146 (27.8) | |
| 65–69 | 192 (32.4) | 177 (33.7) | |
| ≥70 | 129 (21.8) | 107 (20.3) | |
| Sex | —† | ||
| Female | 212 (35.8) | 168 (31.9) | |
| Male | 380 (64.2) | 358 (68.1) | |
| Smoking status | —† | ||
| Never | 99 (16.7) | 33 (6.3) | |
| Former | 294 (49.7) | 294 (55.9) | |
| Current | 199 (33.6) | 199 (37.8) | |
| Pack-years smoked | —† | ||
| Never smokers | 99 (16.7) | 33 (6.3) | |
| <30 | 124 (21.0) | 125 (23.8) | |
| 30–40 | 128 (21.6) | 128 (24.3) | |
| 40–50 | 42 (7.1) | 41 (7.8) | |
| ≥50 | 199 (33.6) | 199 (37.8) | |
| Years since quitting smoking | —† | ||
| Not applicable | 298 (50.3) | 232 (44.1) | |
| <15 | 187 (31.6) | 187 (35.6) | |
| ≥15 | 107 (18.1) | 107 (20.3) | |
| Race | .11 | ||
| White | 538 (90.9) | 467 (88.8) | |
| Black | 28 (4.7) | 40 (7.6) | |
| Other | 26 (4.4) | 19 (3.6) | |
| Education | .02 | ||
| ≤12 years/completed high school | 199 (33.6) | 211 (40.1) | |
| >12 years | 393 (66.4) | 315 (59.9) | |
| Body mass index, kg/m2 | .53 | ||
| <25 | 198 (33.5) | 195 (37.1) | |
| 25–29.9 | 270 (45.6) | 224 (42.6) | |
| ≥30 | 114 (19.3) | 101 (19.2) | |
| Missing | 10 (1.7) | 6 (1.1) | |
| History of emphysema or chronic bronchitis | <.0001 | ||
| No | 532 (89.9) | 428 (81.4) | |
| Yes | 60 (10.1) | 98 (18.6) | |
| History of coronary heart disease or heart attack | .14 | ||
| No | 519 (87.7) | 445 (84.6) | |
| Yes | 73 (12.3) | 81 (15.4) | |
| Family history of lung cancer | <.0001 | ||
| No | 508 (85.8) | 398 (75.7) | |
| Yes | 77 (13.0) | 123 (23.4) | |
| Missing | 7 (1.2) | 5 (1.0) | |
| Regularly uses aspirin/ibuprofen | .66 | ||
| No | 218 (36.8) | 187 (35.6) | |
| Yes | 374 (63.2) | 339 (64.5) |
* Two-sided P value was estimated with the χ2 test.
† Matching variable. The distribution of case patients and control subjects appear different because of matching three control subject per case patient for never smokers and one control subject per case patient for former and current smokers.
Of the 68 markers, 11 were statistically significantly associated with lung cancer risk (Table 2; Supplementary Table 2, available online). These 11 markers included acute-phase proteins (CRP, serum amyloid A [SAA]), proinflammatory cytokines (soluble tumor necrosis factor receptor 2 [sTNFRII]), anti-inflammatory cytokines (interleukin 1 receptor antagonist [IL-1RA]), lymphoid differentiation cytokines (interleukin 7 [IL-7]), growth factors (transforming growth factor alpha [TGF-A]), and several chemokines (epithelial neutrophil-activating peptide 78 [ENA 78/CXCL5], monokine induced by gamma interferon [MIG/CXCL9], B cell–attracting chemokine 1 [BCA-1/CXCL13], thymus activation regulated chemokine [TARC/CCL17], macrophage-derived chemokine [MDC/CCL22]). For a majority of these markers, elevated levels were associated with statistically significantly increased lung cancer risk, with odds ratios (comparing highest vs lowest category) ranging from 1.47 for IL-7 to 2.27 for CRP (P trend across marker categories = .05 to <.001). In contrast, elevated levels of IL-1RA were associated with statistically significantly decreased lung cancer risk (OR = 0.71; 95%CI = .51 to 1.00). Associations for CRP and SAA retained statistical significance after Bonferroni correction.
Table 2.
Circulating inflammation markers statistically significantly associated with lung cancer risk*
| Years from sample until selection | ||||||
|---|---|---|---|---|---|---|
| Marker and level, pg/mL | Lung cancer case patients | Control subjects | OR (95% CI) | <2 years | ≥2 years | P interaction‡ |
| Acute-phase proteins | ||||||
| CRP | ||||||
| <1845800 | 84 | 147 | 1.0 | 1.0 | 1.0 | .92 |
| 1845800–4316899 | 131 | 148 | 1.58 (1.06 to 2.35) | 1.32 (0.66 to 2.65) | 1.76 (1.07 to 2.91) | |
| 4316900–9611899 | 134 | 148 | 1.69 (1.14 to 2.51) | 1.65 (0.81 to 3.38) | 1.79 (1.10 to 2.91) | |
| ≥9611900 | 176 | 147 | 2.27 (1.51 to 3.41) | 2.47 (1.18 to 5.17) | 2.20 (1.33 to 3.65) | |
| P trend† | <.001 | .01 | .005 | |||
| SAA | ||||||
| <1780800 | 100 | 147 | 1.0 | 1.0 | 1.0 | .86 |
| 1780800–3339199 | 111 | 148 | 1.21 (0.83 to 1.77) | 1.39 (0.71 to 2.73) | 1.19 (0.73 to 1.93) | |
| 3339200–6591899 | 136 | 148 | 1.59 (1.08 to 2.33) | 1.73 (0.85 to 3.51) | 1.49 (0.92 to 2.40) | |
| ≥6591900 | 178 | 147 | 2.18 (1.48 to 3.22) | 2.80 (1.45 to 5.41) | 2.04 (1.22 to 3.42) | |
| P trend† | <.001 | .002 | .004 | |||
| Proinflammatory cytokines | ||||||
| sTNFRII | ||||||
| <3770 | 114 | 148 | 1.0 | 1.0 | 1.0 | .29 |
| 3770–4785 | 127 | 148 | 1.27 (0.89 to 1.81) | 2.64 (1.33 to 5.23) | 0.88 (0.57 to 1.37) | |
| 4786–6191 | 138 | 148 | 1.35 (0.93 to 1.97) | 2.30 (1.14 to 4.64) | 1.01 (0.64 to 1.61) | |
| ≥6192 | 147 | 148 | 1.50 (1.01 to 2.21) | 2.32 (1.11 to 4.84) | 1.18 (0.73 to 2.91) | |
| P trend† | .05 | .03 | .41 | |||
| Anti-inflammatory cytokines | ||||||
| IL-1RA | ||||||
| Undetectable | 453 | 474 | 1.0 | 1.0 | 1.0 | .63 |
| Detectable | 73 | 118 | 0.71 (0.51 to 1.00) | 0.74 (0.40 to 1.37) | 0.68 (0.45 to 1.02) | |
| P | .05 | .34 | .06 | |||
| Lymphoid differentiation cytokines | ||||||
| IL-7 | ||||||
| Undetectable | 420 | 497 | 1.0 | 1.0 | 1.0 | .56 |
| Detectable | 106 | 95 | 1.47 (1.05 to 2.06) | 1.37 (0.76 to 2.48) | 1.55 (1.02 to 2.36) | |
| P | .02 | .30 | .04 | |||
| Growth factors | ||||||
| TGF-A | ||||||
| <0.67 | 100 | 148 | 1.0 | 1.0 | 1.0 | .67 |
| 0.67–1.67 | 125 | 148 | 1.26 (0.86 to 1.84) | 1.33 (0.64 to 2.75) | 1.25 (0.79 to 1.98) | |
| 1.68–3.20 | 143 | 149 | 1.40 (0.96 to 2.05) | 0.95 (0.48 to 1.88) | 1.71 (1.07 to 2.72) | |
| ≥3.21 | 158 | 147 | 1.56 (1.07 to 2.27) | 1.45 (0.74 to 2.82) | 1.60 (1.00 to 2.54) | |
| P trend† | .02 | .42 | .02 | |||
| Chemokines | ||||||
| ENA78/CXCL5 | ||||||
| <554 | 100 | 148 | 1.0 | 1.0 | 1.0 | .85 |
| 554–882 | 148 | 148 | 1.43 (1.01 to 2.03) | 1.99 (1.01 to 3.90) | 1.28 (0.84 to 1.94) | |
| 883–1265 | 115 | 148 | 1.13 (0.79 to 1.63) | 1.19 (0.62 to 2.31) | 1.09 (0.70 to 1.71) | |
| ≥1266 | 163 | 148 | 1.68 (1.15 to 2.45) | 1.97 (1.00 to 3.87) | 1.59 (1.00 to 2.53) | |
| P trend† | .03 | .15 | .10 | |||
| MIG/CXCL9 | ||||||
| <364 | 123 | 148 | 1.0 | 1.0 | 1.0 | .33 |
| 364–495 | 102 | 148 | 0.92 (0.63 to 1.34) | 1.32 (0.67 to 2.57) | 0.76 (0.47 to 1.21) | |
| 496–765 | 140 | 148 | 1.38 (0.95 to 2.00) | 3.52 (1.65 to 7.51) | 1.02 (0.65 to 1.60) | |
| ≥766 | 161 | 148 | 1.63 (1.12 to 2.36) | 2.29 (1.17 to 4.52) | 1.39 (0.87 to 2.20) | |
| P trend† | .003 | .007 | .09 | |||
| BCA-1/CXCL13 | ||||||
| <13.6 | 113 | 148 | 1.0 | 1.0 | 1.0 | .53 |
| 13.6–18.5 | 112 | 148 | 0.97 (0.67 to 1.40) | 1.03 (0.52 to 2.06) | 0.88 (0.55 to 1.38) | |
| 18.6–25.6 | 123 | 149 | 0.99 (0.68 to 1.45) | 0.89 (0.43 to 1.84) | 0.96 (0.60 to 1.52) | |
| ≥25.7 | 178 | 147 | 1.59 (1.10 to 2.29) | 1.89 (0.96 to 3.70) | 1.45 (0.93 to 2.27) | |
| P trend† | .01 | .08 | .08 | |||
| TARC/CCL17 | ||||||
| <73.5 | 106 | 148 | 1.0 | 1.0 | 1.0 | .79 |
| 73.5–115.0 | 121 | 148 | 1.16 (0.80 to 1.67) | 0.98 (0.51 to 1.89) | 1.17 (0.76 to 1.83) | |
| 115.1–168.6 | 119 | 148 | 1.09 (0.76 to 1.57) | 1.03 (0.55 to 1.96) | 1.05 (0.69 to 1.65) | |
| ≥168.7 | 180 | 148 | 1.50 (1.06 to 2.13) | 1.24 (0.66 to 2.36) | 1.55 (1.06 to 2.36) | |
| P trend† | .03 | .46 | .06 | |||
| MDC/CCL22 | ||||||
| <899.0 | 111 | 148 | 1.0 | 1.0 | 1.0 | .49 |
| 899.0–1137.2 | 114 | 148 | 1.13 (0.78 to 1.65) | 1.17 (0.59 to 2.31) | 1.13 (0.71 to 1.79) | |
| 1137.3–1387.0 | 106 | 148 | 0.96 (0.66 to 1.41) | 1.04 (0.53 to 2.04) | 0.94 (0.59 to 1.50) | |
| ≥1,387.1 | 195 | 148 | 1.72 (1.18 to 2.50) | 1.42 (0.73 to 2.74) | 1.89 (1.19 to 3.00) | |
| P trend† | .009 | .36 | .01 | |||
* Odds ratios (ORs) and 95% confidence intervals (CIs) were estimated in conditional logistic regression models. Models were adjusted for matching criteria, personal history of bronchitis/emphysema, history of coronary heart disease or heart attack, family history of lung cancer, use of aspirin/ibuprofen, body mass index, race, and education. BCA-1/CXCL13 = B cell–attracting chemokine 1; CRP = C-reactive protein; ENA78/CXCL5 = epithelial neutrophil-activating peptide 78; IL-1RA = interleukin 1 receptor antagonist; IL-7 = interleukin 7; MDC/CCL22 = macrophage-derived chemokine; MIG/CXCL9= monokine induced by gamma interferon; SAA = serum amyloid A; sTNFRII = soluble tumor necrosis factor receptor 2; TARC/CCL17 = thymus activation regulated chemokine; TGF-A = transforming growth factor alpha.
† Two-sided P values for trend across marker categories were assessed with the Wald test using marker levels as an ordinal variable with 1 degree of freedom.
‡ Two-sided P values for interactions were estimated with a cross-product term between latency and categories of markers treated as an ordinal variable and assessed with the Wald test.
No statistically significant difference was observed in the associations between marker levels and lung cancer across latency (Table 2) (all P interaction > .05), histology (Supplementary Table 3, available online) (all P heterogeneity > .05), or smoking (Figure 1, A–C) (all P interaction > .05), with the exception of statistically significant differences by smoking for IL-1RA and by histology for TGF-A. Notably, despite a small number of lung cancers among never smokers (n = 30), nine markers were statistically significantly associated with lung cancer among never smokers, including ENA-78/CXCL5 and IL-7, which were both associated with lung cancer overall, and GCP2/CXCL6, G-CSF, IL-6, MIP-1B/CCL4, MCP-2/CCL8, MCP-4/CCL13, and stromal cell–derived factor-1 (SDF-1 A-B/CXCL12), which were not associated with lung cancer overall (Supplementary Table 4, available online).
Figure 1.
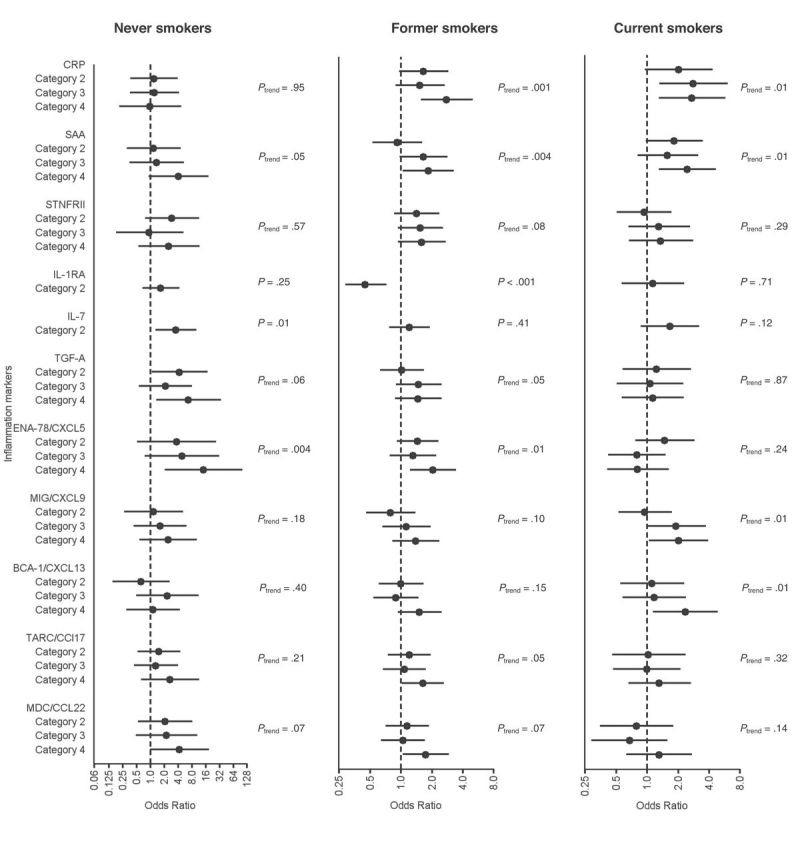
Associations of 11 inflammation markers with lung cancer risk stratified by smoking status. Odds ratios and 95% confidence intervals comparing the highest level vs lowest level from conditional logistic regression models are shown. All models were adjusted for matching factors (age, sex, follow-up time, randomization year, pack-years of smoking, and time since quitting) as well as for personal history of bronchitis/emphysema, history of coronary heart disease or heart attack, family history of lung cancer, use of aspirin/ibuprofen, body mass index, race, and education. P values for trend across marker categories were estimated through the use of markers as ordinal variables with 1 degree of freedom. Multiplicative statistical interactions of inflammation marker associations across smoking status were evaluated using the Wald test. Statistically significant differences by smoking status were only observed for the association between interleukin 1 receptor antagonist (IL-1RA) and lung cancer. All P values were two-sided. BCA-1/CXCL13 = B cell–attracting chemokine 1; CRP = C-reactive protein; ENA78/CXCL5 = epithelial neutrophil-activating peptide 78; IL-7 = interleukin 7; MDC/CCL22 = macrophage-derived chemokine; MIG/CXCL9= monokine induced by gamma interferon; SAA = serum amyloid A; sTNFRII = soluble tumor necrosis factor receptor 2; TARC/CCL17 = thymus activation regulated chemokine; TGF-A = transforming growth factor alpha.
Of the 11 markers statistically significantly associated with lung cancer risk, four (CRP, BCA-1/CXCL-13, MDC/CCL22, and IL-1RA) retained statistical significance in backward step-wise regression. Elevated levels of the cross-validated inflammation score based on these four markers were associated with a 2.8-fold increased lung cancer risk (Table 3) (OR quartile [Q] 4 vs Q1 = 2.79; 95%CI = 1.88 to 4.14; P trend < .001). When analyses were reweighted to the screening arm cohort, 10-year cumulative risks of lung cancer increased from 1.1% for former smokers with inflammation score in Q1 to 3.1% for former smokers in Q4 and from 2.3% for current smokers in Q1 to 7.9% for current smokers in Q4, after accounting for smoking exposures (Figure 2).
Table 3.
Association of inflammation score with lung cancer risk*
| Category | Q1 | Q2 OR (95% CI) | Q3 OR (95% CI) | Q4 OR (95% CI) | P trend† | P interaction/ P hererogeneity‡ |
|---|---|---|---|---|---|---|
| Overall | 1.0 | 1.51 (1.03 to 2.22) | 1.28 (0.85 to 1.93) | 2.79 (1.89 to 4.15) | <.001 | NA |
| Latency | ||||||
| <2 years | 1.0 | 1.34 (0.66 to 2.71) | 1.48 (0.70 to 3.16) | 2.61 (1.27 to 5.37) | .005 | |
| ≥2 years | 1.0 | 1.65 (1.03 to 2.65) | 1.19 (0.73 to 1.96) | 2.98 (1.84 to 4.82) | <.001 | .80 |
| Smoking | ||||||
| Never smokers | 1.0 | 1.88 (0.68 to 5.20) | 1.09 (0.26 to 4.59) | 0.95 (0.19 to 4.79) | .96 | |
| Former smokers | 1.0 | 1.54 (0.93 to 2.53) | 1.30 (0.76 to 2.21) | 3.14 (1.87 to 5.30) | <.001 | |
| Current smokers | 1.0 | 2.17 (0.85 to 5.53) | 1.93 (0.77 to 4.84) | 4.07 (1.72 to 9.62) | <.001 | .12 |
| Histology | ||||||
| AC | 1.0 | 1.67 (0.98 to 2.86) | 1.18 (0.66 to 2.11) | 3.00 (1.68 to 5.37) | .001 | |
| SCC | 1.0 | 0.86 (0.32 to 2.35) | 0.70 (0.25 to 2.02) | 1.45 (0.61 to 3.47) | .25 | |
| Small cell | 1.0 | 1.54 (0.49 to 4.81) | 0.86 (0.24 to 3.15) | 2.28 (0.66 to 7.85) | .21 | |
| Large cell | 1.0 | —§ | —§ | —§ | —§ | |
| Other/unspecified | 1.0 | 2.08 (0.51 to 8.45) | 2.94 (0.87 to 9.96) | 8.83 (2.28 to 34.3) | .001 | .68 |
* The inflammation score was estimated through fivefold cross-validation and was based on four markers: C-reactive protein (CRP), B cell–attracting chemokine 1 (BCA-1/CXCL13), macrophage-derived chemokine (MDC/CCL22), and interleukin 1 receptor antagonist (IL-1RA). Odds ratios (ORs) and 95% confidence intervals (CIs) were estimated in conditional logistic regression models. Models were adjusted for matching criteria, personal history of bronchitis/emphysema, history of coronary heart disease or heart attack, family history of lung cancer, use of aspirin/ibuprofen, body mass index, race, and education. NA = not applicable.
† Two sided P values for trend across inflammation score categories were assessed with the Wald test using score categories as an ordinal variable with 1 degree of freedom.
‡ Two sided P values for interaction were assessed with the Wald test through product terms for the respective covariate with inflammation score (modeled as an ordinal variable). Two-sided P values for heterogeneity were assessed using the Wald test.
§ Not estimable.
Figure 2.

Ten-year standardized cumulative risks of lung cancer separately among never smokers (NS; black lines), former smokers (FS; blue lines), and current smokers (CS; red lines) across an inflammation score which was based on four independent markers (C-reactive protein [CRP], B cell–attracting chemokine 1 [BCA-1/CXCL13], MDC/CCL22 [macrophage-derived chemokine], and interleukin 1 receptor antagonist [IL-1RA]). Cumulative risks were estimated using weighted Cox regression models and were standardized to the age, sex, and smoking (pack-years and time since quit) distributions of the 50862 Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial screening arm participants eligible for selection into our case–control study. The figure also shows relative risks of lung cancer for each inflammation score quartile (Q) and smoking status compared with never smokers with inflammation score in the fourth quartile. The cumulative risks of lung cancer estimated for never smokers in the first, second, and third quartiles of the inflammation score were 0.17%, 0.26%, and 0.12%, respectively. Overall estimated 10-year cumulative risks of lung cancer were 0.16% in never smokers, 1.8% in former smokers, and 5.0% in current smokers (data not shown in figure).
Among control subjects, compared with never smokers, seven markers were statistically significantly elevated in former or current smokers (including CRP, TGF-A, MIG/CXCL9, MDC/CCL22, and TARC/CCL17), and 13 markers were statistically significantly decreased in former or current smokers (including IL-1RA) (Supplementary Table 5, available online). Of the markers associated with lung cancer risk, CRP, SAA, and TGF-A were statistically significantly elevated among individuals with bronchitis/emphysema (Supplementary Table 6, available online). Levels of the inflammation score were statistically significantly higher among former (OR = 2.17; 95% CI = 1.51 to 3.13) and current smokers (OR = 5.10; 95% CI = 3.43 to 7.57) and among those with a history of chronic bronchitis or emphysema (OR = 1.62; 95% CI = 1.18 to 2.22).
Discussion
In the first comprehensive investigation of circulating inflammation markers and prospective lung cancer risk, our key observation was that 11 markers that represent several components of the inflammation process— acute-phase proteins, pro- and anti-inflammatory cytokines, chemokines, and growth factors— were associated with lung cancer risk. Additionally, an inflammation score based on four independent markers (CRP, BCA-1/CXCL13, MDC/CCL22, and IL-1RA) provided good separation in 10-year cumulative risks of lung cancer. Importantly, these associations were present after careful adjustment for smoking, and elevated marker levels preceded lung cancer diagnosis by several years. Our results go beyond previously reported single-marker associations by implicating several classes of inflammation markers in lung carcinogenesis.
The markers we found to be independently associated with increased lung cancer risk included CRP, an acute-phase protein whose levels rapidly increase in response to most forms of inflammation, and BCA-1/CXCL13 and MDC/CCL22, chemokines that regulate the migration of B- and T lymphocytes and natural killer cells to sites of inflammation. Additionally, IL-1RA, an anti-inflammatory cytokine that antagonizes the proinflammatory effects of IL-1, was associated with decreased lung cancer risk. Previously reported associations of CRP with lung cancer (20), laboratory studies of increased cancer progression among IL-1RA–deficient mice (32), and studies of high expression of BCA-1/CXCL13 and MDC/CCL22 and their respective receptors in lung tumors (33,34) all lend biologic support for our observed associations. The other markers found to be associated with lung cancer, albeit not independently of the four markers noted above, included acute-phase proteins (SAA), proinflammatory cytokines (sTNFRII), lymphocyte differentiation cytokines (IL-7), chemokines (ENA-78/CXCL5, MIG/CXCL9, TARC/CCL17), and regulators of tumor growth and angiogenesis (TGF-A, ENA-78/CXCL5, MIG/CXCL9). The observation that several classes of inflammation markers produced by different cell types (epithelial, endothelial, and innate and adaptive immune cells) are associated with lung cancer risk collectively underscores the tumor-promoting role of inflammation and the role of the tumor microenvironment in lung carcinogenesis (22–24,35).
Our use of serum specimens collected before lung cancer diagnosis argues against reverse causation (ie, that the presence of undiagnosed lung cancer influenced inflammation marker levels). We did not observe statistically significant differences in our associations with time since serum collection, and several markers (CRP, SAA, IL-7, TGF-A, TARC/CCL17, and MDC/CCL22) and the inflammation score were associated with lung cancers diagnosed 2 or more years after serum collection. Nevertheless, because the serum specimens we used were collected a median of 2.9 years before lung cancer diagnosis, the markers we found to be associated with lung cancer risk perhaps reflect a mix of etiologically relevant factors that promote lung cancer as well as markers that reflect the presence of lung cancer precursor states or preclinical lung cancer. This consideration of etiologic markers vs early disease markers notwithstanding, we found good separation in 10-year cumulative risks of lung cancer across the inflammation score, providing preliminary evidence for the potential utility of inflammation markers for lung cancer risk stratification.
Although smoking is a known cause of pulmonary inflammation, our study design and observations argue against confounding by smoking (36). We matched control subjects to case patients on several metrics—smoking status, pack-years, and time since quitting—ensuring comparable smoking exposures between case patients and control subjects. Additionally, for several markers and the inflammation score, we found statistically significant associations with lung cancer risk even among former smokers who quit more than 15 years before lung cancer diagnosis. Therefore, we believe our results indicate that interindividual variability in inflammatory responses to similar smoking exposures is associated with lung cancer risk.
We found nine inflammation markers associated with lung cancer risk among never smokers, including some very strong associations (ORs = 2.5–14.4). Although these results should be interpreted with caution because of the limited number of never smokers in our study, given the lack of identified risk factors/biomarkers for lung cancer among never smokers (37), they warrant follow-up in larger studies.
The main strengths of our study include the population-based, prospective design of the PLCO cohort, measurement of inflammation markers using a validated technology, and careful control for cigarette smoking and other confounders.
We also note several limitations. Despite strong biologic plausibility for the association of inflammation markers with lung cancer, our observations need replication given the large number of markers evaluated. We identified 11 statistically significant associations in contrast with four expected associations by chance (type I error rate of 5% for 68 markers). Nonetheless, only CRP and SAA retained statistical significance at conservative Bonferroni thresholds. Further reflecting the need for replication, using the same case patients and control subjects, we previously reported that elevated levels of CRP [measured using a chemiluminescence assay (16)], and IL-6 and IL-8 [measured using high-sensitivity enzyme-linked immunosorbent assay (19)] were associated with increased lung cancer risk. Although our current study replicated the CRP association, we failed to find statistically significant associations for IL-6 and IL-8, likely because of low detectability (26.1% for IL-6) and/or poor reproducibility in this study (coefficients of variation >20%). Further, we measured markers at only one time point. There are little data on the stability of these marker levels over time, with the exception of CRP, which has been shown to be moderately stable (38). To the extent that the temporal stability of inflammation marker levels is moderate or low, we may have underestimated or missed associations of inflammation markers with lung cancer risk. Despite the large number of lung cancers, our stratified analyses by latency, histology, and smoking were underpowered. Finally, the degree to which pulmonary inflammation contributes to elevated circulating inflammation marker levels is unknown. Nonetheless, we found that control subjects with bronchitis/emphysema had statistically significantly higher levels of CRP, SAA, TGF-A, and a higher inflammation score, lending support to the idea that circulating inflammation marker levels partly reflect pulmonary inflammation (39).
In conclusion, our study extends observations from laboratory studies and single-marker association studies by providing epidemiologic evidence for the association of acute-phase proteins, pro- and anti-inflammatory cytokines, chemokines, and growth factors with prospective lung cancer risk. The separation in lung cancer cumulative risks across the inflammation score among current and former smokers provides preliminary evidence for the potential utility of inflammation markers in lung cancer risk stratification. Indeed, despite substantial reductions in lung cancer mortality through low-dose computed tomography screening (40), the high false-positivity rate of this screening modality underscores the need for risk stratification factors beyond smoking behaviors. Pending replication of our results, it remains to be seen whether the addition of inflammation markers to lung cancer risk prediction models (41–44) aids in the identification of individuals at highest lung cancer risk for targeted prevention and screening efforts.
Funding
This research was supported by the Intramural Research Program of the Division of Cancer Epidemiology and Genetics and by contracts from the Division of Cancer Prevention, National Cancer Institute, National Institutes of Health, Department of Health and Human Services.
Supplementary Material
The sponsor reviewed and approved final submission, but did not have a role in design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation of the manuscript or the decision to submit for publication.
We acknowledge Craig Williams, Michael Furr, and Michael Curry of Information Management Services, Inc. who were compensated for statistical programming and Marcus Williams who was compensated for laboratory support.
References
- 1. American Cancer Society Cancer Facts and Figures 2012. American Cancer Society; 2012 [Google Scholar]
- 2. American Cancer Society “Cigarette Smoking.” Available at: http://www.cancer.org/cancer/cancercauses/tobaccocancer/cigarettesmoking/cigarette-smoking-who-and-how-affects-health Last updated January 17, 2013. Accessed on October 11, 2013
- 3. Harris JM, Johnston ID, Rudd R, Taylor AJ, Cullinan P. Cryptogenic fibrosing alveolitis and lung cancer: the BTS study. Thorax. 2010;65(1):70–76 [DOI] [PubMed] [Google Scholar]
- 4. Hubbard R, Venn A, Lewis S, Britton J. Lung cancer and cryptogenic fibrosing alveolitis. A population-based cohort study. Am J Respir Crit Care Med. 2000;161(1):5–8 [DOI] [PubMed] [Google Scholar]
- 5. Le JI, Gribbin J, West J, et al. The incidence of cancer in patients with idiopathic pulmonary fibrosis and sarcoidosis in the UK. Respir Med. 2007;101(12):2534–2540 [DOI] [PubMed] [Google Scholar]
- 6. Shiels MS, Chaturvedi AK, Katki HA, et al. Circulating markers of interstitial lung disease and subsequent risk of lung cancer. Cancer Epidemiol Biomarkers Prev. 2011;20(10):2262–2272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Koshiol J, Rotunno M, Consonni D, et al. Chronic obstructive pulmonary disease and altered risk of lung cancer in a population-based case-control study. PLoS One. 2009;4(10):e7380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schwartz AG, Cote ML, Wenzlaff AS, et al. Chronic obstructive lung diseases and risk of non-small cell lung cancer in women. J Thorac Oncol. 2009;4(3):291–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Littman AJ, White E, Jackson LA, et al. Chlamydia pneumoniae infection and risk of lung cancer. Cancer Epidemiol Biomarkers Prev. 2004;13(10):1624–1630 [PubMed] [Google Scholar]
- 10. Engels EA, Shen M, Chapman RS, et al. Tuberculosis and subsequent risk of lung cancer in Xuanwei, China. Int J Cancer. 2009;124(5):1183–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shiels MS, Albanes D, Virtamo J, Engels EA. Increased risk of lung cancer in men with tuberculosis in the alpha-tocopherol, beta-carotene cancer prevention study. Cancer Epidemiol Biomarkers Prev. 2011;20(4):672–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brenner AV, Wang Z, Kleinerman RA, et al. Previous pulmonary diseases and risk of lung cancer in Gansu Province, China. Int J Epidemiol. 2001;30(1):118–124 [DOI] [PubMed] [Google Scholar]
- 13. Engels EA, Wu X, Gu J, et al. Systematic evaluation of genetic variants in the inflammation pathway and risk of lung cancer. Cancer Res. 2007;67(13):6520–6527 [DOI] [PubMed] [Google Scholar]
- 14. Shiels MS, Engels EA, Shi J, et al. Genetic variation in innate immunity and inflammation pathways associated with lung cancer risk. Cancer. 2012; 118(22):5630–5636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Spitz MR, Gorlov IP, Amos CI, et al. Variants in inflammation genes are implicated in risk of lung cancer in never smokers exposed to second-hand smoke. Cancer Discov. 2011;1(5):420–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chaturvedi AK, Caporaso NE, Katki HA, et al. C-reactive protein and risk of lung cancer. J Clin Oncol. 2010;28(16):2719–2726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Il’yasova D, Colbert LH, Harris TB, et al. Circulating levels of inflammatory markers and cancer risk in the health aging and body composition cohort. Cancer Epidemiol Biomarkers Prev. 2005;14(10):2413–2418 [DOI] [PubMed] [Google Scholar]
- 18. Allin KH, Bojesen SE, Nordestgaard BG. Baseline C-reactive protein is associated with incident cancer and survival in patients with cancer. J Clin Oncol. 2009;27(13):2217–2224 [DOI] [PubMed] [Google Scholar]
- 19. Pine SR, Mechanic LE, Enewold L, et al. Increased levels of circulating interleukin 6, interleukin 8, C-reactive protein, and risk of lung cancer. J Natl Cancer Inst. 2011;103(14):1112–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhou B, Liu J, Wang ZM, Xi T. C-reactive protein, interleukin 6 and lung cancer risk: a meta-analysis. PLoS One. 2012;7(8):e43075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bigbee WL, Gopalakrishnan V, Weissfeld JL, et al. A multiplexed serum biomarker immunoassay panel discriminates clinical lung cancer patients from high-risk individuals found to be cancer-free by CT screening. J Thorac Oncol. 2012;7(4):698–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420(6917):860–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Moss SF, Blaser MJ. Mechanisms of disease: Inflammation and the origins of cancer. Nat Clin Pract Oncol 2005;2(2):90–7 [DOI] [PubMed] [Google Scholar]
- 25. Prorok PC, Andriole GL, Bresalier RS, et al. Design of the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Trial. Control Clin Trials. 2000;21(6 Suppl):273S–309S [DOI] [PubMed] [Google Scholar]
- 26. Oken MM, Hocking WG, Kvale PA, et al. Screening by chest radiograph and lung cancer mortality: the Prostate, Lung, Colorectal, and Ovarian (PLCO) randomized trial. JAMA. 2011;306(17):1865–1873 [DOI] [PubMed] [Google Scholar]
- 27. Chaturvedi AK, Kemp TJ, Pfeiffer RM, et al. Evaluation of multiplexed cytokine and inflammation marker measurements: a methodologic study. Cancer Epidemiol Biomarkers Prev. 2011;20(9):1902–1911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chaturvedi AK, Gaydos CA, Agreda P, et al. Chlamydia pneumoniae infection and risk for lung cancer. Cancer Epidemiol Biomarkers Prev. 2010;19(6):1498–1505 [DOI] [PubMed] [Google Scholar]
- 29. Harrell FE. Regression Modeling Strategies: with Applications to Linear Models, Logistic Regression, and Survival Analysis. New York, NY: Springer-Verlag; 2001 [Google Scholar]
- 30. Katki HA, Mark SD. Survival Analysis for Cohorts with Missing Covariate Information. R News. 2008;8:14–19 [Google Scholar]
- 31. Mark SD, Katki HA. Specifying and implementing nonparametric and semiparametric survival estimators in two-stage (sampled) cohort studies with missing case data. J Am Stat Assoc. 2006;101(474):460–471 [Google Scholar]
- 32. Bunt SK, Yang L, Sinha P, et al. Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res. 2007;67(20):10019–10026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nakanishi T, Imaizumi K, Hasegawa Y, et al. Expression of macrophage-derived chemokine (MDC)/CCL22 in human lung cancer. Cancer Immunol Immunother. 2006;55(11):1320–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. de CL, Goc J, Damotte D, et al. Characterization of chemokines and adhesion molecules associated with T cell presence in tertiary lymphoid structures in human lung cancer. Cancer Res. 2011;71(20):6391–6399 [DOI] [PubMed] [Google Scholar]
- 35. de Visser KE, Coussens LM. The inflammatory tumor microenvironment and its impact on cancer development. Contrib Microbiol. 2006;13: 118–137 [DOI] [PubMed] [Google Scholar]
- 36. Ballaz S, Mulshine JL. The potential contributions of chronic inflammation to lung carcinogenesis. Clin Lung Cancer. 2003;5(1):46–62 [DOI] [PubMed] [Google Scholar]
- 37. Subramanian J, Govindan R. Lung cancer in never smokers: a review. J Clin Oncol. 2007;25(5):561–570 [DOI] [PubMed] [Google Scholar]
- 38. Glynn RJ, MacFadyen JG, Ridker PM. Tracking of high-sensitivity C-reactive protein after an initially elevated concentration: the JUPITER Study. Clin Chem. 2009;55(2):305–312 [DOI] [PubMed] [Google Scholar]
- 39. Sin DD, Man SF, McWilliams A, Lam S. Progression of airway dysplasia and C-reactive protein in smokers at high risk of lung cancer. Am J Respir Crit Care Med. 2006;173(5):535–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Aberle DR, Adams AM, Berg CD, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med. 2011;365(5):395–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tammemagi CM, Pinsky PF, Caporaso NE, et al. Lung cancer risk prediction: Prostate, Lung, Colorectal And Ovarian Cancer Screening Trial models and validation. J Natl Cancer Inst. 2011;103(13):1058–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bach PB, Kattan MW, Thornquist MD, et al. Variations in lung cancer risk among smokers. J Natl Cancer Inst. 2003;95(6):470–478 [DOI] [PubMed] [Google Scholar]
- 43. Cassidy A, Duffy SW, Myles JP, Liloglou T, Field JK. Lung cancer risk prediction: a tool for early detection. Int J Cancer. 2007;120(1):1–6 [DOI] [PubMed] [Google Scholar]
- 44. Spitz MR, Hong WK, Amos CI, et al. A risk model for prediction of lung cancer. J Natl Cancer Inst. 2007;99(9):715–726 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.