

Abstract
Ubiquinone (UQ), a.k.a. coenzyme Q, is a redox-active lipid that participates in several cellular processes, in particular mitochondrial electron transport. Primary UQ deficiency is a rare but severely debilitating condition. Mclk1 (a.k.a. Coq7) encodes a conserved mitochondrial enzyme that is necessary for UQ biosynthesis. We engineered conditional Mclk1 knockout models to study pathogenic effects of UQ deficiency and to assess potential therapeutic agents for the treatment of UQ deficiencies. We found that Mclk1 knockout cells are viable in the total absence of UQ. The UQ biosynthetic precursor DMQ9 accumulates in these cells and can sustain mitochondrial respiration, albeit inefficiently. We demonstrated that efficient rescue of the respiratory deficiency in UQ-deficient cells by UQ analogues is side chain length dependent, and that classical UQ analogues with alkyl side chains such as idebenone and decylUQ are inefficient in comparison with analogues with isoprenoid side chains. Furthermore, Vitamin K2, which has an isoprenoid side chain, and has been proposed to be a mitochondrial electron carrier, had no efficacy on UQ-deficient mouse cells. In our model with liver-specific loss of Mclk1, a large depletion of UQ in hepatocytes caused only a mild impairment of respiratory chain function and no gross abnormalities. In conjunction with previous findings, this surprisingly small effect of UQ depletion indicates a nonlinear dependence of mitochondrial respiratory capacity on UQ content. With this model, we also showed that diet-derived UQ10 is able to functionally rescue the electron transport deficit due to severe endogenous UQ deficiency in the liver, an organ capable of absorbing exogenous UQ.
INTRODUCTION
Ubiquinone (UQ), also known as Coenzyme Q (CoQ), is a lipid composed of a redox-active benzoquinone ring conjugated to an isoprenoid side chain. It is found in all cells, from bacteria to mammals, and in the membranes of most or all organelles where it participates in a variety of cellular processes. The best-known function of UQ is to act as an electron carrier in the mitochondrial respiratory chain, where it serves to transport electrons from Complexes I and II as well as from other mitochondrial dehydrogenases to Complex III (1,2). Moreover, reduced UQ is an important antioxidant in cell membranes and lipoproteins (3). UQ has also been shown to play a role in plasma membrane electron transport, regulation of the mitochondrial permeability transition pore and pyrimidine nucleotide biosynthesis (4–6). Furthermore, an effect of UQ administration to improve endothelial dysfunction has been reported in human patients (7,8). Presently, 11 genes (COQ1-9, YAH1 and ARH1) are known to act in the UQ biosynthetic pathway of the budding yeast Saccharomyces cerevisiae (9,10). UQ biosynthesis in animal cells is similar to that in yeast, although many details remain to be worked out.
In the last two decades, a growing number of human patients with mitochondrial myopathy showing deficiencies of UQ10 have been identified (11–21) (the subscript denotes the number of isoprenoid units in the side chain; UQ10 is the main species in humans but UQ9 is the main species in mice). Primary UQ10 deficiency caused by an inherited defect in UQ biosynthesis, as opposed to secondary complication of other diseases, is a rare and devastating disease that often presents with multisystem disorders and has a high mortality rate if not treated effectively. To this time, mutations in seven of the nine genes encoding proteins required for the final phase of UQ10 biosynthesis inside mitochondria have been reported (reviewed in 22) and more can be expected to follow. Despite these advances, some fundamental questions about the disease remain unanswered. In particular, primary UQ deficiency, like most mitochondrial disorders, often presents with very heterogeneous clinical manifestations (reviewed in 22–24), for which little other than speculations are offered. Moreover, its precise pathogenic mechanisms remain to be fully understood. Under UQ deficient states, diverse biochemical alteration, including impaired energy production, oxidative stress, impaired pyrimidine biosynthesis and increased mitophagy, have been observed and implied as possible pathogenic mechanisms (15,25–27). Endogenous UQ deficiency is a potentially treatable condition and some clinical cases have been reported to respond to UQ supplementation treatments (11,13,17–19). However, findings on the effectiveness of UQ supplementation have been inconsistent (14,16,19,21,28). Development of effective UQ replacement therapies and a proper investigation of their efficacy are still important but challenging tasks. Furthermore, given the antioxidant and respiratory functions of UQ and the implication of mitochondrial dysfunction and oxidative stress in aging, UQ has been marketed as an anti-aging supplement, in spite of very limited scientific evidence to support such use.
The conserved gene that encodes the mitochondrial enzyme that catalyzes the penultimate step of the UQ biosynthetic pathway, the hydroxylation of 6-demethoxyubiquinone (DMQ) to form 6-hydroxyubiquinone, is called COQ7 in yeast, clk-1 in nematodes, Mclk1 or Coq7 in mice and COQ7 in humans (29–32). Contrary to yeast coq7 null mutants, which accumulate the product of an early step of UQ synthesis (33), the losses of CLK-1 in nematode and MCLK1 in mice produce accumulation of the actual substrate of the mutated enzyme, DMQ9 (30,34,35). We previously have shown that mutations in clk-1 and Mclk1 give rise to a wide range of phenotypes in both organisms, including extended longevity when viable (26,36,37). Interestingly, clk-1 mutants are the only UQ biosynthesis-deficient C. elegans mutants that can be maintained as a homozygous line if fed a UQ-replete bacterial diet, all other null mutations in coq genes described so far are lethal (38–42). The key factors that distinguish clk-1 mutants from other coq nematode mutants and the relation between clk-1 phenotypes and the accumulation of DMQ9 have not yet been fully elucidated. In the mouse, full knockout of Mclk1 results in prenatal death (30,31). Mice heterozygous for Mclk1 (Mclk1+/−) look superficially normal but on closer examination demonstrate a variety of biochemical and phenotypic abnormalities (26,37,43). In the most recent study of Mclk1+/− mice, UQ levels in whole liver mitochondria were found to be unaltered, but UQ levels in purified inner mitochondrial membranes (IMM), where the last eight steps of UQ biosynthesis are carried out, were found to be lowered (44). Mclk1+/− mitochondria were also found to have unexpected elevated levels of UQ in the outer mitochondrial membrane. These findings led to the conclusion that even a partial loss of MCLK1 function has a significant effect on UQ production and that MCLK1 is a rate-limiting enzyme in the UQ biosynthetic pathway (44). Most likely, a variety of biochemical alterations observed in Mclk1+/− mice, such as respiratory chain deficiency, decreased ATP production, elevated mitochondrial oxidative stress and increased expression of HIF-1α, stem from decreased levels of UQ in the IMM which is the site of oxidative phosphorylation (OXPHOS) in eukaryotes.
COQ9 is one of the genes in which mutations have been identified in patients with primary UQ10 deficiency (21). The exact biochemical function of COQ9 protein in UQ biosynthesis is unknown, but, intriguingly, loss of COQ9 expression appears to impair UQ biosynthesis by reducing COQ7/MCLK1 levels (45). Yeast coq9 mutants (46,47), a human patient with a lesion in the COQ9 gene (21), as well as a recently described Coq9 mouse mutants carrying a mutation similar to that found in humans (45), display DMQ accumulation along with a severe deficit in UQ levels.
The structure of UQ is conserved from bacteria to vertebrates. It is unknown how its structure exactly determines its functions and whether some of the UQ biosynthesis intermediates have an activity and hence may contribute to the clinical manifestations of a given genetic defect in the biosynthetic route of UQ. Rhodoquinone which has an amino group on the benzoquinone ring in place of one of the methoxy groups of UQ, and which is not found in vertebrates, can function as an electron carrier between Complex I and fumarate reductase in some parasitic species under low oxygen conditions (48). Vitamin K2, which contains a naphthoquinone ring and an isoprenoid side chain, has recently been proposed to be capable of acting as an electron carrier in Drosophila mitochondria (49). DMQ differs from UQ by missing one of the two methoxy groups on the benzoquinone ring. It is yet uncertain how lacking the C6-methoxy group of the ring affect UQ functions, in other words, whether DMQ is capable of fulfilling or interfering with some of the functions of UQ. We previously found that cultured embryonic stem (ES) cells from Mclk1 knockout mice in which DMQ9 is the only detectable quinone are viable and possess significant respiratory activity, suggesting a possible functionality of DMQ9 as a respiratory substrate (30). Supporting this notion is the observation that synthetic DMQ2 can serve as an electron acceptor for respiratory complexes after addition to a mitochondrial preparation from clk-1 worms (34). However, findings that conflict with this notion have also been reported. Yeast coq7-1 mutants lacking UQ6 and producing DMQ6 are respiration-defective (50), arguing against the ability of DMQ to functionally substitute for UQ in the respiratory chain, at least in the yeast S. cerevisiae, which possesses no Complex I. In worms, clk-1 mutant mitochondria have only mildly decreased mitochondrial respiration (51,52). However, in addition to DMQ9, other quinone species (dietary UQ8 and endogenous rhodoquinone) have been observed to be present in clk-1worms (53,54) and they could be responsible for at least some of the mitochondrial electron transport in those mutants. Interestingly, addition of a pentane extract from clk-1 mitochondria (containing DMQ9) to UQ9-replete mitochondria partially inhibits Complex I + III activity, but not Complex II + III (55), indicating a possible site-specific interference of DMQ with UQ function. It is not yet known if any such effect exists in vivo.
RESULTS
Generation of a mouse line with a floxed Mclk1 allele for conditional knockout
As germline deletion of Mclk1 leads to prenatal lethality (30,31), we generated conditional Mclk1 knockout mice in which exons 2 and 3 are flanked by loxP sequences to allow for excision with Cre recombinase (Fig. 1A). Mice homozygous for the loxP-flanked Mclk1 allele (Mclk1loxP/loxP) develop normally and are indistinguishable from wild-type littermates. We obtained total deletion of the loxP-flanked (floxed) region by crossing to a transgenic line in which Cre is expressed in the germline (56). This generated mice having a full knockout of one copy of Mclk1 (Mclk1+/Δ). As expected from the embryonic lethal phenotype of the published Mclk1-null mutants (30,31), no viable Mclk1Δ/Δ pups were obtained from intercrosses of Mclk1+/Δ heterozygotes (data not shown).
Figure 1.
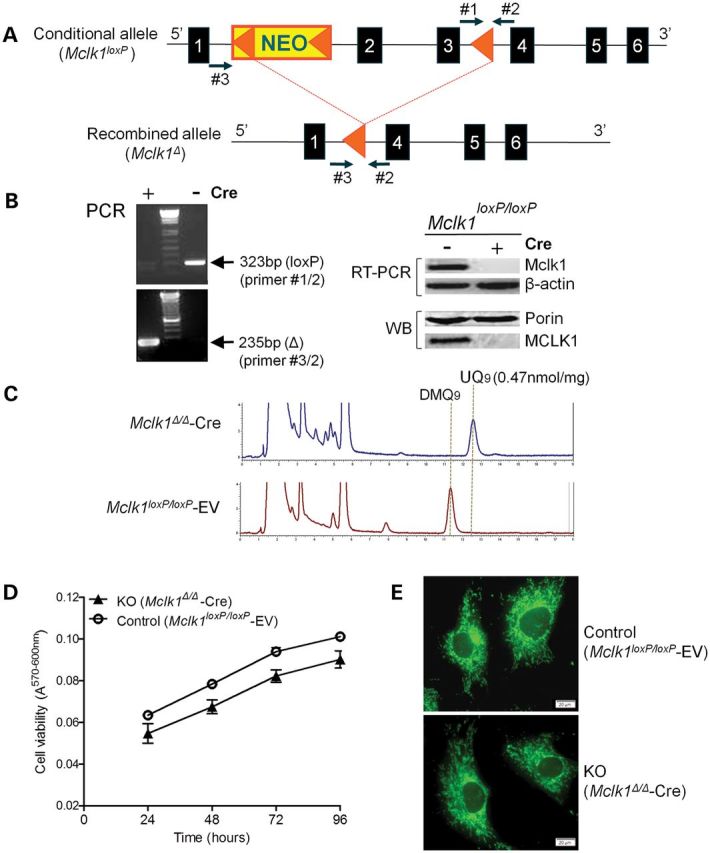
Generation of MEFs lacking MCLK1. (A) Schematic illustration of the conditional Mclk1 allele. A loxP/FRT-flanked Neo cassette (NEO, yellow) is inserted upstream of exon 2. A third loxP site is inserted downstream of exon 3. Exons are shown as blue boxes, and loxP sites are shown as orange triangles. Primers #1 and 2 were used to distinguish the conditional (Mclk1loxP) allele and wild-type allele, whereas PCR using primers #2 and 3 reveal the presence or absence of the recombined allele (Mclk1Δ). (B) Deletion efficiency in Mclk1loxP/loxP MEFs infected with pBabe-puro-Cre retrovirus. The recombined allele (Δ) but not the floxed allele (loxP) was detected by PCR in Mclk1loxP/loxP MEFs infected with Cre-expressing retrovirus (+). The results of the RT-PCR show loss of Mclk1 mRNA expression in Cre-expressing MEFs. β-Actin was used as RT-PCR control. Western blot (WB) indicates that MCLK1 protein is absent in Cre-expressing MEFs. Mitochondrial outer membrane protein Porin served as loading control. (C) HPLC traces of quinone extracts recorded at 275 nm. DMQ9 is the only detectable quinone in Mclk1 knockout MEFs (Mclk1Δ/Δ-Cre). The UQ9 concentration in control cells (Mclk1loxP/loxP MEF lines infected with empty retroviral vector; Mclk1loxP/loxP -EV) is shown (normalized to protein content). (D) Growth curves of the indicated MEFs. Cells were seeded in triplicates in 48-well plates and viability was determined at the indicated time points with alamarBlue Assay. Cell viability is reported as the difference in absorbance at the two wavelengths (A570 nm–A600 nm) which changes with the number of living cells. Each growth curve represents three Mclk1loxP/loxP MEF lines infected with either Cre-expressing retrovirus or retrovirus alone. No significant difference in proliferation rate is observed between Mclk1 knockout and control MEFs. (E) MitoTracker Green staining shows a typical reticular network of mitochondria in knockout MEFs. Scale bars = 20 µm.
Generation and viability of Mclk1 knockout mouse embryonic fibroblasts
We generated Mclk1-null mouse embryonic fibroblasts (MEFs) by retroviral expression of Cre recombinase (pBabe-Puro-Cre) in MEFs derived from conditional Mclk1loxP/loxP mice. As controls, we used the same MEF lines infected with an empty pBabe-Puro vector (Mclk1loxP/loxP-EV). Inactivation of the floxed Mclk1 allele upon infection with Cre-expressing retrovirus was analyzed by PCR amplification of intact or recombined Mclk1. Figure 1B shows that the recombined but not the floxed Mclk1 allele was detected in Cre-expressing MEF cells. Moreover, RT-PCR analysis and an anti-MCLK1 immunoblot verified the absence of MCLK1 expression in those cells (Fig. 1B). These results confirmed that the conversion of the floxed Mclk1 allele (‘loxP’) into the deleted form (‘Δ’) was virtually complete (hereinafter referred to as Mclk1Δ/Δ-Cre MEFs). As anticipated, UQ9, which is the primary isoform of UQ produced in the mouse, was undetectable in Mclk1Δ/Δ-Cre MEFs. Instead, they showed accumulation of the substrate of the missing enzyme, demethoxyubiquinone-9 (DMQ9), at levels very similar to those of UQ9 in control cells (Fig. 1C). Reintroduction of wild-type Mclk1 into Mclk1Δ/Δ-Cre cells restored the biosynthesis of UQ9, while mutated Mclk1 cDNA containing a point mutation equivalent to the Glu-to-Lys substitution in the C. elegans clk-1(e2519) allele was found to produce a mutant MCLK1 protein lacking DMQ hydroxylation activity (Supplementary Material, Fig. S1).
Without added exogenous UQ, Mclk1Δ/Δ-Cre MEFs were fully viable and exhibited no growth defect when cultured in standard culture media [Dulbecco's modified Eagle's medium (DMEM) with 4.5 g/l glucose and 10% fetal bovine serum (FBS)] (Fig. 1D). We also did not observe any obvious morphologic differences between Mclk1Δ/Δ-Cre and control fibroblasts. In order to rule out the possibility that the viability of Mclk1Δ/Δ-Cre MEFs relies on a trace amount of UQ that may be present in FBS, we cultured them in medium supplemented with lipid-stripped FBS and a chemically defined lipid mixture containing no UQ. No apparent difference was noted between the cells grown in the presence of normal FBS and those cultured in the media containing chemically more defined lipids. Thus, we conclude that Mclk1-null MEFs producing no UQ are indeed viable and can proliferate in vitro.
Impaired respiratory chain function in knockout fibroblasts
Staining of Mclk1Δ/Δ-Cre fibroblasts with the mitochondrial dye MitoTracker Green revealed a typical reticular network of mitochondria, indicating the mutant cells contain intact mitochondria (Fig. 1E). To determine the overall activity of the mitochondrial respiratory chain, we measured oxygen consumption of intact Mclk1Δ/Δ-Cre and control vector-transfected fibroblasts. The knockout cells were found to respire oxygen but at a slower rate than their counterparts. Both basal and uncoupler (FCCP: carbonylcyanide-p-trifluoromethoxyphenylhydrazone)-stimulated oxygen consumption rates (OCR) reached to about 50% of the control level (Fig. 2A). Addition of the Complex IV inhibitor cyanide to cell suspension resulted in a complete inhibition of oxygen consumption, confirming that the detected oxygen consumption was predominantly of mitochondrial origin. In order to localize the respiratory defect, we next measured the activities of respiratory chain complexes. The activities of Complex II and Complex II + III in Mclk1Δ/Δ-Cre mitochondria were found significantly decreased, while Complex I + III activity appeared no different from that in control cells (Fig. 2C and D). These measurements were performed on MEFs after 10 passages following retroviral infection (with 5-fold to 10-fold dilutions at each passage). At that time, UQ molecules present in the cells prior to the CRE-dependent Mclk1 knockout should be diluted out of existence. Hence, we interpret these observations to indicate that the accumulated DMQ9 can functionally replace UQ9 in the mammalian mitochondrial respiratory chain, with a higher efficiency for Complex I-dependent respiration than for Complex II-dependent respiration. Measurement of citrate synthase (CS) activity in whole-cell lysates revealed that total CS activity is lower in Mclk1Δ/Δ-Cre fibroblasts, suggesting less mitochondrial content in those cells (Fig. 2B). Despite respiring slowly, Mclk1Δ/Δ-Cre MEFs showed no decrease in cellular ATP levels compared with controls (Supplementary Material, Fig. S2A). After incubation with the ATP-synthase inhibitor oligomycin, a dramatic drop in ATP levels was noted, consistent with the above conclusion that Mclk1Δ/Δ-Cre MEFs have a functional respiratory chain.
Figure 2.

Oxygen consumption and activities of mitochondrial respiratory enzymes. (A) OCR of intact cells measured polarographically. OCR is normalized to total cellular protein. Both basal and uncoupler (FCCP)-stimulated respiration rates are lower in Mclk1 knockout (KO) MEFs in comparison with controls. Results are the mean ± SEM of three paired MEF lines. (B) CS activity in whole-cell extracts normalized to total protein. Results are the mean ± SEM of five paired MEF lines. A lower activity of CS in Mclk1 KO MEFs indicates reduced mitochondrial content. (C and D) Activities of mitochondrial respiratory complexes. Mitochondria prepared from Mclk1 KO MEFs have normal activity of Complexes I–III and decreased activities of Complexes II–III and Complex II. Results are the mean ± SEM of four to five pairs of MEF lines. Paired t-tests were conducted to compare the measurements between control and Mclk1 KO MEFs. The asterisk (*) denotes a statistically significant difference at P < 0.05.
We next examined the ability of Mclk1Δ/Δ-Cre MEFs to grow in glucose-free medium containing galactose, where cells are forced to rely mostly on mitochondrial oxidative phosphorylation to produce ATP to maintain their viability (57). While control fibroblasts continued to grow well after sub-culture in galactose medium, Mclk1Δ/Δ-Cre MEFs failed to survive with galactose alone (Fig. 3A). Provision of exogenous UQ9 in the medium can fully rescue the galactose-induced lethality (Fig. 3B and C), suggesting the absence of UQ solely accounts for the death in galactose medium. The absence of glycolysis stimulates the rate of respiration (57,58) which is often associated with a high production rate of radical oxygen species (ROS). To determine whether the loss of the antioxidant function of UQ could be at least a part of the cause of the cell death in galactose medium, we tested whether treatment with antioxidants N-acetylcysteine (NAC) or Trolox (a vitamin E analogue) could increase the viability of Mclk1Δ/Δ-Cre MEFs in galactose-containing medium. As shown in Figure 3D, their survival in galactose was not improved significantly by the addition of NAC or Trolox, strongly suggesting that the rescue by exogenous UQ9 is ascribed to its ability to enhance respiratory function in Mclk1Δ/Δ-Cre cells. Thus, it seems that, compared with UQ9, DMQ9 is a poor substrate for the respiratory chain and incapable of supporting a relatively high OXPHOS activity. This is also consistent with the observation that Mclk1Δ/Δ-Cre MEFs appeared more sensitive to glycolysis inhibition by 2-deoxy-d-glucose (2-DG) than controls (Supplementary Material, Fig. S2B), indicating a greater reliance on glycolysis for survival. Together, these findings suggest that Mclk1Δ/Δ-Cre fibroblasts still carry out mitochondrial respiration but at a lower rate and with a greater dependence on glycolysis for survival.
Figure 3.

Non-viability of Mclk1 knockout MEFs in galactose medium and rescue by UQ9. (A) Growth curves of Mclk1 knockout (KO) and control MEFs cultured in galactose medium. Cells seeded in 48-well plates were first grown in glucose-containing DMEM overnight, and then culture medium was replaced into glucose-free DMEM containing galactose. Total cellular protein per well was determined using the BCA protein assay (Pierce) at the indicated time points. Most Mclk1 KO MEFs died after 4 days in galactose medium, while control fibroblasts showed continued growth. Results are presented as the mean ± SEM of three independent MEF lines. (B) UQ9 supplementation rescues the viability of Mclk1 KO MEFs grown in galactose-containing medium. Mclk1 KO MEFs were first grown in glucose medium overnight, and then culture medium was replaced into galactose medium added with varying concentrations of UQ9. Cell viability was determined by the alamarBlue Cell Viability Assay 4 days later. Data are expressed as percentage of viability relative to that of glucose-grown KO MEFs (viability = 100%) and presented as the mean ± SEM of three independent MEF lines. The Student's t-test was used to compare the difference in viability between the KO MEFs grown in the presence or absence of UQ9 (*P < 0.05; **P < 0.01). (C) Phase-contrast images of Mclk1 KO MEFs grown in galactose medium supplemented with or without UQ9 (magnification, ×100). Conditions were the same as described for (B). Images were taken after 4 days of growth in galactose. (D) N-Acetylcysteine (NAC) and Trolox (vitamin E analog) fail to rescue the survival of Mclk1 KO MEFs in galactose medium. Experimental conditions were similar to those described for (B). Results are expressed as percentage of viability compared with untreated glucose-grown KO MEFs (set to 100% viability) and are the mean ± SEM of three independent MEF lines. No difference was seen between untreated Mclk1 KO MEFs and those treated with the testing antioxidants (ANOVA/Dunnett test).
Effects of UQ analogues and Vitamin K2 on the respiratory deficiency of knockout fibroblasts
In addition to UQ9, we evaluated the effectiveness of several other UQ analogues in improving mitochondrial electron transport and therefore rescuing the survival of Mclk1Δ/Δ-Cre MEFs in galactose. In our experiments, UQ with isoprenoid tails, UQ4, UQ6 and UQ10, greatly enhanced the survival of Mclk1Δ/Δ-Cre cells in galactose media, whereas idebenone [6-(10-hydroxydecyl) 2,3-dimethoxy-5-methyl-1,4-benzoquinone] was ineffective and only a small rescue effect was seen in the cells treated with decylubiquinone (2,3-dimethoxy-5-methyl-6-decyl-1, 4-benzoquinone) (Fig. 4). More recently, Vitamin K2 has been proposed as a mitochondrial electron carrier in Drosophila (49), so we tested whether the respiratory defect in Mclk1Δ/Δ-Cre MEFs can be rescued by exogenous application of Vitamin K2. As shown in Figure 4, we observed no rescue of the cell lethality when supplementing galactose growth medium with Vitamin K2.
Figure 4.

Rescuing ability of various UQ analogues on galactose-induced lethality. Conditions were the same as described for Figure 3B. Cell viability is expressed as relative percentage compared with that of wild-type control fibroblasts (Mclk1loxP/loxP-EV) treated at the same time with ethanol (vehicle). UQ4, UQ6 and UQ10, but not idebenone, decylubiquinone and Vitamin K2, greatly improve the survival of Mclk1 KO fibroblasts (Mclk1Δ/Δ-Cre) in galactose medium. Indicated doses are the highest doses that did not show significant toxicity on the control fibroblasts (idebenone, decylubiquinone and Vitamin K2) or were the most effective dose among those used in this work (UQ4, UQ6, UQ9 and UQ10). Data are the mean values ± SEM for at least three MEF lines. In each group (wild-type control or KO), compound treatments are compared with the ethanol-treated culture using one-way ANOVA with Dunnet's multiple comparison test. ***P < 0.001.
Generation of liver-specific Mclk1 knockout mice
Embryonic lethality of Mclk1−/− mice is most likely due to a complete absence of UQ in all tissues (30,31). To investigate the consequence of UQ deficiency in an individual tissue, we generated liver-specific Mclk1 knockout mice by crossing Mclk1-floxed mice to Albumin–Cre (AlbCre) transgenic mice which express Cre specifically in hepatocytes (59). To ensure a high efficiency of generating full Mclk1 knockout cells, we used mice that carry one conditional allele of Mclk1 (‘loxP’) and one constitutive-null allele (‘-’). PCR analysis for the recombined knockout allele confirmed that the recombination occurred in the liver but not in other tissue samples analyzed (Fig. 5A), indicating that disruption of Mclk1 was restricted to the liver. RT-PCR and western blot analysis showed that MCLK1 expression was almost completely lost in the liver of Mclk1loxP/−, AlbCre+ mice, confirming successful ablation of Mclk1 gene expression in the liver (Fig. 5A). The remaining trace expression is likely confined to other, minor, liver cell types. As expected, MCLK1 protein levels were not reduced in the kidneys (Fig. 5A). To our surprise, liver-specific deletion of Mclk1 produced no gross abnormalities. Mclk1loxP/−,AlbCre+ mice (hereafter termed Mclk1liver-KO mouse) grew normally and were indistinguishable from their control littermates (Mclk1loxP/− and Mclk1loxP/+). At 15 months of age, Mclk1liver-KO mice had body weights similar to those of controls (Fig. 5B). At present, the oldest Mclk1liver-KO mice have reached ≈21 months. Up to this age, there was no significant difference in mortality between Mclk1liver-KO and control mice. Knockout livers also appeared normal at necropsy (data not shown).
Figure 5.

Conditional knockout of Mclk1 in liver. (A) Confirmation of Mclk1 deletion in the liver of Mclk1liver-KO mice (Mclk1loxP/−, AlbCre+). Upper panel: on PCR analysis, the band of excised allele is observed in the liver of Mclk1liver-KO mice but not in other tissues or the liver of AlbCre-negative mice (Mclk1loxP/−). PCR was performed on DNA isolated from tissues of 4-month-old mice (I, intestine; Lu, lung; B, brain; M, skeletal muscle; S, spleen; K, kidney; H, heart; L, liver; T, tail) or from the liver of Mclk1loxP/− mice. RT-PCR analysis detected mRNA expression of Mclk1 in the liver not expressing the AlbCre transgene, but barely in the liver of Mclk1liver-KO mice. Expression of β-actin, used as RT-CPR control, is identical in AlbCre positive and negative mouse livers. NC is negative control. Lower panel: western blotting analysis for the level of MCLK1 protein in liver mitochondria shows that MCLK1 expression is lost in the liver of Mclk1liver-KO mice, but not changed in the kidney in comparison with AlbCre-negative Mclk1loxP/− controls. The mitochondrial outer membrane protein, porin, was used as loading control. (B) Fifteen-month-old Mclk1liver-KO mice have a similar body weight to AlbCre-negative littermate controls (Mclk1loxP/−). Each column represents the mean ± SEM of nine mice. (C) Quinone content in Mclk1 knockout liver. The columns show the mean ± SEM of the peak areas for DMQ9 and UQ9 on HPLC chromatographs normalized to protein content. UQ9 concentrations expressed also as nmol/mg protein, are reduced by an average of 85% in the liver of Mclk1liver-KO mice relative to the level in Mclk1loxP/− controls. In addition, the knockout liver has substantial accumulation of DMQ9 and it increases with age. n = 5 littermate pairs for 4-month data and 9 littermate pairs for 15-month data. Statistical analysis was performed by the Student's paired t-test.
Altered UQ levels in Mclk1 knockout liver
The results of quinone measurements showed that UQ9 levels were greatly reduced in the liver homogenates from Mclk1liver-KO mice when compared with AlbCre-negative Mclk1loxP/− controls. These livers also accumulated a substantial amount of DMQ9 (Fig. 5C). The UQ9 content in the knockout liver was decreased by ∼85%. This was expected given that the AlbCre transgene is only expressed in hepatocytes and hepatocytes make up ∼90% of the total cell mass in the mouse liver (60). In other words, the small amount of UQ detected in the knockout liver likely originates from a minor population of AlbCre non-expressing cells such as Kupffer cells and endothelial cells. However, our current data do not rule out the possibility that there remained a trace amount of UQ in Mclk1-null hepatocytes. Consistent with liver-specific deletion, Mclk1liver-KO mice showed no discernible change in kidney UQ content (Supplementary Material, Fig. S3).
Mild reduction in respiratory capacity in Mclk1 knockout liver mitochondria
Liver mitochondria isolated from Mclk1liver-KO mice showed a similar degree of UQ loss as in liver homogenates. The level of UQ9 in the knockout liver mitochondria in comparison with controls (Mclk1loxP/−) was 15 ± 4% at 15 months of age. DMQ9 accumulation was observed but to a lesser degree than in liver homogenates (Fig. 6A). Quite strikingly, the mitochondrial preparation from Mclk1 knockout liver exhibited only a mild impairment of respiratory chain function. State 3 (maximal ADP-stimulated) respiration rates with Complex I-linked substrates were decreased by 8 (8-month-old) to 13% (15-month-old), whereas the rates of Complex II-supported respiration appeared to be more affected, but still was only 14 (8-month-old) to 18% (15-month-old) lower than that in Mclk1loxP/−controls (Fig. 6B). In the knockout mitochondria, the activities of Complexes II and III were essentially normal, while the rate of electron transport between the two complexes was significantly decreased, thus confirming the observed respiratory phenotype is due to inadequate UQ levels (Fig. 6C). Other respiratory parameters measured, including state 4 respiration rate, respiratory control ratio, ADP/O ratio (Supplementary Material, Table S1) and mitochondrial aconitase activity (Supplementary Material, Fig. S4), were found not to differ between knockouts and controls. From 8 months to 15 months of age, there appeared to be a decreasing trend in state 3 respiratory rates for both control and Mclk1liver-KO mice (Fig. 6B) as was previously observed in the wild-type (61). However, the difference between the two age groups did not reach statistical significance.
Figure 6.

Mitochondrial respiratory chain function in Mclk1 knockout liver. (A) Liver mitochondria of Mclk1liver-KO mice (Mclk1loxP/−, AlbCre+) exhibit accumulation of DMQ9 along with a dramatic reduction in UQ9 content compared with that in Mclk1loxP/− controls. Columns show the mean ± SEM of peak areas on HPLC chromatographs normalized to protein content. Mitochondrial UQ9 concentrations expressed also as nmol/mg mitochondrial protein, are reduced by an average of 85% in the KO livers compared with controls. n = 8 littermate pairs for 8-month data and 10 littermate pairs for 15-month data. (B) Mclk1 knockout liver displays a mild reduction in mitochondrial state 3 respiration rates. State 3 respiration rates with Complex I-linked substrates, glutamate plus malate, are 8% (8-month-old) to 13% (15-month-old) lower than their respective littermate controls (Mclk1loxP/−). Complex II-dependent respiration, measured with succinate plus rotenone, is decreased by 14% (8-month-old) to 18% (15-month-old). In both control and knockout liver, there seems to be a trend toward a decrease with age, but it is not statistical significant. Columns represent mean values ± SEM of eight to nine mice per group. (C) Enzymatic activities of respiratory complexes in isolated liver mitochondria. The integrated activity of Complex II + III is significantly decreased in the liver mitochondrial preparation from Mclk1liver-KO mice (15-month-old), whereas the activities of individual complexes are unaltered compared with Mclk1loxP/− controls. Columns represent the mean enzymatic activities ±SEM of 11–14 mice. Significant differences were compared with controls by paired t-test. *P < 0.05, **P < 0.01 and ***P < 0.001.
Effect of dietary UQ supplementation on respiratory chain function
To determine whether the mild respiratory defect in Mclk1 knockout hepatocytes could be corrected by supplementation of dietary UQ, we fed Mclk1liver-KO mice with UQ10, beginning at 6 months of age. We scored quinone contents after 7 months of feeding. Both Mclk1liver-KO mice and Mclk1loxP/+ littermates showed an increase of total mitochondrial UQ10 content in the liver, and no difference was evident in UQ10 intake comparing liver mitochondrial preparations from the two groups of mice (Fig. 7A). In both groups of mice, endogenous UQ9 levels in liver mitochondria were not affected by UQ10 administration (Fig. 7A). In the liver mitochondria from untreated Mclk1liver-KO mice versus Mclk1loxP/+ controls, we again observed decreased state 3 respiration rates (Fig. 7B). More importantly, compared with un-treated Mclk1liver-KO mice, state 3 respiration rates of isolated liver mitochondria were significantly higher in UQ10-supplemented knockout mice, whether measured in the presence of Complex I or II substrates (Fig. 7B). Finally, UQ10 intake had no apparent effect on state 3 respiration in phenotypically wild-type Mclk1loxP/+ mice (Fig. 7B).
Figure 7.
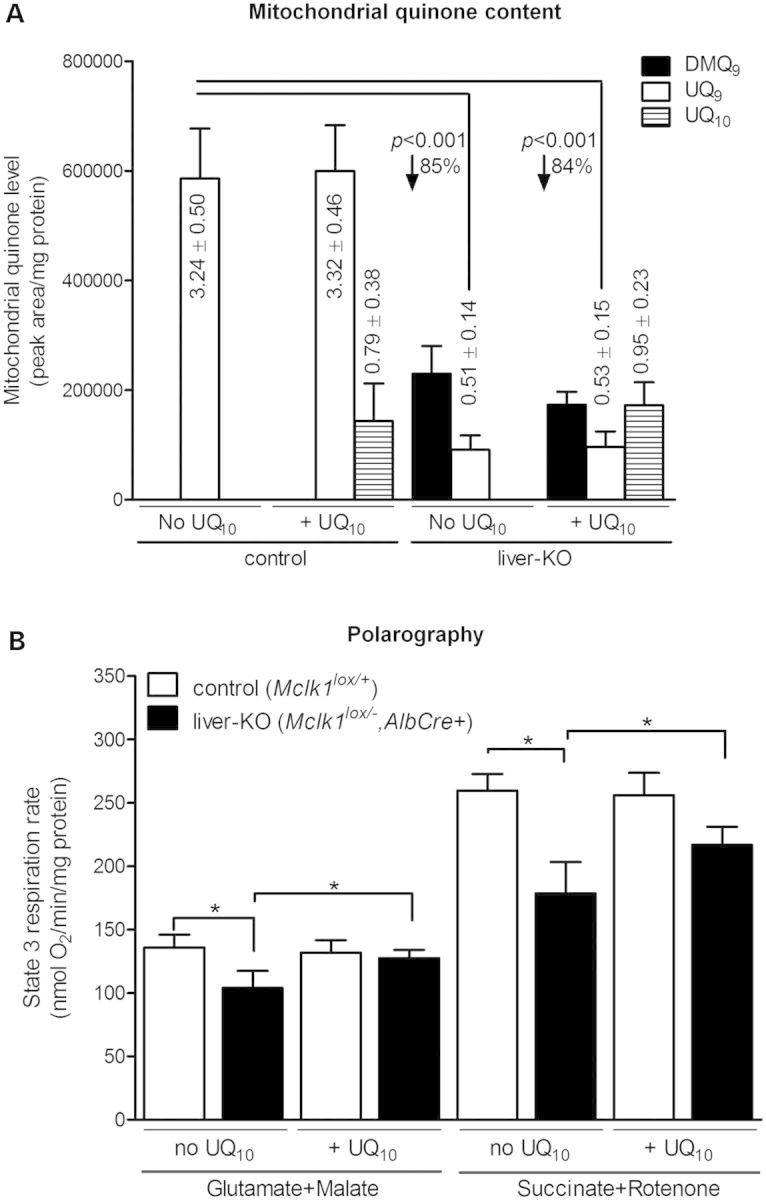
Effects of oral UQ10 supplementation on mitochondrial UQ levels and state 3 respiration rate in the liver. (A) UQ10 administration increases the levels of UQ10 in liver mitochondria. Similar to that shown in Figure 6A, liver mitochondria isolated from Mclk1liver-KO mice have ∼15% UQ9 relative to controls (Mclk1loxP/+) and accumulate DMQ9. Mitochondrial UQ10 uptake is similar in Mclk1 knockout livers and controls. UQ10 supplementation has no effect on the endogenous content of DMQ9 and UQ9. The columns represent the mean ± SEM of quinone peak area on HPLC chromatographs normalized to protein amounts. Mitochondrial UQ concentrations are expressed nmol/mg mitochondrial protein. n = 6 mice per group. (B) Liver mitochondria of UQ10-fed Mclk1liver-KO mice show a higher state 3 respiration rate (21%–23% more) for both Complex I and II substrates than those of untreated Mclk1liver-KO mice. Such effect is not seen in control group (Mclk1loxP/+). Columns represent mean value ± SEM of six mice per group. Statistical evaluation was performed using paired t-test. *P < 0.05.
DISCUSSION
We have described the generation and characterization of conditional Mclk1 knockout mutants in which normal UQ biosynthesis is abolished and which do not contain measurable UQ. However, viability and lower levels of electron transport are sustained by the UQ biosynthetic intermediate DMQ. We have also shown that a severe, but not a complete, loss of UQ in mouse liver (triggered by loss of Mclk1) produced only a mild impairment in respiratory chain function, indicating that in animal tissues where a relatively high respiration rate is expected, such as in mouse liver, very little UQ is required and DMQ does not interfere with UQ-mediated mitochondrial electron transport.
Mclk1 knockout fibroblasts are viable despite the complete absence of UQ
We previously reported that Mclk1−/− mouse ES cells can be grown in vitro without exogenous supply of UQ and possessed appreciable respiratory chain activity (30). Unfortunately, these findings were not conclusive, considering the unique characteristics of ES cells and that only one line had ever been obtained. In the present work, we show that the UQ biosynthetic precursor DMQ is capable of functioning in the mitochondrial respiratory chain of Mclk1 knockout MEFs fully devoid of UQ, albeit inefficiently, in particular in Complex II to III electron transport. We further found that Mclk1 knockout MEFs are not viable in glucose-free galactose-containing medium (Fig. 3A). Galactose is metabolized through the glycolytic pathway only very slowly. For this reason, in medium containing only galactose, cells are forced to use mitochondrial oxidative phosphorylation to sustain ATP production and maintain their viability (57,62). Thus, we interpret the inability of Mclk1 knockout MEFs to grow in galactose-only medium to a reduced rate of ATP production by oxidative phosphorylation. This galactose-induced lethality can be rescued by provision of exogenous UQ (Figs 3B, C and 4), indicating that DMQ is a less efficient respiratory substrate than UQ and that the C6-methoxy group in the UQ ring is important for its electron transport function in the respiratory chain. Of note, studies on skin fibroblasts from patients with UQ deficiency have demonstrated that the viability of these cells is essentially preserved under forced oxidative metabolism in galactose medium (25,63), likely because their UQ deficiency is not as profound as that in Mclk1 knockout MEF. In the future, it would be of interest to carry out further research to identify the threshold of UQ deficiency-induced respiratory depression below which cells will cease to function and die. Furthermore, in vitro studies with a combination of null mutants of UQ biosynthetic genes will be necessary to completely rule out any contribution of potential residual endogenous UQ biosynthesis to the active respiration of Mclk1 knockout cultures.
Mclk1 knockout fibroblasts is a new tool for testing the functionality of UQ analogues in the mammalian respiratory chain
Mclk1 knockout MEFs in galactose provide an excellent model system to evaluate the efficacy of various UQ analogues in promoting the function of the mammalian respiratory chain. We show that exogenously applied UQs with isoprenoid side chains effectively improve the respiratory chain activity of Mclk1 knockout MEFs (Figs 3B and 4) and among the isoprenoid UQs tested, UQ9, which is the main naturally occurring form of UQ in mice, appears to be most effective (Fig. 4), demonstrating a great sensitivity to side chain length. However, the mechanistic significance for this sensitivity is unknown. Idebenone, a synthetic UQ analog which carries exactly the same benzoquinone moiety as UQ and a short hydroxyalkyl side chain, fails to rescue the respiratory growth of Mclk1 knockout fibroblasts, indicating that it cannot replace endogenous UQ in the respiratory chain. This is consistent with studies on human patient fibroblasts with reduced UQ levels (64) and clinical reports that showed a poor efficacy of idebenone in treatment of profound UQ10 deficiency (28,65). Idebenone has been suggested to penetrate better than UQ into tissues and mitochondria (66). However, it is a poor electron acceptor for Complex I activity (67). Its therapeutic effects reported on some disease conditions (68,69) are most probably due to its antioxidant activity. Similar to idebenone, decylUQ has an alkyl side chain, but ends with a methyl group, conferring a relatively high hydrophilicity. It has been shown to be ineffective at restoring aerobic respiration in UQ6-deficient yeast (70), but in cultured human cells with decreased synthesis of UQ10, an increase in cellular respiration was seen after treatment with dycelUQ (71). In our experiments, we observed only a very small rescue effect when decylUQ was added to galactose-grown Mclk1 knockout culture (Fig. 4). Given the ability of decylUQ to serve as an efficient electron transporter in mitochondrial homogenate, the poor rescue observed with decylUQ is likely due to its limited accumulation in the inner membrane of mitochondria when applied exogenously. We further tested whether Vitamin K2 is able to rescue the respiratory chain deficiency of Mclk1 knockout fibroblasts. Vitamin K2 is a quinone having a naphthoquinone structure and an isoprenoid chain. In Drosophila, it has recently been proposed that Vitamin K2 can improve the mitochondrial electron transport leading to increased ATP production by acting as an electron carrier (49). However, we found that addition of Vitamin K2 to galactose medium failed to rescue Mclk1 knockout MEFs from death (Fig. 4). This finding suggests that Vitamin K2 cannot (at least not efficiently) substitute for UQ to mediate electron transport in mammalian mitochondria.
Liver mitochondrial function has a high tolerance to severe UQ deficiency
To our surprise, we found that liver mitochondria from Mclk1liver-KO mice exhibited only a mild reduction (<20%) in state 3 respiration for both Complex I and Complex II substrates (Fig. 6B), although only ∼15% of the cells in the liver are not hepatocytes. Thus, we measure much more respiration (∼80% of wild-type) than could be accounted for by the 15% of non-hepatocytes. This means Mclk1 knockout hepatocytes still respire at fairly high level. In line with this, Mclk1liver-KO mice showed no overt abnormalities. Our finding is in agreement with an earlier study that showed that livers from animals with liver-specific loss of Pdss2 (UQ biosynthetic gene required for the synthesis of isoprenoid side chain) had no detectable UQ, but exhibited >60% state 3 respiratory capacity (72). Although we cannot rule out the possibility that the accumulated DMQ9 contributes to the mitochondrial electron transport activities in Mclk1 knockout hepatocytes, the very low respiratory capacity of Mclk1 knockout MEFs that are supported solely by DMQ9, suggests that the DMQ9 in the knockout liver can only have a minor contribution to the high respiratory capacity observed.
This apparent insensitivity of hepatic mitochondrial respiration to severe UQ deficiency is very intriguing. Recent work from our laboratory showed that a mild decrease of UQ in the IMM in the liver of Mclk1 heterozygous mice (Mclk1+/−) was sufficient to induce mitochondrial dysfunction (26,44). Given that much more severe depletion of UQ is not followed by a much greater inhibition of the respiratory chain function, it seems that there is a nonlinear dependence of mitochondrial respiratory capacity upon UQ content. One reason might be a nonlinear relationship between UQ levels and ROS production. Mclk1+/− mitochondria showed increased mitochondrial generation of ROS (26), which may cause damages to respiratory complexes and hence further handicap the respiratory chain function, whereas both Mclk1 (Supplementary Material, Fig. S4) and Pdss2 knockout livers (73) showed no overproduction of ROS as indicated by the normal activity of mitochondrial aconitase. A nonlinear relation between the degree of UQ deficiency and mitochondrial ROS production has been described for patient fibroblasts with varying severity of UQ10 deficit. In particular, mutant fibroblasts with moderate deficiency (30–50% UQ10) produce maximal oxidative stress, while <20% residual UQ10 is not associated with increased oxidative stress (25,63,74). The complexes of the mitochondrial respiratory chain can assemble into supermolecular structures called supercomplexes which allow more efficient and rapid electron transfer as opposed to a random collision model (75,76). Supercomplex formation is thought to be a very dynamic process and of great importance for regulation of mitochondrial bioenergetics and controlling ROS production (77). It is reasonable to speculate that when UQ deficit is too severe, mitochondrial respiratory chain augments electron transport via the respiratory supercomplexes, enabling more efficient transport of electrons and less production of ROS. However, in both Mclk1 (Fig. 6B) and Pdss2 liver knockouts (72), Complex II respiration which does not involve supercomplexes is only slightly more affected than respiration via complex I for which the I–III–IV supercomplex is an important unit. From these findings, it is unlikely that a compensatory increase in supercomplex levels accounts for the high level of respiratory function in those UQ deficient mutants. Future studies comparing the tissues that are sensitive to UQ depletion to non-sensitive tissues, such as the liver, may reveal the composition and/or functional features of the respiratory chain that govern the variation in its requirement for UQ. It is noteworthy that in general there is a low prevalence of liver involvement in primary UQ10 deficiencies (22) which could be explained by a relatively low sensitivity to UQ deficiency in the liver.
It is very interesting that at 15 months of age (the last time point examined), Mclk1 knockout hepatocytes still appeared to contain residual UQ. Similarly, Pdss2 conditional knockout livers also seemed to not have lost UQ completely at the age of 8 months, as judged by their mitochondria's ability to still respire at 60% of normal level (72). These observations are in stark contrast to our in vitro observation on Mclk1 knockout MEFs in which UQ is rapidly depleted after excision of the floxed Mclk1 alleles. One likely explanation is a difference in cell division rate. When compared with Mclk1 knockout mouse liver, in vitro cultured Mclk1 knockout fibroblasts proliferate more quickly which can result in fast dilution of the initial UQ pool. Moreover, UQ degradation rates may vary greatly in different cell types and conditions. We also cannot exclude the possibility that most of the remaining UQ in mutant livers is derived from the circulation.
UQ10 supplementation restores defective respiration of Mclk1 knockout liver
UQ replacement treatment so far has offered a safe, easy and the only available therapeutic intervention for UQ deficiencies. However, the data on the effectiveness of UQ supplementation in UQ deficiency are not conclusive (13,14,16,17,19,21,28,78). We recently demonstrated that dietary UQ10 (supplemented in base rodent diet) indeed can reach mitochondrial inner membrane and there function in the electron transport chain (44). In the present study, following UQ10 supplementation through drinking water, we observed significant uptake of dietary UQ10 by liver mitochondria along with increased state 3 respiration rates in UQ10-fed Mclk1liver-KO mice compared with untreated knockout mice (Fig. 7). This finding provides another piece of evidence for that dietary UQ is able to functionally rescue endogenous UQ deficiency at the respiratory chain level. There are reports showing that dietary feeding of mice with UQ10 can elevate tissue concentrations of endogenous UQ by an unknown mechanism (79,80). In our experiment, the endogenous content of UQ9 in the liver mitochondria was not affected by oral UQ10 supplementation (Fig. 7A), as we previously observed in wild-type and Mclk1+/− mice (44). UQ10 intake appears to have no effect on state 3 respiration capacities of phenotypically wild-type Mclk1loxP/+mitochondria (Fig. 7B). A similar finding was reported for wild-type mice (80). A plausible interpretation is that normal UQ levels in the liver are saturating for state 3 respiratory rates.
DMQ does not interfere with UQ function in the mammalian mitochondrial respiratory chain
Both Mclk1 knockout ES cells (30) and MEFs (Fig. 2B and C) producing DMQ9 instead of UQ9 showed a Complex II-specific defect, suggesting that DMQ9 is a less efficient substrate for Complex II than for Complex I. This propensity for poor function at Complex II is in good agreement with two previous reports. One is that DMQ8 in the membrane from UbiF mutants is able to function as an electron acceptor for NADH, not for succinate (81). The other study compares UQ2 to synthetic DMQ2, which is found to promote electron transport from Complex I fairly efficiently but produces a much smaller stimulation of succinate oxidation (34). These observations have been interpreted to indicate that Complex II possess a high degree of selectivity for use of quinone species as its electron acceptor (34). Future studies will be needed to probe the molecular basis for the poor activity of DMQ at Complex II.
clk-1 mutant worms have a specific defect in Complex I-dependent respiration (52). The ability of DMQ9 to mediate electron transfer from Complex I more efficiently than from Complex II may provide a mechanistic explanation for the Complex I-specific respiratory dysfunction in clk-1 mitochondria. clk-1 mutants contain a large amount of endogenously produced DMQ9 as well as exogenous UQ8 obtained from the bacterial food source (54). As discussed above, DMQ9 is a less efficient respiratory substrate than UQ9. However, as it is still able to function somewhat adequately at Complex I, it might compete with UQ8 for quinone binding sites, thus inhibiting the activity of UQ8 at Complex I, whereas competitive inhibition on UQ8-mediated Complex II respiration would be less marked because of the relatively inactivity of DMQ9 at Complex II. In contrast to worm clk-1 mitochondria, the liver mitochondria from Mclk1liver-KO mice showed an impairment in both Complex I- and Complex II-dependent respiration, and Complex II-dependent respiration was slightly more severely affected (Fig. 6B). We discussed above that the high OXPHOS activity in Mclk1 knockout hepatocytes could be attributable to the remaining UQ. As the liver knockout in Mclk1 displayed similar respiratory phenotype as described for Pdss2 liver knockout mice (72), accumulation of DMQ is unlikely to significantly influence the respiratory function of endogenous UQ. We further found that oral UQ10 intake (1) elevated total mitochondrial UQ10 content in the liver, (2) had no discernible effect on the endogenous content of DMQ and UQ (Fig. 7A), and (3) increased the mitochondrial state 3 respiration rates in Mclk1 knockout liver (Fig. 7B). These results suggest that DMQ does not significantly inhibit exogenous UQ10-mediated electron transport. Thus, it seems that in the mammalian respiratory chain, which in general has a high electron flux, accumulation of DMQ does not significantly interfere with UQ function, probably because of a greater degree of difference in relative efficiency between DMQ and UQ. These findings suggest that the accumulation of DMQ in the described COQ9 patient (MIM 607426) (21) is likely not clinically meaningful and has no pathogenic role.
MATERIALS AND METHODS
Reagents and chemicals
Cell culture media and reagents were purchased from Invitrogen. All other reagents used in this study were of molecular biology grade or better and were obtained from Sigma-Aldrich unless noted otherwise.
Mice
Mice carrying a conditional Mclk1 knockout allele (loxP, Fig. 1A) were generated by InGenious Targeting Laboratory (Stony Brook, NY, USA). The gene-targeting vector is composed of a 1.8 kb C57BL/6 genomic fragment upstream of the mouse Mclk1 exon 2 followed by a loxP/FRT-flanked Neo cassette, the target region of Mclk1 which is ∼1.7 kb including exons 2 and 3, a single loxP site inserted downstream of exon 3, and an ∼8.8 kb fragment downstream of the third loxP site. NotI-linearized targeting vector was transfected into C57BL/6×129/SvEv hybrid embryonic stem cells. Neomycin-resistant colonies were selected in G418, expanded and screened by PCR for ones that had undergone homologous recombination. Positive colonies were then microinjected into 129/SvEv blastocysts for chimera generation. Chimeric mice were bred to 129/SvEv mice and germline transmission of the floxed Mclk1 allele (loxP) in F1 heterozygous mice was confirmed by PCR analysis of tail DNA. The resulting heterozygotes were interbred to generate homozygous Mclk1loxP/loxP mice. To generate liver-specific Mclk1 knockout mice, AlbCre mice (purchased from The Jackson Laboratory) were bred with Mclk1+/− mice [described in Ref. (30)] to obtain Mclk1+/−, AlbCre+ mice which were then crossed with Mclk1loxP/loxP mice to yield the genotypes of this study: Mclk1loxP/−, AlbCre+, Mclk1loxP/− and Mclk1loxP/+. All experimental mice were housed in the pathogen-free animal facility of McGill University under standard conditions. Studies were conducted under an animal protocol approved by the Animal Care and Use Committee of McGill University.
Genotyping and detection of Cre/lox recombination
Mice were genotyped by PCR with genomic DNA extracted from tail clippings. The floxed Mclk1 allele was amplified using primer #1 (5′-TTTGATGCCCTTGTGGAACGTG-3′) and #2 (5′-ACGATCCTAGCTTCCTTCTGTGAG-3′), producing a 323 bp fragment (Fig. 1A). PCR amplification of the wild-type allele using the same primer set yielded a 261 bp band. To detect Cre-mediated excision of Mclk1 floxed alleles, primers #3 (5′-CTCAGGCTTGTCTTTCATTCTGTC-3′) and #2 were used (Fig. 1A). Primers used to amplify the AlbCre transgene were as follows: forward: 5′-GCCAGCTAAACATGCTTCATC-3′; reverse: 5′-ATTGCCCCTGTTTCACTATCC-3′. PCR reactions were performed using NEB's OneTaq DNA polymerase with 30 amplification cycles of 94°C for 30 s, 60°C for 30 s and 68°C for 60 s. The same PCR conditions were used for detection of the recombined Mclk1loxP allele in genomic DNA isolated from Mclk1loxP/loxP-Cre MEFs and tissues of Mclk1loxP/−, AlbCre+ mice.
Construction of retroviral vector and retroviral infection
The pBabe-Cre-puro retroviral vector was constructed by subcloning a Cre recombinase cDNA into the EcoRI and SalI sites of pBabe-puro retroviral vector. Phoenix™-Eco virus packaging cells (Cedarlane Labs) were used to produce retroviruses. At 60–80% confluence, Phoenix™-Eco cells were transfected with pBabe-Cre-puro or empty pBabe-puro vector (20 µg of plasmid DNA/100 mm plate) using Lipofectamine 2000 following the manufacturer's instructions (Invitrogen). The virus-containing supernatants were harvested after 48 h, filtered and used to replace the culture medium of Mclk1loxP/loxP MEFs, which were prepared from day 13.5 Mclk1loxP/loxP embryos by trypsin digestion according to standard procedures (82). The infection procedure was repeated the next day. Infected MEFs were then incubated in media supplemented with 10 µg/ml of puromycin for 2 days in order to select the cells that stably express viral DNA. Five independent MEF lines were infected with the described retroviral vectors and used at a passage number below 15.
Cells and tissue culture
All cells were maintained in DMEM containing high glucose (25 mm) and supplemented with 10% FBS and 1% penicillin/streptomycin at 37°C in 5% CO2. Cell proliferation assay was performed by plating 5 × 103 cells into 48-well plates in triplicates, following by daily measurement of cell concentration with alamarBlue Cell Viability Assay (Invitrogen). For growth in galactose, cells seeded into 48-well plates were first grown in high glucose DMEM medium overnight and then switched to glucose-free DMEM media supplemented with 10 mm galactose, 10 mm HEPES, 1 mm sodium pyruvate, 10% dialyzed FBS and 1% penicillin/streptomycin. For rescue experiment, various chemicals were added into MEFs culture when medium was changed to galactose medium. Four days after culture in galactose medium, alamarBlue assay was performed for estimation of cell viability. Concentrations between 2.5 and 20 µm were used for all compounds tested. The most effective dose among those tested is reported for the compounds that rescued the galactose growth defect of Mclk1Δ/Δ-Cre MEFs. For those that showed no significant rescue effect, the maximum non-toxic dose, defined as the highest tested concentration that does not produce evidence of lethality in control MEF lines (Mclk1loxP/loxP-EV), is presented.
Microscopy
For phase-contrast microscopy, cells were plated onto glass cover slips coated with poly-d-lysine, fixed with 3.7% paraformaldehyde in PBS for 10 min and then washed by PBS twice. The cover slips were mounted onto slides with one drop of Prolong anti-fade reagent (Invitrogen) and images were acquired on an Olympus BX63 microscope. To label mitochondria, cells were incubated with 200 nm of MitoTracker Green FM (Invitrogen) in growth medium for 30 min. The cells were then fixed in 3.7% paraformaldehyde, washed with PBS and subjected to fluorescence microscopy on an Olympus BX63 microscope using FITC filter set.
Quantification of quinones
Quinone content was quantified as previously described (35). Briefly, cell lysis, liver homogenates or isolated mitochondria were mixed with an equal volume of hexane/ethanol for 10 min by vortexing, centrifuged for 5 min at 8000g and the hexane layer was collected. After evaporation to dryness using a vacuum centrifuge, the quinone residue was dissolved in ethanol and then analyzed by HPLC with UV detection at 275 nm (Beckman System Gold). A reverse phase C18 column (25.0 × 0.46 cm, 5 μm, Highchrom) was used with an isocratic elution at a flow rate of 1.8 ml/min. The mobile phase was methanol/ethanol (70:30 v/v). The concentrations of UQs were estimated by comparison of the peak area with those of standard solutions of known concentration. Quinone levels data were finally normalized to protein content determined by the Bio-Rad Protein Assay (Bio-Rad).
Respiration measurements
Whole-cell oxygen consumption was measured polarographically as previously described (30). Briefly, exponentially growing MEFs cultures were harvested and re-suspended in fresh DMEM medium. The cell suspensions were immediately added to a magnetically stirred Clark-type oxygen electrode chamber thermostated to 37°C, containing DMEM, to a final volume of 2 ml.
After recording the basal respiratory rate, Carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP) and potassium cyanide (KCN) were added successively (final concentration of 1 µm and 2 mm, respectively) and measurement continued for additional 2 min. OCR were normalized to total protein content determined by the Bio-Rad Protein Assay.
Oxygen consumption of isolated liver mitochondria was measured with a high-resolution respirometry system (Oxygraph-2k, Oroboros Instruments) at 30°C in a buffer containing 0.5 mm EGTA, 3 mm MgCl2·6H2O, 60 mm potassium lactobionate, 20 mm taurine, 10 mm KH2PO4, 20 mm HEPES, 110 mm sucrose and 1 g/l fatty acid free BSA, pH 7.1. Glutamate (5 mm) plus malate (2.5 mm) was used as substrates for NADH-linked respiration initiated at Complex I, whereas 10 mm succinate (plus 1.25 µm rotenone) was used to induce Complex II-mediated respiration. The rate of maximal coupled (state 3) respiration was determined in the presence of 0.5 mm ADP, and state 4 was recorded after ADP exhaustion. Results were expressed as pmol oxygen/s/mg mitochondrial protein.
Determination of mitochondrial enzyme activities
Isolation of MEF and liver mitochondria was performed by differential centrifugation as described previously (26,30). Purified mitochondria were subjected to three rounds of freeze/thaw. ETC enzyme activities were measured as donor–acceptor oxidoreductase activities and expressed as nmoles of electron acceptor reduced/min/mg mitochondrial protein. Assays were carried out spectrophotometrically at 30°C using a double-wavelength Beckman DU 640 spectrophotometer. Sensitivity to enzymatic inhibitors was used to confirm assay specificity. Complex II activity was analyzed by UQ1-stimulated dichlorophenolindophenol (DCPIP) reduction with succinate as substrate. The assay was carried out in 1 ml of assay buffer containing 25 mm potassium phosphate (pH 7.4), 20 mm sodium succinate, 2 µg antimycin, 2 µg rotenone, 2 mm KCN, 150 μm DCPIP and 40 µg of mitochondrial protein. The reaction was started with 100 µm UQ1, and the enzyme activity was measured at 600 nm. Inhibition of reaction by 10 mm of malonate was used to confirm the specificity of the assay. The extinction coefficient used for DCIP was 21 per mmcm. Complex III activity was assayed by following the reduction of cytochrome c (50 µm) by decylubiquinol (60 µm) at 550 nm. The assay was carried out in 25 mm potassium phosphate buffer (pH 7.4) supplemented with 1 mm KCN, 2 mg/ml rotenone and 0.1% BSA. The reaction was started with 25 µg of mitochondrial protein, and a mix of antimycin (10 µg/ml) and myxothiazol (2 µg/ml) was used to correct the background rate for non-enzymatic reduction of cytochrome c. The extinction coefficient used for cytochrome c was 19.1 per mm cm. The activities of Complex I + III and Complex II + III were measured by following the reduction of cytochrome c at 550 nm in the presence of the Complex I (0.1 mm NADH) or II (20 mm succinate) substrates, respectively. The assay of Complex I + III activity was performed in 1 ml of reaction mix containing 25 mm potassium phosphate (pH 7.4), 100 µm NADH, 2 mm KCN and 20 μg of mitochondrial protein. The reaction mixture for the Complex II + III activity assay consisted of 25 mm potassium phosphate (pH 7.4), 20 mm succinate, 2 μg/ml rotenone, 0.5 mm EDTA, 2 mm KCN and 40 μg/ml of mitochondrial protein and was pre-incubated at 30°C for 10 min to fully activate Complex II. Reactions were initiated with addition of 30 µm cytochrome c. The increase in absorbance was monitored before and after the addition of 5 µg/ml rotenone (Complex I inhibitor) or 10 mm malonate (Complex II inhibitor). Activities are finally expressed as inhibitor-sensitive cytochrome c reduction rates (nmoles per minute) per mg mitochondrial protein. The Bradford assay was used to quantify the protein concentration.
Total CS activity in MEFs was determined by following the formation of DTNB [5,5′-dithio-bis-(2-nitrobenzoic acid)]-CoA, which is coupled with citrate formation catalyzed by citrated synthase, at 412 nm. Whole-cell extracts were prepared using Cellytic M cell lysis reagent (Sigma) following the manufacturer's instructions. Fifteen micrograms of cellular protein was added to 1 ml of reaction mixture containing 100 mm Tris (pH 8.0), 0.1% Triton X-100 and 0.1 mm acetyl CoA. The reaction was started by adding 10 µl of oxaloacetate (0.25 mm), and linear rate of change in absorbance was followed for 2–3 min. Data are expressed as nmoles per minute per mg cellular protein on the basis of the molar extinction coefficient, 13.6 per mM cm.
RNA preparation and RT-PCR analysis
Total RNA was extracted using Trizol reagent (Invitrogen) and 1 µg of RNA was reverse-transcribed into cDNA using QuantiTect Reverse Transcription Kit (Qiagen) following the manufacturers' protocols. RT-PCR was performed using SYBR Green (Qiagen) on a Bio-Rad CFX 96 real-time PCR thermocycler (Bio-Rad). Primers for the Mclk1 mRNA were forward 5′-CCTGCGCACTGGTGTCCGGA-3′ and reverse 5′-GCACTGCATCCGGCCTGGATAA-3′; and primers for the mRNA of the housekeeping gene β-actin were forward 5′-GGAGCACCCTGTGCTGCTCA-3′ and reverse 5′-GGATTCCATACCCAAGAAGGAAGGC-3′. PCR products were separated on 2% agarose gels and visualized under UV light after staining with ethidium bromide.
Western blotting
Mitochondrial protein samples were loaded onto 12% SDS–PAGE gels, electrophoresed at 120 V and transferred to 0.45 µm polyvinylidene fluoride membranes (Bio-Rad). Membranes were blocked in 5% milk-PBS for 2 h and blotted overnight at 4°C with anti-MCLK1 [dilution 1:500, developed in our laboratory (30)] and anti-porin (dilution 1:2000, purchased from CambioChem) antibodies. After three washes with PBS-0.05% Tween 20, membranes were incubated with horseradish peroxidase-conjugated anti-mouse (dilution 1:10 000, Pierce) and anti-rabbit (dilution 1:2000, Sigma) secondary antibodies, and then developed with an ECL Plus western blotting detection system (GE Healthcare). Signals were visualized on a chemiluminescence scanner (TyphoonTM 9400, GE Healthcare) and analyzed using ImageQuant software (version 5.2, GE Healthcare).
Dietary UQ10 supplementation
Mice received UQ10 supplementation with LiQsorb (Tishcon) added into their drinking water at the concentration of 1 mg/ml, beginning at 6 months of age. At this dose, mice received ∼300–400 mg UQ10/kg body weight/day.
Statistical analysis
All data were expressed as the mean ± SEM. Statistical analyses were performed with Prism (version 5.0, GraphPad Software) using paired or unpaired Student's t-tests, or ANOVA/Dunnett's test as appropriate. Probability value of P < 0.05 was considered significant.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
FUNDING
The work was funded by a grant from the Canadian Institutes of Health Research to S.H. (MOP-97869) and McGill University.
Supplementary Material
ACKNOWLEDGEMENTS
We are thankful to Dr David Dankort for providing reagents and sharing his protocol for retroviral vector-mediated gene transfer.
REFERENCES
- 1.Crane F.L. Discovery of ubiquinone (coenzyme Q) and an overview of function. Mitochondrion. 2007;7(suppl):S2–S7. doi: 10.1016/j.mito.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 2.Lenaz G. A critical appraisal of the mitochondrial coenzyme Q pool. FEBS Lett. 2001;509:151–155. doi: 10.1016/s0014-5793(01)03172-6. [DOI] [PubMed] [Google Scholar]
- 3.Bentinger M., Brismar K., Dallner G. The antioxidant role of coenzyme Q. Mitochondrion. 2007;7(suppl):S41–S50. doi: 10.1016/j.mito.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 4.Walter L., Miyoshi H., Leverve X., Bernard P., Fontaine E. Regulation of the mitochondrial permeability transition pore by ubiquinone analogs. A progress report. Free Rad. Res. 2002;36:405–412. doi: 10.1080/10715760290021252. [DOI] [PubMed] [Google Scholar]
- 5.Evans D.R., Guy H.I. Mammalian pyrimidine biosynthesis: fresh insights into an ancient pathway. J. Biol. Chem. 2004;279:33035–33038. doi: 10.1074/jbc.R400007200. [DOI] [PubMed] [Google Scholar]
- 6.Sun I.L., Sun E.E., Crane F.L., Morre D.J., Lindgren A., Low H. Requirement for coenzyme Q in plasma membrane electron transport. Proc. Natl Acad. Sci. USA. 1992;89:11126–11130. doi: 10.1073/pnas.89.23.11126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Watts G.F., Playford D.A., Croft K.D., Ward N.C., Mori T.A., Burke V. Coenzyme Q(10) improves endothelial dysfunction of the brachial artery in type II diabetes mellitus. Diabetologia. 2002;45:420–426. doi: 10.1007/s00125-001-0760-y. [DOI] [PubMed] [Google Scholar]
- 8.Tiano L., Belardinelli R., Carnevali P., Principi F., Seddaiu G., Littarru G.P. Effect of coenzyme Q10 administration on endothelial function and extracellular superoxide dismutase in patients with ischaemic heart disease: a double-blind, randomized controlled study. Eur. Heart J. 2007;28:2249–2255. doi: 10.1093/eurheartj/ehm267. [DOI] [PubMed] [Google Scholar]
- 9.Tran U.C., Clarke C.F. Endogenous synthesis of coenzyme Q in eukaryotes. Mitochondrion. 2007;7(suppl):S62–S71. doi: 10.1016/j.mito.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pierrel F., Hamelin O., Douki T., Kieffer-Jaquinod S., Muhlenhoff U., Ozeir M., Lill R., Fontecave M. Involvement of mitochondrial ferredoxin and para-aminobenzoic acid in yeast coenzyme Q biosynthesis. Chem. Biol. 2010;17:449–459. doi: 10.1016/j.chembiol.2010.03.014. [DOI] [PubMed] [Google Scholar]
- 11.Salviati L., Trevisson E., Rodriguez Hernandez M.A., Casarin A., Pertegato V., Doimo M., Cassina M., Agosto C., Desbats M.A., Sartori G., et al. Haploinsufficiency of COQ4 causes coenzyme Q10 deficiency. J. Med. Genet. 2012;49:187–191. doi: 10.1136/jmedgenet-2011-100394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quinzii C., Naini A., Salviati L., Trevisson E., Navas P., Dimauro S., Hirano M. A mutation in para-hydroxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency. Am. J. Hum. Genet. 2006;78:345–349. doi: 10.1086/500092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mollet J., Giurgea I., Schlemmer D., Dallner G., Chretien D., Delahodde A., Bacq D., de Lonlay P., Munnich A., Rotig A. Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders .J. Clin. Invest. 2007;117:765–772. doi: 10.1172/JCI29089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mollet J., Delahodde A., Serre V., Chretien D., Schlemmer D., Lombes A., Boddaert N., Desguerre I., de Lonlay P., de Baulny H.O., et al. CABC1 Gene mutations cause ubiquinone deficiency with cerebellar ataxia and seizures. Am. J. Hum. Genet. 2008;82:623–630. doi: 10.1016/j.ajhg.2007.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lopez-Martin J.M., Salviati L., Trevisson E., Montini G., DiMauro S., Quinzii C., Hirano M., Rodriguez-Hernandez A., Cordero M.D., Sanchez-Alcazar J.A., et al. Missense mutation of the COQ2 gene causes defects of bioenergetics and de novo pyrimidine synthesis. Hum. Mol. Genet. 2007;16:1091–1097. doi: 10.1093/hmg/ddm058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lopez L.C., Schuelke M., Quinzii C.M., Kanki T., Rodenburg R.J., Naini A., Dimauro S., Hirano M. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am. J. Hum. Genet. 2006;79:1125–1129. doi: 10.1086/510023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lagier-Tourenne C., Tazir M., Lopez L.C., Quinzii C.M., Assoum M., Drouot N., Busso C., Makri S., Ali-Pacha L., Benhassine T., et al. ADCK3, An ancestral kinase, is mutated in a form of recessive ataxia associated with coenzyme Q10 deficiency. Am. J. Hum. Genet. 2008;82:661–672. doi: 10.1016/j.ajhg.2007.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heeringa S.F., Chernin G., Chaki M., Zhou W., Sloan A.J., Ji Z., Xie L.X., Salviati L., Hurd T.W., Vega-Warner V., et al. COQ6 Mutations in human patients produce nephrotic syndrome with sensorineural deafness. J. Clin. Invest. 2011;121:2013–2024. doi: 10.1172/JCI45693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Diomedi-Camassei F., Di Giandomenico S., Santorelli F.M., Caridi G., Piemonte F., Montini G., Ghiggeri G.M., Murer L., Barisoni L., Pastore A., et al. COQ2 nephropathy: a newly described inherited mitochondriopathy with primary renal involvement. J. Am. Soc. Nephrol. 2007;18:2773–2780. doi: 10.1681/ASN.2006080833. [DOI] [PubMed] [Google Scholar]
- 20.Artuch R., Salviati L., Jackson S., Hirano M., Navas P. Coenzyme Q10 deficiencies in neuromuscular diseases. Adv. Exp. Med. Biol. 2009;652:117–128. doi: 10.1007/978-90-481-2813-6_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duncan A.J., Bitner-Glindzicz M., Meunier B., Costello H., Hargreaves I.P., Lopez L.C., Hirano M., Quinzii C.M., Sadowski M.I., Hardy J., et al. A nonsense mutation in COQ9 causes autosomal-recessive neonatal-onset primary coenzyme Q10 deficiency: a potentially treatable form of mitochondrial disease. Am. J. Hum. Genet. 2009;84:558–566. doi: 10.1016/j.ajhg.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Emmanuele V., Lopez L.C., Berardo A., Naini A., Tadesse S., Wen B., D'Agostino E., Solomon M., DiMauro S., Quinzii C., et al. Heterogeneity of coenzyme Q10 deficiency: patient study and literature review. Arch. Neurol. 2012;69:978–983. doi: 10.1001/archneurol.2012.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Quinzii C.M., Hirano M. Coenzyme Q and mitochondrial disease. Dev. Disabil. Res. Rev. 2010;16:183–188. doi: 10.1002/ddrr.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hirano M., Garone C., Quinzii C.M. Coq(10) deficiencies and MNGIE: two treatable mitochondrial disorders. Biochim. Biophys. Acta. 2012;1820:625–631. doi: 10.1016/j.bbagen.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quinzii C.M., Lopez L.C., Von-Moltke J., Naini A., Krishna S., Schuelke M., Salviati L., Navas P., DiMauro S., Hirano M. Respiratory chain dysfunction and oxidative stress correlate with severity of primary CoQ10 deficiency. FASEB J. 2008;22:1874–1885. doi: 10.1096/fj.07-100149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lapointe J., Hekimi S. Early mitochondrial dysfunction in long-lived Mclk1+/- mice. J. Biol. Chem. 2008;283:26217–26227. doi: 10.1074/jbc.M803287200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rodriguez-Hernandez A., Cordero M.D., Salviati L., Artuch R., Pineda M., Briones P., Gomez Izquierdo L., Cotan D., Navas P., Sanchez-Alcazar J.A. Coenzyme Q deficiency triggers mitochondria degradation by mitophagy. Autophagy. 2009;5:19–32. doi: 10.4161/auto.5.1.7174. [DOI] [PubMed] [Google Scholar]
- 28.Aure K., Benoist J.F., Ogier de Baulny H., Romero N.B., Rigal O., Lombes A. Progression despite replacement of a myopathic form of coenzyme Q10 defect. Neurology. 2004;63:727–729. doi: 10.1212/01.wnl.0000134607.76780.b2. [DOI] [PubMed] [Google Scholar]
- 29.Ewbank J.J., Barnes T.M., Lakowski B., Lussier M., Bussey H., Hekimi S. Structural and functional conservation of the Caenorhabditis elegans timing gene clk-1. Science. 1997;275:980–983. doi: 10.1126/science.275.5302.980. [DOI] [PubMed] [Google Scholar]
- 30.Levavasseur F., Miyadera H., Sirois J., Tremblay M.L., Kita K., Shoubridge E., Hekimi S. Ubiquinone is necessary for mouse embryonic development but is not essential for mitochondrial respiration. J. Biol. Chem. 2001;276:46160–46164. doi: 10.1074/jbc.M108980200. [DOI] [PubMed] [Google Scholar]
- 31.Nakai D., Yuasa S., Takahashi M., Shimizu T., Asaumi S., Isono K., Takao T., Suzuki Y., Kuroyanagi H., Hirokawa K., et al. Mouse homologue of coq7/clk-1, longevity gene in Caenorhabditis elegans, is essential for coenzyme Q synthesis, maintenance of mitochondrial integrity, and neurogenesis. Biochem. Biophys. Res. Commun. 2001;289:463–471. doi: 10.1006/bbrc.2001.5977. [DOI] [PubMed] [Google Scholar]
- 32.Marbois B.N., Clarke C.F. The COQ7 gene encodes a protein in saccharomyces cerevisiae necessary for ubiquinone biosynthesis. J. Biol. Chem. 1996;271:2995–3004. doi: 10.1074/jbc.271.6.2995. [DOI] [PubMed] [Google Scholar]
- 33.Jonassen T., Proft M., Randez-Gil F., Schultz J.R., Marbois B.N., Entian K.D., Clarke C.F. Yeast Clk-1 homologue (Coq7/Cat5) is a mitochondrial protein in coenzyme Q synthesis. J. Biol. Chem. 1998;273:3351–3357. doi: 10.1074/jbc.273.6.3351. [DOI] [PubMed] [Google Scholar]
- 34.Miyadera H., Amino H., Hiraishi A., Taka H., Murayama K., Miyoshi H., Sakamoto K., Ishii N., Hekimi S., Kita K. Altered quinone biosynthesis in the long-lived clk-1 mutants of Caenorhabditis elegans. J. Biol. Chem. 2001;276:7713–7716. doi: 10.1074/jbc.C000889200. [DOI] [PubMed] [Google Scholar]
- 35.Wang Y., Branicky R., Stepanyan Z., Carroll M., Guimond M.P., Hihi A., Hayes S., McBride K., Hekimi S. The anti-neurodegeneration drug clioquinol inhibits the aging-associated protein CLK-1. J. Biol. Chem. 2009;284:314–323. doi: 10.1074/jbc.M807579200. [DOI] [PubMed] [Google Scholar]
- 36.Wong A., Boutis P., Hekimi S. Mutations in the clk-1 gene of Caenorhabditis elegans affect developmental and behavioral timing. Genetics. 1995;139:1247–1259. doi: 10.1093/genetics/139.3.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu X., Jiang N., Hughes B., Bigras E., Shoubridge E., Hekimi S. Evolutionary conservation of the clk-1-dependent mechanism of longevity: loss of mclk1 increases cellular fitness and lifespan in mice. Genes Dev. 2005;19:2424–2434. doi: 10.1101/gad.1352905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jonassen T., Larsen P.L., Clarke C.F. A dietary source of coenzyme Q is essential for growth of long-lived Caenorhabditis elegans clk-1 mutants. Proc. Natl Acad. Sci. USA. 2001;98:421–426. doi: 10.1073/pnas.021337498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hihi A.K., Gao Y., Hekimi S. Ubiquinone is necessary for Caenorhabditis elegans development at mitochondrial and non-mitochondrial sites. J. Biol. Chem. 2002;277:2202–2206. doi: 10.1074/jbc.M109034200. [DOI] [PubMed] [Google Scholar]
- 40.Asencio C., Navas P., Cabello J., Schnabel R., Cypser J.R., Johnson T.E., Rodriguez-Aguilera J.C. Coenzyme Q supports distinct developmental processes in Caenorhabditis elegans. Mech. Ageing Dev. 2009;130:145–153. doi: 10.1016/j.mad.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 41.Gavilan A., Asencio C., Cabello J., Rodriguez-Aguilera J.C., Schnabel R., Navas P. C. elegans knockouts in ubiquinone biosynthesis genes result in different phenotypes during larval development. Biofactors. 2005;25:21–29. doi: 10.1002/biof.5520250104. [DOI] [PubMed] [Google Scholar]
- 42.Earls L.R., Hacker M.L., Watson J.D., Miller D.M., 3rd Coenzyme Q protects Caenorhabditis elegans GABA neurons from calcium-dependent degeneration. Proc. Natl Acad. Sci. USA. 2010;107:14460–14465. doi: 10.1073/pnas.0910630107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang D., Wang Y., Argyriou C., Carriere A., Malo D., Hekimi S. An enhanced immune response of Mclk1(+)/(-) mutant mice is associated with partial protection from fibrosis, cancer and the development of biomarkers of aging. PLoS ONE. 2012;7:e49606. doi: 10.1371/journal.pone.0049606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lapointe J., Wang Y., Bigras E., Hekimi S. The submitochondrial distribution of ubiquinone affects respiration in long-lived Mclk1+/- mice. J. Cell Biol. 2012;199:215–224. doi: 10.1083/jcb.201203090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garcia-Corzo L., Luna-Sanchez M., Doerrier C., Garcia J.A., Guaras A., Acin-Perez R., Bullejos-Peregrin J., Lopez A., Escames G., Enriquez J.A., et al. Dysfunctional Coq9 protein causes predominant encephalomyopathy associated with CoQ deficiency. Hum. Mol. Genet. 2013;22:1233–1248. doi: 10.1093/hmg/dds530. [DOI] [PubMed] [Google Scholar]
- 46.Johnson A., Gin P., Marbois B.N., Hsieh E.J., Wu M., Barros M.H., Clarke C.F., Tzagoloff A. COQ9, a new gene required for the biosynthesis of coenzyme Q in Saccharomyces cerevisiae. J. Biol. Chem. 2005;280:31397–31404. doi: 10.1074/jbc.M503277200. [DOI] [PubMed] [Google Scholar]
- 47.Xie L.X., Ozeir M., Tang J.Y., Chen J.Y., Jaquinod S.K., Fontecave M., Clarke C.F., Pierrel F. Overexpression of the Coq8 kinase in Saccharomyces cerevisiae coq null mutants allows for accumulation of diagnostic intermediates of the coenzyme Q6 biosynthetic pathway. J. Biol. Chem. 2012;287:23571–23581. doi: 10.1074/jbc.M112.360354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brajcich B.C., Iarocci A.L., Johnstone L.A., Morgan R.K., Lonjers Z.T., Hotchko M.J., Muhs J.D., Kieffer A., Reynolds B.J., Mandel S.M., et al. Evidence that ubiquinone is a required intermediate for rhodoquinone biosynthesis in Rhodospirillum rubrum. J. Bacteriol. 2009;192:436–445. doi: 10.1128/JB.01040-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vos M., Esposito G., Edirisinghe J.N., Vilain S., Haddad D.M., Slabbaert J.R., Van Meensel S., Schaap O., De Strooper B., Meganathan R., et al. Vitamin K2 is a mitochondrial electron carrier that rescues pink1 deficiency. Science. 2012;336:1306–1310. doi: 10.1126/science.1218632. [DOI] [PubMed] [Google Scholar]
- 50.Padilla S., Jonassen T., Jimenez-Hidalgo M.A., Fernandez-Ayala D.J., Lopez-Lluch G., Marbois B., Navas P., Clarke C.F., Santos-Ocana C. Demethoxy-Q, an intermediate of coenzyme Q biosynthesis, fails to support respiration in Saccharomyces cerevisiae and lacks antioxidant activity. J. Biol. Chem. 2004;279:25995–26004. doi: 10.1074/jbc.M400001200. [DOI] [PubMed] [Google Scholar]
- 51.Branicky R., Benard C., Hekimi S. clk-1, mitochondria, and physiological rates. Bioessays. 2000;22:48–56. doi: 10.1002/(SICI)1521-1878(200001)22:1<48::AID-BIES9>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 52.Kayser E.B., Sedensky M.M., Morgan P.G., Hoppel C.L. Mitochondrial oxidative phosphorylation is defective in the long-lived mutant clk-1. J. Biol. Chem. 2004;279:54479–54486. doi: 10.1074/jbc.M403066200. [DOI] [PubMed] [Google Scholar]
- 53.Jonassen T., Marbois B.N., Faull K.F., Clarke C.F., Larsen P.L. Development and fertility in Caenorhabditis elegans clk-1 mutants depend upon transport of dietary coenzyme Q8 to mitochondria. J. Biol. Chem. 2002;277:45020–45027. doi: 10.1074/jbc.M204758200. [DOI] [PubMed] [Google Scholar]
- 54.Arroyo A., Santos-Ocana C., Ruiz-Ferrer M., Padilla S., Gavilan A., Rodriguez-Aguilera J.C., Navas P. Coenzyme Q is irreplaceable by demethoxy-coenzyme Q in plasma membrane of Caenorhabditis elegans. FEBS Lett. 2006;580:1740–1746. doi: 10.1016/j.febslet.2006.02.025. [DOI] [PubMed] [Google Scholar]
- 55.Yang Y.Y., Vasta V., Hahn S., Gangoiti J.A., Opheim E., Sedensky M.M., Morgan P.G. The role of DMQ(9) in the long-lived mutant clk-1. Mech. Ageing Dev. 2011;132:331–339. doi: 10.1016/j.mad.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leneuve P., Colnot S., Hamard G., Francis F., Niwa-Kawakita M., Giovannini M., Holzenberger M. Cre-mediated germline mosaicism: a new transgenic mouse for the selective removal of residual markers from tri-lox conditional alleles. Nucleic Acids Res. 2003;31:e21. doi: 10.1093/nar/gng021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marroquin L.D., Hynes J., Dykens J.A., Jamieson J.D., Will Y. Circumventing the Crabtree effect: replacing media glucose with galactose increases susceptibility of HepG2 cells to mitochondrial toxicants. Toxicol. Sci. 2007;97:539–547. doi: 10.1093/toxsci/kfm052. [DOI] [PubMed] [Google Scholar]
- 58.Aguer C., Gambarotta D., Mailloux R.J., Moffat C., Dent R., McPherson R., Harper M.E. Galactose enhances oxidative metabolism and reveals mitochondrial dysfunction in human primary muscle cells. PLoS ONE. 2011;6:e28536. doi: 10.1371/journal.pone.0028536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Postic C., Magnuson M.A. DNA Excision in liver by an albumin-Cre transgene occurs progressively with age. Genesis. 2000;26:149–150. doi: 10.1002/(sici)1526-968x(200002)26:2<149::aid-gene16>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 60.Alison M.R. Regulation of hepatic growth. Physiol. Rev. 1986;66:499–541. doi: 10.1152/physrev.1986.66.3.499. [DOI] [PubMed] [Google Scholar]
- 61.Lapointe J., Stepanyan Z., Bigras E., Hekimi S. Reversal of the mitochondrial phenotype and slow development of oxidative biomarkers of aging in long-lived Mclk1+/- mice. J. Biol. Chem. 2009;284:20364–20374. doi: 10.1074/jbc.M109.006569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Reitzer L.J., Wice B.M., Kennell D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J. Biol. Chem. 1979;254:2669–2676. [PubMed] [Google Scholar]
- 63.Quinzii C.M., Lopez L.C., Gilkerson R.W., Dorado B., Coku J., Naini A.B., Lagier-Tourenne C., Schuelke M., Salviati L., Carrozzo R., et al. Reactive oxygen species, oxidative stress, and cell death correlate with level of CoQ10 deficiency. FASEB J. 2010;24:3733–3743. doi: 10.1096/fj.09-152728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lopez L.C., Quinzii C.M., Area E., Naini A., Rahman S., Schuelke M., Salviati L., Dimauro S., Hirano M. Treatment of CoQ(10) deficient fibroblasts with ubiquinone, CoQ analogs, and vitamin C: time- and compound-dependent effects. PLoS ONE. 2010;5:e11897. doi: 10.1371/journal.pone.0011897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rotig A., Appelkvist E.L., Geromel V., Chretien D., Kadhom N., Edery P., Lebideau M., Dallner G., Munnich A., Ernster L., et al. Quinone-responsive multiple respiratory-chain dysfunction due to widespread coenzyme Q10 deficiency. Lancet. 2000;356:391–395. doi: 10.1016/S0140-6736(00)02531-9. [DOI] [PubMed] [Google Scholar]
- 66.Geromel V., Darin N., Chretien D., Benit P., DeLonlay P., Rotig A., Munnich A., Rustin P. Coenzyme Q(10) and idebenone in the therapy of respiratory chain diseases: rationale and comparative benefits. Mol. Genet. Metab. 2002;77:21–30. doi: 10.1016/s1096-7192(02)00145-2. [DOI] [PubMed] [Google Scholar]
- 67.Lenaz G. Quinone specificity of complex I. Biochim. Biophys. Acta. 1998;1364:207–221. doi: 10.1016/s0005-2728(98)00028-0. [DOI] [PubMed] [Google Scholar]
- 68.Napolitano A., Salvetti S., Vista M., Lombardi V., Siciliano G., Giraldi C. Long-term treatment with idebenone and riboflavin in a patient with MELAS. Neurol. Sci. 2000;21:S981–S982. doi: 10.1007/s100720070015. [DOI] [PubMed] [Google Scholar]
- 69.Haginoya K., Miyabayashi S., Kikuchi M., Kojima A., Yamamoto K., Omura K., Uematsu M., Hino-Fukuyo N., Tanaka S., Tsuchiya S. Efficacy of idebenone for respiratory failure in a patient with Leigh syndrome: a long-term follow-up study. J. Neurol. Sci. 2009;278:112–114. doi: 10.1016/j.jns.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 70.James A.M., Cocheme H.M., Smith R.A., Murphy M.P. Interactions of mitochondria-targeted and untargeted ubiquinones with the mitochondrial respiratory chain and reactive oxygen species. Implications for the use of exogenous ubiquinones as therapies and experimental tools. J. Biol. Chem. 2005;280:21295–21312. doi: 10.1074/jbc.M501527200. [DOI] [PubMed] [Google Scholar]
- 71.Forsman U., Sjoberg M., Turunen M., Sindelar P.J. 4-Nitrobenzoate Inhibits coenzyme Q biosynthesis in mammalian cell cultures. Nat. Chem. Biol. 2010;6:515–517. doi: 10.1038/nchembio.372. [DOI] [PubMed] [Google Scholar]
- 72.Peng M., Falk M.J., Haase V.H., King R., Polyak E., Selak M., Yudkoff M., Hancock W.W., Meade R., Saiki R., et al. Primary coenzyme Q deficiency in Pdss2 mutant mice causes isolated renal disease. PLoS Genet. 2008;4:e1000061. doi: 10.1371/journal.pgen.1000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Falk M.J., Polyak E., Zhang Z., Peng M., King R., Maltzman J.S., Okwuego E., Horyn O., Nakamaru-Ogiso E., Ostrovsky J., et al. Probucol ameliorates renal and metabolic sequelae of primary CoQ deficiency in Pdss2 mutant mice. EMBO Mol. Med. 2011;3:410–427. doi: 10.1002/emmm.201100149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Quinzii C.M., Tadesse S., Naini A., Hirano M. Effects of inhibiting CoQ10 biosynthesis with 4-nitrobenzoate in human fibroblasts. PLoS ONE. 2012;7:e30606. doi: 10.1371/journal.pone.0030606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Genova M.L., Bianchi C., Lenaz G. Supercomplex organization of the mitochondrial respiratory chain and the role of the Coenzyme Q pool: pathophysiological implications. Biofactors. 2005;25:5–20. doi: 10.1002/biof.5520250103. [DOI] [PubMed] [Google Scholar]
- 76.Lenaz G., Genova M.L. Structure and organization of mitochondrial respiratory complexes: a new understanding of an old subject. Antioxid. Redox Signal. 2010;12:961–1008. doi: 10.1089/ars.2009.2704. [DOI] [PubMed] [Google Scholar]
- 77.Lenaz G., Genova M.L. Structural and functional organization of the mitochondrial respiratory chain: a dynamic super-assembly. Int. J. Biochem. Cell Biol. 2009;41:1750–1772. doi: 10.1016/j.biocel.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 78.Salviati L., Sacconi S., Murer L., Zacchello G., Franceschini L., Laverda A.M., Basso G., Quinzii C., Angelini C., Hirano M., et al. Infantile encephalomyopathy and nephropathy with CoQ10 deficiency: a CoQ10-responsive condition. Neurology. 2005;65:606–608. doi: 10.1212/01.wnl.0000172859.55579.a7. [DOI] [PubMed] [Google Scholar]
- 79.Kamzalov S., Sumien N., Forster M.J., Sohal R.S. Coenzyme Q intake elevates the mitochondrial and tissue levels of Coenzyme Q and alpha-tocopherol in young mice. J. Nutr. 2003;133:3175–3180. doi: 10.1093/jn/133.10.3175. [DOI] [PubMed] [Google Scholar]
- 80.Sohal R.S., Kamzalov S., Sumien N., Ferguson M., Rebrin I., Heinrich K.R., Forster M.J. Effect of coenzyme Q10 intake on endogenous coenzyme Q content, mitochondrial electron transport chain, antioxidative defenses, and life span of mice. Free Radic. Biol. Med. 2006;40:480–487. doi: 10.1016/j.freeradbiomed.2005.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wallace B.J., Young I.G. Aerobic respiration in mutants of Escherichia coli accumulating quinone analogues of ubiquinone. Biochim. Biophys. Acta. 1977;461:75–83. doi: 10.1016/0005-2728(77)90070-6. [DOI] [PubMed] [Google Scholar]
- 82.Nagy A. Manipulating the Mouse Embryo : A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2003. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.