

Abstract
The basic-helix–loop–helix-leucine zipper (bHLHZip) protein MITF (microphthalmia-associated transcription factor) is a master regulator of melanocyte development. Mutations in the MITF have been found in patients with the dominantly inherited hypopigmentation and deafness syndromes Waardenburg syndrome type 2A (WS2A) and Tietz syndrome (TS). Additionally, both somatic and germline mutations have been found in MITF in melanoma patients. Here, we characterize the DNA-binding and transcription activation properties of 24 MITF mutations found in WS2A, TS and melanoma patients. We show that most of the WS2A and TS mutations fail to bind DNA and activate expression from melanocyte-specific promoters. Some of the mutations, especially R203K and S298P, exhibit normal activity and may represent neutral variants. Mutations found in melanomas showed normal DNA-binding and minor variations in transcription activation properties; some showed increased potential to form colonies. Our results provide molecular insights into how mutations in a single gene can lead to such different phenotypes.
INTRODUCTION
Microphthalmia-associated transcription factor (MITF) is a bHLHZip transcription factor which belongs to the MYC supergene family. MITF plays an important role in melanocytes, where it has been shown to be involved in regulating many diverse events, including proliferation, differentiation, apoptosis, DNA replication, mitosis and genome stability (reviewed in 1 and 2). Mice which lack MITF have white coat color due to the absence of melanocytes (reviewed in 3).
Mutations in the human MITF gene have been found in patients with the hypopigmentation and deafness syndromes Waardenburg (WS) and Tietz (TS) (reviewed in 3–5). Both syndromes are listed as rare diseases by the Office of Rare Diseases Research (USA) and Orphanet (Europe). WS is an auditory-pigmentary syndrome named after the Dutch ophthalmologist Petrus Waardenburg (6). Four types of WS have been described, all with similar clinical features including sensorineural hearing loss, hypopigmentation of skin (usually patches of hypopigmentation) and hair and pigmentary disturbances of the irises (hypoblastic blue irides and/or heterochromia). In contrast to WS type 1 (WS1), WS2 patients do not show dystopia canthorum (eyelid anomaly; lateral displacement of the inner canthi of the eye) (6–8). The phenotype of WS3 and WS4 patients is similar to WS1 but with additional hypoplasia of limb muscles (WS3) or Hirschprung's disease (WS4). WS2 is inherited in an autosomal-dominant fashion (8). Mutations in MITF have been found in a subset of WS2 patients, categorized as type 2A. WS1 and WS3 have been shown to be due to mutations in PAX3, and WS4 to be due to mutations in the receptor–ligand pair EDN3/EDNRB or in the transcription factor SOX10 (reviewed in 9). SOX10 mutations have also been found in some cases of WS2, termed WS2E (reviewed in 9). TS is another rare hypopigmentation disorder characterized by profound congenital hearing loss and generalized hypopigmentation and is inherited in a fully penetrant autosomal-dominant fashion. MITF mutations have been found in TS patients (5,10–12).
In addition to being essential during melanocyte development, MITF also plays an important role in melanoma. Overexpression of MITF has been shown in cutaneous melanomas (13,14) and MITF gene amplification has been observed in about 30% of metastatic melanoma (15). Somatic mutations in MITF have been found in metastatic melanoma samples (16) and recently, a germline mutation was found in families where several members had melanoma (17,18). MITF has been proposed to fulfill all the criteria of a lineage-survival oncogene (reviewed in 19). As human MITF mutations are found in both auditory-pigmentary disorders and melanomas, it is of importance to understand how mutations in the same transcription factor can lead to such different phenotypes.
Here, we analyzed the DNA-binding and transcription activation ability of 18 MITF mutations found in WS2 and TS patients and 6 MITF mutations found to date in melanoma patients. Our results show that 11 of the 18 mutations associated with WS2 and TS failed to bind DNA and were unable to activate expression from melanocyte-specific promoters. On the other hand, the MITF mutations found in melanoma patients were able to bind DNA and activate expression from melanocyte-specific promoters; some showed increased potential to form colonies. Our functional analysis of human MITF mutations reveals novel insights into their ability to bind DNA, activate transcription and form colonies.
RESULTS
The human MITF mutations studied
A summary of all known human missense MITF mutations found in WS2A and TS patients as well as in melanomas is provided in Table 1, together with information on their location within the protein, the phenotypes assigned to the mutations, the mode of inheritance and the reference describing their characterization. All these mutations were originally identified in genomic DNA. Sometimes, the same mutation has been described in different families (such as R214X, R217del and R217I) and is therefore mentioned accordingly in Table 1. Figure 1A shows the location of the mutations in the protein. In addition to missense mutations, we included the R217del mutation which deletes three nucleotides leading to a deletion of arginine 217 and two stop mutations which result in truncated MITF proteins (R214X and R259X). The R214X mutation results in a protein which truncates in the basic region (BR), whereas R259X truncates in the helix–loop–helix domain. These mutations were included in the study as negative controls as we expected them to be functionally inactive. MITF mutations associated with WS2A and TS (indicated in blue) are located in the basic (11 of 18), helix–loop–helix (4 of 18) and zipper (2 of 18) domains of the protein; one mutation (S298P) is located in the carboxy terminal region of MITF (Fig. 1A). Mutations associated with melanoma (indicated in red) are primarily located in the amino (three of six) and carboxyl (two of six) ends of MITF, but also in the bHLHZip region (one of six) (Fig. 1A); all but one are somatic mutations. The only germline mutation associated with melanoma is E318K. Figure 1B shows where mutations, which affect the bHLHZip region, are located in the MITF structure (20).
Table 1.
Summary of Mitf mutations in WS and TS and melanomas used in this study
| Mutation | Location | Disease | Authors | Phenotype/remarks | Inheritance |
|---|---|---|---|---|---|
| E87R | AD | Melanoma | Cronin et al. (16) | Metastatic melanomas with BRAF (V600E) mutation | Somatic |
| L135V | AD | Melanoma | Cronin et al. (16) | Metastatic melanomas | Somatic |
| L142F | AD | Melanoma | Cronin et al. (16) | Metastatic melanomas | Somatic |
| R203K | BR | WS2 | Tassabehji et al. (5) | Typical WS2. Mutation found in proband does not segregate with disease | Neutral variant |
| K206Q | BR | WS2 | Leger et al. (12) | 36-year female; SNHL, white forelock blue irides | Familial |
| N210K | BR | Tietz | Smith et al. (10) | Deafness, blonde hair, white eyelashes & eyebrows, cutaneous hypopigmentation | Familial |
| I212M | BR | Tietz/WS2 | Welch et al. (43) | Blue eyes, hypopigmentation of hair and skin, 14 of 28 affected members have SNHL | Familial |
| I212S | BR | WS2 | Leger et al. (12) | 9-year male; generalized hypopigmentation, freckles, blue irides, premature graying | Familial |
| E213D | BR | WS2 | Leger et al. (12) | 33-year female; SNHL, white forelock, premature graying, blue irides | De novo |
| R214X | BR | WS2 | Lalwani et al. (42) | SNHL, heterochromia of the irides, premature graying, white forelock, a three-generation WS2 family | Familial |
| - | - | WS2 | Nobukuni et al. (40) | SNHL, heterochromia of the irides, white forelock, early graying | Familial |
| R216K | BR | WS2 | Leger et al. (12) | 21-year female; SNHL, white forelock, fair skin, blue irides | De novo |
| R217del | BR | Tietz | Leger et al. (12) | 3-year female; SNHL, generalized hypopigmentation, blue irides | Familial |
| - | - | WS2 | Chen et al. (41) | Three affected families | Familial |
| - | - | Tietz | Tassabehji et al. (5) | SNHL, blonde/red hair color, pale skin, blue irides; dominant pedigree | Familial |
| -l | - | Tietz | Shigemura et al. (11) | 15-year male, generalized hypopigmentation, blue irides, blonde hair and eyebrows | Familial |
| R217I | BR | Tietz | Léger et al. (12) | 3-year female; SNHL, generalized hypopigmentation, blue irides and white forelock | Familial |
| - | - | WS2 | Chen et al. (41) | SNHL, blue irides, premature graying | De novo |
| R217G | BR | WS2 | Chen et al. (41) | No data available | n/a |
| I224S | HLH | WS2 | Pingault et al. (9) | 6-year female; SNHL, white forelock, heterochromia of the irides | Familial |
| G244R | HLH | Melanoma | Cronin et al. (16) | Metastatic melanomas with BRAF (V600E) mutation | Somatic |
| S250P | HLH | WS2 | Tassabehji et al. (5) | Female patient; unilateral hearing loss, premature graying | Familial |
| Y253C | HLH | WS2 | Read et al. (8) | No data available | n/a |
| R259X | HLH | WS2 | Nobukuni et al. (40) | SNHL, heterochromia of the irides, white forelock, early graying | Familial |
| N278D | ZD | WS2 | Tassabehji et al. (5) | Female patient; SNHL, inherited from unaffected father | Familial |
| L283P | ZD | WS2 | This study | 30-year female; SNHL, heterochromia of the irides | Familial |
| S298P | AD | WS2 | Tassabehji et al. (5) | Affected patient comes from a three-generation history of WS2 | Neutral variant |
| E318K | AD | Melanoma | Bertolotto et al. (17) | Five of 62 melanoma and renal cell carcinoma patients harbor the E318K mutation | Germline |
| D380N | AD | Melanoma | Cronin et al. (16) | Metastatic melanomas with a BRAF (V600E) mutation | Somatic |
AD, activation domain; BR, basic region; HLH, helix–loop–helix; ZD, zipper domain; SNHL, sensorineural hearing loss, n/a not available.
Figure 1.
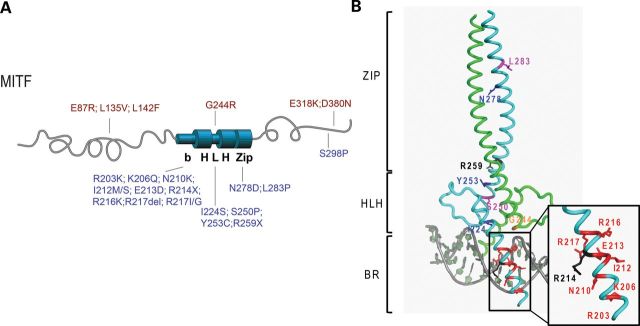
MITF mutations found in melanoma, WS2A and TS patients. (A) A graphic depiction of the human MITF protein with the location of the mutations indicated. The mutations found in melanoma patients are indicated in red (on top). These mutations are located at the amino- and carboxyl ends, and only one mutation is located in the helix–loop–helix domain (G244R). The mutations found in WS2A and TS patients are indicated in blue (on the bottom). Most of these mutations are located in the BR of MITF and in the HLH and zipper domains. The WS2A mutation S298P is located at the carboxyl end of MITF. (B) Structure of the bHLHZip region of MITF showing the location of the mutant residues. To separate mutations leading to TS and WS2A syndromes and those causing melanoma, the respective mutations are mapped on two different molecules of the dimeric MITF structure. The TS/WS2A mutations on the first protomer in cyan are highlighted in the following colors: residues mutated into stop codons, black; residues mutated into prolines, magenta; residues when mutated affecting direct DNA contacts, red; residues when mutated affecting protein–protein interactions, blue. In sharp contrast, only one melanoma-inducing mutation has been found within the bHLHZip region for which the structure of MITF was determined. This mutation (G244) has been highlighted in orange on the second MITF protomer, colored in green.
For functional analysis, all the mutations were re-created in a mouse Mitf expression construct which produces the melanocyte-specific M-MITF protein (reviewed in 3). Sequence alignment of the mouse and human MITF proteins showed 94% identity; all the mutant sites are completely conserved between the two species (Supplementary Material, Fig. S1). In addition, we created two different E213D mutations [the original c.639A>C mutation identified by (21) and a c.639A>T mutation (not found in WS2A patients) both of which result in a substitution for aspartic acid] and the N278P mutation which introduces a helical breaker in the leucine zipper segment of the protein.
Eleven out of 18 WS2A and TS mutations failed to bind DNA
To determine DNA-binding ability of the wild-type and mutant MITF proteins, electrophoretic mobility shift assays (EMSAs) were performed using a double-stranded oligonucleotide with a central E-box (CACGTG) as a probe. The proteins were expressed using an in vitro translation system; expression analyzed using western blot analysis and protein levels adjusted to eliminate differences in DNA-binding studies (Supplementary Material, Fig. S2).
As shown in Figure 2, 6 of the 11 mutations which affect the BR of MITF (E213D, R214X, R216K, R217Del, R217I, R217G) failed to bind the E-box DNA sequence (CACGTG). The R203K and N210K mutant proteins were able to bind to the E-box probe equal to wild-type MITF, whereas the K206Q mutant protein showed some but decreased binding ability (Fig. 2A). Changing residue I212 to either methionine (I212M) or serine (I212S) did not affect the ability to bind the E-box sequence (Fig. 2B). Mutations in the HLH domain (I224S, S250P and R259X) induced loss of DNA binding (Fig. 2B and C). Interestingly, the tyrosine-to-cysteine substitution at residue 253 was able to bind DNA, albeit weakly (Fig. 2C). Mutations affecting the zipper domain (N278D/P and L283P) also failed to bind DNA. The L283P mutation is a novel non-truncating MITF mutation found in a WS2A patient (Table 1). The only WS2A mutation located at the C-terminal region of MITF, S298P, did not affect the DNA-binding ability. In summary, 11 out of the 18 WS2A and TS mutations investigated failed to bind DNA.
Figure 2.
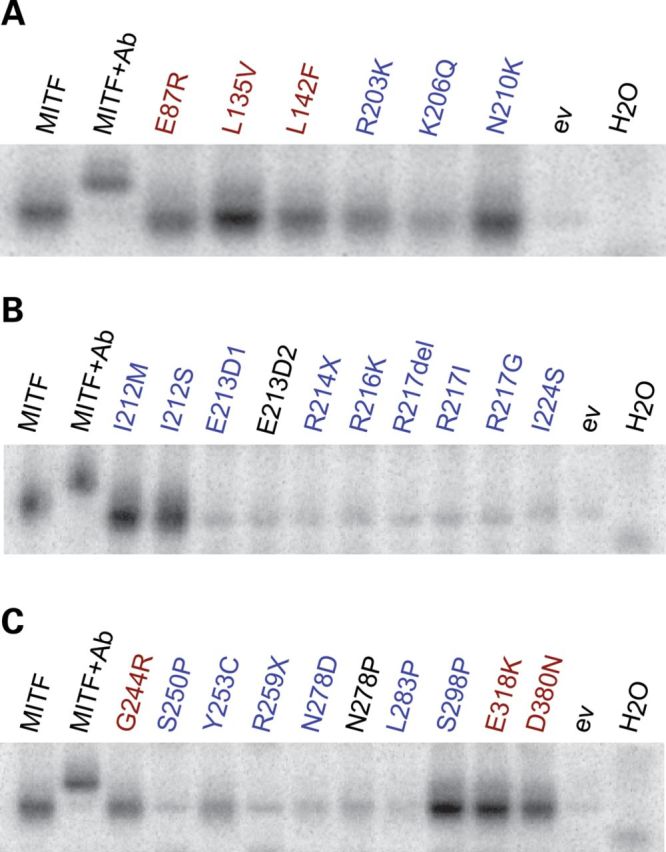
MITF mutations and their ability to bind the E-box. (A–C) EMSAs showing the binding of wild-type and mutant MITF proteins to the E-box sequence (CACGTG). In each case, Lane 1 shows binding of the wild-type MITF protein, and Lane 2 shows a supershift where the C5 MITF antibody was added. Empty vector pcDNA3.1 (ev) and water (H2O) were loaded as negative controls. Melanoma mutations are indicated in red, whereas WS2A and TS mutations are indicated in blue. The synthetic E213D2 and N278P mutations are indicated in black. Representative data from three separate experiments are shown.
MITF also binds strongly to the TCATGTG sequence, sometimes referred to as the M-box sequence (20). In order to investigate if the MITF mutant proteins which showed E-box binding (R203K, K206Q, N210K, I212M, I212S and S298P) can also bind this sequence, gel shift assays were performed using TCATGTG as a probe. As was seen with the E-box probe, the R203K, N210K and S298P mutations can bind the M-box sequence similar to wild–type, whereas K206Q has severely impaired DNA-binding ability (Supplementary Material, Fig. S3). Consistent with previous results (20), the two I212 mutations (I212M/S) showed less binding to the M-box than to the E-box (Supplementary Material, Fig. S3). In summary, our results show that most mutations associated with WS2A and TS failed to bind DNA. The exceptions are R203K, N210K and S298P which show normal-level DNA binding, and the Y253C and K206Q mutations which show reduced but significant DNA binding. The two I212 mutations, I212M and I212S, affect the ability of MITF to bind the M-box but not the E-box. This position has been suggested to determine the ability of MITF to bind the M-box sequence (20).
DNA-binding ability of MITF mutations found in melanoma is similar to wild-type MITF
Gel shift assays were also used to determine the DNA-binding ability of MITF mutations found in melanoma patients. The E87R, L135V, L142F, G244R, E318K and D380N mutations did not affect the DNA-binding ability of MITF to either the E-box (Fig. 2A and C) or the M-box (Supplementary Material, Fig. S3). Interestingly, the L135V protein showed increased DNA binding when compared with wild-type MITF (Fig. 2A and Supplementary Material, Fig. S3).
Reduced transcription activation potential of WS2A and TS mutants
In order to investigate whether the MITF mutants were able to activate gene expression, we performed reporter gene assays with several different MITF target promoters. MITF has been shown to activate expression from the melanocyte-specific tyrosinase (TYR), tyrosinase-related protein 1 (TYRP1) and dopachrome tautomerase (DCT) promoters (22–24) (Supplementary Material, Fig. S4). HEK293T cells, which do not express MITF endogenously, were cotransfected with the respective promoter constructs and MITF expression constructs (wild type or mutant) and effects on expression determined using luciferase reporter assays. Supplementary Material, Figure S5 shows that all the transfected cells express the MITF protein. Figure 3A shows fold activation by the mutant proteins when compared with wild-type MITF for the four different promoters used. Consistent with the results of the DNA-binding assays, 11 of the 18 mutations which affect the basic (E213D1/2, R214X, R216K, R217Del, R217I and R217G), the HLH (I224S, S250P and R259X) and zipper domains (N278D/P and L283P) of MITF failed to activate expression from the TYR promoter (P < 0.05). The R206Q and the Y253C mutant proteins showed a significantly reduced ability to activate tyrosinase expression [48 and 71% compared with wild-type MITF (P < 0.05)], consistent with the reduced ability to bind DNA. The two I212 mutations (I212M and I212S) also showed a significantly reduced ability to activate expression from the TYR promoter (P < 0.05). The TYR promoter contains one E-box and one M-box. Since both mutant proteins bound the E-box at similar levels as the wild-type MITF protein but showed reduced binding to the M-box, this suggests either that the M-box is more important for activating TYR expression or that these sites act cooperatively. Alternatively, the I212 mutations may affect transactivation ability of MITF. Surprisingly, the R203K, N210K and S298P mutations, which are all able to bind the E-box, showed significantly increased transcription activity compared with wild-type MITF (P < 0.05). Indeed, the activation of tyrosinase by the R203K and N210K mutations was 1.5- and 2.2-fold compared with wild-type MITF (P = ≤5 × 10−4 and <9.3 × 10−6, respectively), whereas the S298P mutation activated TYR 1.3-fold compared with wild-type MITF (P = 0.042).
Figure 3.
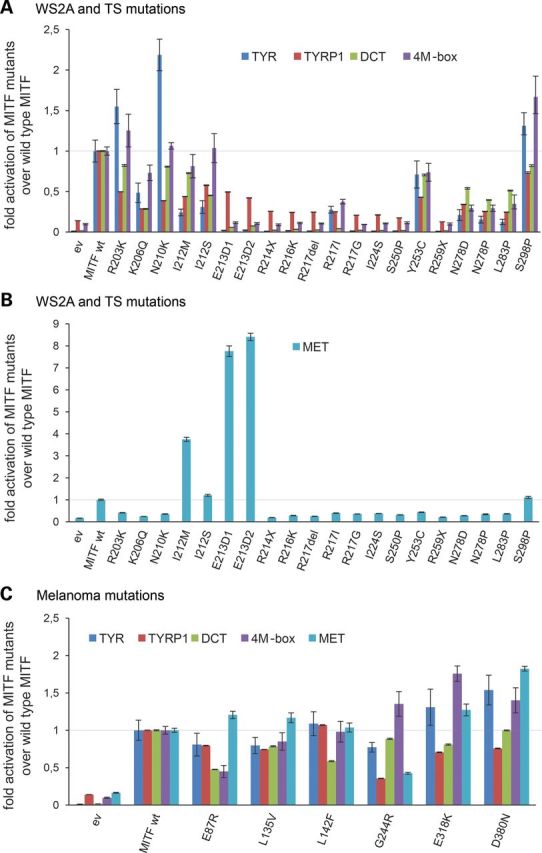
Transcriptional activity of MITF mutations associated with WS2A, TS and melanoma. (A) Transcription activation ability of WS2A and TS mutant proteins from the TYR, TYRP1, DCT and 4M-box promoters. The data are shown as fold activation of MITF mutants over wild-type MITF. Error bars indicate the standard error of mean. (B) Transcription activation ability of WS2A and TS mutations from the MET promoter. The I212M and E213D mutations show hyperactivation from the MET promoter. The data are shown as fold activation of MITF mutants over wild-type MITF. Error bars indicate the standard error of mean. (C) Transcription activation ability of the melanoma mutations from the TYR, TYRP1, DCT, 4M-box and MET promoters. All cotransfection assays were repeated twice in six replicates and fold induction calculated over MITF wild type.
We also determined the ability of the mutations to activate expression of the pigment-cell specific TYRP1 promoter (Fig. 3A). The mutations either completely failed to activate expression from this promoter (R214X, R216K, R217Del, R217I, R217G, I224S, S250P, R259X, N278D, N278P and L283P) or showed significant reduction (R203K, K206Q, N210K, I212M/S, E213D and Y253C) in activation when compared with wild type (P < 0.05). The hyperactivation observed with the R203K, N210K and S298P mutant proteins from the TYR promoter was not observed using the TYRP1 reporter nor with the other reporters used in the study (see below). In fact, all the three mutations showed reduced TYRP1 activation when compared with wild-type MITF (Fig. 3A).
Similar to what was observed with the TYR promoter, MITF mutations which affect the basic and HLH regions failed to activate expression from the DCT promoter (Fig. 3A). The K206Q, I212S, N278D, N278P and L283P mutations showed reduced activation, whereas the E213D, R214X, R216K, R217Del, R217I, R217G, I224S and R259X mutations showed a complete lack of activation (P < 0.05). The R203K, N210K and S298P mutations showed somewhat reduced transcription activation potential (82.4, 80.7 and 81%, respectively) when compared with the wild-type MITF protein, but the difference was not statistically significant. As these mutations lead to increased TYR activation, this suggests that the mutant proteins can have promoter-specific effects on transcription. The I212M and Y253C proteins showed slightly but significantly reduced transcription activation of DCT (72.7 and 70.5%, respectively, compared with wild-type MITF, P < 0.05).
MITF robustly activates expression of a synthetic promoter consisting of four tandem M-box sequences (23). The R203K, N210K and I212M/S mutations were able to activate expression from this promoter construct at levels similar to wild-type MITF (Fig. 3A), whereas the K206Q mutation showed reduced activation (73.1% compared with wild-type MITF, P = 0.0075). The only mutation which showed hyperactivation from this synthetic promoter was S298P, which showed a 1.67-fold increase (P < 0.05). The remaining mutations (E213D, R214X, E216K, R217Del, R217I, R217G, I224S, S250P, Y253C, R259X, N278D, N278P and L283P) failed to activate the 4M-box construct (P < 0.05).
The proto-oncogene MET, which encodes the cell-surface receptor for hepatocyte growth factor (25), has been implicated in renal cell carcinoma (26) and in late stages of melanoma (27). Chromatin-immunoprecipitation coupled to high throughput sequencing of a human melanoma cell line (501mel) revealed that MET is a target of MITF and that MITF is able to activate expression from the MET promoter (17,28) (Supplementary Material, Fig. S4b). Most of the MITF mutations associated with WS2A and TS (R203K, K206Q, N210K, R214X, R216K, R217Del, R217I, R217G, I224S, S250P, Y253C, R259X, N278D/P and L183P) were unable to activate expression from the MET promoter (P < 0.05) (Fig. 3B). The I212S and S298P mutations showed activation potential similar to wild-type MITF (1.2- and 1.1-fold, respectively). Although the R203K, K206Q, N210K and Y253C proteins were able to bind DNA, they were unable to activate expression from the MET promoter. Surprisingly, the I212M and E213D mutations showed strong activation of the MET promoter, 3.7- and 8-fold activation compared with wild-type MITF, respectively (Fig. 3B). The I212M mutation alters the DNA-binding profile of MITF, such that it binds less to the M-box than to E-box sequences (20), whereas the E213D mutation cannot bind the E-box sequence.
To summarize, our results suggest that 11 of the 18 mutations associated with WS2A and TS located within the BR (E213D, R214X, R216K, R217Del, R217I and R217G), the HLH domain (I224S, S250P and R259X), and the three mutations affecting the zipper domain (N278D and L283P) fail to activate expression from melanocyte-specific promoters such as TYR TYRP1, DCT and the synthetic 4M-box promoter (Fig. 3A and B). However, mutations at the beginning of the BR (R203K and N210K) and the S298P mutation at the C-terminus can still activate expression from many pigment-cell specific promoters. Importantly, 2 of the 18 mutations lead to a promoter-specific increase in expression, most notably the MET promoter.
Mutations associated with melanoma have promoter-specific effects on transcription
We also tested the transcription activation ability of MITF mutations associated with melanoma. Interestingly, these mutations had promoter-specific effects on transcription activation, and the effects depended on where in the protein the mutations are located (Fig. 3C). The E87R mutation showed statistically significant reduced activation from the TYR, TYRP1, DCT and 4M-box promoters and L135V for TYR, TYRP1 and DCT (P < 0.05). However, both proteins activated MET at normal levels. The L142F mutant protein showed a normal level of transcription activation from all promoters tested, except DCT, which was reduced to ∼50% of the levels seen in wild-type MITF (Fig. 3C). The G244R mutation, located in the HLH domain of MITF, showed varied promoter-specific effects with reduced activation of TYR, TYRP1 and MET promoters, normal activation of DCT and increased expression of the synthetic 4M-box promoter (1.35-fold induction; P < 0.05). Interestingly, the two mutations located in the carboxyl end of MITF, E318K and D380N showed significantly increased activation potential from the TYR (1.3- and 1.5-fold, respectively), 4M-box (1.7- and 1.4-fold, respectively) and MET (1.3- and 1.82-fold, respectively) promoters; activation of TYRP1 was reduced in both cases whereas activation of DCT was not statistically different from wild type (Fig. 3C).
Nuclear localization
Since the results of the transcription activation assays were performed in cell-based assays using HEK293T cells, it was possible that the effects observed in those assays were due to altered nuclear localization of the mutant proteins. Thus, we determined the nuclear localization of all the MITF mutations by transfecting HEK293T cells with each of the mutant proteins and then used the 1D2 monoclonal anti-MITF antibody (29) to determine subcellular localization. The results show that all the mutations are primarily located in the nuclei (Supplementary Material, Fig. S6), except the two deletion mutations R214X and R259X which also have a major cytoplasmic component as seen by staining with the C5 anti-MITF antibody (the 1D2 antibody recognizes the C-end of MITF which is missing from the two mutants). Surprisingly, the R217del mutation which has been shown to affect nuclear localization of MITF in NIH3T3 and 293T cells (30) was nuclear in our assay using either the 1D2 or C5 antibodies.
Clonogenicity
In order to investigate whether the MITF mutants associated with melanoma have clonogenic properties that differ from the wild-type protein, we created stable HEK293 cell lines carrying the MITF mutations E87R, L142F, G244R and D380N. As controls, we also created stable lines expressing wild-type MITF and empty vector. All the lines created expressed the respective proteins at similar levels (Supplementary Material, Fig. S7). In order to investigate the clonogenic potential of these mutant proteins, we seeded 2500 cells in three replicates and cultured the cells for 1 week. Cells transfected with an empty vector or wild-type MITF showed similar clonogenic potential as HEK293 cells alone (Fig. 4). Although there were slight variations in the colony formation of empty vector transfected cells and untransfected HEK293 cells, these differences were not statistically significant (Fig. 4). As shown in Figure 4, the melanoma mutations E87R and L142F showed significantly higher clonogenicity than wild-type MITF (34 and 20% increase, respectively). However, the G244R and D380N mutations showed the same clonogenic behavior as wild-type MITF.
Figure 4.

Clonogenic behavior of MITF mutations associated with melanoma. Untransfected HEK293 cells, empty vector and wild-type MITF transfected cells showed similar clonogenic potential. The slight differences observed were not statistically significant. The melanoma mutations E87R and L142F showed significantly (P < 0.05, indicated by asterisks) higher clonogenic potential than wild-type MITF. The G244R and D380N mutations behaved like wild-type MITF.
DISCUSSION
Our analysis of the DNA-binding and transcription activation potential of human MITF mutations associated with melanoma and WS2A and TS is summarized in Table 2. Eleven of 18 WS2A and TS mutations showed no DNA-binding or transcription activation potential. Many of the mutations affect residues in the DNA-binding domain which make specific contacts with DNA, including E213 and R217 as well as R216, which does not make direct contacts with DNA but is conserved in the MITF subfamily of bHLHZip proteins (20). The exact role of R216 is not clear, and it is surprising that changing this residue for lysine, another basic amino acid, has this large effect on DNA binding. The R217del, R214X and R259X mutations have no affinity for DNA (Fig. 2) (20) and, therefore, serve as negative controls. The R214X mutation is missing a significant portion of the DNA-binding domain and all the rest of the protein, and can thus not bind DNA. The R259X mutation is missing everything following the HLH domain and presumably cannot form DNA-binding homodimers. Consistent with that conclusion, other mutations affecting the structure of the HLH and ZIP regions also affect the DNA-binding ability of MITF. This includes the S250P, L283P, I224S and N278D mutations. In the structure of MITF, N278 plays an important role in forming hydrogen bonds in one of the heptad repeats of the leucine zipper, thus connecting the two MITF protomers (20). Consequently, DNA-binding ability is abolished when this residue is mutated to aspartate (Fig. 2C). Mutating N278, S250 and L283 to proline, an amino acid that cannot be accommodated in an α-helix, disrupts proper folding of the respective regions and, therefore, impairs the dimerization ability of MITF. Due to its location in the hydrophobic core of the HLH domain, we expect the I224S mutation to disrupt the nearby three-dimensional structure as well. The fact that mutations affecting dimerization of MITF lead to dominant phenotypes suggests that haploinsufficiency can lead to WS2A and TS.
Table 2.
Summary of DNA-binding assays and transcriptional activation analysis of MITF and MITF mutants; WS2A and TS mutations indicated in blue and melanoma mutations are indicated in red [shown in percentages which represent densitometric scans of EMSA gels (DNA-binding) and transcriptional activation ratios]
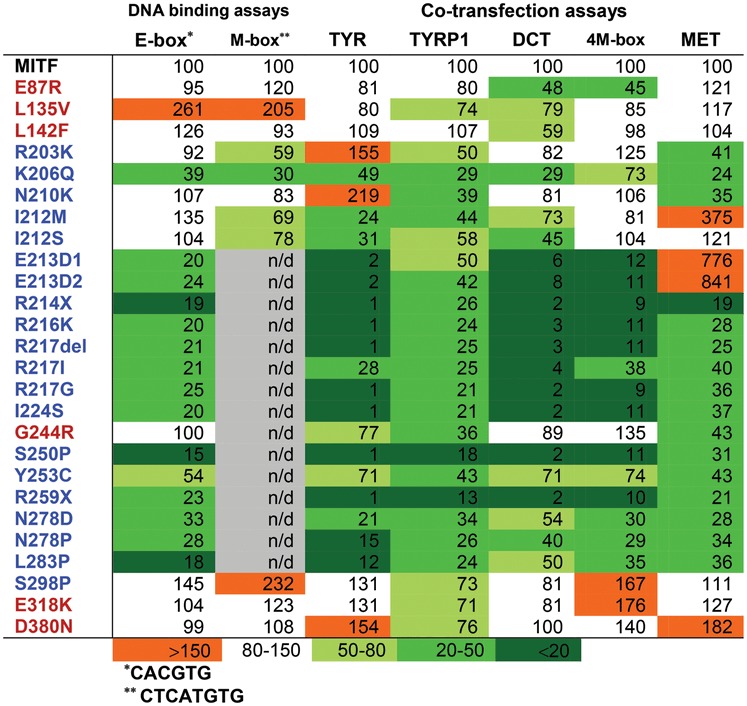 |
Surprisingly, a third of the WS2A and TS mutations showed normal or near-normal DNA-binding and transcription activation ability. The R203K and K206Q mutations showed somewhat reduced DNA binding, whereas the N210K, I212M, I212S and S298P mutations all showed normal-level binding to the E-box sequence. All six mutations showed significant transcription activation ability from some of the promoters tested, especially R203K, N210K and S298P, which activate expression of some promoters at normal levels and some at increased levels. The R203K mutation was originally described in a patient who inherited it from his unaffected mother (5). The family had a four-generation history of WS2, but only one member harbored the R203K mutation (5). This, together with the fact that the R203K mutant protein behaves like the wild-type MITF protein suggests that this is a neutral variant and not a disease-causing mutation. The side chain of the R203 residue was not visible in the structure of the MITF protein (20), and at present its role is not entirely clear. Residues 210–221 of MITF have been suggested to contain a nuclear localization signal (30). The I212N mutation is able to enter the nucleus (30) and so is the R217I mutation (31). The nuclear localization potential of the other mutations affecting this region has not been tested; all fail to bind DNA in vitro, except N210K.
The N210K mutation was originally identified in a family where affected family members were reported as born ‘snow-white’ but gradually gained some pigmentation; they had blue eye color and bilateral congenital sensorineural and profound hearing loss (10). Surprisingly, the N210K mutant protein retains significant DNA-binding and transcriptional activity. Interestingly, this position has been shown to be mutated to serine (N210S) in macchiato Franches-Montagnes horses (also called Freiberger). MITF is 97.9% conserved between humans and horses; the N210 position is conserved (Supplementary Material, Fig. S8). An EMSA showed that the N210S mutation had reduced ability to bind the M-box probe (32). The N210S mutation thus has more severe effects on protein function in vitro than the N210K mutation. Consistent with this, lysine is a positively charged and long-chained residue that has the ability to maintain the hydrogen bond with DNA in which the wild-type residue is involved, whereas serine, a neutral and short-chained residue, does not. Both the human and horse mutations are inherited in a dominant fashion and both have severe effects on the phenotype, particularly with respect to effects on hearing. The different effects on function are therefore surprising and call for further investigations.
The S298P mutation is located at the carboxyl end of the leucine zipper of MITF. It has been suggested that S298 might be phosphorylated by GSK3β, and that substituting serine for either alanine or proline destroys MITF as a substrate for GSK3β (33). GSK3β phosphorylates many of its substrates via the optimal consensus site S/T-X-X-X-S/T, where S and T are phosphoserine and phosphothreonine and X any amino acid (34,35). Interestingly, S298 does not fulfill all the criteria of a GSK3β substrate (36). Takeda et al. (2000) showed that the S298A or S298P MITF mutations severely affected binding to the M-box sequence and ability to activate expression from the TYR promoter, whereas substituting serine 302 with alanine had no effect on the ability to activate TYR (33). Our results, showing that S298P has normal DNA binding, are not consistent with the results of Takeda et al. (33). Similarly, substituting S298 with alanine and performing cotransfection assays with the TYR, 4M-box, TYRP1 and MET promoter constructs revealed no statistically significant difference from wild-type MITF (Supplementary Material, Fig. S9). Analysis of the Exome Variant Server database (http://evs.gs.washington.edu/EVS/) shows that a C/T polymorphism (rs104893747) which changes serine to proline at residue 298 in MITF is prevalent in European Americans and has a minor allele frequency of 0.046%. This polymorphism was not found in sequenced African Americans. The prevalence of all WSs combined is estimated to be 1/42 000 of the population (8), suggesting that rs104893747 is much more frequent than all WSs. This, together with our results showing that the S298P mutation has no major effect on function, suggests that this is a rare polymorphism in Caucasian populations that is functionally equivalent to the wild-type protein. The phenotype assigned to the S298P polymorphism must therefore be due to another non-identified non-coding mutation in the MITF gene, or to a mutation in another gene associated with WS.
The I212M and I212S mutations showed normal ability to bind the E-box whereas ability to bind the M-box was reduced (Fig. 2, Supplementary Material, Fig. S3). This is consistent with the results of Pogenberg et al. who showed that the I212 position is critical for allowing MITF to bind the M-box sequence (20). Mutating this site alters the target gene profile of MITF, leading to an imbalance in the activation of target genes, as observed by the hyperactivation of the MET promoter by the I212M mutation and the reduced ability to activate the TYR, TYRP1 and DCT promoters. Surprisingly, both mutations are able to activate expression of the 4M-box promoter. An imbalance in target gene selection is a novel model for how mutations can lead to WS2A and TS.
Paradoxically, despite severely affecting DNA binding of MITF and reducing activation from TYR, TYRP1, DCT and 4M-box promoters, the E213D mutations lead to hyperactivation of the MET promoter. It is possible that this mutant protein can bind sequences other than the E-box to activate expression of the MET promoter. It is also possible that this mutation activates MET through another mechanism that does not involve direct binding to DNA. Interestingly, the I212M and E213D mutations, both of which hyperactivate the MET promoter, affect adjacent residues of the basic DNA-binding domain. The hyperactivation data suggest that mutations in MITF can have specific effects on gene expression or function, despite severely affecting the ability of MITF to bind the E-box.
We also analyzed the functional effects of somatic MITF mutations identified in melanoma tumor tissues; E87R, L135V, L142F, G244R and D380N (16) and a recently described germline mutation, E318K (17,18). Our results show that all the melanoma mutations were able to bind the E-box probe at normal levels, and most were capable of activating expression from melanocyte-specific promoters, although minor differences were observed, depending on both the mutation involved and the promoter investigated. Critically, four mutations (E87R, L135V, E318K and D380N) tended to increase activation from the MET promoter; this increase was statistically significant only for the D380N mutation (Fig. 3C). One of these mutations, L135V, showed increased DNA-binding ability, but this was not reflected in effects on transcription activation. The two mutations located in the C-terminus of MITF, E318K and D380N significantly increased transactivation potential of the tyrosinase and 4M-box promoters, but only D380N showed statistically significant activation of the MET promoter, suggesting that these mutations lead to increased but promoter-dependent transcription activation potential of MITF. This is consistent with the results of Bertolotto et al. (2011) who showed that the E318K mutation led to increased transcription activation of the HIF1α and synthetic 3M-box reporters (17). Consistent with our results, Bertolotto et al. did not observe any difference between wild-type MITF and the E318K mutant with respect to activation of the MET promoter (17). Although the E87R and L135V mutations tend to increase activation from the MET promoter, both mutations lead to reduced transcription activation potential from the pigment-specific promoters. This indicates that the effects of these two mutations are more subtle than the effects of the E318K and D380N mutations and may affect target genes differently. The L135V and L142F mutations are located within a proposed activation domain of MITF (37,38). Consistent with that both mutants showed significantly decreased transcriptional activation of the TYRP, TYRP1 and DCT promoters. The germline mutation E318K has recently been associated with melanoma and occurs within a SUMOylation consensus motif, IKQE. Since SUMOylation is generally reported to have inhibitory effects with respect to transcriptional activation, a mutation inhibiting SUMOylation may lead to activation of protein function (17,18,23).
The clonogenic assays showed that the E87R and L142F mutations increased clonogenic potential of MITF, whereas the D380N mutation did not. Unfortunately, very little is known about the functional role of the N and C domains of MITF. However, our results suggest that these domains have distinct function, and that they can lead to melanoma through different pathways, either leading to increased proliferation through mechanisms depending on the N-end of MITF, or by affecting transcription activation potential through features of the C-end of MITF. How these effects are mediated is not known at present.
In summary, we have shown that 11 of 18 MITF mutations associated with WS2A and TS, located in the bHLH-ZIP domains of the MITF protein, fail to bind DNA and activate melanocyte-specific promoters in cotransfection assays. The remaining seven mutations either have normal or reduced DNA-binding and transcription activity. The MITF mutations found in melanoma behave similar to the wild-type MITF protein in terms of DNA binding. However, these mutations affect transactivation potential and possess the ability to form colonies in clonogenic assays. Clearly, the amino- and carboxyl domains of MITF, where these mutations are located, play an important role in transactivation ability of MITF and the oncogenic role of MITF. The roles that these domains play in MITF function need further analysis.
MATERIALS AND METHODS
Cell culture
HEK 293 (ATTC; CRL-1573) and HEK293T cells were maintained in Dulbeccós modified Eagle Medium (GIBCO), supplemented with 10% fetal bovine serum, 100 U of penicillin/ml and 100 μg of streptomycin/ml and cultured in a humidified incubator at 37°C with 5% CO2. HEK293T cells are derived from HEK293 cells but contain the large T antigen and are resistant to Geneticin.
Cotransfection assays
HEK 293T cells (1 × 105) were seeded in 96-well plates and transfected with 35 ng of Luciferase reporter construct (TYR, TYRP1, DCT, 4M-box and MET) and 15 ng wild-type or mutant mouse MITF constructs using ExGen 500 in vitro transfection reagent (Fermentas). pRenilla (1.5 ng) was used to normalize luciferase activity. Twenty-four hours after transfection, cells were lysed with passive lysis buffer (Promega) and assayed for firefly and Renilla luciferase activity according to manufacturer's instructions (Promega). Transfection experiments were carried out in six replicates each time and repeated at least twice. Measurements were taken with a Modulus II microplate reader (Turner Biosystems). Luciferase signals were normalized to corresponding Renilla signals and results expressed as fold activation over wild-type MITF. Error bars show the standard error of mean and significance was calculated via Student's t-test (P < 0.05 have been considered statistically significant).
Mutagenesis
Mutations were created in an expression vector (pcDNA3.1) containing the wild-type mouse MITF gene using the QuickChange Lightning site-directed mutagenesis kit from Stratagene and verified by Sanger sequencing. The QuickChange primer design program provided by Agilent Technologies was used to design the primers (listed in Supplementary Material, Table S1).
Protein expression
Wild-type and mutant MITF proteins were synthesized using the TNT quick coupled transcription/translation system according to manufacturer's protocol (Promega). TNT proteins were analyzed by electrophoresis on 8% SDS–polyacrylamide gels. After electroblotting the proteins on a polyvinylidine difluoride membrane, membranes were incubated with an MITF antibody (C5, Thermo Scientific) and β-actin (13E5, Cell Signaling) overnight at 4°C in phosphate buffered saline (PBS) with 5% milk. The membranes were scanned on an Odyssey reader (Li-Core), and the bands quantitated and compared by intensity comparison to wild-type MITF (Supplementary Material, Fig. S2). To ensure equal protein loading on the EMSA gel, protein amounts were adjusted accordingly. For determining expression of transfected MITF proteins in cotransfection assays, constructs expressing wild-type and mutant MITF proteins were transfected into HEK293T cells and the cells lysed in RIPA buffer. Electrophoresis and electroblotting were performed as described above followed by incubation with the 1D2 antibody overnight at 4°C in PBS with 5% milk.
EMSA
The double stranded oligonucleotides encompassing MITF-binding sequences E-box 5′-AAAGAGTAGCACGTGCTACTCAGA-3′ and M-box 5′-AAAGAGGCTCATGTGCTA-ACCAGA-3′ were 32P-dCTP-end-labeled (PerkinElmer) using the Klenow fragment (Fermentas) and subsequently purified using Sephadex G50. TNT expressed proteins were incubated in 2 mm spermidine, 2 mm MgCl2, 10% fetal calf serum, 2 ng/μl poly(dI)(dC)) for 15 min on ice. For supershift assays, 0.5 μl Mitf antibody (C5, Thermo Scientific) was added and incubated for 30 min on ice. The labeled probe was incubated with binding buffer (39) for a couple of minutes at room temperature and added to the TNT protein. After a 10 min incubation period, the DNA–protein complexes were resolved by electrophoresis on a 4.2% non-denaturating polyacrylamide gel in 0.25×TBE buffer for 2–2.5 h at 130 V. In addition to the empty vector (pcDNA3.1) control, a water negative control was also included. The gel was incubated overnight on a storage phosphor screen and the gel scanned on a Typhoon 8610 imager (Molecular Dynamics). Intensity of bands was determined using ImageQuant.
Nuclear localization
HEK293T cells were seeded in 8-well chamber slides (4 × 105 cells/well) and transfected with 1 µg DNA using Fugene HD transfection reagent (Promega). The following day, the cells were fixed with 4% formalin for 15 min at room temperature, permeabilized in 0.1% Triton-x100 for 8 min, washed 3 times in PBS before blocking in PBS containing 0.05% Triton-X100, 0.25% BSA (PBT) and 5% normal goat serum. Cells were incubated with the C5 (Thermo scientific) or 1D2 anti-MITF antibodies overnight at 4°C, washed three times in PBT, incubated with a Cy3-labeled secondary antibody for 1 h at room temperature, washed three times in PBT and incubated for 15 min in TO-PRO-3 (Invitrogen). Cells were rinsed with PBS, slides mounted with Fluoroshield (Sigma) and imaged on a Zeiss 510 confocal microscope.
Clonogenic assays
HEK 293 cells (ATTC; CRL-1573) were transfected with wild-type and mutant MITF proteins using ExGene (Fermentas) transfection reagent according to manufacturer's protocol. After 48 h, cells were treated with 1–2 mg/ml Geneticin (G418). After several weeks under constant Geneticin selection, cells were counted (Countess) and reseeded in six-well plates at a density of 2500 cells/well. To ensure that the proteins are expressed, western blot analysis of whole cell lysates was performed (Supplementary Material, Fig. S5). After 1 week of incubation, plates were stained with 0.5% crystal violet. Plates were scanned and colonies counted via the open access program Clonocounter. Clonogenic assays were performed in triplicates and repeated twice. Error bars show the standard error of mean, and the significance was calculated using Student's t-test. P-values <0.05 were considered statistically significant.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by grants from the Icelandic Research Fund and the Research Fund of the University of Iceland (E.S.).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Colin Goding, Heinz Arnheiter and Lionel Larue for thoughtful comments on the manuscript. We acknowledge Dr Sandrine Marlin (Hospital Trousseau, Paris, France) for communicating clinical data. We thank Richard Frederickson for providing the template for Figure 1A. The authors thank the NHLBI GO Exome Sequencing Project and its ongoing studies which produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the WHI Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926) and the Heart GO Sequencing Project (HL-103010).
Conflict of Interest statement: None declared.
REFERENCES
- 1.Cheli Y., Ohanna M., Ballotti R., Bertolotto C. Fifteen-year quest for microphthalmia-associated transcription factor target genes. Pigment Cell Melanoma Res. 2009;23:27–40. doi: 10.1111/j.1755-148X.2009.00653.x. [DOI] [PubMed] [Google Scholar]
- 2.Strub T., Giuliano S., Ye T., Bonet C., Keime C., Kobi D., Le Gras S., Cormont M., Ballotti R., Bertolotto C., et al. Essential role of microphthalmia transcription factor for DNA replication, mitosis and genomic stability in melanoma. Oncogene. 2011;30:2319–2332. doi: 10.1038/onc.2010.612. [DOI] [PubMed] [Google Scholar]
- 3.Steingrimsson E., Copeland N.G., Jenkins N.A. Melanocytes and the microphthalmia transcription factor network. Annu. Rev. Genet. 2004;38:365–411. doi: 10.1146/annurev.genet.38.072902.092717. [DOI] [PubMed] [Google Scholar]
- 4.Hughes A.E., Newton V.E., Liu X.Z., Read A.P. A gene for Waardenburg syndrome type 2 maps close to the human homologue of the microphthalmia gene at chromosome 3p12-p14.1. Nat. Genet. 1994;7:509–512. doi: 10.1038/ng0894-509. [DOI] [PubMed] [Google Scholar]
- 5.Tassabehji M., Newton V.E., Liu X.Z., Brady A., Donnai D., Krajewska-Walasek M., Murday V., Norman A., Obersztyn E., Reardon W., et al. The mutational spectrum in Waardenburg syndrome. Hum. Mol. Genet. 1995;4:2131–2137. doi: 10.1093/hmg/4.11.2131. [DOI] [PubMed] [Google Scholar]
- 6.Waardenburg P.J. A new syndrome combining developmental anomalies of the eyelids, eyebrows and nose root with pigmentary defects of the iris and head hair and with congenital deafness. Am. J. Hum. Genet. 1951;3:195–253. [PMC free article] [PubMed] [Google Scholar]
- 7.Tietz W. A syndrome of deaf-mutism associated with albinism showing dominant autosomal inheritance. Am. J. Hum. Genet. 1963;15:259–264. [PMC free article] [PubMed] [Google Scholar]
- 8.Read A.P., Newton V.E. Waardenburg syndrome. J. Med. Genet. 1997;34:656–665. doi: 10.1136/jmg.34.8.656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pingault V., Ente D., Dastot-Le Moal F., Goossens M., Marlin S., Bondurand N. Review and update of mutations causing Waardenburg syndrome. Hum. Mutat. 2010;31:391–406. doi: 10.1002/humu.21211. [DOI] [PubMed] [Google Scholar]
- 10.Smith S.D., Kelley P.M., Kenyon J.B., Hoover D. Tietz syndrome (hypopigmentation/deafness) caused by mutation of MITF. J. Med. Genet. 2000;37:446–448. doi: 10.1136/jmg.37.6.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shigemura T., Shiohara M., Tanaka M., Takeuchi K., Koike K. Effect of the mutant microphthalmia-associated transcription factor found in Tietz syndrome on the in vitro development of mast cells. J. Pediatr. Hematol. Oncol. 2010;32:442–447. doi: 10.1097/MPH.0b013e3181d9da5d. [DOI] [PubMed] [Google Scholar]
- 12.Leger S., Balguerie X., Goldenberg A., Drouin-Garraud V., Cabot A., Amstutz-Montadert I., Young P., Joly P., Bodereau V., Holder-Espinasse M., et al. Novel and recurrent non-truncating mutations of the MITF basic domain: genotypic and phenotypic variations in Waardenburg and Tietz syndromes. Eur. J. Hum. Genet. 2012;20:584–587. doi: 10.1038/ejhg.2011.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.King R., Weilbaecher K.N., McGill G., Cooley E., Mihm M., Fisher D.E. Microphthalmia transcription factor. A sensitive and specific melanocyte marker for MelanomaDiagnosis. Am. J. Pathol. 1999;155:731–738. doi: 10.1016/S0002-9440(10)65172-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.King R., Googe P.B., Weilbaecher K.N., Mihm M.C., Jr, Fisher D.E. Microphthalmia transcription factor expression in cutaneous benign, malignant melanocytic, and nonmelanocytic tumors. Am. J. Surg. Pathol. 2001;25:51–57. doi: 10.1097/00000478-200101000-00005. [DOI] [PubMed] [Google Scholar]
- 15.Garraway L.A., Widlund H.R., Rubin M.A., Getz G., Berger A.J., Ramaswamy S., Beroukhim R., Milner D.A., Granter S.R., Du J., et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature. 2005;436:117–122. doi: 10.1038/nature03664. [DOI] [PubMed] [Google Scholar]
- 16.Cronin J.C., Wunderlich J., Loftus S.K., Prickett T.D., Wei X., Ridd K., Vemula S., Burrell A.S., Agrawal N.S., Lin J.C., et al. Frequent mutations in the MITF pathway in melanoma. Pigment Cell Melanoma Res. 2009;22:435–444. doi: 10.1111/j.1755-148X.2009.00578.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bertolotto C., Lesueur F., Giuliano S., Strub T., de Lichy M., Bille K., Dessen P., d'Hayer B., Mohamdi H., Remenieras A., et al. A SUMOylation-defective MITF germline mutation predisposes to melanoma and renal carcinoma. Nature. 2011;480:94–98. doi: 10.1038/nature10539. [DOI] [PubMed] [Google Scholar]
- 18.Yokoyama S., Woods S.L., Boyle G.M., Aoude L.G., Macgregor S., Zismann V., Gartside M., Cust A.E., Haq R., Harland M., et al. A novel recurrent mutation in MITF predisposes to familial and sporadic melanoma. Nature. 2011;480:99–103. doi: 10.1038/nature10630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garraway L.A., Sellers W.R. Lineage dependency and lineage-survival oncogenes in human cancer. Nat. Rev. Cancer. 2006;6:593–602. doi: 10.1038/nrc1947. [DOI] [PubMed] [Google Scholar]
- 20.Pogenberg V., Ogmundsdottir M.H., Bergsteinsdottir K., Schepsky A., Phung B., Deineko V., Milewski M., Steingrimsson E., Wilmanns M. Restricted leucine zipper dimerization and specificity of DNA recognition of the melanocyte master regulator MITF. Genes Dev. 2012;26:2647–2658. doi: 10.1101/gad.198192.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leger S., Balguerie X., Goldenberg A., Drouin-Garraud V., Cabot A., Amstutz-Montadert I., Young P., Joly P., Bodereau V., Holder-Espinasse M., et al. Novel and recurrent non-truncating mutations of the MITF basic domain: genotypic and phenotypic variations in Waardenburg and Tietz syndromes. Eur. J. Hum. Genet. 2012;20:584–587. doi: 10.1038/ejhg.2011.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bentley N.J., Eisen T., Goding C.R. Melanocyte-specific expression of the human tyrosinase promoter: activation by the microphthalmia gene product and role of the initiator. Mol. Cell Biol. 1994;14:7996–8006. doi: 10.1128/mcb.14.12.7996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murakami H., Arnheiter H. Sumoylation modulates transcriptional activity of MITF in a promoter-specific manner. Pigment Cell Res. 2005;18:265–277. doi: 10.1111/j.1600-0749.2005.00234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lowings P., Yavuzer U., Goding C.R. Positive and negative elements regulate a melanocyte-specific promoter. Mol. Cell Biol. 1992;12:3653–3662. doi: 10.1128/mcb.12.8.3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bottaro D.P., Rubin J.S., Faletto D.L., Chan A.M., Kmiecik T.E., Vande Woude G.F., Aaronson S.A. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science. 1991;251:802–804. doi: 10.1126/science.1846706. [DOI] [PubMed] [Google Scholar]
- 26.Schmidt L., Duh F.M., Chen F., Kishida T., Glenn G., Choyke P., Scherer S.W., Zhuang Z., Lubensky I., Dean M., et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat. Genet. 1997;16:68–73. doi: 10.1038/ng0597-68. [DOI] [PubMed] [Google Scholar]
- 27.Natali P.G., Nicotra M.R., Di Renzo M.F., Prat M., Bigotti A., Cavaliere R., Comoglio P.M. Expression of the c-Met/HGF receptor in human melanocytic neoplasms: demonstration of the relationship to malignant melanoma tumour progression. Br. J. Cancer. 1993;68:746–750. doi: 10.1038/bjc.1993.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Strub T., Giuliano S., Ye T., Bonet C., Keime C., Kobi D., Le Gras S., Cormont M., Ballotti R., Bertolotto C., et al. Essential role of microphthalmia transcription factor for DNA replication, mitosis and genomic stability in melanoma. Oncogene. 2011;30:2319–2332. doi: 10.1038/onc.2010.612. [DOI] [PubMed] [Google Scholar]
- 29.Bharti K., Liu W., Csermely T., Bertuzzi S., Arnheiter H. Alternative promoter use in eye development: the complex role and regulation of the transcription factor MITF. Development. 2008;135:1169–1178. doi: 10.1242/dev.014142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takebayashi K., Chida K., Tsukamoto I., Morii E., Munakata H., Arnheiter H., Kuroki T., Kitamura Y., Nomura S. The recessive phenotype displayed by a dominant negative microphthalmia-associated transcription factor mutant is a result of impaired nucleation potential. Mol. Cell Biol. 1996;16:1203–1211. doi: 10.1128/mcb.16.3.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang H., Luo H., Chen H., Mei L., He C., Jiang L., Li J.D., Feng Y. Functional analysis of MITF gene mutations associated with Waardenburg syndrome type 2. FEBS Lett. 2012;586:4126–4131. doi: 10.1016/j.febslet.2012.10.006. [DOI] [PubMed] [Google Scholar]
- 32.Hauswirth R., Haase B., Blatter M., Brooks S.A., Burger D., Drogemuller C., Gerber V., Henke D., Janda J., Jude R., et al. Mutations in MITF and PAX3 cause ‘splashed white’ and other white spotting phenotypes in horses. PLoS Genet. 2012;8:e1002653. doi: 10.1371/journal.pgen.1002653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takeda K., Takemoto C., Kobayashi I., Watanabe A., Nobukuni Y., Fisher D.E., Tachibana M. Ser298 of MITF, a mutation site in Waardenburg syndrome type 2, is a phosphorylation site with functional significance. Hum. Mol. Genet. 2000;9:125–132. doi: 10.1093/hmg/9.1.125. [DOI] [PubMed] [Google Scholar]
- 34.Fiol C.J., Wang A., Roeske R.W., Roach P.J. Ordered multisite protein phosphorylation. Analysis of glycogen synthase kinase 3 action using model peptide substrates. J. Biol. Chem. 1990;265:6061–6065. [PubMed] [Google Scholar]
- 35.Wang Q.M., Park I.K., Fiol C.J., Roach P.J., DePaoli-Roach A.A. Isoform differences in substrate recognition by glycogen synthase kinases 3 alpha and 3 beta in the phosphorylation of phosphatase inhibitor 2. Biochemistry. 1994;33:143–147. doi: 10.1021/bi00167a018. [DOI] [PubMed] [Google Scholar]
- 36.Frame S., Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochem. J. 2001;359:1–16. doi: 10.1042/0264-6021:3590001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sato S., Roberts K., Gambino G., Cook A., Kouzarides T., Goding C.R. CBP/p300 as a co-factor for the microphthalmia transcription factor. Oncogene. 1997;14:3083–3092. doi: 10.1038/sj.onc.1201298. [DOI] [PubMed] [Google Scholar]
- 38.Vachtenheim J., Drdova B. A dominant negative mutant of microphthalmia transcription factor (MITF) lacking two transactivation domains suppresses transcription mediated by wild type MITF and a hyperactive MITF derivative. Pigment Cell Res. 2004;17:43–50. doi: 10.1046/j.1600-0749.2003.00108.x. [DOI] [PubMed] [Google Scholar]
- 39.Beuret L., Flori E., Denoyelle C., Bille K., Busca R., Picardo M., Bertolotto C., Ballotti R. Up-regulation of MET expression by alpha-melanocyte-stimulating hormone and MITF allows hepatocyte growth factor to protect melanocytes and melanoma cells from apoptosis. J. Biol. Chem. 2007;282:14140–14147. doi: 10.1074/jbc.M611563200. [DOI] [PubMed] [Google Scholar]
- 40.Nobukuni Y., Watanabe A., Takeda K., Skarka H., Tachibana M. Analyses of loss-of-function mutations of the MITF gene suggest that haploinsufficiency is a cause of Waardenburg syndrome type 2A. Am. J. Hum. Genet. 1996;59:76–83. [PMC free article] [PubMed] [Google Scholar]
- 41.Chen H., Jiang L., Xie Z., Mei L., He C., Hu Z., Xia K., Feng Y. Novel mutations of PAX3, MITF, and SOX10 genes in Chinese patients with type I or type II Waardenburg syndrome. Biochem. Biophys. Res. Commun. 2010;397:70–74. doi: 10.1016/j.bbrc.2010.05.066. [DOI] [PubMed] [Google Scholar]
- 42.Lalwani A.K., Attaie A., Randolph F.T., Deshmukh D., Wang C., Mhatre A., Wilcox E. Point mutation in the MITF gene causing Waardenburg syndrome type II in a three-generation Indian family. Am. J. Med. Genet. 1998;80:406–409. [PubMed] [Google Scholar]
- 43.Welch K.O., Smith S.D., Hoover D., Arnos K.S., Kelley P.M., Pandya A. Baltimore, MD, USA: 2002 ASHG Annual Meeting; 2002. A variant of Tietz syndrome caused by a mutation in the basic domain of the MITF gene. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.