

Abstract
Objective:
To conclusively test for a specific association between the biological marker 25-hydroxy-vitamin D3, a transcriptionally active hormone produced in human skin and liver, and the prevalence and severity of Parkinson disease (PD).
Methods:
We used liquid chromatography/tandem mass spectrometry to establish an association specifically between deficiency of 25-hydroxy-vitamin D3 and PD in a cross-sectional and longitudinal case-control study of 388 patients (mean Hoehn and Yahr stage of 2.1 ± 0.6) and 283 control subjects free of neurologic disease nested in the Harvard Biomarker Study.
Results:
Plasma levels of 25-hydroxy-vitamin D3 were associated with PD in both univariate and multivariate analyses with p values = 0.0034 and 0.047, respectively. Total 25-hydroxy-vitamin D levels, the traditional composite measure of endogenous and exogenous vitamin D, were deficient in 17.6% of patients with PD compared with 9.3% of controls. Low 25-hydroxy-vitamin D3 as well as total 25-hydroxy-vitamin D levels were correlated with higher total Unified Parkinson’s Disease Rating Scale scores at baseline and during follow-up.
Conclusions:
Our study reveals an association between 25-hydroxy-vitamin D3 and PD and suggests that thousands of patients with PD in North America alone may be vitamin D–deficient. This finding has immediate relevance for individual patients at risk of falls as well as public health, and warrants further investigation into the mechanism underlying this association.
Our interest in vitamin D in Parkinson disease (PD) was sparked when an unbiased genome-wide expression screen indicated increased expression of the vitamin D receptor (VDR) gene in the blood of patients with PD.1 Reduced serum levels of the major vitamin D metabolite, total 25-hydroxy-vitamin D (25[OH]D), were observed in several small cross-sectional studies of patients with PD2 and were more pronounced than in Alzheimer disease.3 These studies, however, disagreed on whether vitamin D deficiency in PD relates to disease severity.2,3 A tantalizing signal linking serum vitamin D to risk of developing PD emerged in a population-based cohort study, although the number of incident cases was small.4 It was found that 25[OH]D3 attenuates 6-hydroxydopamine-induced neurotoxicity,5 and polymorphisms in the VDR gene have been associated with PD in some studies,6,7 but not others (see www.pdgene.org).
Endogenous vitamin D3 is produced through photochemical action of sunlight on 7-dehydrocholesterol in human skin. Hydroxylation steps produce the major vitamin D hormones, 25-hydroxy-vitamin D3 and the biologically more active 1,25-hydroxy-vitamin D3, respectively. These hormones transactivate target genes in the brain and other organs through the nuclear receptor VDR (reviewed in ref. 8). Vitamin D2, by contrast, is produced in yeast and mushrooms, not humans. Very few foods naturally contain meaningful levels of vitamin D, but milk, orange juice, and cereal fortified with either vitamin D2 or D3 are widely available.9
Does vitamin D matter for patients with PD? In this study, we used liquid chromatography/tandem mass spectrometry to investigate an association specifically between deficiency of the transcriptionally active human hormone 25[OH]D3 and PD.
METHODS
Study population.
Three hundred eighty-eight patients with PD and 283 control subjects without neurologic disease nested in the Harvard Biomarker Study population who met eligibility criteria as of 2010 were included. The Harvard NeuroDiscovery Center Biomarker Study (HBS) is a Harvard-wide longitudinal case-control study designed to accelerate the discovery and validation of molecular diagnostics that track or predict progression of early-stage PD. Inclusion criteria for PD cases were age 21 years or older, diagnosis of PD according to UK PD Society Brain Bank criteria (modified as described in ref. 10) or according to movement disorders specialist assessment, and Mini-Mental State Examination scores >21 or next of kin present to provide informed consent. Exclusion criteria for PD cases were diagnosis of a blood or bleeding disorder, known hematocrit <30%, known active ulcer, or active colitis. Inclusion criteria for healthy controls were no current diagnosis or history of a neurologic disease, and age 21 years or older. Exclusion criteria for controls were the same as for cases. Controls were comparable to the PD cases in that they were drawn from the same source population and could be identified as cases, if they had disease. Diagnosis of cases and controls was monitored during follow-up to ensure high diagnostic accuracy. Clinical disease severity was assessed in an operationally defined “on state” based on expert judgment at the time of the visit as in refs. 11–13. Patients thus were not necessarily at the peak of optimal response (true on state) or in their true off state (at least 24 hours and as much as 4 weeks after the last dose). Of the 388 patients with PD, 296 had a second study visit (mean time from enrollment ± SD, 12.6 ± 2.4 months), and 295 had a third study visit (25.81 ± 5.0 months).
Standard protocol approvals and patient consents.
Informed consent was obtained from all participants. The study protocol was approved by the institutional review boards of Brigham and Women's Hospital and Massachusetts General Hospital.
Liquid chromatography/tandem mass spectrometry.
Using liquid chromatography/tandem mass spectrometry levels of 25[OH]D3, a transcriptionally active hormone produced in human skin and liver, as well as the frequently tested composite measure of endogenous and food- or supplement-derived total 25[OH]D (25[OH]D3 plus 25[OH]D2) were assayed at the Clinical Laboratory Research Core at Massachusetts General Hospital similar to the method described in ref. 14. This method involves the addition of deuterated stable isotope (d3-25-hydroxy-vitamin D) to a 200-μL plasma specimen as an internal standard. The 25[OH]D2, 25[OH]D3, and internal standard are purified by a liquid chromatography system, ionized, injected into a tandem mass spectrometer, and quantified relative to calibrators prepared in charcoal-stripped human plasma. In our analysis, the mean coefficient of variation was 14.2% for 25[OH]D3 and 14.7% for total 25[OH]D. The limit of quantitation for 25[OH]D2 is 2 ng/mL and for 25[OH]D3 is 3 ng/mL. Results are shown in nanograms/milliliter (1 ng/mL = 2.496 nmol/L).
Data analysis.
Vitamin D measures and total Unified Parkinson's Disease Rating Scale (UPDRS) scores were log-transformed for purposes of the significance tests to reduce skewness and violation of normality assumptions. Baseline characteristics of cases and controls were examined (table 1), including factors known to affect vitamin D status such as age, sex, race, body mass index, smoking status, vitamin D supplementation, season at blood draw, and latitude of the participants' residence (the latter 2 serve as proxies for sunshine exposure). Less than 5% of cases and <9% of controls had missing values for any of these covariates. Baseline inequalities were found for age, sex, race, and vitamin D supplementation and adjusted for in all multivariate analyses. There was no material baseline inequality for season of blood draw and latitude of the participants' residence. For the cross-sectional analysis, multivariate logistic regression was used to test for associations between vitamin D and PD. Values of 25[OH]D3 (total 25[OH]D) were missing for 5 (2) cases and 2 (2) controls; subjects with missing values were excluded. General linear regression models were used for testing associations of vitamin D with UPDRS scores, duration of PD illness, and Hoehn and Yahr (HY) stage. For the longitudinal analysis, mixed intercept and slope models were used to relate vitamin D levels to UPDRS or HY. Analyses were performed using SAS 9.2 (SAS Institute, Cary, NC), and statistical significance was defined as p < 0.05.
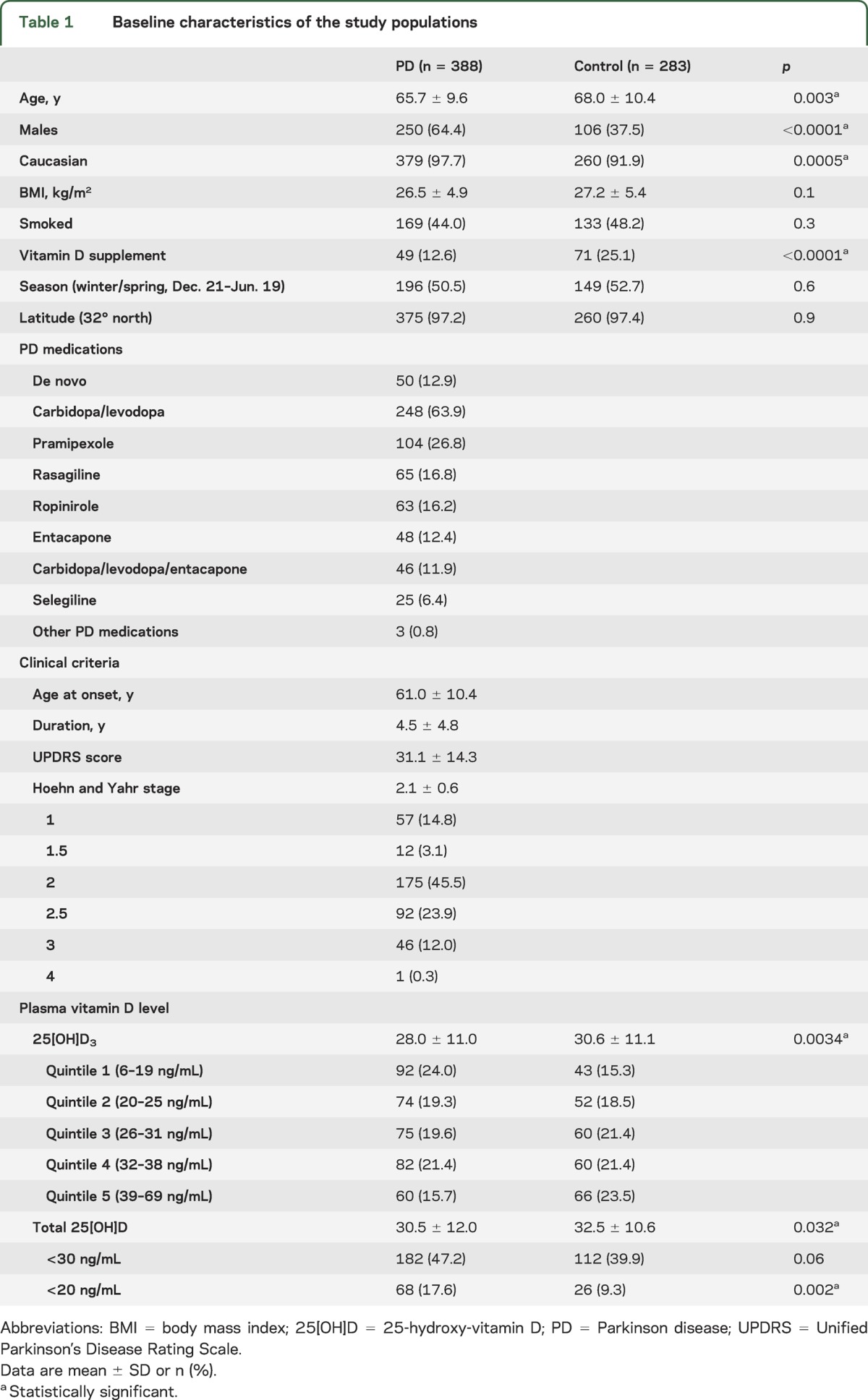
Table 1.
Baseline characteristics of the study populations
RESULTS
Cases were somewhat younger (mean age ± SD, 65.7 ± 9.6 years for cases vs 68.0 ± 10.4 years for controls), included a higher proportion of men (64.4% vs 37.5%), and of Caucasians (97.7% vs 91.9%). Cases reported vitamin D supplementation less frequently than controls (12.6% vs 25.1%). No material differences in body mass index, smoking status, season at blood draw, or latitude of the participants' residence were found. Clinically, cases were early in the disease course with an average modified HY stage of 2.1 ± 0.6 and an average total UPDRS score of 31.1 ± 14.3.
Plasma levels of 25[OH]D3 were associated with the prevalence of PD in both univariate and multivariate analyses adjusting for pertinent baseline inequalities such as age, sex, race, and vitamin D supplementation. In the univariate analysis (table 1), specifically 25[OH]D3 as well as total 25[OH]D levels were lower in plasma of cases compared with controls (28.0 ± 11.0 vs 30.6 ± 11.1 ng/mL with p = 0.0034; and 30.5 ± 12.0 vs 32.5 ± 10.6 ng/mL with p = 0.032). Although there is no consensus on optimal levels of 25[OH]D as measured in serum, vitamin D deficiency is defined by most experts as a 25[OH]D level of <20 ng/mL (50 nmol/L).9 A level of 25[OH]D <30 ng/mL can be considered to indicate a relative insufficiency of vitamin D, and a level of ≥30 ng/mL can be considered to indicate sufficient vitamin D.9 There was vitamin D deficiency in 17.6% of cases (68/388) compared with 9.3% of controls (26/283; p = 0.002). Furthermore, 47.2% of cases (182/388) were vitamin D insufficient compared with 39.9% of controls (112/283; p = 0.06). Thus, a substantial number of controls and an even larger proportion of cases had abnormal levels of this essential hormone.
Multivariate cross-sectional analysis (table 2) adjusting for pertinent baseline inequalities of age, sex, race, and vitamin D supplementation indicated that low levels of 25[OH]D3 were associated with an increased disease prevalence and severity. Individuals with levels in the lowest quintile of 25[OH]D3 values observed in our population had the highest prevalence of PD with an odds ratio of 1.8 with p = 0.03 (95% confidence interval, 1.06–3.2) compared with individuals with values in the highest quintile (table 2). After adjusting for these covariates, deficiency of total 25[OH]D (levels <20 ng/mL) remained enriched in PD with p = 0.03, although the overall relation between total 25[OH]D and PD no longer reached significance (p = 0.27; table 2 and figure). Exploratory subgroup analyses in male and female study participants confirmed strong and significant associations between 25[OH]D3 and total 25[OH]D levels and PD in male study participants, whereas in female subjects, concordant trends were observed that did not reach statistical significance. Previous studies examining the relation between measures of clinical disease severity and vitamin D levels proved inconsistent and controversial.15 The results obtained in our study showed a relation between higher (worse) total UPDRS scores and specifically low 25[OH]D3 as well as low total 25[OH]D levels with p = 0.0096 and 0.02, respectively (table 2, figure). Importantly, even in the prospective, longitudinal analysis, low baseline 25[OH]D3 as well as total 25[OH]D levels were associated with higher UPDRS scores during the clinical follow-up (table 3). Thus, lower vitamin D levels are found in individuals with more advanced disease severity as measured by the “gold standard,” the clinical total UPDRS scale. Associations with the HY scale, an alternate measure of disease severity, failed to reach statistical significance (tables 2 and 3, figure).
Table 2.
Cross-sectional analysis for associations between 25[OH]D3 and total 25[OH]D with PD, total UPDRS score, HY stage, and disease duration at the enrollment visit
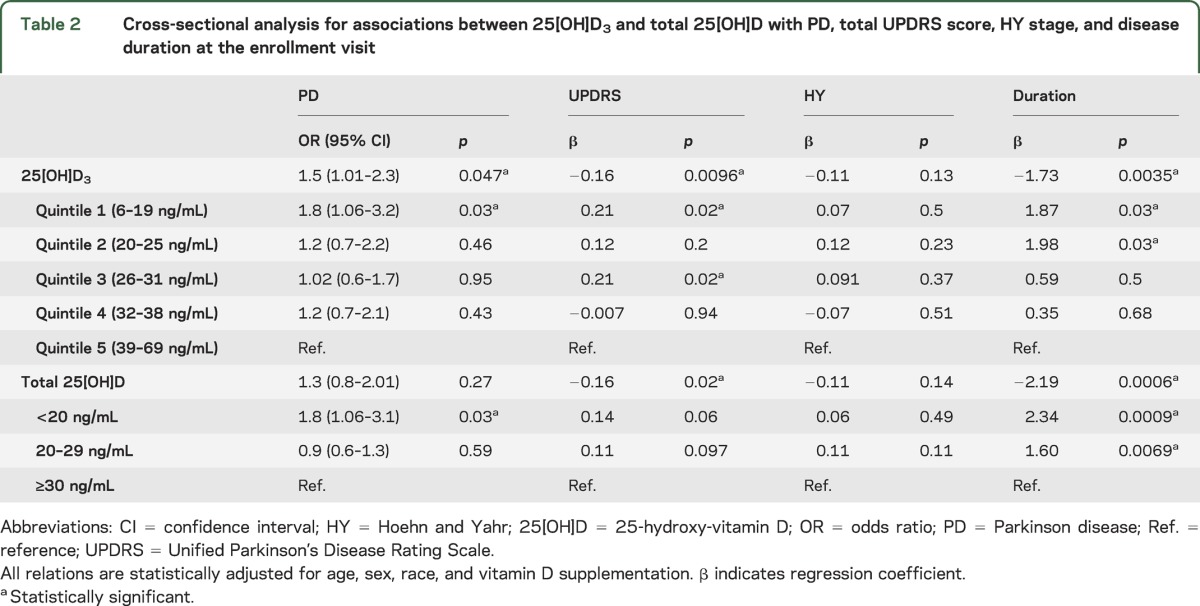
Figure. Plasma levels of 25[OH]D3 and total 25[OH]D in PD.

Relations between 25[OH]D3 (upper panels) or total 25[OH]D plasma levels (lower panels) and PD, HY stage (<3 or ≥3), total UPDRS score, and disease duration are visualized. Raw data are visualized, curves in the scatter plots are fitted by Lowess regression, and p values from the corresponding multivariate analyses adjusted for age, sex, race, and vitamin D supplementation are shown on the top of each panel. See text and table 2 for details. HY = Hoehn and Yahr; 25[OH]D = 25-hydroxy-vitamin D; PD = Parkinson disease; UPDRS = Unified Parkinson’s Disease Rating Scale.
Table 3.
Longitudinal analysis for associations between 25[OH]D3 and total 25[OH]D with total UPDRS scores or HY stage

DISCUSSION
Fractures from falls are a major and common complication of PD. While fractures always lead to a decline in function and quality of life in patients with PD, the most threatening potential consequences are a downward spiral of hospitalization, decompensation, and even death. Endogenous vitamin D enhances bone mineralization. Vitamin D supplementation (at doses higher than 400 IU/d) reduces fractures by at least 20% in individuals older than age 65.16 Should vitamin D management be part of a personalized medicine approach for patients with PD?
This large cross-sectional and longitudinal study of 388 cases and 283 controls nested in the HBS population, combined with consistent results from multiple small, independent association studies,2,3 provides compelling evidence that at least 17% of all patients with PD seen at 2 academic tertiary care centers are vitamin D deficient—almost twice the prevalence of vitamin D deficiency in controls of similar ages without PD. Furthermore, vitamin D deficiency was associated with total UPDRS score (indicating more advanced severity of disease) after adjusting for confounding variables in the cross-sectional analysis, and prospectively, during the follow-up period.
Beyond the rather clear-cut immediate clinical implications of vitamin D deficiency in patients with PD, vitamin D–mediated transactivation of VDR target genes is poised for an important role in dopamine neuron biology and may offer clues for disease modification in PD.8 Both the vitamin D receptor and 1α-hydroxylase (which activates 25[OH]D3 conversion into the active hormone) are expressed in the human brain, with the strongest immunohistochemical staining observed in dopaminergic neurons within the substantia nigra and in the hypothalamus.17 Pretreatment with 1,25[OH]D3 partially restores tyrosine hydroxylase expression and protects against dopamine depletion in substantia nigra of 6-hydroxydopamine–lesioned rats.5,18
This study has several strengths and limitations. Distinct vitamin D metabolites were for the first time specifically assayed in PD. Potential confounding factors (age, sex, race, body mass index, season at blood draw, latitude of residence, smoking, as well as vitamin D supplementation) were examined and adjusted for as indicated. The HBS was specifically designed to minimize bias from sample processing by collecting, handling, and analyzing specimens of cases and controls in a standardized and blinded manner according to rules of evidence (reviewed in ref. 19). Approximately 90% of cases enrolled in the HBS diagnosed with PD by a neurology board-certified, movement disorders fellowship-trained neurologist met modified UK PD Society Brain Bank criteria,10 with accuracy further enhanced by longitudinal annual movement disorders specialist reassessments. Controls were comparable to the PD cases in that they were drawn from the same source population and could be identified as a case, if they had disease. There are several limitations. Despite these provisions, lurking bias is a key threat to all biomarker studies, including ours. Second, our study represents patients and controls receiving care at Massachusetts General Hospital and Brigham and Women's Hospital, not the general US population. Evaluation in minority populations that may be at even higher risk of vitamin D deficiency will be necessary. Third, a previous study of 157 early-stage de novo patients (not treated with dopamine replacement medications) found 21% total 25[OH]D deficiency15 compared with the 17.6% found in the current study. The vast majority of patients in our study were also at an early stage (HY of ≤2.5), but only 12.9% were not receiving PD medications. Thus, however unlikely in the context of previous work, a subtle medication bias can not be completely ruled out. Fourth, association does not imply causation. The goal of our study was to evaluate vitamin D status in patients with PD who are vulnerable to falls and fractures, not to investigate the cause of vitamin D deficiency. It remains to be seen whether PD predisposes to vitamin D deficiency (conceivably by limiting outdoor activities) or whether vitamin D deficiency modifies the disease as suggested by preclinical studies5,8,17,18 and a small randomized placebo-controlled clinical trial20 reported while this manuscript was in preparation. Fifth, this study did not directly evaluate the effect of vitamin D deficiency (or supplementation) on clinical outcomes such as fracture risk in patients with PD, a goal for further investigation.
These data suggest that patients with PD should be included among the categories of individuals at high risk of vitamin D deficiency who warrant vitamin D biomarker measurement and vitamin D treatment. Vitamin D clearly is an excellent target for therapies designed to improve the quality of life of thousands of vitamin D–deficient patients with PD in North America alone—with a potentially major impact on public health and health care cost savings. Moreover, the tantalizing, but inconclusive, epidemiologic, biochemical, transcriptional, and genetic links among vitamin D, vitamin D target genes, and dopamine neuropathology warrant mechanistic and further prospective exploration.
ACKNOWLEDGMENT
The authors thank all the patients and their families and friends for their support and participation. Harvard NeuroDiscovery Center Biomarker Study codirectors: Harvard NeuroDiscovery Center: Clemens R. Scherzer, Bradley T. Hyman, Adrian J. Ivinson. Investigators and study coordinators: Harvard NeuroDiscovery Center: Ana Trisini-Lipsanopoulos, Kaltra Dhima, Stephen Bayer, Kaitlin C. Lockhart; Brigham and Women's Hospital: Lewis R. Sudarsky, Michael T. Hayes, Reisa Sperling; Massachusetts General Hospital: John H. Growdon, Michael A. Schwarzschild, Albert Y. Hung, Alice W. Flaherty, Deborah Blacker, Anne-Marie Wills, U. Shivraj Sohur, Vivek K. Unni, Nicte I. Mejia, Anand Viswanathan, Stephen N. Gomperts, Vikram Khurana, Mark W. Albers, Kyleen E. Swords, Rebecca K. Rudel; University of Ottawa: Michael G. Schlossmacher. Scientific advisory board: Massachusetts General Hospital: John H. Growdon; Brigham and Women's Hospital: Dennis J. Selkoe, Reisa Sperling; Harvard School of Public Health: Alberto Ascherio. Data coordination: Harvard NeuroDiscovery Center: Thomas Yi; Massachusetts General Hospital: Joseph J. Locascio. Biobank management staff: Harvard NeuroDiscovery Center: Zhixiang Liao, Ashley N. Hoesing, Karen Duong, Sarah Roderick.
GLOSSARY
- HBS
Harvard NeuroDiscovery Center Biomarker Study
- HY
Hoehn and Yahr
- 25[OH]D
25-hydroxy-vitamin D
- PD
Parkinson disease
- UPDRS
Unified Parkinson’s Disease Rating Scale
- VDR
vitamin D receptor
AUTHOR CONTRIBUTIONS
Dr. Ding: research project (conception, organization, and execution), statistical analysis (design and execution), manuscript (writing of the first draft). Ms. Dhima and Ms. Lockhart: research project (organization and execution), manuscript (review and critique). Dr. Locascio: statistical analysis (design, execution, supervision), manuscript (review and critique). Ms. Hoesing and Ms. Duong: research project (organization and execution), manuscript (review and critique). Ms. Trisini-Lipsanopoulos: research project (organization, supervision, execution), manuscript (review and critique). Dr. Hayes, Dr. Sohur, and Dr. Wills: research project (execution), manuscript (review and critique). Dr. Mollenhauer: research project (organization and execution), manuscript (review and critique). Dr. Flaherty, Dr. Hung, Dr. Mejia, Dr. Khurana, and Dr. Gomperts: research project (execution), manuscript (review and critique). Dr. Selkoe: research project (organization and execution), manuscript (review and critique). Dr. Schwarzschild: research project (execution), manuscript (review and critique). Dr. Schlossmacher and Dr. Hyman: research project (organization and execution), manuscript (review and critique). Dr. Sudarsky: research project (execution), manuscript (review and critique). Dr. Growdon: research project (conception, organization, and execution), manuscript (review and critique). Dr. Scherzer: research project (conception, design, supervision), analysis and interpretation, manuscript (revision and review).
STUDY FUNDING
Supported by NIH grants R01 NS064155 (C.R.S.), U01 NS082157 (C.R.S.), K24 NS060991 (M.A.S.), the Harvard NeuroDiscovery Center (to C.R.S. and B.T.H.), and the M.E.M.O. Hoffman Foundation (C.R.S.).
DISCLOSURE
H. Ding, K. Dhima, K. Lockhart, J. Locascio, A. Hoesing, K. Duong, A. Trisini-Lipsanopoulos, M. Hayes, and U. Shivraj Sohur report no disclosures. A.-M. Wills participates in the Schering-Plough–sponsored clinical trials of Preladenant, has consulted for Accordant (a CVS/Caremark disease management company), for Asubio Pharmaceuticals, and for NanoDerma Ltd., and has been funded by Schering-Plough/Merck, NIH (National Institute of Neurological Disorders and Stroke) grant U01 NS053369, and the Muscular Dystrophy Association. B. Mollenhauer is a speaker honoraria for Orion Corporation and GlaxoSmithKline, received funding for travel from GlaxoSmithKline, and for teaching presentations at Orion Corporation, and Teva Pharmaceutical Industries Ltd., was an Associate Editor for the Journal of Alzheimer Disease, owns patent WO2008058760 (method of differentially diagnosing dementias), and has pending patents 6612-60001 (novel ELISA-based quantification of α-synuclein proteins in CSF and peripheral blood products using 384-well plates) and 7570/17300PC (microRNA expression profiling of CSF), is a consultant for Bayer Schering Pharma AG, and is funded by Teva Pharmaceutical Industries Ltd., Desitin Pharmaceuticals, GmbH, Boehringer Ingelheim, GE Healthcare, the Michael J. Fox Foundation, the American Parkinson's Disease Association, and the Stifterverband für die Deutsche Wissenschaft (Dr. Werner Jackstädt-Stipend). A. Flaherty reports no disclosures. A. Hung received support from the National Parkinson Foundation. N. Mejia has received funding from the American Academy of Neurology, the Rappaport Foundation, Harvard Medical School, and NIH (National Institute of Neurological Disorders and Stroke) grant T32 NS048005. V. Khurana reports no disclosures. S. Gomperts is funded by Fidelity Biosciences, NIH (National Institute of Mental Health) grant K08 MH81207, the Michael J. Fox Foundation, and the Harvard NeuroDiscovery Center, and was funded by NIH grants from National Institute on Aging (NIA) (National Alzheimer's Coordinating Center) and National Institute of Neurological Disorders and Stroke R21 NS060310. D. Selkoe consults for and holds stock for Elan Corporation PLC. M. Schwarzschild is funded by NIH grants NS060991, NS054978, and NS0558324, DOD grant W81XWH-11-1-0150, the Michael J. Fox Foundation, and the RJG Foundation. M. Schlossmacher has collaborated with Genzyme-Sanofi, has received honoraria as a scientific advisor for Biogen Idec and Teva Neuroscience, and is funded by the Michael J. Fox Foundation, Government of Canada (CIHR), and the Parkinson Society Canada. B. Hyman is a consultant for Neurophage Pharmaceuticals, Pfizer, Inc., Siemens, and Takeda Pharmaceuticals, and is funded by Bristol-Myers Squibb, Biogen Idec, Inc., iPierian, Inc., Neotope, NIH grants P50 AG005134-27, R01 AG026249-06A1, R01 AG041507-01, R21 AG038835-01A1, P01 AG036694-01, R01 AG040530-01, U01 AG016976-13, and U01 AG016976-14, the American Health Assistance Foundation, and the Harvard NeuroDiscovery Center. L. Sudarsky is funded by NIH grant U01 NS053369. J. Growdon serves on the Scientific Advisory Board of Neurimmune Therapeutics, is funded by the Michael J. Fox Foundation and NIH (NIA-ADRC) grant P50 AG005134, and was funded by NIH grants from NIA (National Alzheimer's Coordinating Center) and National Institute of Neurological Disorders and Stroke R21 NS060310. C. Scherzer has collaborated with DiaGenic, Pfizer, Opko, and Proteome Sciences, and is funded by NIH grants R01 NS064155, R01 AG044113, U01 NS082157, U01 AT000613, and P01 NS058793, DOD grant W81XWH-13-1-0115, the Harvard NeuroDiscovery Center, the Michael J. Fox Foundation, and the M.E.M.O. Hoffman Foundation. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Scherzer CR, Eklund AC, Morse LJ, et al. Molecular markers of early Parkinson's disease based on gene expression in blood. Proc Natl Acad Sci USA 2007;104:955–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sato Y, Kikuyama M, Oizumi K. High prevalence of vitamin D deficiency and reduced bone mass in Parkinson's disease. Neurology 1997;49:1273–1278 [DOI] [PubMed] [Google Scholar]
- 3.Evatt ML, Delong MR, Khazai N, Rosen A, Triche S, Tangpricha V. Prevalence of vitamin D insufficiency in patients with Parkinson disease and Alzheimer disease. Arch Neurol 2008;65:1348–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Knekt P, Kilkkinen A, Rissanen H, Marniemi J, Saaksjarvi K, Heliovaara M. Serum vitamin D and the risk of Parkinson disease. Arch Neurol 2010;67:808–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang JY, Wu JN, Cherng TL, et al. Vitamin D(3) attenuates 6-hydroxydopamine-induced neurotoxicity in rats. Brain Res 2001;904:67–75 [DOI] [PubMed] [Google Scholar]
- 6.Butler MW, Burt A, Edwards TL, et al. Vitamin D receptor gene as a candidate gene for Parkinson disease. Ann Hum Genet 2011;75:201–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim JS, Kim YI, Song C, et al. Association of vitamin D receptor gene polymorphism and Parkinson's disease in Koreans. J Korean Med Sci 2005;20:495–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eyles DW, Burne TH, McGrath JJ. Vitamin D, effects on brain development, adult brain function and the links between low levels of vitamin D and neuropsychiatric disease. Front Neuroendocrinol 2013;34:47–64 [DOI] [PubMed] [Google Scholar]
- 9.Holick MF. Vitamin D deficiency. N Engl J Med 2007;357:266–281 [DOI] [PubMed] [Google Scholar]
- 10.Ding H, Sarokhan AK, Roderick SS, et al. Association of SNCA with Parkinson: replication in the Harvard NeuroDiscovery Center Biomarker Study. Mov Disord 2011;26:2283–2286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jankovic J, Kapadia AS. Functional decline in Parkinson disease. Arch Neurol 2001;58:1611–1615 [DOI] [PubMed] [Google Scholar]
- 12.Louis ED, Tang MX, Cote L, Alfaro B, Mejia H, Marder K. Progression of parkinsonian signs in Parkinson disease. Arch Neurol 1999;56:334–337 [DOI] [PubMed] [Google Scholar]
- 13.Alves G, Wentzel-Larsen T, Aarsland D, Larsen JP. Progression of motor impairment and disability in Parkinson disease: a population-based study. Neurology 2005;65:1436–1441 [DOI] [PubMed] [Google Scholar]
- 14.Maunsell Z, Wright DJ, Rainbow SJ. Routine isotope-dilution liquid chromatography-tandem mass spectrometry assay for simultaneous measurement of the 25-hydroxy metabolites of vitamins D2 and D3. Clin Chem 2005;51:1683–1690 [DOI] [PubMed] [Google Scholar]
- 15.Evatt ML, DeLong MR, Kumari M, Auinger P, McDermott MP, Tangpricha V. High prevalence of hypovitaminosis D status in patients with early Parkinson disease. Arch Neurol 2011;68:314–319 [DOI] [PubMed] [Google Scholar]
- 16.Bischoff-Ferrari HA, Willett WC, Wong JB, et al. Prevention of nonvertebral fractures with oral vitamin D and dose dependency: a meta-analysis of randomized controlled trials. Arch Intern Med 2009;169:551–561 [DOI] [PubMed] [Google Scholar]
- 17.Eyles DW, Smith S, Kinobe R, Hewison M, McGrath JJ. Distribution of the vitamin D receptor and 1 alpha-hydroxylase in human brain. J Chem Neuroanat 2005;29:21–30 [DOI] [PubMed] [Google Scholar]
- 18.Sanchez B, Relova JL, Gallego R, Ben-Batalla I, Perez-Fernandez R. 1,25-Dihydroxyvitamin D3 administration to 6-hydroxydopamine-lesioned rats increases glial cell line-derived neurotrophic factor and partially restores tyrosine hydroxylase expression in substantia nigra and striatum. J Neurosci Res 2009;87:723–732 [DOI] [PubMed] [Google Scholar]
- 19.Hennecke G, Scherzer CR. RNA biomarkers of Parkinson's disease: developing tools for novel therapies. Biomark Med 2008;2:41–53 [DOI] [PubMed] [Google Scholar]
- 20.Suzuki M, Yoshioka M, Hashimoto M, et al. Randomized, double-blind, placebo-controlled trial of vitamin D supplementation in Parkinson disease. Am J Clin Nutr 2013;97:1004–1013 [DOI] [PubMed] [Google Scholar]