

Abstract
Mutations in lamin A/C result in a range of tissue-specific disorders collectively called laminopathies. Of these, Emery–Dreifuss and Limb-Girdle muscular dystrophy 1B mainly affect striated muscle. A useful model for understanding both laminopathies and lamin A/C function is the Lmna−/− mouse. We found that skeletal muscle growth and muscle satellite (stem) cell proliferation were both reduced in Lmna−/− mice. Lamins A and C associate with lamina-associated polypeptide 2 alpha (Lap2α) and the retinoblastoma gene product, pRb, to regulate cell cycle exit. We found Lap2α to be upregulated in Lmna−/− myoblasts (MBs). To specifically test the contribution of elevated Lap2α to the phenotype of Lmna−/− mice, we generated Lmna−/−Lap2α−/− mice. Lifespan and body mass were increased in Lmna−/−Lap2α−/− mice compared with Lmna−/−. Importantly, the satellite cell proliferation defect was rescued, resulting in improved myogenesis. Lmna−/− MBs also exhibited increased levels of Smad2/3, which were abnormally distributed in the cell and failed to respond to TGFβ1 stimulation as in control cells. However, using SIS3 to inhibit signaling via Smad3 reduced cell death and augmented MB fusion. Together, our results show that perturbed Lap2α/pRb and Smad2/3 signaling are important regulatory pathways mediating defective muscle growth in Lmna−/− mice, and that inhibition of either pathway alone or in combination can ameliorate this deleterious phenotype.
INTRODUCTION
The eukaryotic nucleus is encased in the nuclear envelope (NE), a structure consisting of the outer nuclear membrane, the inner nuclear membrane and the underlying nuclear lamina (1). Lamins are type V intermediate filaments that form the proteinaceous network underlying the nuclear lamina. There are several lamin isoforms, including A-type lamins (lamin A, C, AΔ10 and C2), encoded by the LMNA gene and the lamins B1 and B2, each encoded by its own gene. More than 250 different mutations have been identified in the LMNA gene which result in disorders termed the laminopathies with a broad range of tissue-specific phenotypes affecting nerves, fat, bone and muscle, and include Hutchinsons-Gilford Progeria, lipodystrophies, dilated cardiomyopathy and Emery–Dreifuss muscular dystrophy (2,3).
Mutations within the LMNA gene affecting skeletal and cardiac muscles are usually autosomal-dominant missense mutations and lead to Limb-Girdle muscular dystrophy type 1B (LGMD-1B) (4), autosomal-dominant Emery–Dreifuss muscular dystrophy (AD-EDMD) (5), congenital muscular dystrophy (6) and dilated cardiomyopathy with conduction defect (DCM-CD) (7). Notably, in man, loss of A-type lamins is embryonic lethal and characterized by pre-term death due to respiratory insufficiency, growth retardation, dysmorphic face, severe joint contractures and severe generalized muscular dystrophy with loss of fibers in intercostal muscles (8). Although the exact mechanisms by which mutations in lamins result in the varied clinical features remain to be elucidated, increasingly compelling evidence suggests that lamins play an important role in regulating nucleoplasmic positioning of chromatin and nuclear shuttling of proteins, and these activities are mediated by interactions between lamins and integral and associated NE proteins (9). Thus, the characterization of tissue-specific lamin-binding proteins may elucidate the clinical pathogenesis of different mutations within the LMNA gene.
Several animal models of laminopathies have now been developed (10,11). Complete deletion of the murine Lmna gene results in muscle weakness and progressive wasting, dystrophy in the peri-vertebral and femoral muscles, irregular fiber diameters, degeneration of the cardiac muscle and death at 4–6 weeks (12). Lmna−/− muscle cells have been shown to have irregular transcriptional activity and, by electron microscopy, to display nuclear atypia including the redistribution of heterochromatin from the nuclear periphery to the nucleoplasm (13). Mice heterozygous for the complete deletion are overtly normal but show AV conduction defects by 1 year of age (14). Several point mutations within the Lmna locus that cause disease in man have also been created in the mouse. These mutations include H222P, which exhibits muscular dystrophy, and N195K, which shows skeletal muscle pathology and cardiomyopathy with a lifespan of 3–6 months (15,16).
Skeletal muscle contains a population of resident stem cells, termed satellite cells, responsible for postnatal muscle growth, hypertrophy and repair (17,18). We have recently shown that bone morphogenic protein signaling operating through Smad4 and Smad1, 5 and 8 controls the balance between satellite cell proliferation and differentiation (19). An important function of lamin A/C is to regulate nucleoplasmic shuttling of Smad transcription factors, particularly those activated by TGFβ1 signaling (20). TGFβ1 is a pleiotropic cytokine expressed in many cell types and involved in mediating cell proliferation and differentiation. Canonical TGFβ1 signaling activates Smad2/3 transcription factors through complex phosphorylation cascades. Upon activation, Smad2/3 translocate from the cytoplasm to the nucleus and transcriptionally activate gene targets (21). Recently, Smad2/3 have been implicated in the regulation of muscle growth and differentiation. Smad2/3 are expressed in myoblasts (MBs) and are activated in response to myostatin signaling to suppress the activity of muscle-specific transcription factors such as MyoD (22). However, muscle-specific deletion of Smad4 and complete ablation of Smad3 result in defective muscle formation (23,24), indicating that Smad2/3 play a complex yet important role in muscle development.
A-type lamins also interact with other NE proteins including lamina-associated polypeptides (LAPs), a family of proteins that are integral to the NE. The LAP family member lamina-associated polypeptide 2 alpha (LAP2α) is nucleoplasmic as it lacks a transmembrane domain and interacts with nucleoplasmic lamin A/C. Mutations in LAP2α cause autosomal-dominant DCM, featuring negative coronary angiogram and dilation of the left ventricle (25). Mice deficient in Lap2α (Lap2α−/−) are viable and fertile. However, cells derived from Lap2α−/− mice are hyperproliferative due to defects in cell cycle exit (26,27). Therefore, gene ablation of Lap2α in Lmna−/− mice could ameliorate their phenotype sufficiently to offer a therapeutic strategy.
Here, we set out to investigate the functional relationship between lamin A/C, Lap2α and Smad2/3 signaling. We found defective postnatal muscle growth in Lmna−/− mice, which was accompanied by reduced satellite cell proliferation. Crucially, Lmna−/− satellite cells showed upregulated Lap2α and pRb signaling. Since A-type lamins interact with Lap2α and pRb to regulate cellular proliferation, we determined whether downregulating Lap2α could ameliorate the proliferation defect. Lmna−/−Lap2α−/− mice had increased lifespan and body weight compared with Lmna−/− mice and enhanced satellite cell proliferation. Lmna−/− MBs also exhibited increased Smad2/3 levels, which was abnormally distributed in the cell and failed to respond correctly to TGFβ1 stimulation. However, inhibiting Smad3 signaling by use of a specific inhibitor reduced cell death and augmented MB fusion. Together, our results show that perturbed Lap2α/pRb and Smad2/3 signaling are important regulatory pathways mediating defective muscle growth in Lmna−/− mice, and that inhibition of either pathway alone or in combination can ameliorate this deleterious phenotype.
RESULTS
Lmna−/− mice form smaller muscle fibers
We have previously shown that muscle fibers from Lmna−/− mice contain fewer myonuclei per myofiber than wild-type (WT) (13), suggesting defective growth of Lmna−/− muscle during early postnatal development (up to 3 weeks) when myonuclei are added (28). To study the dynamics of postnatal muscle growth, we conducted histological analysis of gastrocnemius muscle from Lmna−/− and WT mice at 3 weeks of age (Fig. 1A). In WT muscle, diameters ranged from 20 to 100 with the highest percentage of myofibers (mode) lying in the 41–45 µm range. In Lmna−/− mice, however, the range of myofiber diameters remained from 16 to 80 µm and the myofibers were generally smaller with a shift in distribution to a mode lying in the 35–40 µm range (Fig. 1B and C). In addition, we performed a BrdU dilution assay in which postnatal mice are treated with BrdU to label proliferating cells. In WT mice, BrdU label got diluted over time, while over the same time in Lmna−/− mice, the proportion of BrdU+ve cells remained high, indicating that less dilution has occurred and thus activation or proliferation is decreased (Supplementary Material, Fig. S1).
Figure 1.
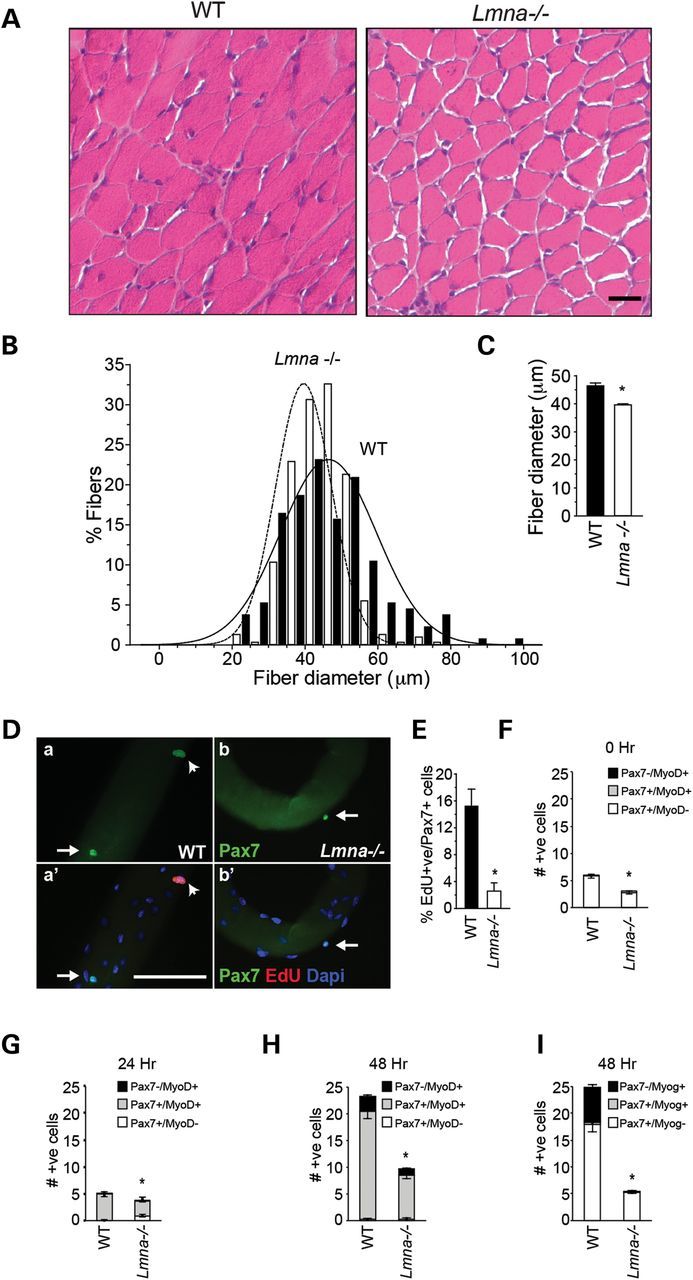
Defective growth of Lmna−/− muscle. (A) H&E sections from gastrocnemius muscles of 3-week-old WT and Lmna−/− mice. (B) Quantitation of fiber diameters shows that the diameter distribution is shifted to the left with smaller fiber diameters predominating. Scale, 50 μm. (C) Quantitation of average fiber diameters shows significantly smaller fibers in Lmna−/− mice compared with WT littermates at 3 weeks. n = 400 fibers, *P < 0.005, SEM. (D). (a–b′) Myofiber-associated proliferating satellite cells detected by staining with Pax7 and EdU. Scale, 100 μm. (E) Fiber-associated EdU+ve /Pax7+ve cells as a percentage of total Pax7+ve satellite cells are significantly decreased in Lmna−/− muscle (n = 3 mice; *P < 0.001) Arrows indicate quiescent (Pax7+ve) satellite cells; arrowheads indicate proliferating (Pax7+ve/EdU+ve) satellite cells. (F) Quantitation of fiber-associated Pax7+ve and MyoD+ve cells on freshly isolated (0 h) single fibers, showing less fiber-associated cells in Lmna−/− mice compared with WT. (G) Quantitation of fiber-associated Pax7- and MyoD-expressing cells on single fibers maintained in culture for 24 h, showing a portion of Lmna−/− cells still MyoD-ve, therefore delayed in their activation. (H and I) Quantitation of fiber-associated MyoD+ve (H) and myogenin+ve (I) satellite cells in Lmna−/− muscle after 48 h in culture (n = 3 mice). Results are shown as means ± SEM. *P < 0.001.
Satellite cell-derived myoblasts from Lmna−/− mice show delayed activation and proliferate at a slower rate than WT
LGMD1B and AD-EDMD are muscle-wasting diseases, which, together with our observation of smaller muscle fibers in the mouse model, suggested that loss of lamin A/C may interfere with the ability of satellite cells to contribute myonuclei. We examined proliferation of satellite cell-derived MBs ex vivo using cultured myofibers isolated from extensor digitorum longus (EDL) muscle. This preparation serves as a model of the satellite cell activation that occurs in response to injury in vivo (29). Isolated muscle fibers were cultured for 24 h in the presence of 5-ethynyl-2′-deoxyuridine (EdU), to label activated cells in the S-phase, and then immunostained for Pax7 to identify satellite cells (Fig. 1Da–b′). Quantitation of proliferating EdU+ve cells as a percentage of total Pax7+ve satellite cells showed that WT fibers contained a mean of 15.3 ± 2.5 (mean ± SEM) % Pax7+ve/EdU+ve satellite cells after 24 h of culture, when compared with 2.3 ± 0.3% Pax7+ve/EdU+ve satellite cells (n = 3 mice, P < 0.001) on Lmna−/− fibers (Fig. 1E).
We further examined activation of satellite cells in WT and Lmna−/− mice by quantifying Pax7 and MyoD immunofluorescence in fiber-associated satellite cells. Freshly isolated fibers contain mostly Pax7+ve/MyoD-ve (quiescent) satellite cells with only occasional Pax7+ve/MyoD+ve (activated) cells (30,31). The number of quiescent fiber-associated satellite cells is lower in Lmna−/− fibers (Fig. 1F, 0 h and (13)) (n = 3, P < 0.001). After 24 h in culture, the majority of Pax7+ve satellite cells also begin to express MyoD in WT mice; the number of Pax7+ve/MyoD+ve satellite cells was reduced from 4.9 ± 0.35 in WT to 2.8 ± 0.18 in Lmna−/− fibers and, importantly, 0.89 ± 0.9 Pax7+ve cells per fiber were still MyoD-ve, compared with 0 in WT (n = 3, P < 0.001) (Fig. 1G), indicating that at least a fraction (∼25%) of Pax7+ve satellite cells had not yet activated. By 48 h, the number of Pax7+ve/MyoD+ve cells was 20.2 ± 1.2 for WT and 8.3 ± 0.7 for Lmna−/− (n = 3 mice, P < 0.001) (Fig. 1H), showing that WT cells had undergone more than two doublings in this time span, while Lmna−/− cells had doubled only once, but had overcome their delay in MyoD expression. At this point (48 h in culture), satellite cells committed to terminal differentiation downregulate Pax7 and start expressing myogenin (32). WT fibers contained 6.5 ± 0.6 Pax7-ve/myog+ve cells, compared with 0.03 ± 0.02 in Lmna−/− fibers (Fig. 1I), confirming the differentiation delay. These data indicate that activation and differentiation, measured as MyoD and myogenin expression, are delayed in Lmna−/− myofiber-associated satellite cells.
To further evaluate activation and proliferation of Lmna−/− primary satellite cells, we generated clonal cultures by trypsinizing satellite cells from freshly isolated EDL myofibers and plating them at clonal density on Matrigel-coated 96-well plates (Fig. 2A) in the absence of bFGF. After 96 h, WT colonies contained on average 25.1 ± 0.9 cells, whereas Lmna−/− colonies contained 14.1 ± 0.8 cells (n = 3, P < 0.001) (Fig. 2B). Accordingly, on extended culture in the presence of bFGF, WT satellite cell-derived MBs yielded several million cells but Lmna−/− cells proliferated poorly, resulting in fewer than million cells over the same period (data not shown).
Figure 2.

Lmna−/− primary and immortalized MBs proliferate slower than WT. (A and B) Clonal cultures of adherent fiber-derived satellite cells. After 96 h in culture, colonies obtained from Lmna−/− single clones contained fewer cells than WT (n = 50 clones from three mice/genotype; *P < 0.001); scale, 100 μm. (C) Lmna−/− H-2K cells proliferate slower than WT (n = 3 clones; *P < 0.05; **P < 0.001). (D) Low-density Lmna−/− MB cultures form fewer myotubes than WT. Primary MBs were plated at low (5 × 104 cells/cm2) density and allowed to differentiate for 5 days, then immunostained with an antibody to myosin heavy chain (red) and counterstained with DAPI (blue). (E) Total number of nuclei is significantly decreased in Lmna−/− low-density cultures. (F) Myogenic fusion quantitated as a percentage of nuclei in myotubes compared with total nuclei is lower in Lmna−/− cultures. (Ga, b) In high-density Lmna−/− MB cultures (3 × 105 cells/cm2), myotubes form to the same extent as WT. Scale, 50 μm. Ga′, b′. High magnification views of a and b insets show nuclear atypia including herniations and chromatin strands in Lmna−/− myotubes. (H) High magnification views show lack of localization of MyoD, myogenin (Myog) and pRbS780 with chromatin in Lmna−/− (arrowheads). Scale bar, 20 μm. Inset, higher magnification view of pRbS780 in Lmna−/− co-localizing with chromatin strands (arrowhead). Scale, 10 μm.
To obtain greater quantities of cells for analysis, we derived immortalized satellite cell lines (H-2K cells) by crossing Lmna−/− mice with ImmortomiceTM (T+) (29). Single clonal satellite cell lines were derived from F2 T+:WT and T+:Lmna−/− littermates of Lmna+/− and T+ crosses. H-2K cells proliferate at 33°C in the presence of IFNγ and differentiate into myotubes at 37°C in the absence of IFNγ (29). When grown under proliferating conditions, Lmna−/− H-2K MBs still proliferated ∼30% slower than WT H-2K MBs (n = 3 clones; P < 0.001) (Fig. 2C). Thus, immortalization by large T antigen produces MB cell lines that exhibit ex vivo proliferation rates characteristic of the genotype of the mouse from which the cells were isolated.
Lmna−/− myoblasts form myotubes with nuclear atypia
We next compared the differentiation potential of both primary satellite cells and H-2K cells derived from Lmna−/− mice. Cells were plated at a low density (5000 cells/cm2) in growth medium, which was replaced after 24 h with low-serum differentiation medium. Under these conditions, the majority of cells withdraw from the cell cycle, but a small proportion continued to proliferate (data not shown). After 5 days of culture, WT MBs efficiently differentiated into myotubes, as shown by immunostaining for myosin heavy chain and counterstaining with DAPI, but Lmna−/− cultures contained fewer cells and in turn formed fewer myotubes (Fig. 2D–F). To determine whether decreased fusion into myotubes in Lmna−/− cultures was due to an inherent inability to differentiate and fuse, or due to the slower proliferation rate, we plated the cells at a high density (28 000 cells/cm2), and immediately switched them into low-mitogen medium to promote cell cycle exit and myogenic differentiation. Under such conditions, both WT- and Lmna−/−-derived MBs efficiently formed myotubes (Fig. 2Ga and b), indicating that Lmna−/− MBs at low density form fewer myotubes because they proliferate more slowly, not because they are defective in fusion. However, the myotubes formed by Lmna−/− MBs showed abnormal myonuclear morphology, for example, having chromatin strands that protruded between neighboring nuclei (Fig. 2Ga′ and b′). These data are consistent with earlier findings that Lmna−/− MBs do not show an inherent inability to differentiate and form myotubes (33), but indicate that the reduced proliferation can result in decreased myotube formation.
Nuclear abnormalities destabilize nuclear localization of transcription factors in Lmna−/− muscle cells
Mutations in lamin A can result in defective targeting and retention of nuclear proteins (34). To determine whether abnormal nuclear protein retention influences localization of transcription factors, we examined expression of MyoD, myogenin and pRbS780 in Lmna−/− myotubes by immunostaining. We found nuclear abnormalities including mislocalization of MyoD and myogenin and protruding chromatin strands between adjoining nuclei, with MyoD and myogenin sometimes co-localizing with these chromatin strands. In particular, some nuclei were devoid of MyoD/myogenin immunostaining, whereas others contained MyoD/myogenin but had low/negligible DAPI staining, indicating low levels of chromatin. In addition, we found mislocalized pRbS780 (Fig. 2H). These data suggest that defective nuclear targeting and retention of proteins in Lmna−/− muscle cells result in misregulated localization of transcription factors activated during myogenesis.
Quantitative proteomic analysis of Lmna−/− myoblasts and myotubes
To identify differences in protein expression between WT and Lmna−/−, we performed stable isotope labeling by amino acids in cell culture (SILAC) quantitative proteomic profiles of proliferating H-2K MBs and differentiated H-2K myotubes. In this approach, proliferating cells are cultured in the presence of lysine and arginine labeled with 13C (‘heavy’) or normal 12C (‘light’). The presence of ‘heavy’ labeled amino acids causes a shift in the mass spectra of signature peptides and the different intensities of the peaks were used to obtain relative quantities in Lmna−/− compared with WT samples (Fig. 3A). In MBs, 1103 proteins were identified of which 39 were downregulated and 201 were upregulated >1.5-fold compared with WT. In myotubes, we identified 979 proteins of which 295 were downregulated and 156 were upregulated >1.5-fold (Fig. 3B).
Figure 3.
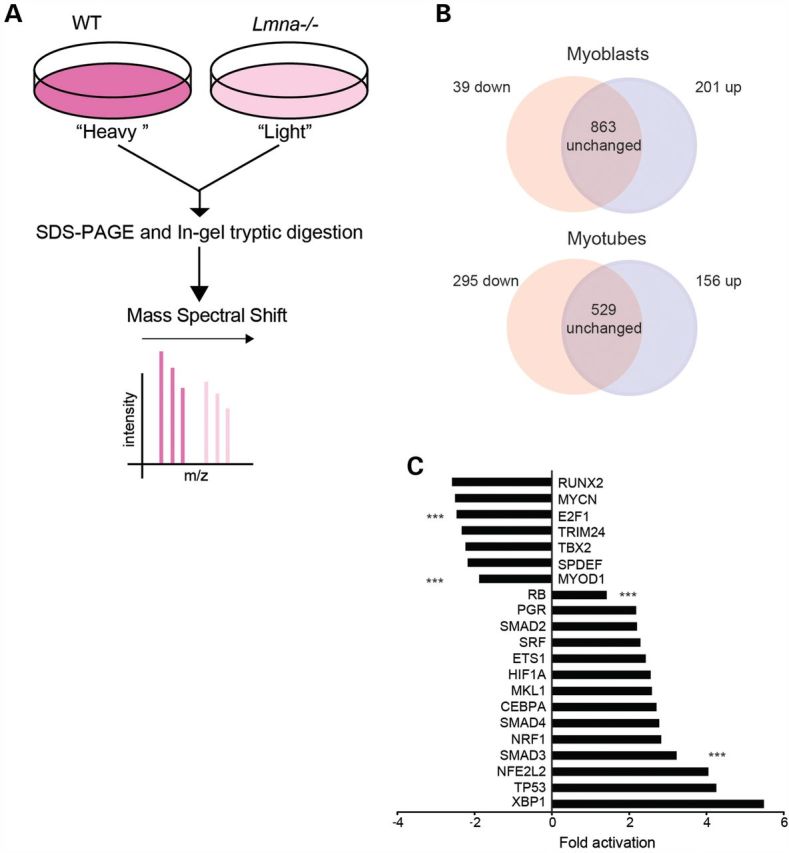
Proteomic analysis of Lmna−/− cells. (A) SILAC protocol. H-2K MBs were in vitro labeled with growth media containing ‘heavy’ and ‘light’ isotopes for three proliferation rounds. Then, pellets were combined for sodium dodecyl sulphate–polyacrylamide gel electrophoresis and in-gel tryptic digestion. Heavy isotopes produce a spectral shift. Peak intensity is used to evaluate relative quantities of peptides. (B) SILAC analysis identified 1103 proteins in MBs and 980 proteins in myotubes. (C) Regulated transcription factor pathways in Lmna−/− H-2K MBs. The X-axis represents activation state: downregulated indicating ‘repressed’ and upregulated indicating ‘activated’ transcription factor pathways. Triple asterisks indicate significantly regulated transcription factor pathways for E2F1, MyoD1, Rb and Smad3 (P < 0.005).
Downregulated MyoD targets in Lmna−/− myoblasts
In Lmna−/− proliferating MBs, decreased proteins included MYH4, MYL1, MYL4, TNNT1 and TNNT3 (Supplementary Material, Table S2), indicating that myogenesis is compromised in Lmna−/− muscle cells. Proteins identified in the SILAC analysis could be targets of activated or inhibited transcription factor pathways. Ingenuity Pathway Analysis (IPA) was used to identify transcriptional activators that were misregulated in Lmna−/− MBs compared with WT (Fig. 3C). The top downregulated myogenesis-associated transcription factors were MyoD (activation z-score −1.88, P = 8.57 E−08) and E2F1 (z-score −2.466, P = 1.21E−10), whereas the top transcriptional activator pathways involved in myogenesis that were upregulated were retinoblastoma (Rb, z-score = 1.62, P = 7.29 E−4) and Smad3 (z-score = 3.2, P = 1.6 E−3) (Fig. 3C).
The Smad2/3 transcription factor pathway is upregulated in Lmna−/− myoblasts
A-type lamins have been implicated in regulating the transcriptional activity of Smad2/3, a signaling pathway important for proliferation, differentiation and cell death (20). Smad2/3, or R-SMADs, are transcription factors activated by members of the TGFβ1 cytokine superfamily (21). The TGFβ1 family cytokines regulate cellular proliferation and differentiation with complex activities that are highly context-dependent. Through its activation of Smad2/3, TGFβ1 represses skeletal muscle-specific gene expression and reduces the activities of MRFs (MyoD, Myf5, myogenin and MRF4) and MEFs (MEF2A-D) (reviewed in 35). TGFβ1 inhibits transcription of the MyoD gene (36), and decreases the transcriptional activity of MRFs without affecting their binding properties, by targeting the bHLH region (37). Furthermore, Smad3 but not Smad2 physically interacts with MyoD, inhibiting its transactivation properties (38,39).
Our proteomic analysis revealed that Smad3 transcriptional targets were significantly upregulated in Lmna−/− MBs (P = 1.6E-3) (Fig. 3C). Among the upregulated proteins were Col2A1, Col1A2, TPM1 and TPM2, known targets of Smad3 (Supplementary Material, Table S2). These data suggest that upregulated Smad3 signaling may have an impact on myogenesis. Interestingly, we also found upregulated Smad3 signaling in Lmna−/− myotubes, and again increased Col2A1, Col1A2, TPM1 and TPM2 (Supplementary Material, Excel file MT).
To address the role of Smad2/3 signaling in Lmna−/− muscle cells, we examined the nucleo-cytoplasmic shuttling of these factors during differentiation. Upon TGFβ1 receptor-mediated activation, Smad2/3 become phosphorylated and translocate to the nucleus to transactivate transcription targets, following which, Smad2/3 become dephosphorylated by protein phosphatase 2A and are exported back to the cytoplasm (18). Thus, activated Smad2/3 are localized to the nucleus, whereas inactive Smad2/3 are cytoplasmic. In proliferating cells, Smad2/3 are activated and thus localized to the nuclei where they repress activation of muscle genes such as MyoD. In contrast, during differentiation, Smad2/3 become inactive and localize to the cytoplasm (39). Western blot analysis showed that the total expression of Smad2/3 was increased in both nuclear and cytoplasmic fractions of Lmna−/− muscle cells compared with WT (Fig. 4A). We assessed activation of Smad2/3 by quantifying the ratio of nuclear to cytoplasmic concentration during the stages of myotube differentiation (Fig. 4B). In WT cells, the activated (nuclear) Smad2/3 peak at the onset of differentiation (days 1 to 2), then become more cytoplasmic (inactive) on days 3 to 4, consistent with published reports (37). In Lmna−/− muscle cells, however, Smad2/3 activity remained upregulated (nuclear) throughout differentiation (days 1–4) (Fig. 4B).
Figure 4.
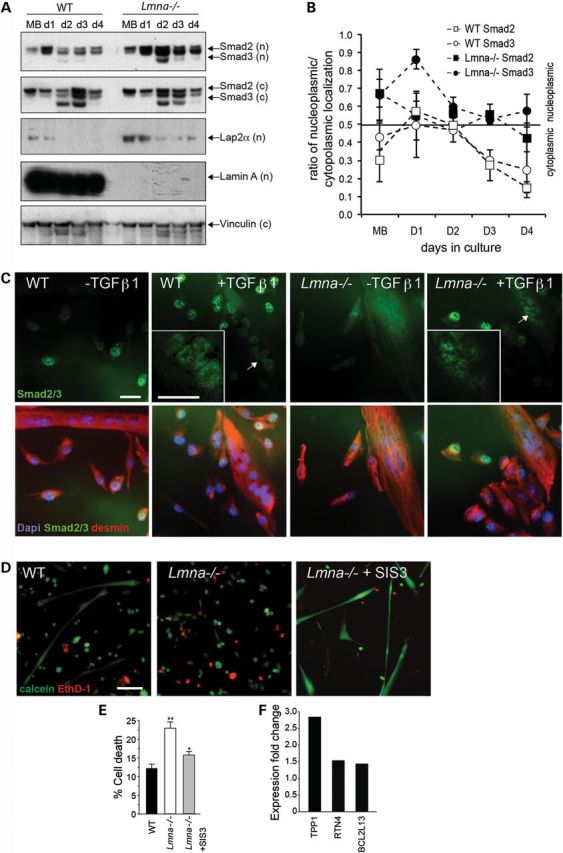
Misregulated Smad2/3 signaling in Lmna−/− muscle cells. (A) Increased Smad2/3 expression in Lmna−/− cells. WT and Lmna−/− muscle cells were collected at different timepoints: proliferating MBs (MB) and days 1–4 of differentiation (d1-d4). Cell pellets were separated into nuclear (n) and cytoplasmic (c) fractions and subjected to immunoblotting using antibodies to Smad2/3. Vinculin was used as a loading control. (B) Quantitation of Smad2/3 in nuclear and cytoplasmic fractions. Values greater than 0.5 indicate a predominantly nucleoplasmic localization (n = 3 experiments per time point ± SEM). (C) WT and Lmna−/− satellite cells were incubated with differentiation medium containing 10 ng/ml of TGFβ1 for 54 h, then immunostained with antibodies to Smad2/3 (green) and desmin (red) and co-stained with DAPI (blue) to visualize nuclei. In untreated WT cells, some Smad2/3 expression can be seen in the nuclei of MBs whereas nuclear localization is decreased in myotubes. In response to treatment with TGFβ1, Smad2/3 localize to the nuclei of both MBs and myotubes (green). In Lmna−/− cells, Smad2/3 are generally upregulated but localization is equally distributed in nuclear and cytoplasmic compartments. In response to TGFβ1 treatment in Lmna−/− MBs, localization in Lmna−/− myotubes remains unclear with more cytoplasmic staining. Scale, 50 μm. Arrow indicates location of inset. Scale, 20 μm. (D) LIVE-DEAD assay showing live calcein+ve (green) and dead EthD-1 (red) staining. WT and Lmna−/− H-2K cells were differentiated for 5 days, then analyzed using the calcein and EthD-1 permeability assay. When Smad3 blocker SIS3 was included in the differentiation media, cell death in Lmna−/− cells was decreased. Scale bar, 100 μm. (E) Lmna−/− myotubes show increased cell death, which is significantly decreased by SIS3 treatment (n = 3 experiments; *, P < 0.05; **, P < 0.01). (F) Cell death-associated proteins TPP1, RTN4 and BCL2L13 identified in proteomic array are upregulated in Lmna−/− myotubes.
Lmna−/− cells do not retain nuclear Smad2/3 in response to TGFβ1 stimulation
Having shown an increase in Smad2/3 levels in untreated Lmna−/− muscle cells, we hypothesized that the absence of interactions between lamin A/C and Smad2/3 may perturb the response of Smad2/3 to TGFβ1 signaling in Lmna−/− MBs.
Treatment of WT MBs with 1 ng/ml TGFβ1 increases cellular proliferation, increases total cell number and decreases differentiation compared with non-treated MBs (unpublished data). We determined the response to TGFβ1 treatment in Lmna−/− primary satellite cells. In untreated WT MBs, immunostaining revealed that Smad2/3 localized to the nuclei, but in myotubes nuclear localization was reduced (Fig. 4C), consistent with our western blot analysis. Treatment with TGFβ1 caused Smad2/3 to localize to the nuclei of both MBs and myotubes in WT. In untreated Lmna−/− MBs, total expression of Smad2/3 was generally upregulated but localization was less clearly nuclear even after treatment with TGFβ1 (Fig. 4C). These data suggest that retention of Smad2/3 is challenged in the nuclei of Lmna−/− muscle cells, making them less able to respond to TGFβ1 stimulation, and we suggest that this results in compensatory upregulation of Smad2/3 signaling, which we were able to show by western blot (Fig. 4A). These data suggest that defective shuttling of Smad2/3 in Lmna−/− muscle results in compensatory activation of Smad2, and especially Smad3. Therefore, a loss of lamin A/C in muscle perturbs Smad2/3 nuclear transport, resulting in compensatory Smad3 target gene upregulation.
Blockade of Smad3 decreases cell death in Lmna−/− myotubes
AD-EDMD is characterized by muscle atrophy and necrosis (40). Since in addition to regulating cell proliferation, Smad2/3 play a role in regulating cell viability, we next determined whether the increased Smad2/3 activity exacerbates death of Lmna−/− myotubes. We examined cell death in myotube cultures using a calcein and EthD-1 permeability assay (LIVE-DEAD) (Fig. 4D). In this approach, membrane-permeant calcein AM enters live cells, which activates a caged green fluorescent conjugate. In contrast, membrane-impermeant EthD-1 (red) labels nucleic acids of only compromised cells. Quantitation of fluorescent calcein and EthD+ve cells showed significantly more dead cells in Lmna−/− myotube cultures than WT cultures. The use of a specific Smad3 blocker, SIS3, increased myotubes formation and decreased cell death in a dose-dependent manner (Supplementary Material, Fig. S2). When Lmna−/− MBs were differentiated in the presence of 5 μm SIS3, cell death was significantly ameliorated from 22.3 ± 1.5% to 15.0 ± 1.0% (mean ± SEM; n = 3 experiments; P < 0.05) (Fig. 4E).
Consistent with this observation, IPA of the proteomic myotube data identified upregulated expression of 14 proteins associated with cell death (Supplementary Material, Table S3, Supplementary Material, Excel MT). Of these, we identified mediators of cell death which were also direct targets of Smad2/3, with TPP1, RTN4 and BCL12L13 upregulated in Lmna−/− myotubes (Fig. 4F). These observations suggest that upregulation of Smad3 results in muscle wasting in Lmna−/−, and blocking Smad3 promotes survival.
Whereas Smad3 signaling is important in mediating the maintenance of Lmna−/− myotubes, Smad3 signaling alone did not appear to explain the proliferation defect we observed in muscle and primary satellite cells.
Upregulated Lap2α/pRb in Lmna−/− myoblasts
We also identified several misregulated targets of pRb/E2F1 signaling in our screen, including DHFR, ERH, VIM and COL2A1 (Supplementary Material, Table S3). This is consistent with our data on the perturbed cell cycle dynamics in Lmna−/− satellite cells: pRb is a transcriptional repressor that, when hypo-phosphorylated (activated), represses E2F1-dependent transcription of cell cycle gene targets, in turn, allowing MBs to exit the cell cycle and differentiate. Alternatively, hyper-phosphorylated pRb does not repress E2F1 and proliferation ensues (41). Consistent with these observations, we found that pRbS780 is reduced and/or mislocalized in Lmna−/− cells (Fig. 2H), indicating that pRb is an activated hypo-phosphorylated state resulting in decreased E2F1-mediated transcription and, in turn, decreased proliferation. We further quantified this using western blot analysis. We found a modest decrease in phospho-pRb, indicative of activation, in proliferating Lmna−/− primary cells compared with WT (Fig. 5A and B) and decreased total pRb in both proliferating MBs and differentiated myotubes. These data indicate that pRb is deregulated in Lmna−/− muscle cells.
Figure 5.
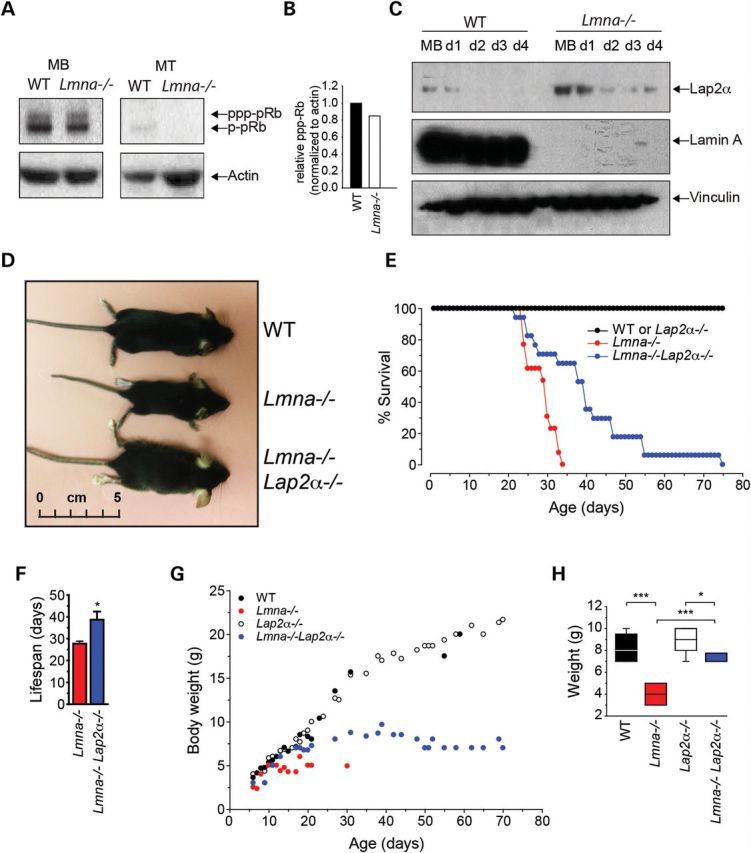
Loss of Lap2α increases body weight and improves lifespan of Lmna−/− mice. (A) Proliferating and differentiating MBs were immunoblotted with an anti-pRb antibody. Total pRb and phospho-pRb are indicated. (B) Quantitation of phospho-pRb in proliferating MBs relative to actin loading control. (C) Changes in Lap2α expression in Lmna−/− MBs. WT and Lmna−/− muscle cells were collected at different timepoints: proliferating MBs and days 1–4 of differentiation (d1-d4). Cell pellets were separated into nuclear and cytoplasmic fractions and subjected to immunoblotting using antibodies to Lap2α and lamin A. Vinculin was used as a loading control. (D) Lmna−/−Lap2α−/− mice at 4 weeks of age are significantly larger than Lmna−/−. (E) Kaplan-Meier survival curve showing that the lifespan of Lmna−/−Lap2α−/− mice is extended compared with Lmna−/−. (F) Average lifespan of Lmna−/−Lap2α−/− mice is significantly increased compared with Lmna−/− (mean ± SEM, n = 15; P < 0.01). (G) Body weights of Lmna−/−Lap2α−/− mice are increased compared with Lmna−/−. Lmna−/−Lap2α−/− mice start showing a decrease in body weight at about 4 weeks. (H) At 3 weeks, the body weights of Lmna−/− mice are significantly lower than WT littermates, but Lmna−/−Lap2α−/− mice are significantly heavier, than Lmna−/− mice (n = 5; *, P < 0.05; ***, P < 0.005).
Nucleoplasmic lamin A/C interacts with pRb in a complex with lamina-associated polypeptide alpha (Lap2α) that maintains pRb in an activated state and allows cell cycle exit (42,43). Since pRb signaling was misregulated in Lmna−/− MBs, we next focused on the role of Lap2α in regulating MB cell cycle in the absence of lamin A/C.
Loss of Lap2α partially rescues the Lmna−/− phenotype
Our proteomic analysis showed an upregulation of Lap2α in Lmna−/− MBs (1.32-fold, Supplementary Material, Data) which was further confirmed by western blotting performed on WT and Lmna−/− primary MBs undergoing differentiation (Fig. 5C), and was consistent with previous reports (33). A loss of Lap2α results in disruption of the lamin A/C-pRb-Lap2α complex, and cells lacking Lap2α do not exit the cell cycle appropriately (26). Lap2α−/− mice are viable, have a normal lifespan and appear to be overtly normal, but Lap2α−/− epithelial cells are hyperproliferative and show decreased pRb activation (26). Importantly, MBs derived from Lap2α−/− mice show an extended proliferative phase and delayed cell cycle exit (27). We reasoned that upregulated Lap2α could cause premature exit from the cell cycle in Lmna−/− cells. To test whether the removal of pRb/Lap2α repressive activity in Lmna−/− cells could alleviate the proliferation defect and improve myogenesis, we generated Lmna−/−Lap2α−/− mice (Fig. 5D).
Lmna−/−Lap2α−/− mice were born at normal Mendelian ratios. Lifespan in Lmna−/−Lap2α−/− mice was significantly longer than that of Lmna−/− mice, increasing from 27.6 ± 1.1 to 38.7 ± 3.8 days (mean ± SEM, n = 15 mice, P < 0.005), although Lmna−/−Lap2α−/− mice had significantly shorter lifespans than WT or Lap2α−/− mice (Fig. 5E and F). Postnatal growth of Lmna−/−Lap2α−/− mice was also dramatically increased, such that while Lmna−/− mice weighed 4.0 ± 0.5 g at 3 weeks, Lmna−/−Lap2α−/− mice weighed 7.3 ± 0.2 g (mean ± SEM), with WT mice weighing 8.2 ± 0.3 g (n = 4; P < 0.005). Lmna−/−Lap2α−/− mice continued to grow until about 6 weeks of age and weighed 9.7 ± 1.2 g at 39 days (Fig. 5G and H). Thus, absence of Lap2α ameliorates the Lmna−/− phenotype by improving growth and longevity in Lmna−/−Lap2α−/− mice.
Muscle size increase and enhanced proliferation in Lmna−/−Lap2α−/− myoblasts
We compared muscle fiber diameters in Lmna−/− and Lmna−/−Lap2α−/− mice (Fig. 6A, quantified in D). Histological analysis showed that average muscle diameters were 46.48 ± 0.79 in WT, 39.85 ± 0.42 in Lmna−/− and 46.57 ± 0.52 μm in Lmna−/−Lap2α−/− mice, indicating that muscle fibers were larger in Lmna−/−Lap2α−/− mice than Lmna−/− (n = 3, P < 0.005) (Fig. 6D). Thus, we further analyzed the muscle sections for satellite cells by immunostaining for Pax7. In WT muscle sections, there was an average of 10 Pax7+ve cells per field. In contrast, Lmna−/− muscle contained an average of 8.5 cells per field and Lmna−/−Lap2α−/− contained ∼15 cells (Fig. 6B), suggesting that a loss of Lap2α increased satellite cell number in Lmna−/− muscle. To further study growth of satellite cells in Lmna−/−Lap2α−/− mice, we derived clonal cultures from the four genotypes: WT, Lmna−/−, Lap2α−/−and Lmna−/−Lap2α−/−. After 72 h, WT cultures produced on average 12.4 ± 0.4 cells per colony while Lmna−/− cultures contained significantly fewer cells (8.3 ± 0.4; P < 0.001) (Fig. 6E). Lap2α−/− colonies contained more cells than WT (16.0 ± 0.5; P < 0.001), consistent with previous reports (27). Lmna−/−Lap2α−/− cultures produced 13.1 ± 0.4 cells, significantly more than Lmna−/− cultures (P < 0.001), and not significantly different from WT (P = 0.24) (Fig. 6E). These data suggest that loss of Lap2α in Lmna−/− cells restores the number of satellite cells per colony to WT levels, indicating a re-balancing of cell cycle dynamics.
Figure 6.
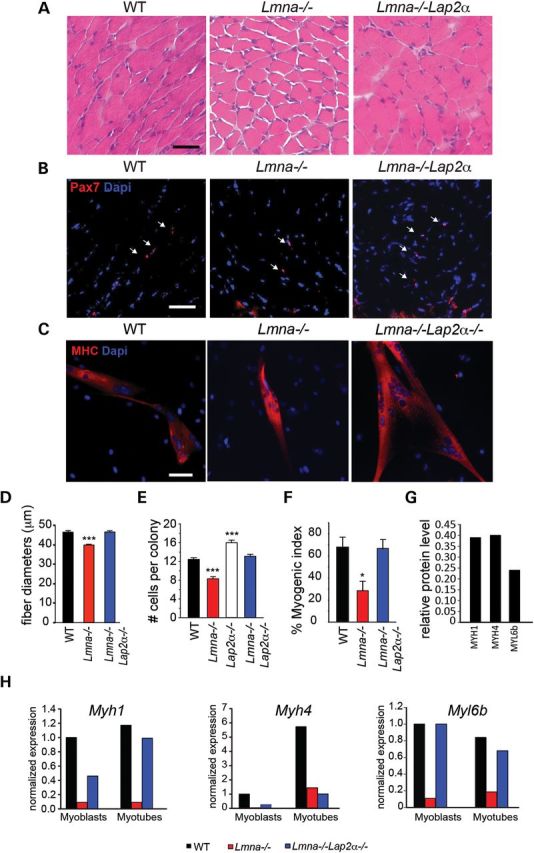
Higher satellite cell numbers and increased MB activation/proliferation in Lmna−/−Lap2α−/− mice. (A) H&E stained gastrocnemius sections of WT, Lmna−/− and Lmna−/−Lap2α−/− 3 week-old mice showing increased muscle fiber diameter size in Lmna−/−Lap2α−/−. (B) Frozen sections were immunostained with an anti-Pax7 antibody to mark satellite cells and co-stained with DAPI. Lmna−/− muscle sections contain less Pax7+ve cells (arrows) than WT. Lmna−/−Lap2α−/− muscle sections contain more satellite cells than Lmna−/−. (C) Primary WT, Lmna−/− and Lmna−/−Lap2α−/− MBs were differentiated for 5 days then immunostained with an antibody to MHC (red) and co-stained with DAPI (blue). Scale bar, 50 μm. Lmna−/−Lap2α−/− cultures contained more myotubes. (D) Quantitation of fiber diameters in 3 week-old WT, Lap2α−/− and Lmna−/−Lap2α−/− mice (n = 3 mice; ***, P < 0.005). (E) Clonal satellite cell cultures were derived from WT, Lmna−/−, Lap2α−/− and Lmna−/−Lap2α−/− mice. After 72 h, cell numbers in the satellite cell colonies were quantified and are shown ± SEM (n>100 colonies/mouse, from at least 3 mice/genotype). Lmna−/−Lap2α−/−-derived cells proliferated at the same rate as WT cells, and significantly more than Lmna−/− cells. (F) Quantitation of myogenesis in primary satellite cell cultures. Myogenic index was calculated as percentage of myonuclei compared with total nuclei (n = 6 non-overlapping fields from 2 independent experiments, ± SEM; *, P < 0.05). (G) Myogenic proteins Myh1, Myh4 and Myl6b are decreased in Lmna−/− MBs compared with WT. (H) Validation of proteomic data by qRT-PCR showing similar decreases of Myh1, Myh4 and Myl6b in Lmna−/− cells compared with WT in proliferating MBs and differentiated myotubes. The expression of Myh1 and Myl6b is restored to WT levels in Lmna−/−Lap2α−/− mice.
Myogenesis is improved in Lmna−/−Lap2α−/− mice
Since we observed increased MB proliferation through loss of Lap2α in Lmna−/− cells, we expected to see more fusion under low density differentiation conditions in vitro. Thus, we compared myotube formation in Lmna−/− and Lmna−/−Lap2α−/− primary MBs using MHC immunostaining. When grown at low density, satellite cells from Lmna−/−Lap2α−/− mice formed bigger myotubes than Lmna−/− MBs (Fig. 6C, quantified in F). Whereas the fusion index in satellite cells cultured from Lmna−/− mice was significantly lower than WT, the index was restored to WT levels in satellite cell cultures from Lmna−/−Lap2α−/−, reflecting the enhanced proliferation (Fig. 6F). Furthermore, proteomic analysis of Lmna−/− H-2K myotubes showed that proteins required for force generation including Myh1, Myh4 and Myl6h were reduced to 0.68, 0.68 and 0.41 of WT, respectively (Supplementary Material, Table S2, Fig. 6G). We validated these proteomic analysis findings by measuring the expression of these genes in primary satellite cells. A quantitative reverse transcription–polymerase chain reaction (qRT–PCR) showed similarly reduced expression of these genes in Lmna−/− primary satellite cells and myotubes, compared with WT. In contrast, expression of Myh1 and Myl6b was restored to WT levels in Lmna−/−Lap2α−/− MBs and myotubes (Fig. 6H). These data suggest that reduction of Lap2α can partially rescue the myogenic deficiency associated with the lack of lamin A/C.
Blockade of Smad3 promotes myogenesis in Lmna−/−Lap2α−/− myotubes
Since neither reducing Smad3 nor Lap2α/pRb completely restored myogenesis, we tested if these two pathways could synergize to rescue differentiation in Lmna−/− MBs. We thus differentiated WT, Lmna−/− and Lmna−/−Lap2a−/− MBs in the presence of the Smad3 inhibitor SIS3, and immunostained cultures for MHC (Fig. 7A). Treatment with SIS3 significantly increased the size of myotubes from a mean ± SEM of 9.14 ± 1.32 to 20.15 ± 3.73 nuclei/myotube in Lmna−/− cultures (Fig. 7B). Interestingly, treatment with SIS3 also increased the size of myotubes in WT and Lmna−/−Lap2α−/−, suggesting that inhibiting Smad3 during differentiation has the added effect of further increasing muscle growth. As the effect on Lmna−/−Lap2α−/− was cumulative, the data suggest that upregulated Smad3 and Lap2α are likely two mechanisms that contribute independently to poor growth and wasting in Lmna−/− muscle. We summarize our findings in Figure 8.
Figure 7.

Blockade of Smad3 increases myotubes size in Lmna−/− Lap2α−/−. (A) MHC immunostaining showing decreased myotubes in Lmna−/− cultures, compared to both WT and Lmna−/−Lap2α−/−. Treatment with SIS3 (bottom panels) increases myotube size. Scale, 50 μm. (B) Quantitation of nuclei per myotubes in SIS3-treated and control (CTRL) cultures (n = 6 non-overlapping fields from 2 independent experiments. *,P < 0.05; **,P < 0.01).
Figure 8.
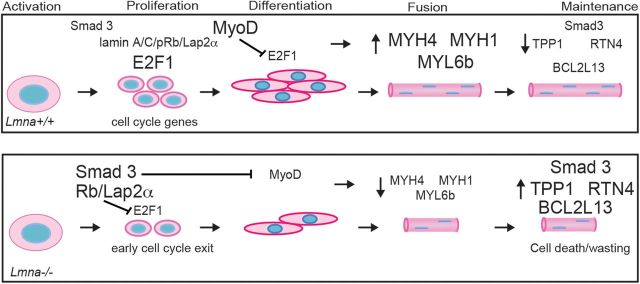
Hypothetical model of lamin A/C interactions. In WT activated satellite cells, levels of Smad3 remain low. Proliferation is driven by nucleoplasmic lamin A/C/Lap2α/pRb complexes which prevent pRb from repressing cell cycle gene activation by E2F1. MyoD increases in differentiating cells resulting in myotube formation. In the absence of lamin A/C, there is compensatory upregulation of Smad3, which represses MyoD. In addition, Smad3 potentiates pRb, which represses E2F1 and also inhibits MyoD activity, including transcriptional activation of MYH1, MYH4 and MYL6b. As a result, cell proliferation is decreased and in turn results in smaller muscle being formed. During the maintenance of the myofiber, upregulated Smad3 signaling in Lmna−/− leads to cell death and muscle wasting, including upregulated cell-death related proteins TPP1, RTN4 and BCL2L13.
DISCUSSION
A-type lamins interact with pRb/Lap2α and Smad3 to mediate muscle formation and maintenance
Nuclear lamins function by providing structure to the nucleus as they form a scaffold connecting the nucleoplasm through the NE LINC complex to the cytoplasm (44). The scaffold is formed through interactions with a growing number of identified NE and nucleoplasmic proteins. Bioinformatic analysis by use of the database analysis software STRING 9.0 and reports by others show that this list includes SYNE1, EMD, LEMD3, LAP2α, pRb, SREBP1 and Smad3 (Supplementary Material, Fig. S3, (45,46, reviewed in 47). Because these interactions further influence the tethering of transcriptionally inactive heterochromatin to the nuclear periphery, they form the basis of the so-called ‘lamin hypothesis’ which postulates a role for lamin A/C and related proteins in the control of gene expression. The interactions between lamin A/C and the other binding partners are determined by the tissue-specific nature of their expression and thus explain how so many different mutations within the LMNA gene can result in the broad spectrum of phenotypes observed in the laminopathies (2).
Here, we focused on the muscle phenotype resulting from the absence of lamin A/C. We describe for the first time the role of the satellite cell in striated muscle forms of the laminopathies by showing delayed activation and decreased proliferation of primary and immortalized MBs from Lmna−/− mice and defective postnatal growth of these animals. Based on our in vivo analysis of muscle fiber diameters, the dilution of BrdU label, reduced numbers of Pax7+ve/EdU+ve fiber-associated satellite cells, and our in vitro analysis of primary and immortalized Lmna−/− satellite cells, we conclude that decreased proliferation contributes to a decreased myofiber size in Lmna−/− mice. Our data are consistent with reports of human diploid fibroblasts lacking LMNA, which grow slowly in culture (48), senesce prematurely (49) and exit from the cell cycle (41). This also parallels the premature senescence that was observed in adult fibroblasts from progeroid mice harboring a deletion within exon 9 of the lamin A/C gene (50). On the other hand, our observations in primary satellite cells and T antigen-immortalized MBs conflict with a report of increased proliferation in paw epidermis of Lmna−/− mice (26) and increased proliferation of Lmna−/− expanded and selected spontaneously immortalized MBs (51). It is possible that lamins A and C have a different activity in embryonic tissues and/or undifferentiated cells, which would help explain why Lmna−/− mice are not smaller than littermates at birth. Thus, A-type lamins appear to play a complex role in cell cycle regulation that may lead to different phenotypes in different tissues and is dependent on the differentiation stage. In this respect, it is of importance that our examination of muscle sections, myofiber-associated satellite cells and of primary MB cultures is consistent with the small size of Lmna−/− mice and muscle, as well as muscle weakness and myopathy in AD-EDMD and LGMD1B patients.
We further used quantitative proteomics to identify MyoD, pRb and Smad2/3 as the main transcription factor pathways misregulated in Lmna−/− muscle. Based on our findings, we propose that at least two separate processes undermine muscle growth and maintenance in the absence of lamin A/C. In activated satellite cells, A-type lamins interact with Lap2α and pRb to control proliferation and self-renewal. In proliferating cells, where pRb is phosphorylated, pRbS780 does not repress E2F1-dependent transcriptional activation, allowing the cell cycle to proceed. One such target identified by our proteomic analysis is ribonucleotide reductase M1, important for DNA synthesis during S-phase (52), which was downregulated in Lmna−/− cells (Supplementary Material, Table S2). These interactions allow activation of E2F1, which maintains the proliferating cell in the cell cycle.
In addition, Smad3 is inhibitory for myotube differentiation; thus the combined compensatory upregulation of pRb/Lap2α and Smad3 in Lmna−/− results in decreased satellite cell activation, as shown by delayed expression of MyoD and myogenin, decreased MB proliferation and decreased expression of muscle-specific genes—Myh1, Myh4 and Myl6b. While MB proliferation is affected in Lmna−/−, resulting in fewer muscle cells and myofibers formation, our data show that the process of myofusion in itself is not affected, as MBs plated at high density efficiently fused into myotubes and muscle fibers form in mice. Although our proteomic profiling data do not establish a causative relationship, others have demonstrated that TGFβ1 induces cell cycle arrest by initiating the recruitment of a complex consisting of Smad2/3, phosphorylated pRb and E2F4 (53), suggesting that this complex may also be active in proliferating MBs, as summarized in our model (Fig. 8). Interestingly, a common target of both pRb and Smad3 pathways, Col2A1, was shown to be upregulated in Lmna−/−. Col2A1 is negatively regulated by E2F1 (54). In addition, Col2A1 transcription is activated by Smad3 (55). Thus, a potential complex forming between Smad2/3 and pRb could play a role in Lmna−/− cells.
Smad2/3 signaling could be upregulated to compensate for loss of responsiveness to TGFβ1
TGFβ1 increases MB proliferation and prevents exit from the cell cycle while simultaneously inhibiting myogenesis by promoting the degradation of MyoD and decreasing myogenin, troponin and MHC expression (56). TGFβ1 also suppresses expression of Sca-1 that may be required for satellite cell activation, and as a result, stem cell-like MB populations are maintained (57,58). Our finding of upregulated Smad2/3 in Lmna−/− MBs suggests that the muscle defect arising from decreased proliferation is, in part, due to misregulated Smad2/3 signaling. An interaction between lamin A/C and Smad2/3 has previously been proposed in fibroblasts. Lamin A/C were shown to interact with protein phosphatase 2A required for nuclear export of Smad2/3 (20). An indirect connection between lamin A/C and Smad2/3 may also arise via the common binding partner Man1/Lemd3 (59; reviewed in 60). On the other hand, proteomic analysis of Smad3 co-immunoprecipitates identified lamin A/C as a direct binding partner of Smad3 (45). Lamins A and C have been shown to regulate general nuclear protein import (34) and localization of NE proteins including emerin and Lap2α (12). Thus, total loss of lamin A/C would be predicted to result in mislocalization of many nuclear proteins including Smad3 and other transcription factors involved in myogenesis.
In addition to disrupting proliferation and myogenesis in Lmna−/− muscle, upregulated Smad3 signaling results in transcription of cell death-associated proteins such as TPP1, RTN4 and BCL2L13, leading to muscle wasting (Fig. 8). Importantly, blocking Smad3 using the specific inhibitor SIS3 promoted both myogenesis and muscle maintenance, as evidenced by both decrease in cell death and increase in myotube size, suggesting that muscle wasting in laminopathies could be improved by TGFβ1 inhibition.
Partial rescue of Lmna−/− phenotype by loss of Lap2α
Lmna−/− mice survive postnatally for 4–6 weeks and show pathologies including myopathy, cardiomyopathy, sarcopenia, osteopenia, decreased bone formation, neuropathy and abnormal neuromuscular junctions (12,61–64). A gene-trap mutation in the Lmna locus was reported to show a similar phenotype with decreased myofiber diameters and cardiac defects (65). Cardiac conduction defects and myopathy were also described in mice that are heterozygotes for loss of lamin A/C (14), as well as homozygous mice carrying the H222P mutation in the Lmna locus (16). Recently, expression of a lamin A/C transgene under a cardiac-specific MHC promoter in Lmna−/− mice improved lifespan by 12% (66). These reports support the notion that a cardiac defect is a major contributor to death in Lmna−/− mice.
Our observation of premature cell cycle arrest in Lmna−/− satellite cells correlates with upregulated Lap2α, deregulated pRbS780 and decreased E2F1 transcription. In view of the reported involvement of Lap2α in pRb-mediated cell cycle arrest, this may be the cause of the proliferation defect in Lmna−/− satellite cells. Alternatively, the cell cycle phenotype may arise from loss of lamin A/C, leading to mislocalization of Lap2α/pRb/E2F1 complexes and/or their binding to chromatin. Here, we have shown that loss of Lap2α in Lmna−/− mice led to a 30% increase in survival rate and a 25–50% increase in body weight. As loss of Lap2α was not sufficient to completely rescue the Lmna−/− phenotype, we speculate that the cardiac defect was still the primary cause of death in both Lmna−/− and Lmna−/−Lap2α−/− mice. However, the data implicate arrested growth as another likely contributor to the reduced lifespan in Lmna−/− mice, such that Lap2α deficiency promoted increased proliferation leading to increased perinatal growth and increased muscle mass, that in itself favors longer survival, perhaps by improving their ability to suckle and feed postnatally.
The observation that Lap2α protein levels are higher in the absence of lamin A/C is paralleled by a recent finding that lamin A loss and progeric LMNA mutations also result in increased Sun1 protein levels, leading, in this latter case to redistribution of Sun1 protein from the NE to the Golgi where it may disrupt Golgi function (67). This parallel extends to the observation that genetic ablation of Sun1 significantly increased longevity and weight gain and ameliorated the muscle and cardiac pathology associated with the loss, or mutation, of lamin A. Although we have not looked at Lap2α distribution (or accumulation/turnover) in our Lmna−/− cells, the fact that Lap2α protein levels are increased suggests that a common molecular mechanism of excess NE- or lamin-associated protein accumulation may underlie the pathology associated with a loss of lamin A.
A-type lamins exert their activity by promoting formation of complexes that mediate transcription of key genes involved in regulating cellular functions. We have provided here at least two distinct mechanisms by which lamins A and C contribute to growth and maintenance of skeletal muscle, through direct or indirect complexes with pRb/Lap2α and Smad3, respectively. Taken together, our observations shed light on the range of molecular mechanisms involved in laminopathies and open the way toward potential new therapeutic approaches.
MATERIALS AND METHODS
Mice
Lmna−/− mice were previously described (12). For derivation of immortalized satellite cells (H-2K cells), Lmna−/− mice were crossed with ImmortomiceTM (Charles River Labs, Wilmington, MA, USA) (T+) which express a temperature-sensitive SV40 large T-antigen gene (tsA58) under the control of an interferon-gamma [IFNγ-inducible promoter (H-2Kb)] (29) and genotyped using indicated primers (Supplementary Material, M&M Primers). Lmna−/−Lap2α−/− mice were generated by crossing the Lmna−/− mice with the Lap2α−/− mice (26) and maintained as heterozygous intercrosses. All mice were maintained according to Children's National Medical Center IACUC guidelines. Mice were euthanized using CO2 asphyxiation followed by cervical dislocation and muscles were flash-frozen in liquid nitrogen cooled in isopentane.
Single myofiber isolation
Freshly removed EDL muscles were incubated in 0.2% collagenase Type I (Sigma, St Louis, MO, USA) in DMEM-GlutaMAX (Life Technologies, Carlsbad, CA, USA) and 1% (v/v) penicillin/streptomycin solution (Sigma) for one and a half hours at 37°C. Collagenase was then inactivated and individual myofibers liberated by trituration, as described in detail elsewhere (68). Myofibers were microscopically examined to ensure that they were free of capillaries or residual connective tissue. Selected myofibers were cultured and fixed for 10 min in 4% paraformaldehyde/PBS (Sigma) at the desired time points.
Myofiber and cell culture
For suspension cultures, myofibers were incubated in DMEM-GlutaMAX (Invitrogen), 10% (v/v) horse serum (Invitrogen), 0.5% (v/v) chick embryo extract (US Biological, Swampscott, MA, USA), and 1% (v/v) penicillin/streptomycin solution (Sigma) (plating medium) at 37°C in 5% CO2. Fiber-associated satellite cells were obtained as previously described (68). Briefly, isolated myofibers were plated in six-well plates (BD Labware) coated with 1 mg/ml Matrigel (BD Biosciences) in DMEM with 20% (v/v) fetal calf serum (Gibco), 10% (v/v) horse serum (Gibco), 1% (v/v) chick embryo extract (US Biological), 1% (v/v) penicillin/streptomycin solution (Sigma) and 5 ng/ml bFGF (PeproTech) (growth medium), and maintained at 37°C in 5% CO2. After 72 h in culture, myofibers were removed, and the satellite cell-derived MBs passaged and re-plated in Matrigel-coated culture dishes for further expansion.
Satellite cells for clonal cultures were derived as previously described (69). Briefly, freshly isolated EDL single myofibers (15–20 per mouse per experiment) from Lmna−/− and WT mice were stripped of satellite cells immediately upon isolation by digestion in 0.05% Trypsin/EDTA (Gibco) at 37°C for 5 min followed by gentle trituration. Myofiber fragments were removed by passing through a 40 μm cell strainer (BD Falcon), and satellite cells collected. Individual cells were plated in Matrigel-coated 96-well plates (BD Falcon) and maintained in the growth medium described above, without bFGF. The cultures were fixed after 72 h or 96 h, for 10 min in 4% paraformaldehyde/PBS (Sigma), washed with PBS and stained with Blue Coomassie. The number of cells per well (thus, per clone) was then counted using an Olympus CK-2 microscope.
Immortalized satellite cells (H-2K cells) were derived from Lmna−/−:T+ mice single fibers as described above. Control H-2K cells were derived from WT:T+ F2 littermates. H-2K were maintained at 33°C in growth medium containing 0.2% IFNγ (Millipore) and differentiated at 37°C in DMEM-GlutaMAX containing 5% horse serum without IFNγ (29). For experimental treatments and immunohistochemistry, MBs were cultured on Matrigel-coated LAB-TEK® eight-well chamber slides (Nunc, Rochester, NY, USA) or glass-bottom culture dishes (MatTek Corporation, Ashland, MA, USA).
Histology and immunofluorescence microscopy
For histology, 10 μm sections were air dried, then stained with hematoxylin and eosin (H&E). Images were acquired using a Nikon Eclipse E800 microscope equipped with a 10×/0.45 and 20×/0.75 objectives and RT Slider Diagnostic SPOT CCD camera using Photoshop (Adobe) acquisition software. Image analysis was performed using ImageJ software (70) and quantified using MinFeret calculation as previously described (71).
For double labeling with Pax7 and EdU, freshly isolated single fibers were pulsed with EdU for 24 h, then fixed in 4% paraformaldehyde/PBS (Sigma). EdU detection was performed using the Click-iT® EdU AlexaFluor® 594 Imaging kit (Molecular Probes/Invitrogen), according to the manufacturer's protocol. Subsequently, fibers were incubated with an anti-Pax7 antibody (Developmental Hybridoma Studies Bank). Percentage of EdU+ve/Pax7+ve cells was derived by dividing by total number of Pax7+ve satellite cells counted on 50 fibers isolated from three individual mice. In parallel, fibers were immunostained with anti-Pax7 and either anti-MyoD or anti-myogenin antibodies and quantitated as above.
Satellite cells and differentiated myotubes cultured on silanized coverslips were fixed with fresh 4% paraformaldehyde/PBS (Sigma), blocked with 5% horse serum and 1% bovine serum albumin, then incubated overnight with anti-myosin heavy chain (Developmental Hybridoma Studies Bank); anti-MyoD (Novus Biologicals, Littleton, CO); anti-myogenin (Santa Cruz); anti-RbS780 (Abcam, Cambridge, MA, USA). Fluorophore-conjugated secondary antibodies were applied for 1 hour. Cells were treated with TGFβ1 (R&D Systems, Minneapolis, MN, USA) using indicated concentrations and incubation times. LIVE-DEAD® cytotoxicity assay was performed following manufacturer's instructions (Molecular Probes).
For Smad3 blocking assay, MBs were treated with indicated concentrations of the Smad3-specific phosphorylation inhibitor SIS3 (EMD Biosciences, Billerica, MA, USA) for 5 days, then incubated with LIVE-DEAD® cytotoxicity assay reagent or fixed and immunostained using an anti-myosin heavy chain antibody. The myogenic fusion index was calculated as the percentage of nuclei in myotubes relative to total nuclei.
Immunofluorescence micrographs were acquired at ambient temperature using a Zeiss Axiovert 200 inverted microscope equipped with 20×/0.3 and 63×/1.4 objectives and an Axiocam Mrm CCD camera. Images were acquired using Axiovision 4.8.2 software and analyzed using Photoshop and ImageJ software.
Stable isotope labeling In culture (SILAC) proteomics
WT H-2K cells were cultured in ‘heavy’ (or ‘labeled’) media in which Arg and Lys were replaced by 13C6-Arg and 13C6, 15N2-Lys (Cambridge Isotopes, Andover, MA, USA), whereas Lmna−/− H-2K cells were grown in ‘light’ (or ‘unlabeled’) media (Atlanta Biologicals, Lawrenceville, GA, USA) containing 12C6-Arg and 12C6, 14N2-Lys (Sigma-Aldrich). Three biological replicates (H-2K clones) were pooled for each condition. MBs were passaged three times to allow for sufficient incorporation of label. This approach has been found to produce >98% incorporation (72). At this point, MBs were plated onto Matrigel-covered dishes and cultured for 3 days in growth medium without IFNγ before harvesting.
Whole cell lysates were prepared using RIPA buffer containing anti-proteases (Mini-Complete, Roche). Protein concentration of each sample was determined using the Bradford assay (Bio-Rad). Fifty microgram ‘labeled’ protein was mixed with 50 μg ‘unlabeled’ protein and separated on a 4–12% NuPAGE gel (Invitrogen). Gels were stained with Bio-Safe Coomassie (Bio-Rad, Hercules, CA, USA) and each lane was sliced into 30 pieces, followed by digestion with trypsin (Promega, Madison, WI, USA) as previously described (73). Resultant peptides from each band were dried by vacuum centrifugation and resuspended in 6 µl of 0.1% TFA for analysis by mass spectrometry. LC-MS/MS was performed on a hybrid Thermo LTQ-Orbitrap-XL mass spectrometer (Thermo Fisher Scientific, USA) coupled online to a nano-LC system (Eksigent, Dublin, CA, USA) (73). Proteins were identified from raw MS and MS/MS data using the Sequest algorithm in the Bioworks Browser 3.3.1 software (Thermo Fisher Scientific) against the Uniprot mouse database (UniProt release-2010_11, 16,333 entries) indexed for fully tryptic, 300–4000 mass range, and two missed cleavages.
Database analysis
Protein annotations were acquired using the Keyword and GO terms in the Uniprot Protein Knowledgebase (http://www.uniprot.org/). The molecular mass values and percentage of the protein covered by the matched peptides were retrieved from Bioworks output files. Known contaminant proteins from processing or serum (e.g. keratin, albumin, hemoglobin) were excluded from the analysis. Biological function analysis was performed using UniProt Gene Ontology, DAVID, IPA and STRING 9.0.
Western blotting
Cultured primary MBs were maintained in proliferation or differentiation media for indicated number of days. Nuclear and cytoplasmic fractions were prepared using the NE-PER kit (Pierce) according to manufacturer's instructions. Lysates (10 μg) were loaded onto 4–12% NuPage Bis-Tris gels (Invitrogen) and transferred onto PVDF membranes. Following blocking in 5% Blocking Grade Reagent (Bio-Rad), membranes were probed overnight with antibodies to Smad2/3 (Cell Signaling), lamin A (Abcam), vinculin (Sigma), actin (Santa Cruz) and pRb (Santa Cruz). The Lap2α monoclonal antibody was previously described (26). Appropriate secondary antibodies were applied for 30 min and following washes, membranes were developed using ECL reagent (Pierce) and processed on a BioMax film (Sigma).
RNA isolation and real-time analysis
Total RNA was isolated from satellite cells using the RNeasy Mini kit (Qiagen). First-strand synthesis was performed using the First Strand cDNA Synthesis kit (Roche) according to manufacturer's instructions. A qRT–PCR was performed on an ABI 7900HT Fast Real-Time PCR system using an SYBR® Green PCR master mix (Applied Biosystems) and indicated PCR primers (Supplementary Material, M&M Primers). Raw CT values were normalized to S18, HPRT and GAPDH and relative gene expression was obtained using the CT method (74).
Statistics
Student's t-test was used in all statistical analyses unless indicated otherwise.
SUPPLEMENTARY METHODS
BrdU dilution assay
The thymidine analog 5-bromo-2′-deoxyuridine (BrdU; Sigma) was injected subcutaneously (250 μg/g body weight) into newborn mice during the first 7 postnatal days. Animals were euthanized and quadriceps muscle was harvested on indicated postnatal days. Frozen sections (10 μm) were fixed in ice-cold acetone for 10 min, denatured in 2 N HCl for 1 h, and incubated with 0.15 m sodium borate solution to neutralize before primary antibody incubation.
For BrdU staining, sections were incubated with biotinylated anti-BrdU (MoBU-1, Life Technologies) and anti-laminin (Sigma) antibodies. The following day, sections were incubated for 1 h with FITC-conjugated anti-streptavidin, Marina Blue-conjugated goat anti-rabbit antibody (Life Technologies) and propidium iodide (Sigma).
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
FUNDING
This work was supported by the Muscular Dystrophy Association to T.V.C. and T.A.P. and partially supported by NIH core grants: NIH 2R24HD050846-06 (National Center for Medical Rehabilitation Research), IDDRC 5P30HD040677-10 (Intellectual and Developmental Disabilities Research Center) and by NIH UL1RR031988 (GWU-CNMC CTSI), and by Austrian Science Research Fund (FWF P22403-B12) to R.F. V.F.G. was previously supported by The Medical Research Council project grant G0700307 (http://www.mrc.ac.uk/) awarded to P.S.Z., J.A.E. and Jennifer Morgan.
Supplementary Material
REFERENCES
- 1.Burke B., Mounkes L.C., Stewart C.L. The nuclear envelope in muscular dystrophy and cardiovascular diseases. Traffic. 2001;2:675–683. doi: 10.1034/j.1600-0854.2001.21001.x. doi:10.1034/j.1600-0854.2001.21001.x. [DOI] [PubMed] [Google Scholar]
- 2.Scharner J., Gnocchi V.F., Ellis J.A., Zammit P.S. Genotype–phenotype correlations in laminopathies: how does fate translate? Biochem. Soc. Trans. 2010;38:257–262. doi: 10.1042/BST0380257. doi:10.1042/BST0380257. [DOI] [PubMed] [Google Scholar]
- 3.Cohen T.V., Hernandez L., Stewart C.L. Functions of the nuclear envelope and lamina in development and disease. Biochem. Soc. Trans. 2008;36:1329–1334. doi: 10.1042/BST0361329. doi:10.1042/BST0361329. [DOI] [PubMed] [Google Scholar]
- 4.Muchir A., Bonne G., van der Kooi A.J., van Meegen M., Baas F., Bolhuis P.A., de Visser M., Schwartz K. Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B) Hum. Mol. Genet. 2000;9:1453–1459. doi: 10.1093/hmg/9.9.1453. doi:10.1093/hmg/9.9.1453. [DOI] [PubMed] [Google Scholar]
- 5.Bonne G., Di Barletta M.R., Varnous S., Becane H.M., Hammouda E.H., Merlini L., Muntoni F., Greenberg C.R., Gary F., Urtizberea J.A., et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery–Dreifuss muscular dystrophy. Nat. Genet. 1999;21:285–288. doi: 10.1038/6799. doi:10.1038/6799. [DOI] [PubMed] [Google Scholar]
- 6.Quijano-Roy S., Mbieleu B., Bonnemann C.G., Jeannet P.Y., Colomer J., Clarke N.F., Cuisset J.M., Roper H., De Meirleir L., D'Amico A., et al. De novo LMNA mutations cause a new form of congenital muscular dystrophy. Ann. Neurol. 2008;64:177–186. doi: 10.1002/ana.21417. doi:10.1002/ana.21417. [DOI] [PubMed] [Google Scholar]
- 7.Fatkin D., MacRae C., Sasaki T., Wolff M.R., Porcu M., Frenneaux M., Atherton J., Vidaillet H.J., Jr, Spudich S., De Girolami U., et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N. Engl. J. Med. 1999;341:1715–1724. doi: 10.1056/NEJM199912023412302. doi:10.1056/NEJM199912023412302. [DOI] [PubMed] [Google Scholar]
- 8.van Engelen B.G., Muchir A., Hutchison C.J., van der Kooi A.J., Bonne G., Lammens M. The lethal phenotype of a homozygous nonsense mutation in the lamin A/C gene. Neurology. 2005;64:374–376. doi: 10.1212/01.WNL.0000149763.15180.00. doi:10.1212/01.WNL.0000149763.15180.00. [DOI] [PubMed] [Google Scholar]
- 9.Zastrow M.S., Vlcek S., Wilson K.L. Proteins that bind A-type lamins: integrating isolated clues. J. Cell Sci. 2004;117:979–987. doi: 10.1242/jcs.01102. doi:10.1242/jcs.01102. [DOI] [PubMed] [Google Scholar]
- 10.Stewart C.L., Kozlov S., Fong L.G., Young S.G. Mouse models of the laminopathies. Exp. Cell Res. 2007;313:2144–2156. doi: 10.1016/j.yexcr.2007.03.026. doi:10.1016/j.yexcr.2007.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cohen T.V., Stewart C.L. Fraying at the edge mouse models of diseases resulting from defects at the nuclear periphery. Curr Topics Developmental Biology. 2008;84:351–384. doi: 10.1016/S0070-2153(08)00607-8. doi:10.1016/S0070-2153(08)00607-8. [DOI] [PubMed] [Google Scholar]
- 12.Sullivan T., Escalante-Alcalde D., Bhatt H., Anver M., Bhat N., Nagashima K., Stewart C.L., Burke B. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J. Cell Biol. 1999;147:913–920. doi: 10.1083/jcb.147.5.913. doi:10.1083/jcb.147.5.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gnocchi V.F., Scharner J., Huang Z., Brady K., Lee J.S., White R.B., Morgan J.E., Sun Y.B., Ellis J.A., Zammit P.S. Uncoordinated transcription and compromised muscle function in the lmna-null mouse model of Emery–Dreyfuss muscular dystrophy. PLoS One. 2011;6:e16651. doi: 10.1371/journal.pone.0016651. doi:10.1371/journal.pone.0016651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wolf C.M., Wang L., Alcalai R., Pizard A., Burgon P.G., Ahmad F., Sherwood M., Branco D.M., Wakimoto H., Fishman G.I., et al. Lamin A/C haploinsufficiency causes dilated cardiomyopathy and apoptosis-triggered cardiac conduction system disease. J. Mol. Cell Cardiol. 2008;44:293–303. doi: 10.1016/j.yjmcc.2007.11.008. doi:10.1016/j.yjmcc.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mounkes L.C., Burke B., Stewart C.L. The A-type lamins: nuclear structural proteins as a focus for muscular dystrophy and cardiovascular diseases. Trends Cardiovasc. Med. 2001;11:280–285. doi: 10.1016/s1050-1738(01)00126-8. doi:10.1016/S1050-1738(01)00126-8. [DOI] [PubMed] [Google Scholar]
- 16.Arimura T., Helbling-Leclerc A., Massart C., Varnous S., Niel F., Lacene E., Fromes Y., Toussaint M., Mura A.M., Keller D.I., et al. Mouse model carrying H222P-Lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Hum. Mol. Genet. 2005;14:155–169. doi: 10.1093/hmg/ddi017. doi:10.1093/hmg/ddi017. [DOI] [PubMed] [Google Scholar]
- 17.Relaix F., Zammit P.S. Satellite cells are essential for skeletal muscle regeneration: the cell on the edge returns centre stage. Development. 2012;139:2845–2856. doi: 10.1242/dev.069088. doi:10.1242/dev.069088. [DOI] [PubMed] [Google Scholar]
- 18.Scharner J., Zammit P.S. The muscle satellite cell at 50: the formative years. Skelet. Muscle. 2011;1:28. doi: 10.1186/2044-5040-1-28. doi:10.1186/2044-5040-1-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ono Y., Calhabeu F., Morgan J.E., Katagiri T., Amthor H., Zammit P.S. BMP signalling permits population expansion by preventing premature myogenic differentiation in muscle satellite cells. Cell Death Differ. 2011;18:222–234. doi: 10.1038/cdd.2010.95. doi:10.1038/cdd.2010.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Berlo J.H., Voncken J.W., Kubben N., Broers J.L., Duisters R., van Leeuwen R.E., Crijns H.J., Ramaekers F.C., Hutchison C.J., Pinto Y.M. A-type lamins are essential for TGF-beta1 induced PP2A to dephosphorylate transcription factors. Hum. Mol. Genet. 2005;14:2839–2849. doi: 10.1093/hmg/ddi316. doi:10.1093/hmg/ddi316. [DOI] [PubMed] [Google Scholar]
- 21.Zi Z., Chapnick D.A., Liu X. Dynamics of TGF-beta/Smad signaling. FEBS Lett. 2012;586:1921–1928. doi: 10.1016/j.febslet.2012.03.063. doi:10.1016/j.febslet.2012.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Langley B., Thomas M., Bishop A., Sharma M., Gilmour S., Kambadur R. Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J. Biol. Chem. 2002;277:49831–49840. doi: 10.1074/jbc.M204291200. doi:10.1074/jbc.M204291200. [DOI] [PubMed] [Google Scholar]
- 23.Ge X., McFarlane C., Vajjala A., Lokireddy S., Ng Z.H., Tan C.K., Tan N.S., Wahli W., Sharma M., Kambadur R. Smad3 signaling is required for satellite cell function and myogenic differentiation of myoblasts. Cell Res. 2012;21:1591–1604. doi: 10.1038/cr.2011.72. doi:10.1038/cr.2011.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han D., Zhao H., Parada C., Hacia J.G., Bringas P., Jr, Chai Y. A TGFbeta-Smad4-Fgf6 signaling cascade controls myogenic differentiation and myoblast fusion during tongue development. Development. 2012;139:1640–1650. doi: 10.1242/dev.076653. doi:10.1242/dev.076653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taylor M.R., Slavov D., Gajewski A., Vlcek S., Ku L., Fain P.R., Carniel E., Di Lenarda A., Sinagra G., Boucek M.M., et al. Thymopoietin (lamina-associated polypeptide 2) gene mutation associated with dilated cardiomyopathy. Hum. Mutat. 2005;26:566–574. doi: 10.1002/humu.20250. doi:10.1002/humu.20250. [DOI] [PubMed] [Google Scholar]
- 26.Naetar N., Korbei B., Kozlov S., Kerenyi M.A., Dorner D., Kral R., Gotic I., Fuchs P., Cohen T.V., Bittner R., et al. Loss of nucleoplasmic LAP2alpha-lamin A complexes causes erythroid and epidermal progenitor hyperproliferation. Nat. Cell Biol. 2008;10:1341–1348. doi: 10.1038/ncb1793. doi:10.1038/ncb1793. [DOI] [PubMed] [Google Scholar]
- 27.Gotic I., Schmidt W.M., Biadasiewicz K., Leschnik M., Spilka R., Braun J., Stewart C.L., Foisner R. Loss of LAP2 alpha delays satellite cell differentiation and affects postnatal fiber-type determination. Stem Cells. 2010;28:480–488. doi: 10.1002/stem.292. doi:10.1002/stem.292. [DOI] [PubMed] [Google Scholar]
- 28.White R.B., Bierinx A.S., Gnocchi V.F., Zammit P.S. Dynamics of muscle fibre growth during postnatal mouse development. BMC Dev. Biol. 2010;10:21. doi: 10.1186/1471-213X-10-21. doi:10.1186/1471-213X-10-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morgan J.E., Beauchamp J.R., Pagel C.N., Peckham M., Ataliotis P., Jat P.S., Noble M.D., Farmer K., Partridge T.A. Myogenic cell lines derived from transgenic mice carrying a thermolabile T antigen: a model system for the derivation of tissue-specific and mutation-specific cell lines. Dev. Biol. 1994;162:486–498. doi: 10.1006/dbio.1994.1103. doi:10.1006/dbio.1994.1103. [DOI] [PubMed] [Google Scholar]
- 30.Zammit P.S., Heslop L., Hudon V., Rosenblatt J.D., Tajbakhsh S., Buckingham M.E., Beauchamp J.R., Partridge T.A. Kinetics of myoblast proliferation show that resident satellite cells are competent to fully regenerate skeletal muscle fibers. Exp. Cell Res. 2002;281:39–49. doi: 10.1006/excr.2002.5653. doi:10.1006/excr.2002.5653. [DOI] [PubMed] [Google Scholar]
- 31.Gnocchi V.F., White R.B., Ono Y., Ellis J.A., Zammit P.S. Further characterisation of the molecular signature of quiescent and activated mouse muscle satellite cells. PLoS One. 2009;4:e5205. doi: 10.1371/journal.pone.0005205. doi:10.1371/journal.pone.0005205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zammit P.S., Golding J.P., Nagata Y., Hudon V., Partridge T.A., Beauchamp J.R. Muscle satellite cells adopt divergent fates: a mechanism for self-renewal? J. Cell Biol. 2004;166:347–357. doi: 10.1083/jcb.200312007. doi:10.1083/jcb.200312007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Melcon G., Kozlov S., Cutler D.A., Sullivan T., Hernandez L., Zhao P., Mitchell S., Nader G., Bakay M., Rottman J.N., et al. Loss of emerin at the nuclear envelope disrupts the Rb1/E2F and MyoD pathways during muscle regeneration. Hum. Mol. Genet. 2006;15:637–651. doi: 10.1093/hmg/ddi479. doi:10.1093/hmg/ddi479. [DOI] [PubMed] [Google Scholar]
- 34.Busch A., Kiel T., Heupel W.M., Wehnert M., Hubner S. Nuclear protein import is reduced in cells expressing nuclear envelopathy-causing lamin A mutants. Exp. Cell Res. 2009;315:2373–2385. doi: 10.1016/j.yexcr.2009.05.003. doi:10.1016/j.yexcr.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 35.Kollias H.D., McDermott J.C. Transforming growth factor-beta and myostatin signaling in skeletal muscle. J. Appl. Physiol. 2008;104:579–587. doi: 10.1152/japplphysiol.01091.2007. doi:10.1152/japplphysiol.01091.2007. [DOI] [PubMed] [Google Scholar]
- 36.Vaidya T.B., Rhodes S.J., Taparowsky E.J., Konieczny S.F. Fibroblast growth factor and transforming growth factor beta repress transcription of the myogenic regulatory gene MyoD1. Mol. Cell Biol. 1989;9:3576–3579. doi: 10.1128/mcb.9.8.3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martin J.F., Li L., Olson E.N. Repression of myogenin function by TGF-beta 1 is targeted at the basic helix–loop–helix motif and is independent of E2A products. J. Biol. Chem. 1992;267:10956–10960. [PubMed] [Google Scholar]
- 38.Liu D., Kang J.S., Derynck R. TGF-beta-activated Smad3 represses MEF2-dependent transcription in myogenic differentiation. EMBO J. 2004;23:1557–1566. doi: 10.1038/sj.emboj.7600179. doi:10.1038/sj.emboj.7600179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu D., Black B.L., Derynck R. TGF-beta inhibits muscle differentiation through functional repression of mitogenic transcription factors by Smad3. Genes Dev. 2001;15:2950–2966. doi: 10.1101/gad.925901. doi:10.1101/gad.925901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sewry C.A., Brown S.C., Mercuri E., Bonne G., Feng L., Camici G., Morris G.E., Muntoni F. Skeletal muscle pathology in autosomal dominant Emery–Dreifuss muscular dystrophy with lamin A/C mutations. Neuropathol. Appl. Neurobiol. 2001;27:281–290. doi: 10.1046/j.0305-1846.2001.00323.x. doi:10.1046/j.0305-1846.2001.00323.x. [DOI] [PubMed] [Google Scholar]
- 41.Pekovic V., Harborth J., Broers J.L., Ramaekers F.C., van Engelen B., Lammens M., von Zglinicki T., Foisner R., Hutchison C., Markiewicz E. Nucleoplasmic LAP2alpha-lamin A complexes are required to maintain a proliferative state in human fibroblasts. J. Cell Biol. 2007;176:163–172. doi: 10.1083/jcb.200606139. doi:10.1083/jcb.200606139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mancini M.A., Shan B., Nickerson J.A., Penman S., Lee W.H. The retinoblastoma gene product is a cell cycle-dependent, nuclear matrix-associated protein. Proc. Natl. Acad. Sci. USA. 1994;91:418–422. doi: 10.1073/pnas.91.1.418. doi:10.1073/pnas.91.1.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ozaki T., Saijo M., Murakami K., Enomoto H., Taya Y., Sakiyama S. Complex formation between lamin A and the retinoblastoma gene product: identification of the domain on lamin A required for its interaction. Oncogene. 1994;9:2649–2653. [PubMed] [Google Scholar]
- 44.Stewart C.L., Roux K.J., Burke B. Blurring the boundary: the nuclear envelope extends its reach. Science. 2007;318:1408–1412. doi: 10.1126/science.1142034. doi:10.1126/science.1142034. [DOI] [PubMed] [Google Scholar]
- 45.Grimsby S., Jaensson H., Dubrovska A., Lomnytska M., Hellman U., Souchelnytskyi S. Proteomics-based identification of proteins interacting with Smad3: SREBP-2 forms a complex with Smad3 and inhibits its transcriptional activity. FEBS Lett. 2004;577:93–100. doi: 10.1016/j.febslet.2004.09.069. doi:10.1016/j.febslet.2004.09.069. [DOI] [PubMed] [Google Scholar]
- 46.Lloyd D.J., Trembath R.C., Shackleton S. A novel interaction between lamin A and SREBP1: implications for partial lipodystrophy and other laminopathies. Hum. Mol. Genet. 2002;11:769–777. doi: 10.1093/hmg/11.7.769. doi:10.1093/hmg/11.7.769. [DOI] [PubMed] [Google Scholar]
- 47.Wilson K.L., Foisner R. Lamin-binding proteins. Cold Spring Harb. Perspect. Biol. 2010;2:a000554. doi: 10.1101/cshperspect.a000554. doi:10.1101/cshperspect.a000554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Muchir A., van Engelen B.G., Lammens M., Mislow J.M., McNally E., Schwartz K., Bonne G. Nuclear envelope alterations in fibroblasts from LGMD1B patients carrying nonsense Y259X heterozygous or homozygous mutation in lamin A/C gene. Exp. Cell Res. 2003;291:352–362. doi: 10.1016/j.yexcr.2003.07.002. doi:10.1016/j.yexcr.2003.07.002. [DOI] [PubMed] [Google Scholar]
- 49.Goldman R.D., Shumaker D.K., Erdos M.R., Eriksson M., Goldman A.E., Gordon L.B., Gruenbaum Y., Khuon S., Mendez M., Varga R., et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA. 2004;101:8963–8968. doi: 10.1073/pnas.0402943101. doi:10.1073/pnas.0402943101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hernandez L., Roux K.J., Wong E.S., Mounkes L.C., Mutalif R., Navasankari R., Rai B., Cool S., Jeong J.W., Wang H., et al. Functional coupling between the extracellular matrix and nuclear lamina by Wnt signaling in progeria. Dev. Cell. 2012;19:413–425. doi: 10.1016/j.devcel.2010.08.013. doi:10.1016/j.devcel.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frock R.L., Kudlow B.A., Evans A.M., Jameson S.A., Hauschka S.D., Kennedy B.K. Lamin A/C and emerin are critical for skeletal muscle satellite cell differentiation. Genes Dev. 2006;20:486–500. doi: 10.1101/gad.1364906. doi:10.1101/gad.1364906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mann G.J., Musgrove E.A., Fox R.M., Thelander L. Ribonucleotide reductase M1 subunit in cellular proliferation, quiescence, and differentiation. Cancer Res. 1988;48:5151–5156. [PubMed] [Google Scholar]
- 53.Yang X., Long L., Reynolds P.N., Morrell N.W. Expression of mutant BMPR-II in pulmonary endothelial cells promotes apoptosis and a release of factors that stimulate proliferation of pulmonary arterial smooth muscle cells. Pulm. Circ. 2009;1:103–110. doi: 10.4103/2045-8932.78100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Furumatsu T., Tsuda M., Taniguchi N., Tajima Y., Asahara H. Smad3 induces chondrogenesis through the activation of SOX9 via CREB-binding protein/p300 recruitment. J. Biol. Chem. 2005;280:8343–8350. doi: 10.1074/jbc.M413913200. doi:10.1074/jbc.M413913200. [DOI] [PubMed] [Google Scholar]
- 55.Scheijen B., Bronk M., van der Meer T., Bernards R. Constitutive E2F1 overexpression delays endochondral bone formation by inhibiting chondrocyte differentiation. Mol. Cell Biol. 2003;23:3656–3668. doi: 10.1128/MCB.23.10.3656-3668.2003. doi:10.1128/MCB.23.10.3656-3668.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schabort E.J., van der Merwe M., Loos B., Moore F.P., Niesler C.U. TGF-beta's delay skeletal muscle progenitor cell differentiation in an isoform-independent manner. Exp. Cell Res. 2009;315:373–384. doi: 10.1016/j.yexcr.2008.10.037. doi:10.1016/j.yexcr.2008.10.037. [DOI] [PubMed] [Google Scholar]
- 57.Long K.K., Montano M., Pavlath G.K. Sca-1 is negatively regulated by TGF-beta1 in myogenic cells. FASEB J. 2011;25:1156–1165. doi: 10.1096/fj.10-170308. doi:10.1096/fj.10-170308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mu X., Li Y. Conditional TGF-beta1 treatment increases stem cell-like cell population in myoblasts. J. Cell Mol. Med. 2011;15:679–690. doi: 10.1111/j.1582-4934.2010.01042.x. doi:10.1111/j.1582-4934.2010.01042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cohen T.V., Kosti O., Stewart C.L. The nuclear envelope protein MAN1 regulates TGFbeta signaling and vasculogenesis in the embryonic yolk sac. Development. 2007;134:1385–1395. doi: 10.1242/dev.02816. doi:10.1242/dev.02816. [DOI] [PubMed] [Google Scholar]
- 60.Bengtsson L. What MAN1 does to the Smads. TGFbeta/BMP signaling and the nuclear envelope. FEBS J. 2007;274:1374–1382. doi: 10.1111/j.1742-4658.2007.05696.x. doi:10.1111/j.1742-4658.2007.05696.x. [DOI] [PubMed] [Google Scholar]
- 61.Mejat A., Decostre V., Li J., Renou L., Kesari A., Hantai D., Stewart C.L., Xiao X., Hoffman E., Bonne G., et al. Lamin A/C-mediated neuromuscular junction defects in Emery–Dreifuss muscular dystrophy. J. Cell Biol. 2009;184:31–44. doi: 10.1083/jcb.200811035. doi:10.1083/jcb.200811035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tong J., Li W., Vidal C., Yeo L.S., Fatkin D., Duque G. Lamin A/C deficiency is associated with fat infiltration of muscle and bone. Mech. Ageing Dev. 2011;132:552–559. doi: 10.1016/j.mad.2011.09.004. doi:10.1016/j.mad.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 63.De Sandre-Giovannoli A., Chaouch M., Kozlov S., Vallat J.M., Tazir M., Kassouri N., Szepetowski P., Hammadouche T., Vandenberghe A., Stewart C.L., et al. Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2) and mouse. Am. J. Hum. Genet. 2002;70:726–736. doi: 10.1086/339274. doi:10.1086/339274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li W., Yeo L.S., Vidal C., McCorquodale T., Herrmann M., Fatkin D., Duque G. Decreased bone formation and osteopenia in lamin a/c-deficient mice. PLoS One. 2011;6:e19313. doi: 10.1371/journal.pone.0019313. doi:10.1371/journal.pone.0019313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kubben N., Voncken J.W., Konings G., van Weeghel M., van den Hoogenhof M.M., Gijbels M., van Erk A., Schoonderwoerd K., van den Bosch B., Dahlmans V., et al. Post-natal myogenic and adipogenic developmental: defects and metabolic impairment upon loss of A-type lamins. Nucleus. 2011;2:195–207. doi: 10.4161/nucl.2.3.15731. doi:10.4161/nucl.2.3.15731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Frock R.L., Chen S.C., Da D.F., Frett E., Lau C., Brown C., Pak D.N., Wang Y., Muchir A., Worman H.J., et al. Cardiomyocyte-specific expression of lamin a improves cardiac function in lmna(-/-) mice. PLoS One. 2012;7:e42918. doi: 10.1371/journal.pone.0042918. doi:10.1371/journal.pone.0042918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen C.Y., Chi Y.H., Mutalif R.A., Starost M.F., Myers T.G., Anderson S.A., Stewart C.L., Jeang K.T. Accumulation of the inner nuclear envelope protein Sun1 is pathogenic in progeric and dystrophic laminopathies. Cell. 2012;149:565–577. doi: 10.1016/j.cell.2012.01.059. doi:10.1016/j.cell.2012.01.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rosenblatt J.D., Lunt A.I., Parry D.J., Partridge T.A. Culturing satellite cells from living single muscle fiber explants. In Vitro Cell Dev. Biol. Anim. 1995;31:773–779. doi: 10.1007/BF02634119. doi:10.1007/BF02634119. [DOI] [PubMed] [Google Scholar]
- 69.Ono Y., Boldrin L., Knopp P., Morgan J.E., Zammit P.S. Muscle satellite cells are a functionally heterogeneous population in both somite-derived and branchiomeric muscles. Dev. Biol. 2010;337:29–41. doi: 10.1016/j.ydbio.2009.10.005. doi:10.1016/j.ydbio.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. doi:10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Briguet A., Courdier-Fruh I., Foster M., Meier T., Magyar J.P. Histological parameters for the quantitative assessment of muscular dystrophy in the mdx-mouse. Neuromuscul. Disord. 2004;14:675–682. doi: 10.1016/j.nmd.2004.06.008. doi:10.1016/j.nmd.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 72.Formolo C.A., Williams R., Gordish-Dressman H., MacDonald T.J., Lee N.H., Hathout Y. Secretome signature of invasive glioblastoma multiforme. J. Proteome Res. 2011;10:3149–3159. doi: 10.1021/pr200210w. doi:10.1021/pr200210w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sharma N., Medikayala S., Defour A., Rayavarapu S., Brown K.J., Hathout Y., Jaiswal J.K. Use of quantitative membrane proteomics identifies a novel role of mitochondria in healing injured muscles. J. Biol. Chem. 2010;287:30455–30467. doi: 10.1074/jbc.M112.354415. doi:10.1074/jbc.M112.354415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pfaffl M.W. A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. doi:10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.