

Abstract
Objectives
Infection with yellow fever virus (YFV), the prototypic mosquito-borne flavivirus, causes severe febrile disease with haemorrhage, multi-organ failure and a high mortality. Moreover, in recent years the Flavivirus genus has gained further attention due to re-emergence and increasing incidence of West Nile, dengue and Japanese encephalitis viruses. Potent and safe antivirals are urgently needed.
Methods
Starting from the crystal structure of the NS3 helicase from Kunjin virus (an Australian variant of West Nile virus), we identified a novel, unexploited protein site that might be involved in the helicase catalytic cycle and could thus in principle be targeted for enzyme inhibition. In silico docking of a library of small molecules allowed us to identify a few selected compounds with high predicted affinity for the new site. Their activity against helicases from several flaviviruses was confirmed in in vitro helicase/enzymatic assays. The effect on the in vitro replication of flaviviruses was then evaluated.
Results
Ivermectin, a broadly used anti-helminthic drug, proved to be a highly potent inhibitor of YFV replication (EC50 values in the sub-nanomolar range). Moreover, ivermectin inhibited, although less efficiently, the replication of several other flaviviruses, i.e. dengue fever, Japanese encephalitis and tick-borne encephalitis viruses. Ivermectin exerts its effect at a timepoint that coincides with the onset of intracellular viral RNA synthesis, as expected for a molecule that specifically targets the viral helicase.
Conclusions
The well-tolerated drug ivermectin may hold great potential for treatment of YFV infections. Furthermore, structure-based optimization may result in analogues exerting potent activity against flaviviruses other than YFV.
Keywords: antiviral drug discovery, flavivirus helicase inhibition, new use of existing drug, in silico docking, structure-based drug design
Introduction
The genus Flavivirus comprises small single-stranded RNA viruses within the Flaviviridae family. The flavivirus group includes several pathogens of global medical importance, namely (i) haemorrhagic fever viruses such as yellow fever virus (YFV) and dengue viruses (DENV), and (ii) encephalitic viruses such as West Nile virus (WNV), Japanese encephalitis (JEV) and tick-borne encephalitis viruses (TBEV).1 Infections by either of the last two neurotropic viruses may result in life-threatening aseptic encephalitis, with a high risk of life-long debilitating neurological sequelae.
YFV is the type member of the emerging and re-emerging vector-borne flaviviruses. Infections with YFV cause a severe febrile disease with haemorrhage, multi-organ failure and shock, and an exceedingly high mortality (up to 50% of cases).2,3 YFV is a zoonotic agent that, even with the availability of a safe and efficient vaccine, continues to be reintroduced from sylvatic animal reservoirs into the human population, causing outbreaks in endemic regions of South America and Africa on a regular, yet poorly predictable, basis with an estimated annual number of cases of >200 000.4,5 Moreover, recent increases in the density and distribution of the urban mosquito vector, Aedes aegypti, as well as the rise in global air travel, increase the risk of introduction and spread of YFV to North and Central America, the Caribbean and Asia.2 A major gap in our knowledge about YFV is how to manage and treat patients. Treatment of YFV by supportive care is essentially ineffective, and even improvements in intensive care have not changed the lethality.3 Likewise, the four DENV serotypes have considerably expanded their geographic distribution in recent years. With billions of people at risk, more than 50 million cases, and ∼12 500–25 000 deaths annually, DENV is considered an emerging pathogen in a growing number of countries.6 In particular, the presence of four DENV serotypes has complicated the design of vaccines because incomplete protection against one serotype may influence the disease outcome once infection is established by a different serotype, through a process referred to as antibody-mediated disease enhancement.7
Annually there are ∼30 000–50 000 cases of JEV reported in Asia. Case-fatality rates range from 0.3% to 60%. In Russia and Europe, TBEV causes ∼10 000–12 000 human cases, which may present with severe clinical presentations and a significant rate (10%–20%) of long-lasting neurological sequelae in survivors. WNV was detected for the first time in the Americas in 1999; the virus is now endemic on this continent.5,8 There is thus an urgent need for antiviral drugs to treat life-threatening infections with flaviviruses.
During the flavivirus replication cycle, the viral genome is transcribed into a negative-strand RNA, which in turn is used as a template for the synthesis of new viral genomic RNA. To maintain viral replication the nascent transcripts must be unwound from their complementary template RNAs by an ATP-dependent helicase activity. In flaviviruses this activity is provided by the C-terminal domain of non-structural (NS) protein 3 (NS3 helicase domain).9–11 Since all flaviviral NS proteins are indispensable for viral replication, any of them may be considered a promising target for selective inhibitors of viral replication for therapeutic intervention.12 Moreover, in addition to the commonly targeted viral polymerase and protease functions, the feasibility of interfering with viral replication by particularly inhibiting RNA helicase activity has been a successful proof of concept for hepatitis C virus (HCV)13 and picornaviruses.14 For flaviviruses, a number of nucleoside 5′ triphosphates (TPs), such as ribavirin-TP15–17 (1-β-d-ribofuranosyl-1,2,4-triazole-3-carboxamide-5′-triphosphate), IDA-TP (4,6-diamino-8-imino-8H-1-β-d-ribofuranosylimidazo [4,5-e] [1,3]diazepine-5′-triphosphate) and ITA-TP18 (5,8-dioxo-5,6,7,8-tetrahydro-4H-1-β-d-ribofuranosylimidazo[4,5-e][1,2,4]triazepine-5′-triphosphate) have been reported as weak inhibitors of the ATPase activity of helicases from HCV, JEV, DENV and WNV.16,18,19 However, since ATP is a key nucleotide of host cell metabolism, ATP-mimetic molecules may result in adverse effects on the host cell. A more innovative helicase inhibitor development strategy (as demonstrated to some extent for HCV20) might be directed specifically against the RNA binding and unwinding mechanisms mediated by NS3, which have recently been unravelled in fine molecular detail.11,21
Based on the above considerations, we performed an in silico docking search targeting a selected region of the ssRNA access site in the crystal structure of the NS3 helicase domain21,22 of Kunjin virus (an Australian variant of WNV, to which we refer as WNV23), using a library of mostly commercial small molecules. We identified the widely used anti-helminthic drug ivermectin as a molecule that not only displayed a high predicted binding affinity towards the modelled NS3 ssRNA binding pocket, but also inhibited the NS3 helicase activity of several flaviviruses in vitro at sub-micromolar concentrations. Most importantly, ivermectin proved to be a selective inhibitor of the replication of several flaviviruses in cell culture, such as JEV, TBEV and DENV (sub-micromolar EC50 values), and a highly potent inhibitor of YFV replication (sub-nanomolar EC50 values). Considering that this well tolerated drug has been licensed for >20years for the treatment of parasitic infections in man, our results provide the prospect of the first specific anti-flavivirus therapy by the off-label use of ivermectin (patent application EP2010/065880).
Materials and methods
Chemical database for virtual screening and reagents
The virtual Library of Pharmacologically Active Compounds (LOPAC) used for the docking analysis was accessed from Sigma-Aldrich, and included 1280 commercially available compounds (www.sigmaaldrich.com). The compounds tested in vitro, paromomycin sulphate, ouabain and ivermectin, were obtained from Sigma-Aldrich; ribavirin (Virazole) was purchased from ICN Pharmaceuticals (Costa Mesa, CA, USA). The compounds were dissolved at 20 mM in DMSO and stored at −20°C. The compounds were used as provided, without further purification.
In silico search for NS3 helicase inhibitors
The AutoDock4 software package24 was used for a docking search using compounds from the LOPAC library, and Python Molecule Viewer 1.4.5 (MGL-tools package, http://mgltools.scripps.edu/) for analysis of the data. The atomic coordinates from the crystal structure of the WNV helicase domain, solved in our laboratory, were chosen as the docking model (PDB 2QEQ21). After the addition of hydrogen atoms (PMV, MGL Tools package, http://mgltools.scripps.edu/), Kollman charges25 were added to the model. A discrete grid with dimensions 23 × 19 × 15 Å3 (AutoGrid4; step size 0.375 Å, 62 × 50 × 40 = 124 000 points26) was then centred on the putative ssRNA access site between helices α2 in subdomain II and α9 in subdomain III. Twenty genetic algorithm searches were run using AutoDock4 for each compound (provided with Gasteiger charges27) in the LOPAC library (with 150 individuals in the population and 27 000 generations24). The docking search produced a ranked list of compounds with predicted free energy of binding (ΔG) ranging between +9.0 and −11.5 kcal/mol. The best three molecules [paromomycin sulphate, ouabain and ivermectin (form B1a from Sigma-Aldrich, modelled with 11 rotatable bonds)], displaying ΔG values between −11.5 and −9.5 kcal/mol, were selected to be tested in in vitro activity assays.
Expression and purification of NS3 and NS5 domains
The WNV, DENV serotype 2 and YFV helicase domains were expressed and purified as previously described.28,29 DENV RNA-dependent RNA polymerase (RdRp), DENV full-length NS5, WNV RdRp and WNV full-length NS5 were produced in an Escherichia coli Rosetta (DE3) pRos expression system and purified through a Ni2+ column and gel filtration (Hi Load 16/60 Superdex 200, GE Healthcare) as described for the WNV helicase domain.28,29
Helicase inhibition assays using radioactive and fluorescent (FRET-based) labels
The helicase activity was assayed using radiolabelled dsRNA substrate in the presence of Mg2+ and ATP. The dsRNA substrate was prepared as described previously.30 Briefly, primer 1 (5′-CACCUCUCUAGAGUCGACCUGCAGGCAUCG-3′) was labelled with [γ-32P]ATP at its 5′ end using T4 polynucleotide kinase and annealed with the complementary primer 2 (5′-CGACUCUAGAGAGGUG-3′). WNV NS3 helicase (200 nM, see below) was preincubated with various concentrations of ivermectin (between 5 and 400 nM) in 50 mM Tris/HCl pH 8.0, 5 mM dithiothreitol, 10 mM KCl, 20 U/mL RiboLock Ribonuclease Inhibitor (Fermentas), 5 mM MnCl2 and 5 mM MgCl2. The reactions were initiated by adding the proteins to the reaction mixture containing 6 fmol of dsRNA, and were quenched after 30 min at 37°C by addition of 6 μL of loading dye (50 mM EDTA, 0.5% SDS, 50% glycerol and 0.1% bromophenol blue). The assay mixtures were resolved by electrophoresis through 17% polyacrylamide gels that were dried and analysed by phosphoimaging (Typhoon; GE Healthcare).
Fluorescence helicase assays were performed as described by Boguszewska-Chachulska et al.31 Briefly, the substrate for the fluorescent helicase test was prepared by annealing, at a 1:1.2 molar ratio, a Cy3-labelled 30-mer (5′-CACCUCUCUAGAGUCGACCUGCAGGCAUCG-3) to a Black Hole Quencher (BHQ)-labelled 16-mer (5′-CGACUCUAGAGAGGUG-3′), by brief heating to 90°C then slow cooling to room temperature. Standard helicase assays were performed in 50 mM Tris/HCl pH 7.5, 5 mM dithiothreitol, 10 mM KCl, 20 U/mL RiboLock Ribonuclease Inhibitor (Fermentas), 5 mM MnCl2 and 5 mM MgCl2 (buffer 1), 20 nM substrate, 1 mM ATP and 250 nM capture strand (5′-CGACUCUAGAGAGGUG-3′) in a reaction volume of 200 μL. The YFV, YFV-double mutant, DENV, DENV-double mutant and WNV, WNV-double mutant helicase domains (200 nM) were pre-incubated with various concentrations of ivermectin. The unwinding reaction was started by addition of helicases or ATP. The fluorescent signal increase was measured using a Fluorescence Reader (Infinite 200; Tecan). Relative fluorescence was calculated by subtracting the mean fluorescence of the blank (assay without protein) from all samples, and the values obtained were converted to percentage of activity.
The HCV helicase activity assay in the absence and presence of eight concentrations of ivermectin (0.78, 1.56, 3.13, 6.25, 12.5, 25, 50 and 100 μM) was performed as described previously.32 In brief, the reaction mixture [25 mM MOPS pH 6.5, 1.25 mM MgCl2, 5 nM Cy5-4-methylbenzhydrylamine hydrochloride (MBHA) substrate (DNA), 12.5 nM HCV NS3 helicase, 0.05 mM dithiothreitol, 0.01% Tween 20, 5 μg/mL BSA and 1 mM ATP] and ivermectin were added to a white half-volume 96-well microplate using EPmotion. Fluorescence was monitored using the VarioSkan. The reaction was incubated for 2 min before VarioSkan injection of ATP.
Helicase kinetics and inhibition by ivermectin
RNA unwinding was measured under the same experimental conditions as those described above, with dsRNA concentrations ranging from 0 to 100 nM, in the absence or presence of ivermectin at various concentrations. After blank subtraction (the same mixture in the absence of the enzyme), the curve was fitted linearly to get the rate of product formation. Different rates measured at varying substrate concentrations were plotted according to the Lineweaver–Burk equation, with amounts of dsRNA (substrate) ranging from 0 to 100 nM, to get VMAX and Km. The same experiment performed at increasing inhibitor concentrations yielded parallel lines in the double reciprocal plot, indicating that ivermectin behaves as an uncompetitive inhibitor. To estimate the inhibition constant (Ki) we used the Lineweaver–Burk equation for an uncompetitive inhibitor extrapolated for infinite substrate concentration:
Double mutation of YFV, DENV and WNV helicase domain
The double YFV (T413I and D414E), DENV helicase mutant (T408I and D409E) and WNV double mutant (T409I and D410E) were produced using the primer design software provided by the Agilent Technologies web site (www.agilent.com/genomics/qcpd). The primers for YFV were as follows: sense (5′-GGGACTTTGTCGTCACAATAGAGATATCTGAGATGGGA GCA-3′); antisense (5′-TGCTCCCATCTCAGATATCTCTATTGTGACGACAAAG TCCC-3′).
The primers for DENV were as follows: sense (5′-GGGACTTCGTGG TCACAATTGAGATTTCAGAAATGGGTGCC-3′); antisense (5′-GGCACCCATT TCTGAAATCTCAATTGTGACCACGAAGTCCC-3′).
The primers for WNV were as follows: sense (5′-GGGACTTTGTCGTCAC AATAGAGATATCTGAGATGGGAGCA-3′); antisense (5′-TGCTCCCATCTCAGAT ATCTCTATTGTGACGACAAAGTCCC-3′).
The proteins were expressed and purified as previously described.28,29
Biophysical characterization of the NS3–ivermectin interaction
Thermofluorimetric (thermal shift) assays for the evaluation of the YFV, WNV and DENV helicase domain melting temperatures (Tm) in the absence/presence of ivermectin were conducted in a MiniOpticon Real Time PCR Detection System (Bio-Rad), using the fluorescent dye Sypro orange. Solutions of 2.5 μL of the helicase domain (final protein concentrations ranged between 0.5 and 5 mg/mL) were diluted in 19 μL of buffer 1 and mixed with 3.5 μL of Sypro orange (Sigma) diluted 60× and 0.5 μL of 2 mM ivermectin. In control samples the inhibitor was replaced by DMSO. The sample plates were heated from 25 to 95°C with a heating rate of 0.2°C/min. Fluorescence intensities were measured within excitation and emission ranges of 470–505 and 540–700 nm, respectively. The experiments were repeated in the presence of 0.5 μL of 50 μM dsRNA, prepared by annealing at a 1:1 molar ratio of primer 1 (5′-CACCUCUCUAGAGUCGACCUGCAGGCAUCG-3′) and complementary primer 2 (5′-CGACUCUAGAGAGGUG-3′).
NS3 ATPase assay
The luciferase/luciferin-based ATP Detection Kit (Sigma–Aldrich) was prepared according to the manufacturer's instructions. Twenty microlitres of luciferase/luciferin was added to 185.5 μL of reaction buffer and the reactions were initiated by adding 10 μg of YFV, DENV or WNV helicase and 2.5 μL of ATP (100 mM). In parallel experiments, the YFV, DENV or WNV helicase domain was incubated with ivermectin to a final concentration of 1 μM at 30°C for 10 min. In control samples, the DENV or WNV helicase domain was omitted from the reaction mixture. All luminescence measurements were performed with a Cary Eclipse Fluorescence Spectrophotometer (Varian) at 25°C. Luminescence was measured continuously for 30 min (1 s reading time).
NS5 RdRp activity assay
In vitro RNA synthesis assays were performed using poly(C) (MP Biomedicals) as template annealed with oligo(G)12 as primer (62.5 nM final concentration) and GTP (100 mM final concentration) as substrate, in a 200 μL reaction mixture containing buffer 1, PicoGreen Quantitation Reagent (Molecular Probes) and 1 μg of DENV RdRp, DENV full-length NS5, WNV RdRp or WNV full-length NS5. Ivermectin was added to a final concentration of 1 μM. Standard assays were performed after enzyme/drug preincubation for 10 min at room temperature. Reactions were started by the addition of GTP and incubated for 15–20 min at 25°C following the fluorescence of the samples in a fluorescence reader (Cary Eclipse Fluorescence Spectrophotometer). Relative fluorescence was calculated by subtracting the mean fluorescence of the blank (assay without protein) from all samples.
Viruses and cells
YFV 17D vaccine strain (Stamaril) [Aventis Pasteur (MSD, Brussels, Belgium)] and DENV serotype 2 New Guinea were passaged once in Vero-B cells (ATCC CCL-81) to prepare a working virus stock and stored at −80°C until further use; JEV strain SA-14 (GenBank accession number U14163), TBEV strain Oshima 5-10 (GenBank accession number AB062063) and WNV strain NY99 (GenBank accession number NC_009942) were passaged once in Vero E6 cells (ATCC C1008) to prepare a working virus stock and stored at −80°C until further use.
Cytotoxic and cytostatic assays
Potential cytotoxic effects of the compounds were evaluated in uninfected quiescent Vero-B and Vero E6 cells. The cells were seeded at 5 × 104 cells/well in a 96-well plate (Becton Dickinson Labware, Franklin Lakes, NJ, USA) in the presence of 2-fold serial dilutions and incubated for 4 days. The culture medium was discarded and 100 μL of 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulphophenyl)-2H-tetrazolium/phenazinemethosulphate (MTS/PMS; Promega, Leiden, the Netherlands) in PBS was added to each well. Following a 2 h incubation at 37°C, the optical density was determined at 498 nm. The cytotoxic activity was calculated using the following formula: percentage host cell metabolism = 100 × (ODCompound/ODCC), where ODCompound and ODCC are the optical density at 498 nm of the uninfected cell cultures treated with the compound and of the uninfected, untreated cell cultures, respectively. The 50% cytotoxic concentration (i.e. the concentration that reduces the total cell number by 50%; CC50) was calculated by logarithmic interpolation.
Antiviral cell-based assays
YFV-17D cytopathic effect (CPE)-based assay
Vero-B cells were seeded at a density of 2 × 104 cells/well (96-well plate, Falcon) in 100 μL of assay medium and allowed to adhere overnight. Subsequently, a compound dilution series was prepared in the assay medium on top of the cells, after which 100 μL of assay medium containing 100 CCID50 (50% cell culture infectious dose) of virus was added. Plates were incubated for 6 days at 37°C (95%–99% relative humidity and 5% CO2). The cells were fixed with 70% ethanol and stained with 1% methylene blue. Pictures were taken at ×100 magnification. Ribavirin was included as a reference compound. The potential cytotoxic effect of the compounds was evaluated in uninfected cells in a parallel assay with the same experimental setup.
Virus yield reduction assays
DENV serotype 2 and YFV-17D Vero-B cells (5 × 104) were seeded in 96-well plates. One day later, culture medium was replaced with 100 μL of assay medium containing a 2× serial dilution of the compounds and 100 μL of virus inoculums (50 CCID50/μL). Following a 2 h incubation period the cell monolayer was washed three times with assay medium to remove non-adsorbed virus and cultures were further incubated for 4 days in the presence of the inhibitor. Supernatant was harvested and viral RNA load was determined by quantitative real-time PCR (qRT-PCR) as described previously.33
TBEV strain Oshima 5-10, JEV strain SA-14 and WNV strain NY99. Antiviral activity was assessed in Vero E6 cells grown in 24-well tissue culture plates. Viruses were used at a dilution of moi <1 (i.e. between 0.1 and 1). The compounds were used at a final concentration ranging between 0 and 5 μM. Pre-incubation in the presence of the antiviral was performed during the 90 min virus adsorption step. The cells were then washed once and incubated for 3 days in the presence of the antiviral compounds. All conditions were assayed in duplicate. Three days (72 h) post-infection, each corresponding cell culture supernatant was removed, clarified by centrifugation and stored at −80°C before analysis using a viral titre reduction assay.
The viral titre reduction assay was performed using BHK21 cells seeded in 96-well plates. When the cells reach 80% confluence, they were infected with 150 μL of 10-fold serial dilutions of the virus (derived from the Vero cell clarified supernatants) for 4 days before microscopic examination and positive cytopathic effect (CPE) well counting. For each supernatant sample, the infectivity titre was expressed as TCID50/mL using the Karber formula. For each virus, titres of viral supernatants were then compared and represented as the percentage of the positive control (viral titre from infected cell supernatant sample without antiviral compound) for the calculation of EC50 values. For WNV, the viral RNA load was also determined by real-time qRT-PCR as described previously.33
Time-of-drug-addition study
One day prior to infection, Vero-B cells were seeded in a 24-well Falcon plate (2 × 105 cells/well). Ivermectin (50 nM) was added at 0, 2, 4, 6, 8 and 24 h after YFV-17D infection (50 CCID50). In parallel experiments, ribavirin (400 μM) was used as a reference compound. Cells from each well were collected at 24 h after virus infection. For RNA extraction, at 24 h post-infection, cells were washed with PBS and lysed with lysis buffer (buffer RLT, RNeasy Mini Kit; Qiagen) to obtain cytoplasmic extracts. Cytoplasmic RNA was then extracted according to the manufacturer's instructions and analysed for the presence of viral RNA by RT-qPCR using a standard curve. Intracellular viral RNA production was monitored during one replication cycle in untreated cells. Confluent Vero-B cells in a 24-well plate were infected and incubated at 37°C for 1 h. After washing, 700 μL of assay medium was added and cells were harvested every 2 h. RNA replication was monitored by measuring intracellular RNA by RT-qPCR following reverse transcription. Time-of-addition studies using full-length dengue reporter virus expressing Renilla luciferase were performed as described previously.33
Results
Identification of ivermectin as putative flavivirus NS3 inhibitor by in silico docking
To identify potential NS3 helicase inhibitors, we chose to target the ssRNA access site of the enzyme in an in silico docking library search. Since the structure of DENV helicase in complex with RNA11 was not available when we initiated this work, we modelled the flaviviral NS3 helicase/RNA interaction by superposition of the structure of WNV NS3 helicase (solved in our laboratory, PDB 2QEQ21) and the DNA-bound bacterial helicase PcrA (PDB 3PJR34). This model was used to locate the putative ssRNA access site within the WNV NS3 helicase domain (Figure 1a). This region is located between helices α2 in subdomain II and α9 in subdomain III (forming an ‘α-helical gate’); this model-based identification was later confirmed by the crystal structure of DENV in complex with ssRNA.11 The selected target site was then explored in silico by docking low molecular weight compounds from the LOPAC library (see the Materials and methods section). This docking search (AutoDock424) resulted in the identification of three molecules with calculated binding free energy (ΔG) for WNV NS3 helicase ranging from −11.5 to −9.5 kcal/mol; the compounds were ivermectin (a macrocyclic lactone antibiotic with broad-spectrum antiparasitic activity, Figure 1b), ouabain (a cardiac glycoside) and paromomycin sulphate (an aminoglycoside antibiotic used to treat, among others, cutaneous leishmaniasis). These molecules were selected for further biochemical evaluation in in vitro helicase activity assays.
Figure 1.
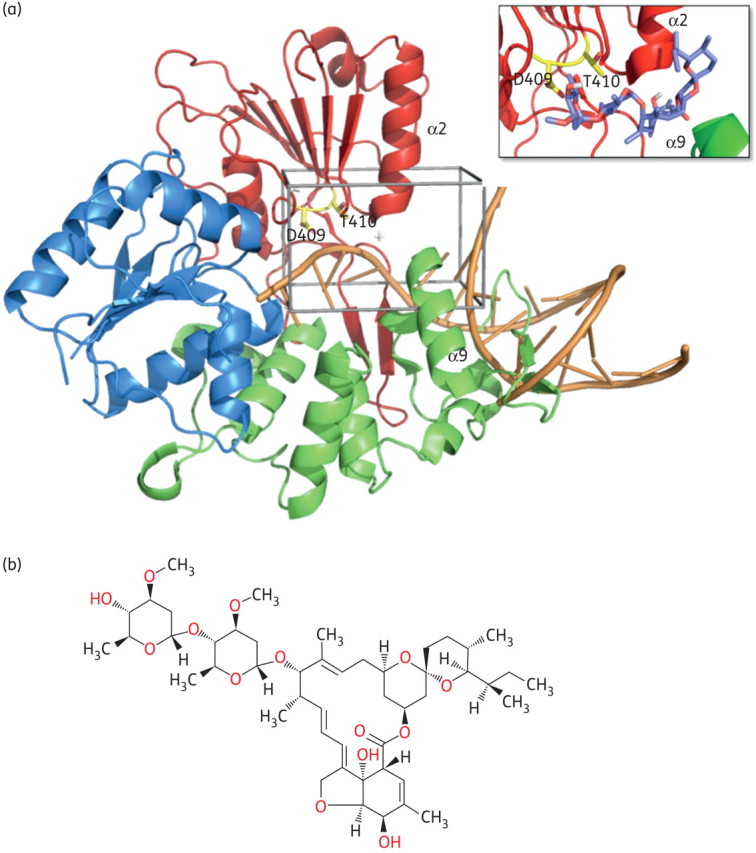
In silico docking of flavivirus helicase. (a) Model of the helicase/RNA interaction for WNV helicase domain. The crystal structure of free WNV helicase (PDB 2QEQ) was used to identify a potential ssRNA access site to the enzyme active site by superposition with the DNA-bound bacterial helicase PcrA (PDB 3PJR). The respective region is located between helices α2 in subdomain II (red) and α9 in subdomain III (green) of the WNV helicase, forming an α-helical gate for the entering RNA substrate (orange worm/sticks). This putative helicase ssRNA access site (enclosed in black) was then chosen as the target site for the in silico ligand search. The amino acids selected for mutation (Asp409 and Thr410) are shown in yellow. The close-up box shows a potential docking conformation of ivermectin in the ssRNA access site. The location of the molecule's macro ring is well established, having the two sugar rings of the compound located inside the protein. The region occupied by the inhibitor is conserved in all binding modes of the different docked conformations. (b) Chemical structure of ivermectin (B1a form; Sigma-Aldrich). Figures created using PyMol (http://www.pymol.org). This figure appears in colour in the online version of JAC and in black and white in the print version of JAC.
Specific inhibition of flaviviral NS3 helicase unwinding activity
The inhibitory effects of the three selected molecules (ivermectin, ouabain and paromomycin sulphate) on flavivirus helicase activity were evaluated against the helicase domain of WNV. Helicase inhibition activity was assayed using a radiolabelled RNA substrate in the presence of Mg2+ and ATP.30 For this purpose, the NS3 helicase domain was pre-incubated with various concentrations of the inhibitors. Ouabain and ivermectin inhibited the dsRNA unwinding activity of WNV helicase with an IC50 between 200 and 400 nM (not shown). Paromomycin sulphate showed strong inhibitory activity at all concentrations tested (as low as 0.5 nM), likely due, however, to its non-specific RNA-binding capacity, reported previously.35
Using a complementary FRET-based helicase assay (see the Materials and methods section), we demonstrated that the specific inhibitory activity of ivermectin extends further to the NS3 helicase of the YFV helicase with an IC50 of 122 ± 10 nM (Figure S1a, available as Supplementary data at JAC Online) and DENV with an IC50 of 500 ± 70 nM (Figure S1b, available as Supplementary data at JAC Online). Using this assay, the activity of ivermectin against the WNV helicase was confirmed, with an IC50 of 350 ± 40 nM (Figure S1c, available as Supplementary data at JAC Online).
To exclude any effect of the inhibitor on the ATP binding site, the influence of ivermectin on the ATPase activity of the YFV, WNV and DENV helicase domains was assessed. Ivermectin at a concentration of 1 μM did not affect the ATPase activity of the YFV, WNV and DENV helicase domains (Figure S2, available as Supplementary data at JAC Online), suggesting that the compound does not restrict the helicase ATP binding site located at a distance of ∼25 Å from its predicted binding site in the helicase domain.
On the other hand, the inhibition of helicase activity does not result from ligand-induced protein destabilization/aggregation/denaturation, as demonstrated by thermofluorometric assays. In fact, YFV, DENV and WNV helicases displayed essentially the same Tm in the presence and absence of ivermectin, with or without dsRNA (not shown).
To rule out any off-target effect of ivermectin on the flaviviral NS5 RdRp, polymerase activity assays were performed using both the RdRp domains and the NS5 full-length proteins from DENV and WNV. The polymerase activity of any of the four proteins was not affected by the presence of 1 μM ivermectin (Figure S3, available as Supplementary data at JAC Online).
Finally, to verify the specificity of ivermectin for the flavivirus helicases, we performed activity assays on HCV helicase. The helicase of HCV, which belongs to the family Flaviviridae, is closely related to flavivirus helicases.30 Tests using HCV helicase in the presence of ivermectin (up to 100 μM) did not show any inhibition of the dsRNA unwinding activity of the enzyme, indicating that ivermectin inhibition is exclusive for flavivirus helicases (not shown).
YFV, DENV and WNV helicase double mutants
In order to characterize extensively the part of the protein involved in ivermectin binding, we analysed, by in silico docking, all the possible helicase/ivermectin interactions in the ssRNA access site. Using all the known coordinates of the different flaviviral helicases, we produced a number of virtual conformations of the ligand in the chosen site using the program AutoDock4 (not shown). This virtual search pointed to possible mutations perturbing the predicted binding mode of the inhibitor to the selected site of the protein. In particular, we identified Thr408 and Asp409 (numeration as in DENV helicase PDB 2JLQ) as high-frequency interactors in the different conformations of ivermectin produced by virtual docking. This analysis suggested that the substitution of the two amino acids with others having slightly bulkier side chains could hamper ligand binding. Since the two amino acids are conserved among flaviviral helicases, in order to preserve activity we chose to make two minor mutations: Thr408Ile and Asp409Glu in the DENV helicase domain, Thr409Ile and Asp410Glu in the WNV helicase domain (Figure 1a) and Thr413Ile and Asp414Glu in the YFV helicase domain.
As expected, the mutated YFV, DENV and WNV helicases maintained good dsRNA unwinding activity. In contrast, the mutant helicases were not inhibited by ivermectin (up to 5 μM), indicating that binding of the inhibitor was strongly hampered by the inserted mutations (Figure S1a–c). This indicates that the two amino acids (or one of the two) selected for mutation strongly interact with the inhibitor and consequently that ivermectin actually interacts with the ssRNA access site as predicted by our structural analysis. Notably, both residues are conserved among all the flaviviruses and belong to the conserved motif V in helicase superfamily II.
Helicase kinetics and inhibition
In order to investigate the mechanism of helicase inhibition exerted by ivermectin we performed kinetic assays. It is possible to describe the helicase kinetics by a simple Michaelis–Menten model when neglecting the inhibitory effect due to ADP production during the first 10 min of reaction. The mechanism of inhibition proved to be uncompetitive for all three viral enzymes, with the inhibition constant being 19 ± 0.2, 354 ± 23 and 175 ± 25 nM for YFV, DENV and WNV, respectively (Figure 2a–c), indicating that ivermectin is able to bind effectively to the protein only when RNA is present (enzyme–substrate complex). The inhibition constants calculated by kinetic analysis are reported in Table 1.
Figure 2.
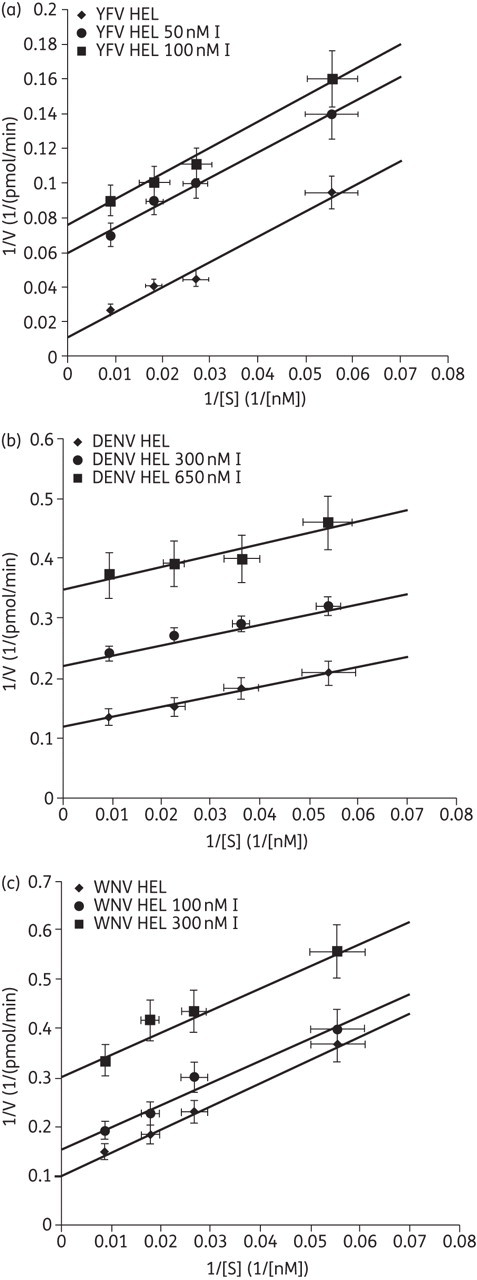
Kinetic studies of the mechanism of helicase inhibition by ivermectin. Double-reciprocal plot of the rate of dsRNA unwinding versus substrate concentration (dsRNA) during the inhibition of (a) YFV helicase, (b) DENV helicase and (c) WNV helicase by an increasing amount of ivermectin (I). HEL, helicase.
Table 1.
Effect of ivermectin on the activity of recombinant flavivirus helicases, viral RNA formation in virus-infected cultures and host cell metabolism
| Virus | Fluorescent helicase inhibition, IC50 (μM)a | Helicase kinetics, Ki (μM)b | qRT–PCR, EC50 (μM)c | CC50 (μM)d |
|---|---|---|---|---|
| YFV | 0.12 ± 0.01 | 0.019 ± 0.002 | 0.0005 | 3.5 |
| DENV | 0.50 ± 0.07 | 0.354 ± 0.023 | 0.7 | 3.8 |
| WNV | 0.35 ± 0.04 | 0.175 ± 0.025 | 4 | 10 |
All cell data are presented as averages of three independent experiments and all standard errors were <10%.
aCompound concentration required to achieve 50% inhibition of the helicase activity using the fluorescent helicase inhibition assay; helicase domain of YFV = amino acids 184–623, helicase domain of DENV = amino acids 168–618 and helicase domain of WNV = amino acids 180–619.
bInhibition constant calculated from helicase kinetic assays.
cCompound concentration required to inhibit viral RNA synthesis by 50% in Vero cells infected with the YFV 17D vaccine strain, the DENV serotype 2 New Guinea C or the WNV strain NY99.
dCompound concentration required to reduce the viability of Vero-B cells (for YFV and DENV) and Vero E6 cells (for WNV) by 50%.
Inhibition of in vitro viral replication
The potential inhibitory effects of the identified molecules on in vitro replication of different flaviviruses (YFV, DENV and WNV) were evaluated. Among the three compounds, only ivermectin inhibited the replication of the selected viruses. In particular, highly potent inhibition of YFV replication was observed. In a CPE reduction assay in Vero-B cell culture, the EC50 for inhibition of YFV replication was between 3.1 and 6.3 nM (Figure 3a), whereas in virus yield reduction assays the EC50 for inhibition of viral progeny formation was in the low sub-nanomolar range (∼0.5 nM, Figure 3b and Table 1). Ivermectin proved less active against DENV in the CPE reduction assay (EC50 >1 μM, not shown), although inhibition in virus yield reduction assays was observed (EC50 0.7 μM, quantified by qRT–PCR; Table 1). Ivermectin inhibited the production of infectious WNV, although with an EC50 of 4 μM (Table 1). Interestingly, the antiviral activity of ivermectin appears to be selective against flaviviruses, as demonstrated by its capability to inhibit the production of infectious TBEV and JEV viruses with EC50 ∼0.2 and 0.3 μM (CC50 of ∼10 μM, not shown), respectively, and its failure to inhibit bovine viral diarrhoea virus (BVDV) and HCV (which belong to the related genera pestivirus and hepacivirus, respectively, within the Flaviviridae family), but also Coxsackievirus B2 (CVB-2) and poliovirus 1 (Sb-1) (Picornaviridae family), herpes simplex virus 1 (HSV-1) and vaccinia virus (VV) (Herpesviridae and Poxviridae family, respectively), vesicular stomatitis virus (VSV) and respiratory syncytial virus (RSV) (Rhabdoviridae and Paramyxoviridae family, respectively) and Reovirus-1 (Reo-1) (Reoviridae family) (data not shown).
Figure 3.
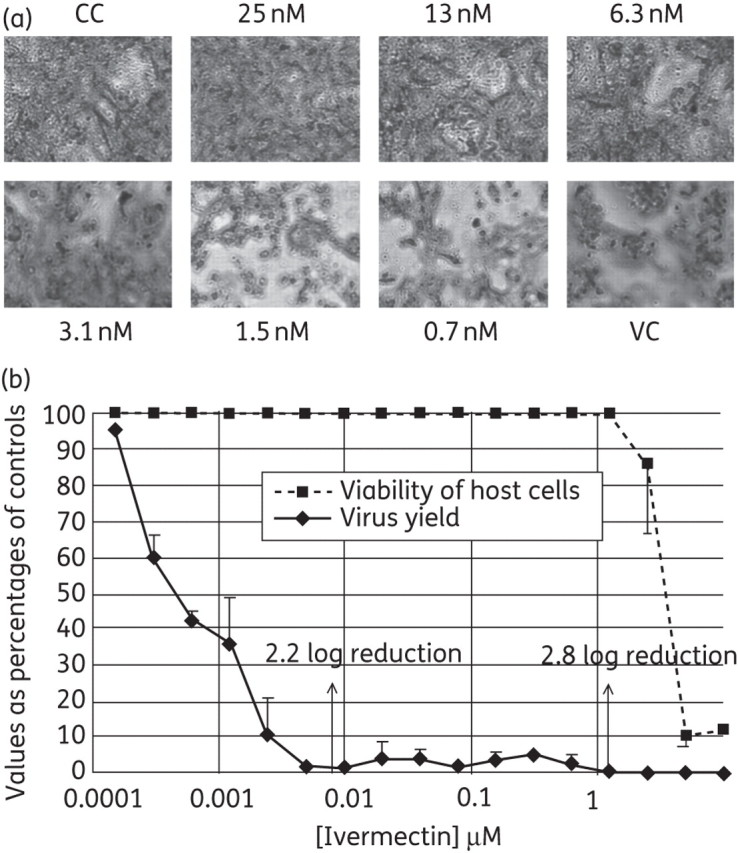
YFV-17D-induced CPE. (a) YFV-17D-induced CPE: concentration-dependent inhibition by ivermectin. At concentrations of >3 nM complete protection against virus-induced CPE formation (at 6 days post-infection) is observed. VC, virus-infected control without drug; CC, cell control (uninfected/untreated). Cells are shown at ×100 magnification. (b) Effect of ivermectin on the viability of uninfected host cells and on virus yield determined by RT-qPCR. Both results are reported as percentages of untreated controls. Arrows indicate the concentrations at which the reduction (‘log reduction’) in viral RNA levels was determined. Mean values of at least three independent experiments ± SD.
Time-of-drug-addition assays
Time-of-drug-addition assays (0–24 h) were performed to examine at which stage in the viral replication cycle (from virus binding to release) ivermectin exerts its antiviral activity on YFV replication. In the virus growth curve without inhibitor, onset of intracellular viral RNA synthesis was at 14 h post-infection (Figure 4a). Ivermectin and the reference molecule ribavirin were highly effective in inhibiting YFV replication when added during the first 14 h after virus infection, but when added at a later stage, i.e. after onset of intracellular viral RNA synthesis (as assessed in the untreated cultures) the molecule lost its antiviral effect (Figure 4b). Comparable results were obtained in the DENV serotype 2 time-of-addition study, performed using the full-length dengue reporter virus expressing Renilla luciferase (not shown).
Figure 4.

Time-dependent inhibition of YFV replication. (a) Kinetics of intracellular YFV-17D RNA synthesis following infection of Vero cells. Onset of viral RNA synthesis is detected at 14 h post-infection. (b) Effect of time of addition of ivermectin (left-hand panel) or ribavirin (right-hand panel) to YFV-17D-infected Vero cells. Both molecules gradually lost their protective activity when added at a timepoint (or later) that coincided with onset of viral replication in the control cultures.
Discussion
Infections with YFV and other emerging and re-emerging pathogenic flaviviruses (such as DENV, WNV and JEV) pose a serious global public health problem.1 Potent and safe antivirals are urgently needed. Such drugs may be used for the treatment of life-threatening infections with YFV, or encephalitis induced by WNV or JEV infections. Substantial progress has been made in recent years in understanding the biology of replication of flaviviruses, in some cases even in great molecular detail,12,36 allowing innovative approaches to drug discovery, such as structure-based drug screening and design.12,37
We undertook an in silico screen for potential inhibitors of the flaviviral NS3 helicase. Starting from the crystal structure of the NS3 helicase domain of WNV,21 we identified the enzyme's putative ssRNA access site, located between helicase subdomains II and III (α-helical gate; Figure 1a). According to our mechanistic analysis this site might be crucial for the helicase enzymatic function, and thus be a target for inhibition of viral replication. Virtual docking of the LOPAC library to this site identified ivermectin (a broad-spectrum antiparasitic agent), ouabain (a cardiac glycoside) and paromomycin sulphate (an aminoglycoside antibiotic) as potential flavivirus helicase binding compounds. Using in vitro enzymatic assays employing recombinant NS3 helicases of flaviviruses from several distantly related serogroups (WNV from the JEV serogroup and YFV and DENV from the YFV serogroup), we could confirm that ivermectin inhibited the dsRNA unwinding activity of different flaviviral helicases (with IC50 values in the sub-micromolar range; Figure 2). For ivermectin, the specificity of this inhibition was corroborated by excluding any off-target effects on the helicase-associated ATPase activity and the overall native protein fold (by thermal denaturation analysis).
Extending our in silico analysis to all the available structures of flaviviral helicases, we simulated a number of possible conformations of ivermectin inside the ssRNA access site. The results allowed the identification of two conserved amino acids (T408 and D409 in DENV) often interacting with the different conformations of ivermectin. This suggests that a quasi-conservative substitution of both amino acids (with slightly bulkier side chains) could hamper ligand binding to the protein. Accordingly, a DENV helicase double mutant (T408I and D409E), a WNV helicase double mutant (T409I and D410E) and a YFV helicase double mutant (T413I and D414E) were generated. The mutated proteins preserved helicase activity but were not inhibited by ivermectin up to a concentration of 5 μM. This provides strong evidence that the two amino acids (or one of the two) strongly interact with the inhibitor, confirming the binding of ivermectin to the ssRNA access site. Notably, both residues are part of the motif V in helicase superfamily II and are conserved among the flaviviruses.
We later showed that ivermectin behaves as an uncompetitive helicase inhibitor, able to bind only to the protein/RNA complex blocking the enzymatic activity. This result is in agreement with the failure to measure binding of ivermectin to the enzyme using micro-calorimetry (not shown) or to obtain crystals of the protein–ligand complex (data not shown). On the basis of the available structures of DENV bound to ssRNA (PDB 2JLU) it is not possible to predict a plausible interaction site or a model of the ternary complex. Reasonably, during activity the helicase/RNA complex changes its structure,38 allowing ivermectin to interact with the identified amino acids to block dsRNA unwinding.
Most importantly, we demonstrated that ivermectin (and not ouabain or paromomycin sulphate) inhibited the in vitro replication of different flaviviruses. Ivermectin proved most potent against YFV, but also inhibited, although less efficiently, the in vitro replication of DENV, JEV, WNV and TBEV. The highly potent antiviral effect of ivermectin against YFV (EC50 ∼0.5 nM) is striking, considering that other recently reported inhibitors of flavivirus replication display activity that is several orders of magnitude lower (NITD008, EC50 2 μM39 and NITD-618, EC50 1–4 μM,40 both against DENV; 2′-C-methylcytidine, EC50 100 μM41 and T-705, EC50 330 μM,42 both against YFV).
We demonstrated in a time-of-drug-addition assay that ivermectin exerts its anti-YFV activity when administered during the first 14 h after virus entry into cells (Figure 4b). In fact, the compound gradually loses its antiviral potency when first added to YFV-infected cultures after onset of intracellular viral RNA synthesis (Figure 4a). Thus, the compound is effective only during that particular phase of the flaviviral replication cycle in which the viral helicase is functionally active.36
Despite the fact that ivermectin exerts in vitro anti-helicase activity (but neither anti-ATPase nor anti-RdRp activity), we cannot exclude the possibility that the molecule exerts its antiviral activity against flaviviruses via additional or other unrelated mechanism(s). In particular for YFV, the net gap between the IC50 value against YFV helicase (120 nM) and the EC50 value in YFV cell cultures (0.5 nM) indicates that yet undiscovered process(es) other than helicase inhibition may contribute to the mechanism of action of ivermectin inhibition in cell culture. Besides, the mechanism behind the very selective and potent YFV activity may differ from that of the other flaviviruses (at least the ones we examined in our work), because of the different EC50 values observed in the three viruses. In fact, the lower EC50 value observed in DENV and, most strikingly, in WNV cell culture experiments could be related to different factors (i.e. compound solubility or permeability, metabolism, non-specific protein binding), including lower susceptibility of the viral replication complex (involving NS3 and NS5) with respect to the isolated helicase domain.
Our data, however, show that the mechanism of inhibition does not involve the early stages of the viral replication cycle (i.e. virus attachment and/or virus entry). The data presented nevertheless stress the key role played by helicase inhibition in the whole antiviral activity. To add independent proof that ivermectin acts in the infected cells by inhibition of the NS3 helicase, we tried to select ivermectin drug-resistant variants by serial passaging of YFV with increasing concentrations of the drug (expecting that adaptive mutations in the helicase domain would be selected). Unfortunately, even following extensive efforts, varying several experimental parameters (among others, multiplicity of infection, drug concentration increments and host cell type), drug-resistant virus variants were not selected after >30 serial passages of YFV, for >6 months in the presence of ivermectin. This may indicate that the barrier to resistance is high and that mutants may not emerge because they may not be viable.
Ivermectin is known as an anti-helminthic agent for oral administration. In the mid-1980s the compound was introduced as probably the most broad-spectrum anti-parasite medication ever,43,44 which interferes with the parasite’s nervous system and muscle function by binding and activating glutamate-gated chloride channels.45 Ivermectin is used in humans mainly for the treatment of onchocerciasis, but is also effective against other worm infestations (such as strongyloidiasis, ascariasis, trichuriasis and enterobiasis). Ivermectin is also effective against most mites and some lice.
Considering that ivermectin has been used for the treatment of a variety of parasitic disease in man for >20 years, assessing its potential for the treatment of life-threatening flavivirus infections in clinical trials may require a minimum effort. Mining of epidemiological records in tropical regions where flaviviruses are endemic and where ivermectin has been administered for decades during population-wide onchocerciasis eradication programmes may offer first insights into the protective roles offered by the new application of this old drug.
Funding
This work was funded by the FP7 HEALTH-2010 Collaborative Project SILVER (No. 260644). A FEBS short-term fellowship also supported part of the activities reported here (M. P.).
Transparency declarations
None to declare.
Supplementary data
Figures S1, S2 and S3 are available as Supplementary data at JAC Online (http://jac.oxfordjournals.org/).
References
- 1.Gould EA, Solomon T. Pathogenic flaviviruses. Lancet. 2008;371:500–9. doi: 10.1016/S0140-6736(08)60238-X. doi:10.1016/S0140-6736(08)60238-X. [DOI] [PubMed] [Google Scholar]
- 2.Monath TP. Yellow fever: an update. Lancet Infect Dis. 2001;1:11–20. doi: 10.1016/S1473-3099(01)00016-0. doi:10.1016/S1473-3099(01)00016-0. [DOI] [PubMed] [Google Scholar]
- 3.Staples JE, Monath TP. Yellow fever: 100 years of discovery. JAMA. 2008;300:960–2. doi: 10.1001/jama.300.8.960. doi:10.1001/jama.300.8.960. [DOI] [PubMed] [Google Scholar]
- 4.Barrett AD, Higgs S. Yellow fever: a disease that has yet to be conquered. Annu Rev Entomol. 2007;52:209–29. doi: 10.1146/annurev.ento.52.110405.091454. doi:10.1146/annurev.ento.52.110405.091454. [DOI] [PubMed] [Google Scholar]
- 5.Ellis BR, Barrett AD. The enigma of yellow fever in East Africa. Rev Med Virol. 2008;18:331–46. doi: 10.1002/rmv.584. doi:10.1002/rmv.584. [DOI] [PubMed] [Google Scholar]
- 6.Vasilakis N, Weaver SC. The history and evolution of human dengue emergence. Adv Virus Res. 2008;72:1–76. doi: 10.1016/S0065-3527(08)00401-6. doi:10.1016/S0065-3527(08)00401-6. [DOI] [PubMed] [Google Scholar]
- 7.Guzman MG, Kouri G. Dengue haemorrhagic fever integral hypothesis: confirming observations, 1987–2007. Trans R Soc Trop Med Hyg. 2008;102:522–3. doi: 10.1016/j.trstmh.2008.03.001. doi:10.1016/j.trstmh.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 8.Petersen LR, Hayes EB. West Nile virus in the Americas. Med Clin North Am. 2008;92:1307–22. doi: 10.1016/j.mcna.2008.07.004. ix doi:10.1016/j.mcna.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 9.Bartholomeusz AI, Wright PJ. Synthesis of dengue virus RNA in vitro: initiation and the involvement of proteins NS3 and NS5. Arch Virol. 1993;128:111–21. doi: 10.1007/BF01309792. doi:10.1007/BF01309792. [DOI] [PubMed] [Google Scholar]
- 10.Caruthers JM, McKay DB. Helicase structure and mechanism. Curr Opin Struct Biol. 2002;12:123–33. doi: 10.1016/s0959-440x(02)00298-1. doi:10.1016/S0959-440X(02)00298-1. [DOI] [PubMed] [Google Scholar]
- 11.Luo D, Xu T, Watson RP, et al. Insights into RNA unwinding and ATP hydrolysis by the flavivirus NS3 protein. EMBO J. 2008;27:3209–19. doi: 10.1038/emboj.2008.232. doi:10.1038/emboj.2008.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bollati M, Alvarez K, Assenberg R, et al. Structure and functionality in flavivirus NS-proteins: perspectives for drug design. Antiviral Res. 2010;87:125–48. doi: 10.1016/j.antiviral.2009.11.009. doi:10.1016/j.antiviral.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manfroni G, Paeshuyse J, Massari S, et al. Inhibition of subgenomic hepatitis C virus RNA replication by acridone derivatives: identification of an NS3 helicase inhibitor. J Med Chem. 2009;52:3354–65. doi: 10.1021/jm801608u. doi:10.1021/jm801608u. [DOI] [PubMed] [Google Scholar]
- 14.De Palma AM, Vliegen I, De Clercq E, et al. Selective inhibitors of picornavirus replication. Med Res Rev. 2008;28:823–84. doi: 10.1002/med.20125. doi:10.1002/med.20125. [DOI] [PubMed] [Google Scholar]
- 15.Huggins JW. Prospects for treatment of viral hemorrhagic fevers with ribavirin, a broad-spectrum antiviral drug. Rev Infect Dis. 1989;11(Suppl 4):S750–61. doi: 10.1093/clinids/11.supplement_4.s750. doi:10.1093/clinids/11.Supplement_4.S750. [DOI] [PubMed] [Google Scholar]
- 16.Borowski P, Mueller O, Niebuhr A, et al. ATP-binding domain of NTPase/helicase as a target for hepatitis C antiviral therapy. Acta Biochim Pol. 2000;47:173–80. [PubMed] [Google Scholar]
- 17.Sbrana E, Xiao SY, Guzman H, et al. Efficacy of post-exposure treatment of yellow fever with ribavirin in a hamster model of the disease. Am J Trop Med Hyg. 2004;71:306–12. [PubMed] [Google Scholar]
- 18.Zhang N, Chen HM, Koch V, et al. Ring-expanded (“fat”) nucleoside and nucleotide analogues exhibit potent in vitro activity against Flaviviridae NTPases/helicases, including those of the West Nile virus, hepatitis C virus, and Japanese encephalitis virus. J Med Chem. 2003;46:4149–64. doi: 10.1021/jm030842j. doi:10.1021/jm030842j. [DOI] [PubMed] [Google Scholar]
- 19.Borowski P, Niebuhr A, Mueller O, et al. Purification and characterization of West Nile virus nucleoside triphosphatase (NTPase)/helicase: evidence for dissociation of the NTPase and helicase activities of the enzyme. J Virol. 2001;75:3220–9. doi: 10.1128/JVI.75.7.3220-3229.2001. doi:10.1128/JVI.75.7.3220-3229.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gozdek A, Zhukov I, Polkowska A, et al. NS3 peptide, a novel potent hepatitis C virus NS3 helicase inhibitor: its mechanism of action and antiviral activity in the replicon system. Antimicrob Agents Chemother. 2008;52:393–401. doi: 10.1128/AAC.00961-07. doi:10.1128/AAC.00961-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mastrangelo E, Milani M, Bollati M, et al. Crystal structure and activity of Kunjin virus NS3 helicase; protease and helicase domain assembly in the full length NS3 protein. J Mol Biol. 2007;372:444–55. doi: 10.1016/j.jmb.2007.06.055. doi:10.1016/j.jmb.2007.06.055. [DOI] [PubMed] [Google Scholar]
- 22.Chu PW, Westaway EG. Replication strategy of Kunjin virus: evidence for recycling role of replicative form RNA as template in semiconservative and asymmetric replication. Virology. 1985;140:68–79. doi: 10.1016/0042-6822(85)90446-5. doi:10.1016/0042-6822(85)90446-5. [DOI] [PubMed] [Google Scholar]
- 23.Scherret JH, Poidinger M, Mackenzie JS, et al. The relationships between West Nile and Kunjin viruses. Emerg Infect Dis. 2001;7:697–705. doi: 10.3201/eid0704.010418. doi:10.3201/eid0704.010418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morris GM, Goodsell DS, Halliday RS, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19:1639–62. doi:10.1002/(SICI)1096-987X(19981115)19:14<1639::AID-JCC10>3.0.CO;2-B. [Google Scholar]
- 25.Singh UC, Kollman PA. An approach to computing electrostatic charges for molecules. J Comput Chem. 1984;5:129–45. doi:10.1002/jcc.540050204. [Google Scholar]
- 26.Goodford PJ. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J Med Chem. 1985;28:849–57. doi: 10.1021/jm00145a002. doi:10.1021/jm00145a002. [DOI] [PubMed] [Google Scholar]
- 27.Gasteiger J, Marsili M. A new model for calculating atomic charges in molecules. Tetrahedron Lett. 1978;34:3181–4. doi:10.1016/S0040-4039(01)94977-9. [Google Scholar]
- 28.Mastrangelo E, Bollati M, Milani M, et al. Preliminary crystallographic characterization of an RNA helicase from Kunjin virus. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2006;62:876–9. doi: 10.1107/S1744309106028776. doi:10.1107/S1744309106028776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De Colibus L, Speroni S, Coutard B, et al. Purification and crystallization of Kokobera virus helicase. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2007;63:193–5. doi: 10.1107/S1744309107005283. doi:10.1107/S1744309107005283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu J, Bera AK, Kuhn RJ, et al. Structure of the flavivirus helicase: implications for catalytic activity, protein interactions, and proteolytic processing. J Virol. 2005;79:10268–77. doi: 10.1128/JVI.79.16.10268-10277.2005. doi:10.1128/JVI.79.16.10268-10277.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boguszewska-Chachulska AM, Krawczyk M, Stankiewicz A, et al. Direct fluorometric measurement of hepatitis C virus helicase activity. FEBS Lett. 2004;567:253–8. doi: 10.1016/j.febslet.2004.04.072. doi:10.1016/j.febslet.2004.04.072. [DOI] [PubMed] [Google Scholar]
- 32.Belon CA, High YD, Lin TI, et al. Mechanism and specificity of a symmetrical benzimidazolephenylcarboxamide helicase inhibitor. Biochemistry. 2010;49:1822–32. doi: 10.1021/bi901974a. doi:10.1021/bi901974a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaptein SJ, De Burghgraeve T, Froeyen M, et al. A derivate of the antibiotic doxorubicin is a selective inhibitor of dengue and yellow fever virus replication in vitro. Antimicrob Agents Chemother. 2010;54:5269–80. doi: 10.1128/AAC.00686-10. doi:10.1128/AAC.00686-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Velankar SS, Soultanas P, Dillingham MS, et al. Crystal structures of complexes of PcrA DNA helicase with a DNA substrate indicate an inchworm mechanism. Cell. 1999;97:75–84. doi: 10.1016/s0092-8674(00)80716-3. doi:10.1016/S0092-8674(00)80716-3. [DOI] [PubMed] [Google Scholar]
- 35.Vicens Q, Westhof E. Crystal structure of paromomycin docked into the eubacterial ribosomal decoding A site. Structure. 2001;9:647–58. doi: 10.1016/s0969-2126(01)00629-3. doi:10.1016/S0969-2126(01)00629-3. [DOI] [PubMed] [Google Scholar]
- 36.Mukhopadhyay S, Kuhn RJ, Rossmann MG. A structural perspective of the flavivirus life cycle. Nat Rev Microbiol. 2005;3:13–22. doi: 10.1038/nrmicro1067. doi:10.1038/nrmicro1067. [DOI] [PubMed] [Google Scholar]
- 37.Sampath A, Padmanabhan R. Molecular targets for flavivirus drug discovery. Antiviral Res. 2009;81:6–15. doi: 10.1016/j.antiviral.2008.08.004. doi:10.1016/j.antiviral.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mastrangelo E, Bolognesi M, Milani M. Flaviviral helicase: insights into the mechanism of action of a motor protein. Biochem Biophys Res Commun. 2012;417:84–7. doi: 10.1016/j.bbrc.2011.11.060. doi:10.1016/j.bbrc.2011.11.060. [DOI] [PubMed] [Google Scholar]
- 39.Yin Z, Chen YL, Schul W, et al. An adenosine nucleoside inhibitor of dengue virus. Proc Natl Acad Sci USA. 2009;106:20435–9. doi: 10.1073/pnas.0907010106. doi:10.1073/pnas.0907010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xie X, Wang QY, Xu HY, et al. Inhibition of dengue virus by targeting viral NS4B protein. J Virol. 2011;85:11183–95. doi: 10.1128/JVI.05468-11. doi:10.1128/JVI.05468-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Julander JG, Jha AK, Choi JA, et al. Efficacy of 2′-C-methylcytidine against yellow fever virus in cell culture and in a hamster model. Antiviral Res. 2010;86:261–7. doi: 10.1016/j.antiviral.2010.03.004. doi:10.1016/j.antiviral.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Julander JG, Shafer K, Smee DF, et al. Activity of T-705 in a hamster model of yellow fever virus infection in comparison with that of a chemically related compound, T-1106. Antimicrob Agents Chemother. 2009;53:202–9. doi: 10.1128/AAC.01074-08. doi:10.1128/AAC.01074-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Omura S. Ivermectin: 25 years and still going strong. Int J Antimicrob Agents. 2008;31:91–8. doi: 10.1016/j.ijantimicag.2007.08.023. doi:10.1016/j.ijantimicag.2007.08.023. [DOI] [PubMed] [Google Scholar]
- 44.Geary TG. Ivermectin 20 years on: maturation of a wonder drug. Trends Parasitol. 2005;21:530–2. doi: 10.1016/j.pt.2005.08.014. doi:10.1016/j.pt.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 45.Cully DF, Vassilatis DK, Liu KK, et al. Cloning of an avermectin-sensitive glutamate-gated chloride channel from Caenorhabditis elegans. Nature. 1994;371:707–11. doi: 10.1038/371707a0. doi:10.1038/371707a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.