

Abstract
The small airway epithelium (SAE), the first site of smoking-induced lung pathology, exhibits genome-wide changes in gene expression in response to cigarette smoking. Based on the increasing evidence that the epigenome can respond to external stimuli in a rapid manner, we assessed the SAE of smokers for genome-wide DNA methylation changes compared with nonsmokers, and whether changes in SAE DNA methylation were linked to the transcriptional output of these cells. Using genome-wide methylation analysis of SAE DNA of nonsmokers and smokers, the data identified 204 unique genes differentially methylated in SAE DNA of smokers compared with nonsmokers, with 67% of the regions with differential methylation occurring within 2 kb of the transcriptional start site. Among the genes with differential methylation were those related to metabolism, transcription, signal transduction and transport. For the differentially methylated genes, 35 exhibited a correlation with gene expression, 54% with an inverse correlation of DNA methylation with gene expression and 46% a direct correlation. These observations provide evidence that cigarette smoking alters the DNA methylation patterning of the SAE and that, for some genes, these changes are associated with the smoking-related changes in gene expression.
INTRODUCTION
DNA methylation, the attachment of methyl groups to cytosine bases followed by guanine (CpG sites), is an epigenetic modification that plays a role in development, regulation of cell type and tissue-specific gene expression (1–3). Hypermethylation of CpG islands around gene promoters is generally correlated with gene silencing, whereas hypomethylation usually is associated with active gene transcription (4,5). Recent evidence suggests that the epigenome is affected by, and can rapidly respond to, external stimuli such as diet and environment (6–25).
With this background, we hypothesized that cigarette smoke, with its >4000 compounds and 1014 oxidants per puff, may have profound effects on the methylome of the small airway epithelium (SAE), the cell population that takes the initial stress of cigarette smoke and is the first site of lung pathology in cigarette smokers (26–32). In humans, the SAE consists of four major cell types: ciliated, secretory, columnar and basal cells (30,33). These cells provide a barrier and innate immunity that protect the airway from environmental stressors, pollutants and pathogens (34–36). Cigarette smoking is associated with disordering of the differentiation of the SAE basal cells, with consequent disordered function of the airway mucociliary barrier (34,35). The ability of the SAE to alter gene expression in response to external stimuli is critical to airway defense and repair mechanisms (30,31,34,36).
To assess whether smoking is associated with changes in the methylome of the SAE, and whether this has consequences to gene expression of this cell population, we evaluated the genome-wide methylation status of the epigenome of the SAE of smokers compared with that of nonsmokers and examined whether the differences in small airway epithelial DNA methylation correlated with the smoking-related genome-wide changes in the small airway epithelial transcriptome. The data demonstrate that smoking is associated with a broad range of genome-wide methylation-related changes of the SAE and that many of these smoking-related epigenetic changes correlate with smoking-associated changes in the small airway epithelial transcriptome. Interestingly, whereas some smoking-related hypermethylation correlated with decreased expression and some smoking-mediated hypomethylation correlated with increased expression, we also observed the opposite, with hypermethylation correlated with up-regulation and hypomethylation associated with down-regulation, highlighting the complex dynamics of DNA methylation and its role in transcriptional regulation.
RESULTS
DNA from the SAE of 19 nonsmokers and 20 smokers was assessed by the HELP (HpaII tiny fragment Enriched by Ligation-mediated PCR) assay for the methylation status of 117 521 HpaII fragments. This generates a normalized fluorescent intensity ratio representing the ratio of unmethylated DNA to total DNA, referred to as the ‘methylation level’, a positive number representing a less methylated state and a negative number a more methylated DNA fragment. For most individual HpaII fragments, the methylation levels among subjects were similar, with a median coefficient of variation in all fragments of 0.16. To assess the validity of the overall assay, the methylation status of genes located on X chromosome was assessed. As expected, of 3871 HpaII fragments located on the X chromosome, 659 (17%) showed a difference in methylation states (P < 0.05, fold-change greater than ±1.5) based on gender. Sex chromosomes were excluded from further analysis.
To assess whether there was a significant effect of smoking on the methylation of any probed HpaII fragment, a quantile–quantile (Q–Q) plot was constructed assessing the impact of smoking after correction for the covariates, batch and gender (Fig. 1A). The data showed a significant deviation from the expected probability distribution, suggesting an effect of smoking on DNA methylation. The chromosomal distribution of the P-values showed a broad distribution of significant signals across all chromosomes with no hot spots of methylation change (Fig. 1B). Using a Benjamini–Hochberg-corrected P-value <0.05, we observed four significantly differentially methylated fragments between smokers and nonsmokers that represented four unique genes (CYP1B1, CYP1A1, ALDH3A1, SFRP2), all of which were hypomethylated in smokers. For multivariate data exploration purposes, we followed the approach of Pascual et al. (8) and considered P < 0.05 and a fold-change greater than ±1.5 as our threshold for the remainder of our analysis. A total of 220 differentially methylated fragments were found on the autosomes, of which 164 (75%) were hypomethylated and 56 (25%) were hypermethylated (Fig. 1C), i.e. on a global genome basis, smoking is associated with ∼3-fold more hypomethylated than hypermethylated fragments. These smoking-dependent HpaII fragments represent only ∼0.2% of those surveyed, suggesting there is no global effect of smoking on epithelial DNA methylation.
Figure 1.

Genome-wide methylation differences of SAE DNA of smokers versus nonsmokers. The data are derived from HELP assay analysis of n = 19 nonsmokers and n = 20 smokers. (A) Assessment of significance of DNA methylation differences by the Q–Q plot comparing smokers with nonsmokers. The Q–Q plot shows the distribution of expected P-values (log10) against the observed distribution. (B) Chromosomal distribution of DNA methylation differences of smokers compared with nonsmokers. For each probe, the significance is displayed on the y-axis as the −log10 of the P-value. The results are ordered along the x-axis by chromosome, with each bar representing a different chromosome. (C) Assessment of differential DNA methylation of the SAE for all probe sets comparing smokers versus nonsmokers; y-axis, negative log10 of P-value; x-axis, log2-transformed fold-change; red dots are probe sets with differential DNA methylation, gray dots are probe sets without differential methylation. Differentially methylated probe sets have a P < 0.05, and a fold-change of greater than ±1.5. (D) Phenotype clustering based on DNA methylation levels. The data were analyzed by Pearson's dissimilarity unsupervised hierarchical analysis with an average linkage of smokers and nonsmokers based on the DNA methylation of 220 differentially methylated probe sets. Genes having more DNA methylation in smokers compared with nonsmokers are represented in blue, less methylation in red and no change in gray. The genes are represented horizontally and the individuals vertically.
The consistency of the methylation response to smoking was assessed by two methods. First, the 220 differentially methylated HpaII fragments were used to construct an unsupervised cluster in which 19 of the 20 smokers were clearly separated from the nonsmokers (Fig. 1D). Second, to assess inter-subject variability, an index was defined measuring conformity of methylation change to the average response in all smokers. The methylation index for nonsmokers varied from 0 to 8.3%, with a median of 0.5%. In contrast, the methylation index for smokers varied from 5.9 to 20.1%, with a median of 13.5%. Only one nonsmoker had an index within the range defined by the smokers (P < 10−12; Fig. 2).
Figure 2.

SAE DNA methylation index. The index was calculated using the 204 unique smoking-responsive genes based on the percentage of smoking-responsive genes each subject expressed outside the normal range defined as the average DNA methylation level of the healthy nonsmokers ± 2 standard deviations. For genes represented by more than one probe set, the probe set with the lowest P-value was used. Y-axis, index; x-axis, subjects (n = 19 nonsmokers, n = 20 smokers) ordered by increasing index values.
The 220 HpaII fragments correspond to 204 unique genes differentially methylated between smokers and nonsmokers. To provide an overview of these 204 unique genes that were differentially methylated between smokers and nonsmokers, the genes were assigned biological categories (Fig. 3). In general, there were more smoking-related hypomethylated genes than hypermethylated genes, but approximately equal ratios of more or less methylation in each category. The exception was for cell cycle and DNA repair which only had hypomethylated genes.
Figure 3.

Biological categories of the 204 unique genes that are differentially methylated between smokers and nonsmokers. Genes were put into categories for biological processes and plotted based on fold-change (log2). Above the x-axis are hypermethylated genes and below are the hypomethylated genes. Each bar represents a gene in the category; categories are separated by gray boxes.
The 204 unique genes differentially methylated between smokers and nonsmokers were divided into hyper- and hypomethylated gene sets. Among the top 25 hypermethylated genes (Table 1), the two most significant based on P-value were in the loci for JAG1 (jagged 1), a ligand that initiates notch signaling and BPIFB1 [bactericidal/permeability-increasing (BPI) fold containing family B, member 1], a protein involved in the innate immune response. Among the top 25 hypomethylated genes (Table 2), the top two loci were CYP1B1 (cytochrome P450, family 1, subfamily B, polypeptide 1) and CYP1A1 (cytochrome P450, family 1, subfamily A, polypeptide 1), enzymes that catalyze many reactions involved in drug metabolism and the synthesis of cholesterol, steroids and other lipids. For the smoking-related hyper- and hypomethylated genes, the most common categories were the metabolism and signal transduction genes.
Table 1.
Top 25 SAE hypermethylated genes of smokers compared with nonsmokersa
| Biological categoryb | Gene | Gene title | Fold-change (smokers/nonsmokers)c | P-value (smokers/nonsmokers) |
|---|---|---|---|---|
| Apoptosis | BCL2L15 | BCL2-like 15 | −1.6 | 3.1 × 10−3 |
| Growth | DNAH5 | Dynein, axonemal, heavy chain 5 | −1.9 | 1.3 × 10−4 |
| PARVA | Parvin, alpha | −1.5 | 7.9 × 10−3 | |
| Immune response | BPIFB1 | BPI fold containing family B, member 1 | −1.8 | 4.2 × 10−6 |
| CFHR3 | Complement factor H-related 3 | −2.1 | 3.0 × 10−5 | |
| Metabolism | ALDH1A3 | Aldehyde dehydrogenase 1 family, member A3 | −1.8 | 8.0 × 10−6 |
| HTRA1 | HtrA serine peptidase 1 | −1.5 | 5.9 × 10−5 | |
| PAPSS2 | 3′-Phosphoadenosine 5′-phosphosulfate synthase 2 | −1.9 | 5.8 × 10−4 | |
| CYP2F1 | Cytochrome P450, family 2, subfamily F, polypeptide 1 | −1.9 | 5.5 × 10−3 | |
| Regulation | E2F2 | E2F transcription factor 2 | −1.6 | 4.4 × 10−3 |
| PRDM1 | PR domain containing 1, with ZNF domain | −1.5 | 5.0 × 10−3 | |
| Scaffolding | USH1G | Usher syndrome 1G (autosomal recessive) | −1.7 | 7.1 × 10−4 |
| Signal transduction | JAG1 | Jagged 1 | −1.6 | 3.0 × 10−6 |
| SCGB1A1 | Secretoglobin, family 1A, member 1 (uteroglobin) | −2.0 | 2.2 × 10−4 | |
| ANKS4B | Ankyrin repeat and sterile alpha motif domain containing 4B | −1.6 | 4.0 × 10−4 | |
| PCDH18 | Protocadherin 18 | −1.5 | 7.6 × 10−4 | |
| RALB | v-ral simian leukemia viral oncogene homolog B (ras related; GTP-binding protein) | −1.8 | 4.8 × 10−3 | |
| OR4N2 | Olfactory receptor 4N2 | −1.7 | 5.2 × 10−3 | |
| CCBE1 | Collagen and calcium-binding EGF domains 1 | −1.6 | 6.6 × 10−3 | |
| Transport | ABCA13 | ATP-binding cassette, subfamily A (ABC1), member 13 | −1.9 | 3.4 × 10−5 |
| SYPL1 | Synaptophysin-like 1 | −1.6 | 3.8 × 10−5 | |
| Unknown | CCDC70 | Coiled-coil domain containing 70 | −1.5 | 1.1 × 10−5 |
| TMEM139 | Transmembrane protein 139 | −1.7 | 4.4 × 10−5 | |
| SH3TC2 | SH3 domain and tetratricopeptide repeats 2 | −1.7 | 2.9 × 10−4 | |
| HNRNPM | Heterogeneous nuclear ribonucleoprotein M | −1.6 | 7.1 × 10−3 |
aThe 25 unique, known genes with the highest smoker/nonsmoker DNA methylation ratio based on P-values for the 220 differentially methylated probe sets, grouped into functional categories.
bThe biological categories were assigned by GO, the Human Protein Reference Data Base (hprd.org) and GeneCards (www.genecards.org).
cWith the HELP assay, positive values represent hypomethylation of smokers compared with nonsmokers.
Table 2.
Top 25 SAE hypomethylated genes of smokers compared with nonsmokersa
| Biological categoryb | Gene | Gene title | Fold-change (smokers/nonsmokers)c | P-value (smokers/nonsmokers) |
|---|---|---|---|---|
| Apoptosis | GRAMD4 | GRAM domain containing 4 | 1.7 | 8.2 × 10−3 |
| DNA repair | REV1 | REV1 homolog (Saccharomyces cerevisiae) | 1.9 | 1.4 × 10−2 |
| Growth | SPTBN5 | Spectrin, beta, non-erythrocytic 5 | 1.7 | 1.2 × 10−2 |
| Metabolism | CYP1B1 | Cytochrome P450, family 1, subfamily B, polypeptide 1 | 3.0 | 7.4 × 10−11 |
| CYP1A1 | Cytochrome P450, family 1, subfamily A, polypeptide 1 | 2.3 | 6.1 × 10−7 | |
| UGT1A6 | UDP glucuronosyltransferase 1 family, polypeptide A6 | 2.1 | 1.5 × 10−4 | |
| ALDH3A1 | Aldehyde dehydrogenase 3 family, member A1 | 1.8 | 4.5 × 10−4 | |
| AKR1C2 | Aldo-keto reductase family 1, member C2 (dihydrodiol dehydrogenase 2; bile acid binding) | 2.0 | 1.2 × 10−3 | |
| LYPLA1 | Lysophospholipase I | 1.5 | 8.8 × 10−3 | |
| GSTM1 | Glutathione S-transferase mu 1 | 1.8 | 1.1 × 10−2 | |
| GSTM5 | Glutathione S-transferase mu 5 | 1.7 | 1.7 × 10−2 | |
| Protein trafficking | ARL17A | ADP-ribosylation factor-like 17A | 1.5 | 3.9 × 10−3 |
| Regulation | PRDM8 | PR domain containing 8 | 1.6 | 1.2 × 10−3 |
| Signal transduction | SFRP2 | Secreted frizzled-related protein 2 | 1.6 | 1.2 × 10−6 |
| DMKN | Dermokine | 1.6 | 4.7 × 10−3 | |
| ALS2CL | ALS2 C-terminal like | 1.5 | 5.1 × 10−3 | |
| STIM2 | Stromal interaction molecule 2 | 1.5 | 1.0 × 10−2 | |
| RIN1 | Ras and Rab interactor 1 | 2.3 | 1.1 × 10−2 | |
| Transcription | INO80D | INO80 complex subunit D | 1.6 | 1.4 × 10−2 |
| RBFOX3 | RNA-binding protein, fox-1 homolog (Caenorhabditis elegans) 3 | 1.6 | 1.7 × 10−2 | |
| Transport | PKD2L1 | Polycystic kidney disease 2-like 1 | 2.0 | 2.8 × 10−3 |
| KCNJ15 | Potassium inwardly-rectifying channel, subfamily J, member 15 | 1.5 | 4.4 × 10−3 | |
| Unknown | CATSPER1 | Cation channel, sperm associated 1 | 1.6 | 1.0 × 10−2 |
| CDRT1 | CMT1A duplicated region transcript 1 | 1.7 | 1.7 × 10−4 | |
| KLHL29 | Kelch-like 29 (Drosophila) | 1.6 | 1.4 × 10−2 |
aThe 25 unique, known genes with the highest smoker/nonsmoker DNA methylation ratio based on P-values for the 220 differentially methylated probe sets grouped into functional categories.
bThe biological categories were assigned by GO, the Human Protein Reference Data Base (hprd.org) and GeneCards (www.genecards.org).
cWith the HELP assay, positive values represent hypomethylation of smokers compared with nonsmokers.
As examples for probe location along a gene, all HpaII fragments for eight loci with smoking-dependent methylation changes were visualized, with none representing the transcriptional start site of the gene, and methylation levels for the HpaII fragments displayed as box and whisker plots (Fig. 4). Each of the eight examples had six to ten HpaII fragments, for which methylation status was assessed by HELP assay, with variation among the fragments at one locus by up to 32-fold (5 units on log2 scale). Overall, for each gene, there was specificity to the placement of methyl groups along the gene and its flanking regions, rather than a diffuse smoking-related phenomenon. For most genes, only one HpaII fragment was differentially methylated between smokers and nonsmokers (Fig. 4A–G), although for CYP1B1, two fragments were differentially methylated (Fig. 4H). The location along the gene of the differentially methylated HpaII site also varied widely from gene to gene, with no consistent pattern of the differentially methylated HpaII fragment being upstream or downstream of the transcription start site (Supplementary Material, Fig. S1). Approximately 67% of the differentially methlyated HpaII fragments were within 2000 bp of the transcription start site of a gene, although not generally within CpG islands. For example, the ALDH3A1 locus showed a hypomethylated HpaII fragment ∼1000 bp downstream of the transcriptional start site with 1.8-fold difference in methylation level (Fig. 4A, Table 2). In contrast, the JAG1 locus showed a hypermethylated HpaII fragment 2000 bp upstream of the transcriptional start site with 1.6-fold difference in methylation between smokers and nonsmokers (Fig. 4G, Table 1).
Figure 4.

Examples of site-specific, smoking-related SAE DNA methylation differences. The data for 20 smokers (smokers, gray boxes) and 19 nonsmokers (nonsmokers, open boxes) for individual probe sets located across each gene. For all panels, the probe locations are mapped along the gene sites indicated (A–J) along each gene; 0 represents the transcription start site. Direction of transcription is indicated by the arrow and CpG islands are indicated by a dashed line. The probe set with significant differential methylation between smokers and nonsmokers is designated with an asterisk. The significance of individual probe location along a gene is assessed using box and whisker plots. Shown are examples of probe locations along genes that are more or less methylated in the SAE of smokers compared with nonsmokers. The y-axis indicates the ratio of unmethylated to methylated DNA (log2), where smaller numbers indicate higher methylation levels. (A) ALDH3A1; (B) PAPSS2; (C) ABCAI3; (D) SRFP2; (E) CYPIA1; (F) DNAH5; (G) JAG1; and (H) CYP1B1. The open boxes represent nonsmokers; the gray boxes represent smokers.
Ingenuity Pathway Analysis was performed on the 204 unique genes that were differentially methylated between smokers and nonsmokers. The most significant pathway was related to xenobiotic metabolism, with 9 of the 204 genes in the pathway differentially methylated, including AKR1C2, ALDH1A3, ALDH3A1, CYP1A1, CYP1B1, CYP2F1, GSTM1, GSTM5 and UGT1A6 (P = 1.06 × 10−6). Ingenuity Pathway Analysis was also performed on the top 25 hyper- and hypomethylated genes. The top 25 hypermethylated genes showed no clear pathways, but the top 25 hypomethylated genes included 7 genes involved in xenobiotic metabolism (P = 5.2 × 10−11), 6 in xenobiotic metabolism signaling (P = 8.6 × 10−7) and 5 genes involved in aryl hydrocarbon receptor signaling (P = 5.8 × 10−7). Interestingly, of genes related to small airway epithelial function, there were seven ciliated cell-related genes that showed smoking-dependent methylation changes: two were hypermethylated and five hypomethylated. There were nine genes that were basal cell-related (four hypermethylated, five hypomethylated), and one secretory cell-related gene that was hypermethylated (Table 3).
Table 3.
Genes related to the SAE function with smoking-related changes in DNA methylation
| Gene categorya | Geneb | Smokers/nonsmokersc | |
|---|---|---|---|
| Fold-change | P-value | ||
| Ciliated cell-related | DNAH5 | −1.9 | 1.3 × 10−4 |
| VWA3B | −1.6 | 1.0 × 10−2 | |
| TPPP | 1.7 | 1.8 × 10−2 | |
| TNC | 1.7 | 3.5 × 10−2 | |
| IRX3 | 1.5 | 4.5 × 10−2 | |
| IQUB | 1.5 | 4.7 × 10−2 | |
| TPPP3 | 1.8 | 4.7 × 10−2 | |
| Basal cell-related | JAG1 | −1.6 | 7.4 × 10−11 |
| ALDH1A3 | −1.8 | 3.0 × 10−6 | |
| ALS2CL | 1.5 | 8.0 × 10−6 | |
| CCBE1 | −1.6 | 5.1 × 10−3 | |
| KLHL29 | 1.6 | 1.0 × 10−2 | |
| MAN2A1 | −1.6 | 1.4 × 10−2 | |
| SLC2A9 | 2.1 | 2.5 × 10−2 | |
| TNC | 1.7 | 2.6 × 10−2 | |
| PRPH | 2.0 | 3.5 × 10−2 | |
| Secretory cell-related | SCGB1A1 | −2.0 | 2.2 × 10−4 |
aBasal cell-related genes (46); ciliated-related genes (51–53); and secretory cell-related gene (45).
bThe probe set with the highest smoker/nonsmoker DNA methylation ratio based on P-value for the 220 differentially methylated probe sets.
cWith the HELP assay, smoker/nonsmoker hypermethylation is represented by negative values, and hypomethylation is represented by positive values.
To assess the relationship between smoking-dependent hypo- and hypermethylation and smoking-dependent increase or decrease in gene expression, a starburst plot was generated examining the 220 HpaII fragments (P < 0.05, fold-change greater than ±1.5), representing 204 unique genes that had smoking-dependent, DNA methylation differences (Fig. 5). Of these 204 genes, 193 had the corresponding HG-U133 Plus 2.0 gene expression data. Of these 193 genes, 35 displayed a change in gene expression. Of the 35 genes, 24 were hypomethylated—14 were associated with up-regulation of gene expression, and 10 associated with down-regulation of gene expression (P < 0.05). For the 11 out of 35 smoking-dependent hypermethylated genes, 6 were associated with up-regulation of gene expression and 5 associated with down-regulation (P < 0.05). There were no consistent correlations between the direction of smoking-dependent methylation change and direction of change in gene expression (P > 0.9), although three genes (CYP1B1, CYP1A1 and ALDH3A1) showed a substantial degree of hypomethylation and increase in gene expression level in response to smoking.
Figure 5.

Correlation of SAE DNA methylation and gene expression. Unique genes that show differences in DNA methylation between smokers and nonsmokers (P < 0.05, fold-change greater than ±1.5) that also have corresponding gene expression (n = 193, i.e. 204 minus the 11 differential DNA methylation unique genes that do not have corresponding gene expression probes on the Affymetrix HG-U133 Plus 2.0 Array). The starburst plots show −log10 P-value of smokers/nonsmokers plotted for DNA methylation (x-axis) versus gene expression (y-axis) for each gene. Dashed lines, P-values = 0.05. White circles, hypermethylated genes with down-regulated gene expression (right lower quadrant) and hypomethylated genes with up-regulated gene expression (left upper quadrant). Black circles, hypermethylated genes with up-regulated gene expression (right upper quadrant) and hypomethylated genes with down-regulated gene expression (left lower quadrant).
Examples of genes with a correlation of change in methylation (P < 0.05, fold-change greater than ±1.5) and gene expression (P < 0.05, fold-change greater than ±1.5) in smokers demonstrated that, for all genes, the smokers and nonsmokers were clearly separated. In five of the examples shown (Fig. 6), there was smoking-induced hypomethylation, with a corresponding increase in gene expression. However, one gene, ABCA13, ATP-binding cassette, subfamily A, member 13, exhibited a smoking-induced hypermethylation and decreased gene expression (Fig. 6C).
Figure 6.

Examples of correlation of SAE DNA methylation with gene expression. Shown are genes that are up- and down-regulated in the SAE of smokers compared with nonsmokers whose expression correlates with an increase or decrease of DNA methylation. Each symbol represents an individual. The data are based on 20 smokers (filled circle) and 16 nonsmokers (open circle). The x-axis represents the log2 ratio of unmethylated to methylated DNA; smaller numbers indicate higher methylation levels. (A and B) The y-axis represents the corresponding log2 HG U133 Plus 2.0 relative gene expression levels. (C–F) The y-axis represents the corresponding HG U133 Plus 2.0 relative gene expression levels for the genes indicated. (A) CYP1A1; (B) CYP1B1; (C) ABCA13; (D) ALDH3A1; (E) SFRP2; and (F) AKR1C2.
To validate the DNA methylation findings, we examined four genes from the study: three genes, ALDH3A1, CYP1B1 and SFRP2, which display an inverse correlation of methylation and gene expression, and one negative control gene, RPL26, which does not show any methylation differences between smokers and nonsmokers in the HELP assay. Primers were designed for the EpiTyper assay to cover the region of the gene found to be differentially methylated by the HELP assay. All CpGs with in this region were analyzed using the Sequenom EpiTyper analysis. A P-value of <0.05 and a fold-change of ±1.5 were used to define differential methylation. For ALDH3A1, 50% of the CpGs analyzed were differentially methylated in smokers compared with nonsmokers. For CYP1B1, 44% of all CpGs analyzed were differentially methylated in smokers compared with nonsmokers. SFRP2 contained 48% of CpGs analyzed that were differentially methylated in smokers compared with nonsmokers. Lastly, for our negative control gene, RPL26, only 5% of CpGs analyzed were differentially methylated in smokers compared with nonsmokers. The results of the HELP assay are compared with a representative sample of the Sequenom EpiTyper data in Figure 7. We observe a similar distribution of DNA methylation levels in smokers compared with nonsmokers when comparing the HELP assay and Sequenom EpiTyper validation. These results confirm our ability to detect variable DNA methylation using the HELP assay.
Figure 7.

Biological validation of the HELP assay by the Sequenom EpiTyper assay. Shown are three genes that display differential DNA methylation between smokers and nonsmokers by the HELP assay (A1, B1 and C1) and corresponding biological validation by Sequemom EpiTyper by two representative CpGs (A2, A3, B2, B3, C2 and C3). The data are based on 12 smokers (black bars) and 14 nonsmokers (gray bars). The x-axis represents the number of subjects found in each grouping. The y-axes for (A1), (B1) and (C1) represent the unmethylated to methylated ratio (log2 smokers/nonsmokers) from the HELP assay, where smaller numbers indicate higher methylation levels. The y-axes for (A2), (A3), (B2), (B3), (C2) and (C3) represent the %methylation at a particular CpG from the Sequenom EpiTyper validation.
DISCUSSION
There is increasing evidence that the environment can have significant effects on the methylation pattern of the genome (7–23,25). The present study adds to this concept, demonstrating that chronic insult to the lung by cigarette smoking is associated with significant changes in methylation patterns of the DNA derived from the SAE, the epithelial population that demonstrates the first evidence of smoking-induced pathology, and the initial site of development of most smoking-related lung disorders (30–32). We assayed these DNA methylation changes using the HELP assay, which relies on the use of methylation-sensitive and -insensitive restriction enzymes; therefore, only CpGs that are recognized by the restriction enzymes will be assayed. The data demonstrate that, genome-wide, smoking causes methylation changes in ∼0.2% of genes surveyed distributed across the genome. The majority of DNA methylation changes we observed were characterized by hypomethylation and the minority by hypermethylation. For those genes affected, the methylation changes were mostly limited to a focal region of the gene, rather than diffuse changes. There was no consistent pattern of differentially methylated regions being up- or downstream of the transcription start site, but most changes were <2 kb from the transcription start. Other than genes related to xenobiotic processes, several of which were hypomethylated in relation to smoking and signal transduction, several genes were also hypermethylated in association with smoking, with the affected genes representing a broad variety of functions. Interestingly, genes related to the function of the SAE, including basal cells (the stem/progenitor cells of the airway epithelium) and cilia and secretory cells (the differentiated airway cells that mediate most of the SAE functions), were also found to be differentially methylated in response to smoking. Together, these data demonstrate smoking has a significant impact on the SAE function, adding another layer of complexity of how smoking disorders the function of this critical cell population.
Comparison of the smoking-related changes in the SAE methylome with SAE gene expression demonstrated hypermethylation linked to down-regulation and hypomethylation to up-regulation, but we also observed the opposite, with hypermethylation associated with gene up-regulation and hypomethylation linked to gene down-regulation, i.e. the data do not support a simple inverse relationship of hypermethylation and reduced gene expression and hypomethylation with increased gene expression. This lack of predicted correlation, however, has also been observed in genome-wide analysis of lung parenchyma of subjects with idiopathic pulmonary fibrosis (37). Methylation along a gene, in a promoter region or in the gene per se, alone or in combination with other epigenetic changes, may directly or indirectly change the transcriptional output of a gene or genomic region (5). Upon further examination of the six genes that display an increase in DNA methylation and an increase in gene expression, three of them contain hypermethylation in the gene body. It has been noted that gene body methylation is not associated with transcriptional repression (5,38), which may explain part of our observations. Of the other three genes that are hypermethylated with increased gene expression, one of them, C2orf58, is chromosomal open reading frame whose function is not well characterized. For the 10 genes that are hypomethylated and display decreased gene expression, it is likely that other repressive mechanisms, such as repressive histone modifications, are controlling the regulation of transcription on these genes and that the decrease in DNA methylation alone is not sufficient to reactivate the transcriptional machinery (39).
Environmental influences on DNA methylation
Recent evidence has suggested that environmental factors can affect DNA methylation patterning as well as gene expression in a variety of tissues in a rapid and robust manner. For example, mice exposed to particulate matter air pollution display hypermethylation of the p16 promoter and the matrix metalloproteinase-2 (MMP2) promoter in DNA extracted from the whole lung (7). Pascual et al. (8) investigated the DNA methylation patterns of B lymphocytes of humans with house dust mite allergic asthma compared with non-allergic controls. A number of differentially methylated loci were identified between allergic and non-allergic controls. CYP26A1 was hypermethylated in allergic asthmatic subjects and this increase in methylation correlated with a decrease in gene expression (8). The effect of heavy metal exposure on DNA methylation has also been explored. Li et al. (9) observed that, in PC12 cell lines, a neurodevelopmental model, a decrease in global methylation of the PC12 cells after treatment with high concentrations of Pb2+ and, specifically, a decrease in methylation at the promoter of the amyloid precursor protein (APP) gene correlated with an increase in APP gene expression and protein levels.
Diet has also been implicated to affect levels of methylation in a variety of genes. Dolinoy et al. (10) found that in utero exposure to dietary genistein, the major phytoestrogen in soy, modified coat color in mice by inducing methylation of CpG sites in a retrotransposon upstream of the transcriptional start site of the Agouti gene responsible for coat color. Similarly, high protein or protein-restricted diets fed to sows affected DNA methylation of individual CpG sites in a number of metabolic genes in offspring compared with controls (11). Adult diet has also been shown to affect the DNA methylation patterning in a variety of tissues. Cynomolgus monkeys transitioned from a high soy diet to a low soy diet had change in global DNA methylation levels in liver and muscle tissue (12). Adult rats fed high-fat sucrose diets supplemented with methyl donors had modified liver DNA methylation levels of specific CpGs on the fatty acid synthase (FASN) gene in a diet-dependent manner (13).
Smoking-associated changes in lung DNA methylation
Cigarette smoking, with its >4000 compounds and 1014 free radicals per puff, represents a major environmental stress (26–29). Consistent with our observations in the SAE, several studies have demonstrated that cigarette smoke in vivo and in vitro can mediate differences in the methylation status of individual genes in lung cells as well as non-lung cells. For example, immortalized human bronchial epithelial cells (HBECs) exposed to cigarette smoke extract display hypermethylation in association with tumor suppressor genes such as (RASSF1A) and retinoic acid receptor β2 (RARβ2) in a dose-dependent manner (14). For RASSF1A, the increase in DNA methylation correlated with a decrease in gene expression. Hypermethylation has also been linked to the RASSF1A, as well as p16, RARβ2 and H-cadherin (CDH13) genes in sputum samples of smokers compared with nonsmokers (15). Smoking has been found to affect the DNA methylation patterns in these genes by a number of studies (15–17); however, in the present study, we do not observe differential methylation of these genes in smokers compared with nonsmokers. These studies surveyed a larger cohort of smokers (107 heavy smokers, 89 lung cancer free current and former smokers and 100 former smokers), which also had a larger pack-year range (30–172, 1–183 and 20–136 pack-years, respectively), whereas our subjects are healthy smokers with relatively low pack-years (11–29 pack-years). Additionally, when studying non-small-cell lung cancer, researchers have observed methylation of these genes in only a subset of subjects, ranging from 16 to 44%, depending on the gene (40,41). This variability, coupled with our smaller subject size (n = 19 nonsmokers and n = 20 healthy smokers) and low pack-year history, may explain why we are observing a different cohort of genes that are differentially methylated. The genes found to be differentially methylated in the present study may reflect changes occurring as a direct response to cigarette smoke on the SAE.
Several studies have examined the effect of smoking on the methylation status of specific genes in alveolar macrophages. Monick et al. (20) examined genome-wide DNA methylation patterning in alveolar macrophages as well as lymphoblasts, observing differences of smokers compared with nonsmokers. This study also noted that the aryl hydrocarbon receptor repressor (AHRR) gene displayed an inverse correlation of DNA methylation levels and gene expression in lymphoblasts. Philibert et al. (21) examined genome-wide DNA methylation levels in alveolar macrophages and observed differences when comparing smokers and nonsmokers, with a number of genes having an inverse correlation between DNA methylation and gene expression. Together with the airway epithelial studies, these observations demonstrate that cigarette smoking has a significant influence on the methylome of both epithelium and resident immune cells, representing a multi-cell type response to environmental stress.
Experimental data show mixed observations for the reversibility of DNA methylation in response to smoking cessation (15–17,19,22,23). Analysis of HBECs or sputum indicates no differences in the methylation status between current and former smokers (15–17). However, analysis of other tissues suggests partial reversibility of the DNA methylation levels of certain genes (19,22,23). These differences may be gene-specific or based on different tissues or location within the lung. Leng et al. (42) examined the sputum of current and former smokers and found that genetic variation, such as SNPs, may play a role as predictors for the acquisition of gene promoter methylation, supporting the concept that genetic variation impacts susceptibility to epigenetic modification. Qiu et al. (43) examined the DNA methylation of blood leukocytes in subjects with and without COPD and found that DNA methylation status at specific CpG loci is associated with both the presence and severity of COPD.
Since, to our knowledge, this study is the first to examine DNA methylation changes of the human SAE in response to cigarette smoking, we are somewhat limited in our interpretation of the data due to a lack of external validation. However, our findings are supported by other publications which have demonstrated in whole-lung tissue as well as macrophages that genes involved in the metabolism of xenobiotics by cytochrome P450 and aryl hydrocarbon signaling are differentially methylated in response to cigarette smoking (18–20). This suggests the importance of epigenetic regulation of these pathways in response to cigarette smoke. Similar to other studies, we observed alterations in DNA methylation levels of cytochrome P450 genes in response to smoking. For example, CYP1A1 displayed lower DNA methylation levels in smokers compared with nonsmokers in lung tissue (18,19). Anttila et al. (19) also observed an increase in DNA methylation levels of CYP1A1 in active smokers who had quit smoking 1 to 7 days earlier. In our study, we observed an inverse correlation of DNA methylation and gene expression for both CYP1A1 and CYP1B1. Finally, Tekpli et al. (18) also found that CYP1A1 was less methylated in normal lung tissue of current and former smokers compared with nonsmokers. This study also demonstrated that CYP1A1 methylation levels were inversely correlated with mRNA levels when comparing NHBE cells, immortalized HBECs and a human lung adenocarcinoma cell line. We also observed changes in DNA methylation that correspond to changes in gene expression in a subset of the genes involved in xenobiotic metabolism and aryl hydrocarbon receptor signaling, suggesting a potential role of transcriptional regulation by DNA methylation for these pathways in response to a severe environmental stressor such as cigarette smoke.
Because the SAE plays such a central role in lung defense, and with the knowledge that smoking alters small airway epithelial differentiation and function in mucociliary clearance, smoking-induced changes to DNA methylation could have profound effects on small airway epithelial function and, hence, host defense. Because the airway epithelium takes the brunt of inhaled cigarette smoke, it is the cell population with the greatest smoking-induced methylation changes. The SAE, although critical to the function of the lung, represents a very small proportion of the lung cell population, likely <1% of the total lung parenchymal cell population (44). The observation that smoking is associated with genome-wide changes in the methylation status of small airway epithelial DNA points out the importance of assessing individual cell types within an organ, rather than the organ as a whole.
MATERIALS AND METHODS
Study population
Healthy nonsmokers and healthy smokers were recruited from the general population in New York City. Individuals were evaluated at the Weill Cornell or Rockefeller University NIH Clinical and Translational Science Center and Department of Genetic Medicine Clinical Research Facility, using Institutional Review Board-approved clinical protocols and signed informed consent prior to any procedures. The criteria for ‘healthy’ was based on a history, physical examination, complete blood count, coagulation studies, liver function tests, urine studies, chest X-ray, EKG and pulmonary function tests as previously described (30,45). All subjects were negative for HIV1 and had normal α1-antitrypsin levels (for full inclusion/exclusion criteria, see Supplementary Material, Methods). Smoking status was verified by urine nicotine and cotinine levels, measured using liquid chromatography-tandem mass spectrometry (ARUP Laboratories, Salt Lake City, UT, USA). ‘Nonsmokers’ (n = 19) were defined as self-reported life-long nonsmokers, with non-detectable urine nicotine (<2 ng/ml) and cotinine (<5 ng/ml); ‘smokers’ (n = 20) were defined as self-reported current smokers with urine nicotine ≥30 ng/ml and/or urine cotinine ≥50 ng/ml (Table 4).
Table 4.
Demographics of the study population and biological samplesa
| Parameter | DNA methylation HELP assay |
HG-U133 Plus 2.0 Microarray | ||
|---|---|---|---|---|
| Healthy nonsmokersb | Healthy smokersb | Healthy nonsmokersb | Healthy smokersb | |
| n | 19 | 20 | 16 | 20 |
| Gender (M/F) | 8/11 | 2/18 | 8/8 | 2/18 |
| Age (years) | 34 ± 11 | 43 ± 7 | 35 ± 11 | 43 ± 7 |
| Race (B/W/H)c | 8/6/5 | 11/3/6 | 6/5/5 | 11/3/6 |
| Smoking history (pack-years) | – | 20.3 ± 9.0 | – | 20.3 ± 9.1 |
| Urine nicotine (mg/ml) | – | 1357 ± 1140 | – | 1357 ± 1140 |
| Urine cotinine (mg/ml) | – | 1865 ± 1102 | – | 1865 ± 1102 |
| Pulmonary function parametersd | ||||
| FVC | 102 ± 10 | 110 ± 8 | 103 ± 9 | 110 ± 8 |
| FEV1 | 101 ± 10 | 104 ± 8 | 99 ± 10 | 104 ± 8 |
| FEV1/FVC | 82 ± 7 | 78 ± 5 | 81 ± 5 | 76 ± 5 |
| TLC | 92 ± 12 | 94 ± 12 | 91 ± 12 | 94 ± 12 |
| DLCO | 92 ± 14 | 87 ± 8 | 93 ± 14 | 87 ± 8 |
| Brushed epitheliume | ||||
| Number recovered × 10f | 0.9 ± 0.4 | 0.9 ± 0.3 | 0.9 ± 0.3 | 0.9 ± 0.3 |
| %Epithelial cells | 99.1 ± 1.0 | 99.3 ± 0.9 | 99.0 ± 1.1 | 99.3 ± 0.9 |
| %Inflammatory cells | 0.9 ± 1.0 | 0.7 ± 0.9 | 1.0 ± 1.1 | 0.7 ± 0.9 |
| Differential cell countf | ||||
| Ciliated (%) | 66.0 ± 16.2 | 64.4 ± 8.5 | 66.5 ± 17.5 | 64.4 ± 8.5 |
| Secretory (%) | 8.2 ± 4.3 | 11.0 ± 7.7 | 8.8 ± 4.4 | 11.0 ± 7.7 |
| Basal (%) | 10.5 ± 7.6 | 6.1 ± 4.5 | 9.6 ± 7.8 | 6.1 ± 4.5 |
| Undifferentiated (%) | 10.6 ± 5.2 | 18.1 ± 9.5 | 9.8 ± 4.8 | 18.1 ± 9.5 |
aData are presented as mean ± standard deviation.
bA total of 39 subjects (n = 19 nonsmokers and 20 smokers) were assessed by the DNA methylation HELP assay; the same 20 smokers and a subset (n = 16) of the 19 nonsmokers were assessed by the HG-U133 Plus 2.0 Microarray. Cells recovered by brushing of the SAE were used for both the DNA methylation assay and HG-U133 Plus 2.0 Microarray.
cB, black; W, white; H, Hispanic.
dPulmonary function testing parameters are given as %predicted value with the exception of FEV1/FVC, which is reported as %observed; FVC, forced vital capacity; FEV1, forced expiratory volume in 1 s; TLC, total lung capacity; DLCO, diffusing capacity of the lung for carbon monoxide.
eSAE.
fAs a percentage of the SAE recovered.
Epithelial sampling, cDNA preparation and microarray processing
The SAE (10–12th order) was collected by fiberoptic bronchoscopy by brushing as described previously (30). After withdrawing the bronchoscope, the cells were dislodged from the brush by flicking the brush tip in 5 ml of ice-cold Bronchial Epithelium Basal Medium (Lonza, Basel, Switzerland). An aliquot of all airway epithelial samples was used to quantify the total number of cells recovered, and to quantify the percentage of epithelial and inflammatory cells and the proportions of epithelial cell subtypes. Cells from a second aliquot were pelleted for DNA extraction by the Qiagen Puregene kit (Germantown, MD, USA). The remaining sample was immediately processed for RNA extraction and for microarray analysis as described previously (30,46). Total RNA was extracted from the SAE of 16 out of the 19 nonsmokers and all 20 smokers, using the TRIzol method (Invitrogen, Carlsbad, CA, USA). RNA quality was assessed by Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). RNA was processed to generate cDNA, and genome-wide gene expression analysis was performed using the HG-U133 Plus 2.0 Array (Affymetrix, Santa Clara, CA, USA) according to Affymetrix protocols. The captured image data from the HG-U133 Plus 2.0 arrays were processed using the MAS5 algorithm in GeneSpring version 7.3 (Affymetrix). Overall microarray quality was verified by the criteria: (i) 3′/5′ ratio for GAPDH ≤3; and (ii) scaling factor ≤10.0. CEL files were processed by Partek Genomics Suite Software version 6.6 (Partek, Inc., St Louis, MO, USA) for quality control, identification of outliers and determination of expression level for all probe sets, using the robust multi-chip average method with Partek default parameters (47). The phenotypes were evaluated in Partek for sources of variation. A two-way ANOVA was performed to assess smoking significance corrected by gender. The raw data and FPKM values are publically available at the Gene Expression Omnibus site (http://www.ncbi.nlm.nih.gov/geo/), accession number GSE43079.
HELP assay
DNA extracted from the SAE was quantitated by spectrophotometry and analyzed by agarose gel electrophoresis to assess integrity. The microarray-based high-resolution HELP assay was performed on a total of 39 samples (19 nonsmokers and 20 smokers) (48). A 720K Roche-NimbleGen custom array capturing 117 521 HpaII fragments was used to assay CpG islands, CpG island shores and reference sequence promoters. Quality control of arrays included assessment of MspI and HpaII intensity distribution and spatial uniformity of the Cy3 and Cy5 signals (49). For all queried HpaII fragments, intensities were processed to determine the Q-centered (Qcent) ratio and the log2 multi-sample, quantile-normalized unmethylated/methylated (HpaII/MspI) ratio. The Qcent parameter was exported to Excel with fragment annotation details for statistical analysis.
Categorization of methylation states was defined using an HpaII/MspI ratio threshold of 0, defining hypomethylated loci with a positive log2 ratio value, where more methylated loci had a negative log2 ratio value. Qcent ratios were adjusted for batch and gender to assess effects of smoking on methylation of the 117 521 HpaII fragments. A three-way ANOVA (Partek) was used and fold-change was determined as: [least square mean smokers/least square mean nonsmokers]. Probe fragments with a smoking versus nonsmoking Benjamini–Hochberg-corrected P-value <0.05 were initially used to assess significance. For multivariate data exploration purposes, we followed the approach of Pascual et al. (8), and a P-value <0.05 calculated by a Student's t-test and a fold-change greater than ±1.5 were designated as our threshold. The closest gene was determined for these fragments and annotation files were used to map individual HpaII fragments relative to transcription start site of the closest gene. In total, 204 probe fragments corresponding to unique genes were found to be differentially methylated in smokers compared with nonsmokers. Methylation in smokers and nonsmokers was visualized by box and whisker plots.
DNA methylation smoking index
A DNA methylation index for the SAE was calculated using 204 unique genes that were differentially methylated in response to smoking as identified by the HELP analysis. For genes represented by more than one HpaII fragment, the fragment with the lowest P-value was used. The DNA methylation index was calculated based on the percentage of differentially methylated genes outside the normal range defined as the average methylation level (as assessed by the Qcent ratio) of the healthy nonsmokers at ±2 standard deviations:
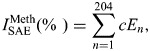 |
where E1 has a value of 1 if the expression level for gene 1 is above or below the average methylation level of healthy nonsmokers ± 2 standard deviations or has a value of 0 if the methylation level is not above or below the average methylation level of healthy nonsmokers ± 2 standard deviations; E2 is the index for gene 2, etc., and the constant (c = 100/204) normalizes the index to the percent of the 204 genes that are outside of the range of healthy nonsmokers (50).
Pathway analysis and biological characterization
The 204 unique genes that were differentially methylated in smokers compared with nonsmokers as well as the top 25 unique, known differentially methylated hypo- and hypermethylated genes were assigned to molecular pathways using the online utility Ingenuity Pathway Analysis (Ingenuity Systems, Redwood City, CA, USA). The biological categories for the global significantly hypo- and hypermethylated genes were assigned using the Affymetrix site (www.affymetrix.com), Gene Ontology (GO), the Human Protein Reference Data Base (www.hprd.org) and GeneCards (www.genecards.org). Biological categories for the top 25 hypo- and hypermethylated genes were assigned by Human Protein Reference Data Base (www.hprd.org) and GeneCards (www.genecards.org).
Correlation of expression with methylation
The correlation of smoking-dependent methylation of SAE DNA and SAE smoking-dependent expression was assessed by two methods. First, starburst plots were used to assess the overall correlation of P-values for smoking-dependent methylation with the P-value for smoking-dependent expression. This was performed for the 220 HpaII probe fragments, which were differentially methylated in smokers compared with nonsmokers (P < 0.05, fold-change greater than ±1.5). These 220 fragments corresponded to 193 unique genes that also had the corresponding HG-U133 Plus 2.0 gene expression data. These 193 unique genes were used to assess the correlation. Second, the methylation level for the differentially methylated (P < 0.05, fold-change greater than ±1.5) smoking-dependent fragments was plotted against the corresponding expression level that were also different between smokers and nonsmokers for the same individuals (P < 0.05, fold-change greater than ±1.5).
Sequemon Epityper assay (MASSArray)
Quantitative analysis of DNA methylation was performed using the Sequenom EpiTyper analysis, which relies on base-specific cleavage followed by MALDI-TOF mass spectrometry. Briefly, DNA extracted from the SAE was quantitated by spectrophotometry and analyzed by agarose gel electrophoresis to assess integrity. Primers were designed using the Sequenom EpiDesigner Beta website (www.epidesigner.com). See Supplementary Material, Table S1, for primer sequences. All amplicons had a size range between 100 and 500 bp. Data were analyzed using the EpiTyper MassArray v1.0 software (Sequenom, Inc., San Diego, CA, USA).
Statistics
Comparison of demographic parameters among groups was performed by two-tailed Student's t-test or chi-square test. A three-way ANOVA was performed on the DNA methylation HELP data to examine the influence of batch and gender on smoking response. For the HG-U133 Plus 2.0 gene expression data, a two-way ANOVA was performed to examine the influence of gender on smoking response.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
FUNDING
This work was supported, in part, by R01HL107882, UL1 RR024996 and UL1 RR024143. L.J.B.-A. is partially supported by Hoffman-La Roche, Nutley, NJ, USA; and M.S.W. is partially supported by the Qatar National Research Fund, NPRP 09-742-3-194.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Satwant Narula and Maria Fuentes, Inflammation Discovery Department, Hoffmann-La Roche, for their support, Rita Shaknovich for technical support and N. Mohamed and D.N. McCarthy for editorial support.
REFERENCES
- 1.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 2.Leeb M., Wutz A. Establishment of epigenetic patterns in development. Chromosoma. 2012;121:251–262. doi: 10.1007/s00412-012-0365-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meister P., Mango S.E., Gasser S.M. Locking the genome: nuclear organization and cell fate. Curr. Opin. Genet. Dev. 2011;21:167–174. doi: 10.1016/j.gde.2011.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang I.V., Schwartz D.A. Epigenetic control of gene expression in the lung. Am. J. Respir. Crit. Care Med. 2011;183:1295–1301. doi: 10.1164/rccm.201010-1579PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones P.A. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 6.Bjornsson H.T., Sigurdsson M.I., Fallin M.D., Irizarry R.A., Aspelund T., Cui H., Yu W., Rongione M.A., Ekstrom T.J., Harris T.B., et al. Intra-individual change over time in DNA methylation with familial clustering. JAMA. 2008;299:2877–2883. doi: 10.1001/jama.299.24.2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soberanes S., Gonzalez A., Urich D., Chiarella S.E., Radigan K.A., Osornio-Vargas A., Joseph J., Kalyanaraman B., Ridge K.M., Chandel N.S., et al. Particulate matter air pollution induces hypermethylation of the p16 promoter via a mitochondrial ROS-JNK-DNMT1 pathway. Sci. Rep. 2012;2:275. doi: 10.1038/srep00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pascual M., Suzuki M., Isidoro-Garcia M., Padron J., Turner T., Lorente F., Davila I., Greally J.M. Epigenetic changes in B lymphocytes associated with house dust mite allergic asthma. Epigenetics. 2011;6:1131–1137. doi: 10.4161/epi.6.9.16061. [DOI] [PubMed] [Google Scholar]
- 9.Li Y.Y., Chen T., Wan Y., Xu S.Q. Lead exposure in pheochromocytoma cells induces persistent changes in amyloid precursor protein gene methylation patterns. Environ. Toxicol. 2012;27:495–502. doi: 10.1002/tox.20666. [DOI] [PubMed] [Google Scholar]
- 10.Dolinoy D.C., Weidman J.R., Waterland R.A., Jirtle R.L. Maternal genistein alters coat color and protects Avy mouse offspring from obesity by modifying the fetal epigenome. Environ. Health Perspect. 2006;114:567–572. doi: 10.1289/ehp.8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Altmann S., Murani E., Schwerin M., Metges C.C., Wimmers K., Ponsuksili S. Dietary protein restriction and excess of pregnant German Landrace sows induce changes in hepatic gene expression and promoter methylation of key metabolic genes in the offspring. J. Nutr. Biochem. 2013;24:484–495. doi: 10.1016/j.jnutbio.2012.01.011. [DOI] [PubMed] [Google Scholar]
- 12.Howard T.D., Ho S.M., Zhang L., Chen J., Cui W., Slager R., Gray S., Hawkins G.A., Medvedovic M., Wagner J.D. Epigenetic changes with dietary soy in cynomolgus monkeys. PLoS One. 2011;6:e26791. doi: 10.1371/journal.pone.0026791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cordero P., Gomez-Uriz A.M., Campion J., Milagro F.I., Martinez J.A. Dietary supplementation with methyl donors reduces fatty liver and modifies the fatty acid synthase DNA methylation profile in rats fed an obesogenic diet. Genes Nutr. 2013;8:105–113. doi: 10.1007/s12263-012-0300-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu F., Killian J.K., Yang M., Walker R.L., Hong J.A., Zhang M., Davis S., Zhang Y., Hussain M., Xi S., et al. Epigenomic alterations and gene expression profiles in respiratory epithelia exposed to cigarette smoke condensate. Oncogene. 2010;29:3650–3664. doi: 10.1038/onc.2010.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zochbauer-Muller S., Lam S., Toyooka S., Virmani A.K., Toyooka K.O., Seidl S., Minna J.D., Gazdar A.F. Aberrant methylation of multiple genes in the upper aerodigestive tract epithelium of heavy smokers. Int. J. Cancer. 2003;107:612–616. doi: 10.1002/ijc.11458. [DOI] [PubMed] [Google Scholar]
- 16.Belinsky S.A., Palmisano W.A., Gilliland F.D., Crooks L.A., Divine K.K., Winters S.A., Grimes M.J., Harms H.J., Tellez C.S., Smith T.M., et al. Aberrant promoter methylation in bronchial epithelium and sputum from current and former smokers. Cancer Res. 2002;62:2370–2377. [PubMed] [Google Scholar]
- 17.Soria J.C., Rodriguez M., Liu D.D., Lee J.J., Hong W.K., Mao L. Aberrant promoter methylation of multiple genes in bronchial brush samples from former cigarette smokers. Cancer Res. 2002;62:351–355. [PubMed] [Google Scholar]
- 18.Tekpli X., Zienolddiny S., Skaug V., Stangeland L., Haugen A., Mollerup S. DNA methylation of the CYP1A1 enhancer is associated with smoking-induced genetic alterations in human lung. Int. J. Cancer. 2012;131:1509–1516. doi: 10.1002/ijc.27421. [DOI] [PubMed] [Google Scholar]
- 19.Anttila S., Hakkola J., Tuominen P., Elovaara E., Husgafvel-Pursiainen K., Karjalainen A., Hirvonen A., Nurminen T. Methylation of cxytochrome P4501A1 promoter in the lung is associated with tobacco smoking. Cancer Res. 2003;63:8623–8628. [PubMed] [Google Scholar]
- 20.Monick M.M., Beach S.R., Plume J., Sears R., Gerrard M., Brody G.H., Philibert R.A. Coordinated changes in AHRR methylation in lymphoblasts and pulmonary macrophages from smokers. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2012;159B:141–151. doi: 10.1002/ajmg.b.32021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Philibert R.A., Sears R.A., Powers L.S., Nash E., Bair T., Gerke A.K., Hassan I., Thomas C.P., Gross T.J., Monick M.M. Coordinated DNA methylation and gene expression changes in smoker alveolar macrophages: specific effects on VEGF receptor 1 expression. J. Leukoc. Biol. 2012;92:621–631. doi: 10.1189/jlb.1211632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wan E.S., Qiu W., Baccarelli A., Carey V.J., Bacherman H., Rennard S.I., Agusti A., Anderson W., Lomas D.A., Demeo D.L. Cigarette smoking behaviors and time since quitting are associated with differential DNA methylation across the human genome. Hum. Mol. Genet. 2012;21:3073–3082. doi: 10.1093/hmg/dds135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Breitling L.P., Yang R., Korn B., Burwinkel B., Brenner H. Tobacco-smoking-related differential DNA methylation: 27K discovery and replication. Am. J. Hum. Genet. 2011;88:450–457. doi: 10.1016/j.ajhg.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fraga M.F., Ballestar E., Paz M.F., Ropero S., Setien F., Ballestar M.L., Heine-Suner D., Cigudosa J.C., Urioste M., Benitez J., et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl Acad. Sci. USA. 2005;102:10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feinberg A.P. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447:433–440. doi: 10.1038/nature05919. [DOI] [PubMed] [Google Scholar]
- 26.Rodgman A., Perfetti T. The Chemical Components of Tobacco and Tobacco Smoke. Boca Raton, FL: CRC Press; 2008. [Google Scholar]
- 27.Pryor W.A., Hales B.J., Premovic P.I., Church D.F. The radicals in cigarette tar: their nature and suggested physiological implications. Science. 1983;220:425–427. doi: 10.1126/science.6301009. [DOI] [PubMed] [Google Scholar]
- 28.Church D.F., Pryor W.A. Free-radical chemistry of cigarette smoke and its toxicological implications. Environ. Health Perspect. 1985;64:111–126. doi: 10.1289/ehp.8564111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pryor W.A., Stone K. Oxidants in cigarette smoke. radicals, hydrogen peroxide, peroxynitrate, and peroxynitrite. Ann. NY Acad. Sci. 1993;686:12–27. doi: 10.1111/j.1749-6632.1993.tb39148.x. [DOI] [PubMed] [Google Scholar]
- 30.Harvey B.G., Heguy A., Leopold P.L., Carolan B.J., Ferris B., Crystal R.G. Modification of gene expression of the small airway epithelium in response to cigarette smoking. J. Mol. Med. 2007;85:39–53. doi: 10.1007/s00109-006-0103-z. [DOI] [PubMed] [Google Scholar]
- 31.Hogg J.C., Macklem P.T., Thurlbeck W.M. Site and nature of airway obstruction in chronic obstructive lung disease. N. Engl. J. Med. 1968;278:1355–1360. doi: 10.1056/NEJM196806202782501. [DOI] [PubMed] [Google Scholar]
- 32.Auerbach O., Forman J.B., Gere J.B., Kassouny D.Y., Muehsam G.E., Petrick T.G., Smolin H.J., Stout A.P. Changes in the bronchial epithelium in relation to smoking and cancer of the lung – a report of progress. N. Engl. J. Med. 1957;256:97–104. doi: 10.1056/NEJM195701172560301. [DOI] [PubMed] [Google Scholar]
- 33.Crystal R.G., Randell S.H., Engelhardt J.F., Voynow J., Sunday M.E. Airway epithelial cells: current concepts and challenges. Proc. Am. Thorac. Soc. 2008;5:772–777. doi: 10.1513/pats.200805-041HR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Puchelle E., Zahm J.M., Tournier J.M., Coraux C. Airway epithelial repair, regeneration, and remodeling after injury in chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2006;3:726–733. doi: 10.1513/pats.200605-126SF. [DOI] [PubMed] [Google Scholar]
- 35.Thompson A.B., Robbins R.A., Romberger D.J., Sisson J.H., Spurzem J.R., Teschler H., Rennard S.I. Immunological functions of the pulmonary epithelium. Eur. Respir. J. 1995;8:127–149. doi: 10.1183/09031936.95.08010127. [DOI] [PubMed] [Google Scholar]
- 36.Knight D.A., Holgate S.T. The airway epithelium: structural and functional properties in health and disease. Respirology. 2003;8:432–446. doi: 10.1046/j.1440-1843.2003.00493.x. [DOI] [PubMed] [Google Scholar]
- 37.Sanders Y.Y., Ambalavanan N., Halloran B., Zhang X., Liu H., Crossman D.K., Bray M., Zhang K., Thannickal V.J., Hagood J.S. Altered DNA methylation profile in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2012;186:525–535. doi: 10.1164/rccm.201201-0077OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jones P.A. The DNA methylation paradox. Trends Genet. 1999;15:34–37. doi: 10.1016/s0168-9525(98)01636-9. [DOI] [PubMed] [Google Scholar]
- 39.Fuks F. DNA methylation and histone modifications: teaming up to silence genes. Curr. Opin. Genet. Dev. 2005;15:490–495. doi: 10.1016/j.gde.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 40.Belinsky S.A. Silencing of genes by promoter hypermethylation: key event in rodent and human lung cancer. Carcinogenesis. 2005;26:1481–1487. doi: 10.1093/carcin/bgi020. [DOI] [PubMed] [Google Scholar]
- 41.Zochbauer-Muller S., Minna J.D., Gazdar A.F. Aberrant DNA methylation in lung cancer: biological and clinical implications. Oncologist. 2002;7:451–457. doi: 10.1634/theoncologist.7-5-451. [DOI] [PubMed] [Google Scholar]
- 42.Leng S., Stidley C.A., Liu Y., Edlund C.K., Willink R.P., Han Y., Landi M.T., Thun M., Picchi M.A., Bruse S.E., et al. Genetic determinants for promoter hypermethylation in the lungs of smokers: a candidate gene-based study. Cancer Res. 2012;72:707–715. doi: 10.1158/0008-5472.CAN-11-3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qiu W., Baccarelli A., Carey V.J., Boutaoui N., Bacherman H., Klanderman B., Rennard S., Agusti A., Anderson W., Lomas D.A., Demeo D.L. Variable DNA methylation is associated with chronic obstructive pulmonary disease and lung function. Am. J. Respir. Crit. Care Med. 2012;185:373–381. doi: 10.1164/rccm.201108-1382OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Crapo J.D., Barry B.E., Gehr P., Bachofen M., Weibel E.R. Cell number and cell characteristics of the normal human lung. Am. Rev. Respir. Dis. 1982;126:332–337. doi: 10.1164/arrd.1982.126.2.332. [DOI] [PubMed] [Google Scholar]
- 45.Hackett N.R., Butler M.W., Shaykhiev R., Salit J., Omberg L., Rodriguez-Flores J.L., Mezey J.G., Strulovici-Barel Y., Wang G., Didon L., Crystal R.G. RNA-Seq quantification of the human small airway epithelium transcriptome. BMC Genomics. 2012;13:82. doi: 10.1186/1471-2164-13-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hackett N.R., Shaykhiev R., Walters M.S., Wang R., Zwick R.K., Ferris B., Witover B., Salit J., Crystal R.G. The human airway epithelial basal cell transcriptome. PLoS One. 2011;6:e18378. doi: 10.1371/journal.pone.0018378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Raman T., O'Connor T.P., Hackett N.R., Wang W., Harvey B.G., Attiyeh M.A., Dang D.T., Teater M., Crystal R.G. Quality control in microarray assessment of gene expression in human airway epithelium. BMC Genomics. 2009;10:493. doi: 10.1186/1471-2164-10-493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Khulan B., Thompson R.F., Ye K., Fazzari M.J., Suzuki M., Stasiek E., Figueroa M.E., Glass J.L., Chen Q., Montagna C., et al. Comparative isoschizomer profiling of cytosine methylation: the HELP assay. Genome Res. 2006;16:1046–1055. doi: 10.1101/gr.5273806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thompson R.F., Reimers M., Khulan B., Gissot M., Richmond T.A., Chen Q., Zheng X., Kim K., Greally J.M. An analytical pipeline for genomic representations used for cytosine methylation studies. Bioinformatics. 2008;24:1161–1167. doi: 10.1093/bioinformatics/btn096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tilley A.E., O'Connor T.P., Hackett N.R., Strulovici-Barel Y., Salit J., Amoroso N., Zhou X.K., Raman T., Omberg L., Clark A., et al. Biologic phenotyping of the human small airway epithelial response to cigarette smoking. PLoS One. 2011;6:e22798. doi: 10.1371/journal.pone.0022798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li J.B., Gerdes J.M., Haycraft C.J., Fan Y., Teslovich T.M., May-Simera H., Li H., Blacque O.E., Li L., Leitch C.C., et al. Comparative genomics identifies a flagellar and basal body proteome that includes the BBS5 human disease gene. Cell. 2004;117:541–552. doi: 10.1016/s0092-8674(04)00450-7. [DOI] [PubMed] [Google Scholar]
- 52.Avidor-Reiss T., Maer A.M., Koundakjian E., Polyanovsky A., Keil T., Subramaniam S., Zuker C.S. Decoding cilia function: defining specialized genes required for compartmentalized cilia biogenesis. Cell. 2004;117:527–539. doi: 10.1016/s0092-8674(04)00412-x. [DOI] [PubMed] [Google Scholar]
- 53.Geremek M., Bruinenberg M., Zietkiewicz E., Pogorzelski A., Witt M., Wijmenga C. Gene expression studies in cells from primary ciliary dyskinesia patients identify 208 potential ciliary genes. Hum. Genet. 2011;129:283–293. doi: 10.1007/s00439-010-0922-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.