

Abstract
Objective:
To determine which vascular pathology measure most strongly correlates with white matter hyperintensity (WMH) accumulation over time, and whether Alzheimer disease (AD) neuropathology correlates with WMH accumulation.
Methods:
Sixty-six older persons longitudinally followed as part of an aging study were included for having an autopsy and >1 MRI scan, with last MRI scan within 36 months of death. Mixed-effects models were used to examine the associations between longitudinal WMH accumulation and the following neuropathologic measures: myelin pallor, arteriolosclerosis, microvascular disease, microinfarcts, lacunar infarcts, large-vessel infarcts, atherosclerosis, neurofibrillary tangle rating, and neuritic plaque score. Each measure was included one at a time in the model, adjusted for duration of follow-up and age at death. A final model included measures showing an association with p < 0.1.
Results:
Mean age at death was 94.5 years (5.5 SD). In the final mixed-effects models, arteriolosclerosis, myelin pallor, and Braak score remained significantly associated with increased WMH accumulation over time. In post hoc analysis, we found that those with Braak score 5 or 6 were more likely to also have high atherosclerosis present compared with those with Braak score 1 or 2 (p = 0.003).
Conclusion:
Accumulating white matter changes in advanced age are likely driven by small-vessel ischemic disease. Additionally, these results suggest a link between AD pathology and white matter integrity disruption. This may be due to wallerian degeneration secondary to neurodegenerative changes. Alternatively, a shared mechanism, for example ischemia, may lead to both vascular brain injury and neurodegenerative changes of AD. The observed correlation between atherosclerosis and AD pathology supports the latter.
Disruption of white matter integrity, frequently observed as white matter hyperintensities (WMH) on T2-weighted MRI sequences, has detrimental effects on cognitive function, motor performance, and functional status in the elderly1–3 and is associated with increased risk of all types of dementia, including Alzheimer disease (AD).4 Furthermore, WMH accumulation over time has been shown to increase risk of cognitive decline.3,5–7 While the general consensus is that the etiology of white matter accumulation is secondary to small-vessel ischemic changes,8–13 it is not well established whether those with faster WMH accumulation may have contributions from other pathologies. Because accumulation of WMH increases risk of cognitive decline, identifying the pathologic correlates of faster WMH accumulation could potentially guide interventions targeting specific risks for prevention and treatment to preserve cognitive function in the elderly. Furthermore, while there are previous observations supporting a link between cerebrovascular disease (CVD) and AD,14 it is not clearly established whether WMH progression is associated with neurodegenerative changes of AD. Thus, our aim was to better understand whether small-vessel ischemic disease is the sole contributor to global WMH progression or whether there is any contribution from neurodegenerative disease. To our knowledge, there are few, if any, other studies examining the association between longitudinal changes in global WMH and pathologic findings measured after death. We hypothesized that accumulation of WMH lesions measured on serial MRI scans would be associated with measures of small-vessel ischemic disease and AD pathology.
METHODS
Participants.
Participants were community-dwelling healthy older adults from the Oregon Brain Aging Study.15 Study inclusion and exclusion criteria and measurement methods have been described previously.16 Initially, healthy older persons with no medical comorbidities were enrolled. Later, to better represent the population, subjects with well-controlled, chronic medical conditions were also enrolled. Of the 375 volunteers evaluated between 1989 and 2005, 305 met inclusion criteria and were enrolled. Attrition rates caused by loss to follow-up other than death were <1% per year.
Standard protocol approvals, registrations, and patient consents.
The Portland Veterans Affairs Medical Center and Oregon Health & Science University Institutional Review Boards (IRBs) approved the study (IRB nos. 00002 and 361). Volunteers signed written informed consent.
Participant assessments.
Volunteers were examined every 6 months with the Mini-Mental State Examination17 and Clinical Dementia Rating (CDR) scale.18 A detailed battery of neuropsychological tests,15 Cognistat,19 neurologic examination, and brain MRI scans20 were performed annually. APOE was genotyped in all volunteers using standard methods. Established clinical criteria were used to make diagnoses of possible or probable AD, vascular dementia, and mixed dementia.21,22 Mild cognitive impairment was defined as a CDR = 0.5 and not meeting diagnostic criteria for dementia. Cognitive impairment (CI) was defined as having a CDR >0 at death, and no CI defined as CDR = 0 at death.
Participant inclusion and exclusion criteria.
Autopsy was performed on 125 of the 193 participants who died. Of the 125, 71 met inclusion for this study for having 1) ≥2 WMH volumes measured, 2) last MRI <36 months before death, and 3) last cognitive evaluation <24 months before death. Five subjects were excluded for WMH volumes that were >3 SD than the mean of the entire group at baseline or not having brain tissue available for reexamination, leaving 66 participants.
MRI acquisition.
Images were acquired with a 1.5-tesla magnet. The protocol consisted of continuous-slice, multiecho, multiplanar image acquisition, with 4-mm-thick coronal slices and a 24-cm2 field of view using a 256 × 256 acquisition matrix with the following pulse sequences: 1) T1-weighted sagittal images (repetition time [TR] = 600 milliseconds, echo time [TE] = 20 milliseconds); and 2) multiecho sequence T2-weighted (TR = 2,800 milliseconds, TE = 80 milliseconds) and proton density (TR = 2,800 milliseconds, TE = 32 milliseconds) coronal images perpendicular to the sagittal plane.
MRI analysis.
Analysts were blinded to clinical and previous imaging data. An imaging software, REGION, was used to quantitatively assess regional brain volumes of interest.20 Tissue types on each coronal image were separated using recursive regression analysis of bi-feature space based on relative tissue intensities. Pixel areas were summed for all slices and converted to volumetric measures by multiplying by the slice thickness for the region of interest. Using REGION's sampling tools, the analysts selected representative, unambiguous pixels of WMH (as well as brain tissue, fluid, and bone) from the multiecho sequence display. WMH was distinguished from brain tissue and fluid based on higher signal on both the proton density and T2 images.16 The main outcome of this study was total WMH volume (cm3), which included both subcortical and periventricular WMH. Median number of MRIs was 4.
Pathologic assessments.
Braak and Consortium to Establish a Registry for Alzheimer's Disease systems were used to stage neurofibrillary tangles (NFTs) and neuritic plaques (NPs).23,24 As described previously, brains were examined grossly and microscopically after fixation in neutral-buffered formaldehyde solution for 2 weeks.25 Samples from bilateral cortical lobes, frontal lobe white matter, anterior cingulate gyrus, hippocampus, amygdala, bilateral striatum and thalamus, midbrain, pons, medulla, and cerebellum were evaluated microscopically. Hematoxylin & eosin with Luxol fast blue stain was used to stain myelin on 6-mm sections from all regions. An occipital cortical section was stained with Congo red–gallocyanin for amyloid angiopathy. A modified Bielschowsky silver impregnation method was used to identify diffuse plaques and NPs. Selected sections of the hippocampus and neocortical regions were immunostained with antibody to tau (tau2; Sigma, St. Louis, MO). Lewy bodies were assessed for histologically in the midbrain and nucleus locus coeruleus by evaluation of hematoxylin & eosin/Luxol fast blue–stained sections and by anti-α-synuclein immunohistochemistry (antibody from Novus Biologicals, Littleton, CO) performed on sections of amygdala and frontal cortex. Each subject underwent additional detailed assessments for CVD and vascular brain injury. Presence of lacunar infarcts and large-vessel strokes was evaluated by visual inspection and microscopic examination bilaterally. Because acute or subacute infarcts close to death were more likely to have occurred after a subject's last MRI scan, only more remote infarcts were considered for this study. Myelin pallor was graded in the frontal white matter. The frontal watershed area is 1 of the 2 areas recommended by the National Institute on Aging–Alzheimer’s Association guidelines for neuropathologic assessment for CVD.26 In addition, previous studies have shown progression of WMH to be greatest in the anterior white matter.27 Additional sections from the deep gray nuclei, caudate head (from the anterior portion of the caudate away from the ventricle), the putamen and globus pallidus (at the level of the anterior commissure), and from the midlevel of the thalamus were examined for arteriolosclerosis and microvascular disease. We defined microvascular disease as microscopic lesions attributed to ischemia, ranging from foci of pallor to neuronal loss, and gliosis without a requirement of frank tissue infarction. Microinfarcts were defined as lesions with neuronal loss and gliosis. Remote tissue necrosis distinguished microinfarcts from milder microvascular lesions. Arteriolosclerosis was defined as thickening of the walls and narrowing of the lumens of small arteries and arterioles. Most arteriolosclerosis observed in evaluated cases was hyaline in nature. Atherosclerosis was established by gross examination of the brain, and defined as thickening of the arteries of the brain through deposition of fatty material. Arteriolosclerosis, atherosclerosis, microvascular disease, myelin pallor, and amyloid angiopathy were rated as none, mild, moderate, or severe by a neuropathologist blinded to clinical data.
Statistical analyses.
Pearson χ2, Fisher exact test, or t tests were used as appropriate to compare subject characteristics between clinical diagnostic categories (CI vs no CI). WMH volume was log-transformed to obtain a normal distribution. To identify variables to be included in the final models, first each pathologic variable was included in separate models. The pathologic measures included in each analysis were NFT Braak stage (0, 1, or 2 vs 3 or 4 vs 5 or 6) and NP category (none or mild vs moderate or severe), presence of microinfarcts, lacunar infarcts, large-vessel infarcts, amyloid angiopathy, and low vs high groups for arteriolosclerosis, microvascular disease, atherosclerosis, and myelin pallor. Individual mixed-effects models, which allow random intercept and age slope, with WMH volume as the outcome, were run for each pathologic measure with a time variable (age at each MRI assessment), adjusting for intracranial volume. A final model was created for variables that had a p value <0.1. Analyses were performed with and without adjusting for age at death, duration of follow-up (time from first MRI to death), and APOE status as potential confounders. In secondary analyses, we examined the impact of clinical diagnosis on our final model. Diagnosis before death and diagnosis at each MRI time point as a time-varying covariate were included in the final model. Additionally, in post hoc analysis, independent univariate analyses, using Pearson χ2 or Fisher exact tests, examined the associations between AD and CVD pathology. JKMP version 5.0.1a and SAS version 9.2 (SAS Institute, Cary, NC) were used for statistical analyses.
RESULTS
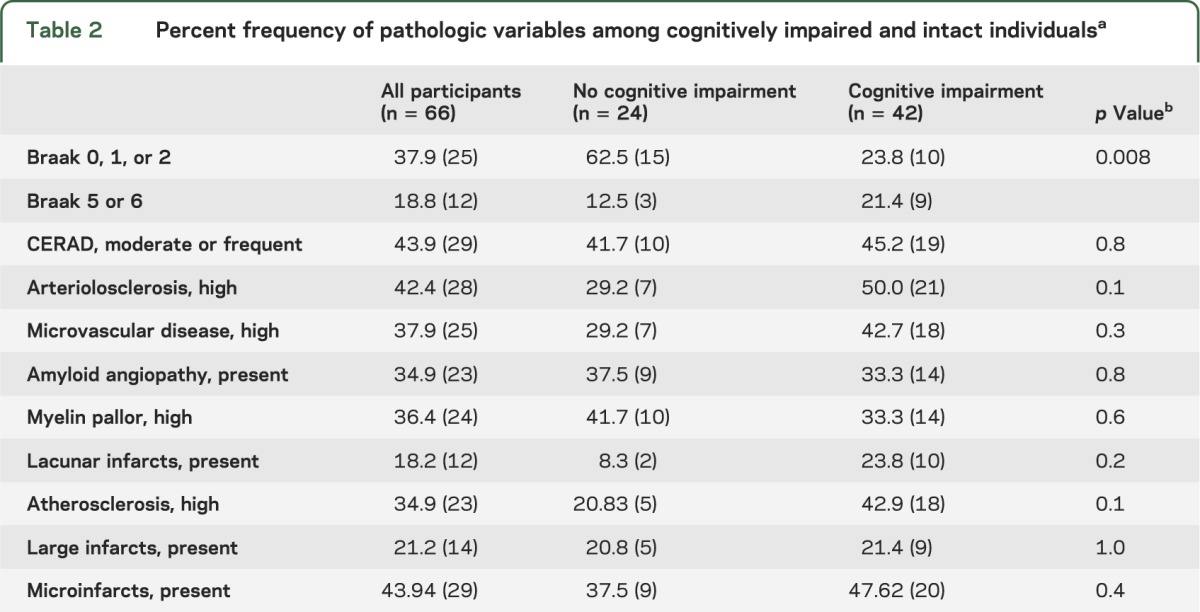
Of the 66 subjects, all of whom had no CI at entry, 42 died with CI (table 1). Clinical diagnoses before death were mild cognitive impairment (n = 16), vascular dementia (n = 3), mixed dementia (n = 1), and AD (n = 22). Of the 66 participants, 86.7% had vascular risk factors including hypertension, diabetes, heart disease, or smoking. Frequency of vascular risk factors was not different between the CI and no CI groups. A higher proportion of individuals in the no CI group had Braak scores 0, 1, or 2 compared with those who had CI (table 2).
Table 1.
Subject characteristicsa
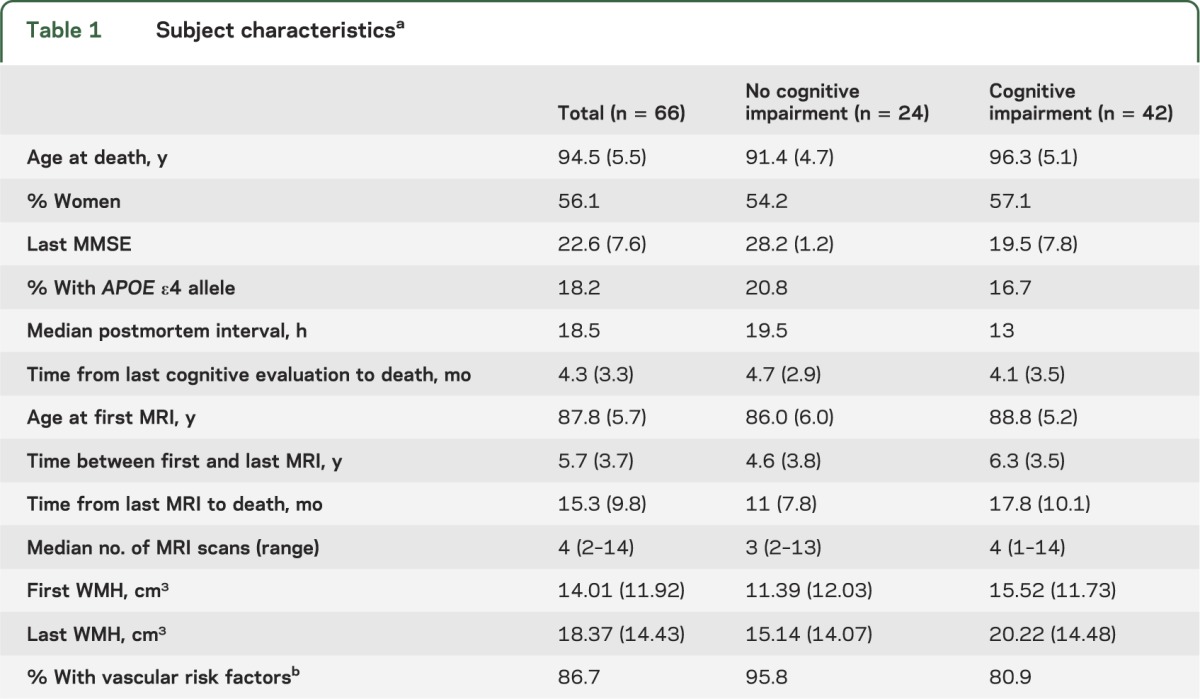
Table 2.
Percent frequency of pathologic variables among cognitively impaired and intact individualsa
Primary analysis.
In individual mixed-effects models, high amounts of arteriolosclerosis (p = 0.002), myelin pallor (p = 0.02), and Braak score (p = 0.07) showed associations with WMH accumulation over time, meeting the criterion to be included in the final model. In the final model, all 3 remained significantly associated with WMH accumulation (table 3, figure). Overall, a 1-year increase in age was associated with a 3% increase in WMH volume. Those with a high degree of arteriolosclerosis had, on average, a 5% larger increase in WMH volume over 1 year compared with those who had a low degree of arteriolosclerosis. Similarly, those with Braak 5 or 6 compared with Braak 1 or 2 had a 5% larger increase in WMH over 1 year (table 3). Those with one or more APOE ε4 alleles had on average 4% more WMH accumulation annually compared with those without the ε4 allele. Findings did not change when adjusted for duration of follow-up and age at death.
Table 3.
Pathologic measures associated with increased WMH accumulation during lifea
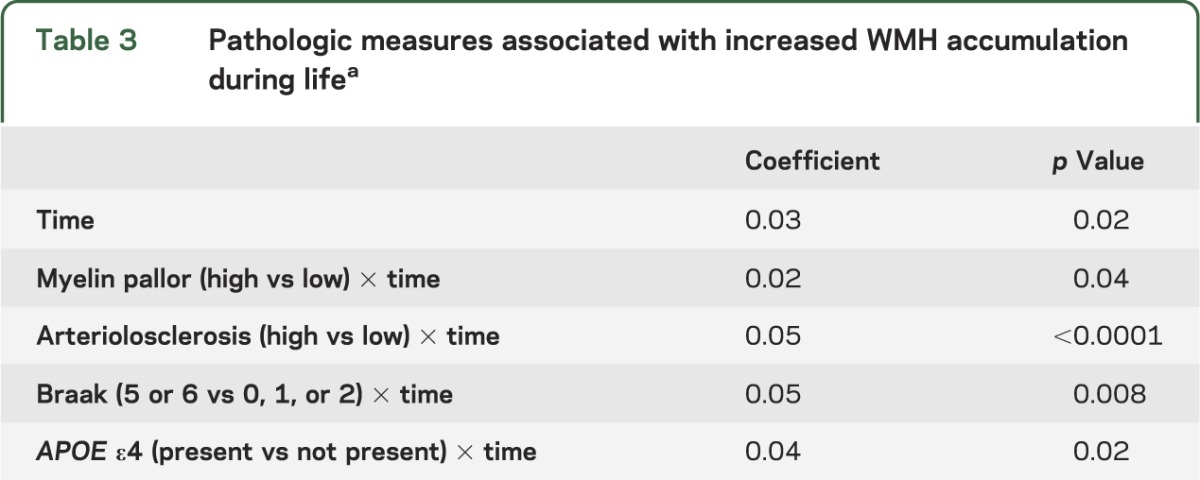
Figure. White matter hyperintensity accumulation over time by arteriolosclerosis group.

(A) Low arteriolosclerosis group. (B) High arteriolosclerosis group. The y-axis shows the white matter hyperintensity volume (numbers are predicted volumes obtained from the mixed-effects models) and the x-axis shows age.
Secondary analysis: Impact of diagnosis.
Inclusion of diagnosis, at death or at each MRI time point as a time-varying covariate, did not change the results, suggesting that the associations are independent of clinical status.
Post hoc analysis: Associations between AD and CVD pathologic measures.
Those with Braak score 5 or 6 were more likely to also have high atherosclerosis present compared with those with Braak score 1 or 2 (p = 0.003). No other significant associations were observed.
DISCUSSION
Our results suggest that arteriolosclerosis is the strongest pathologic correlate of accumulating WMH in aging. This finding is consistent with previous cross-sectional observations demonstrating that small-vessel ischemic disease is one of the main causes of age-associated white matter integrity disruption.8–13 One such recent cross-sectional study examined the pathologic correlates of white matter lesion burden in 93 research participants and found that arteriolosclerosis, when examined alone, was very strongly correlated with WMH volume.28
Equally important, we found that the presence of more extensive NFT pathology (higher Braak stage) is associated with more WMH accumulation over time in very old individuals. There may be a number of plausible explanations for this association. First, wallerian degeneration of the myelinated axons, secondary to the neuronal damage due to NFTs, may lead to white matter disruption that appears as WMH on MRI scans.29 Second, ischemic injury to the axons, manifested as white matter changes on imaging, may eventually lead to tangle formation and neuronal degeneration. There is some indirect evidence from traumatic brain injury research that axonal injury may cause disorganization of the axonal cytoskeleton ultimately triggering tau phosphorylation and aggregation.30 Another plausible mechanism by which NFTs are correlated with white matter changes may be that there is a common mechanism leading to both. In this scenario, ischemia may lead to both changes in the white matter as well as trigger processes that lead to neurodegeneration of AD.31 For example, reduced cerebral blood flow in animal models leads to accumulation of amyloid-β oligomers, and increases both neuronal amyloid-β levels and neuronal tau phosphorylation.32–34 Observational studies in humans also suggest an association between CVD risk factors and AD clinical diagnosis, as well as neuropathologic changes of AD.35 Further supporting the view that ischemia may lead to NFTs in old age, we found that those with high levels of atherosclerosis had more NFTs. This finding replicates previous observations. A recent autopsy study examined the frequency and severity of cerebral atherosclerosis in 1,000 subjects, 410 with AD, 531 with other neurodegenerative diseases (synucleinopathies, TDP-43 [TAR DNA-binding protein 43] proteinopathies, tauopathies), and 59 normal aging subjects. The authors found that the AD group had a significantly higher percentage of individuals with atherosclerosis (77%) compared with normal subjects (47%) or those with other neurodegenerative diseases (43%–67%). Atherosclerosis ratings were highly correlated with NP, NFT, and cerebral amyloid angiopathy ratings in the whole sample and within individual groups.36 Another study examined the correlation between CVD and AD pathology in 1,054 subjects from the National Alzheimer’s Coordinating Center database, and found a strong correlation between atherosclerosis and increased frequency of NPs.37 Both of these studies have found a correlation between atherosclerosis and the neurodegenerative changes of AD. Whereas we found a correlation between atherosclerosis and NFTs, we did not find a correlation between atherosclerosis and NPs. This may be attributable to our relatively small sample size. Similarly, we also did not observe a correlation between atherosclerosis and WMH.
This study has some other limitations. Participants came from a well-described cohort of relatively healthy, mostly Caucasian individuals who were very old, limiting its generalizability to younger cohorts. Our study experienced the common challenges of longitudinal research spanning decades whereby changes in technologies and protocols inevitably occur. To ensure consistency of assessment and analysis of pathologic and imaging data, we retained the same methodology over 2 decades. As a result, the number and types of pathologic measures that we could include were limited. Similarly, because of the spatial resolution of a 1.5-tesla magnet MRI scanner, small regions of pathology may have been missed. Furthermore, our sample size was limited to 66 individuals. All of these limitations may have contributed to a lack of relationship found for a number of pathologic variables. A final limitation is that assessment of our outcome, MRI WMH accumulation, occurred before the autopsy at which time the independent variables including pathologic measures were obtained. Future studies using in vivo methods to measure tau and amyloid burden, vascular integrity, and blood flow, to correlate with changes in WMH in larger populations, are needed to validate our findings. Despite these limitations, to our knowledge, ours is one of the few imaging-pathologic correlation studies that have examined the neuropathologic underpinnings of longitudinal WMH accumulation.
Our study confirms that arteriolosclerosis is the most important pathologic correlate of WMH accumulation over time in aging. Additionally, we validated previous findings suggesting a link between CVD and AD neuropathology. Specifically, our results suggest that cerebral small-vessel disease is the main driver of white matter disruption, which has been shown to contribute to cognitive decline and dementia in old age. Cerebral large-vessel disease, however, may have a dual role, promoting both vascular brain injury as well as triggering mechanisms that lead to neurodegenerative changes of AD. Given the current limited treatments for late-life dementia, interventions throughout the entire life course to reduce both small- and large-vessel CVD are of high priority because reduction in CVD may ultimately reduce the incidence and prevalence of dementia in old age.
GLOSSARY
- AD
Alzheimer disease
- CDR
Clinical Dementia Rating
- CI
cognitive impairment
- CVD
cerebrovascular disease
- NFT
neurofibrillary tangle
- NP
neuritic plaque
- TE
echo time
- TR
repetition time
- WMH
white matter hyperintensity
AUTHOR CONTRIBUTIONS
Deniz Erten-Lyons: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and gave final approval, acquisition of data, statistical analysis, study supervision, obtaining funding. Randall Woltjer: drafting/revising the manuscript, study concept or design, accepts responsibility for conduct of research and gave final approval, acquisition of data. Jeffrey Kaye: drafting/revising the manuscript, study concept or design, accepts responsibility for conduct of research and gave final approval, obtaining funding. Nora Mattek: analysis or interpretation of data, accepts responsibility for conduct of research and gave final approval, statistical analysis. Hiroko H. Dodge: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and gave final approval, statistical analysis. Sarah Green: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and gave final approval, acquisition of data. Huong Tran: drafting/revising the manuscript, analysis or interpretation of data, accepts responsibility for conduct of research and gave final approval, acquisition of data, statistical analysis. Diane B. Howieson: study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and gave final approval. Katherine Wild: drafting/revising the manuscript, accepts responsibility for conduct of research and gave final approval. Lisa C. Silbert: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and gave final approval.
STUDY FUNDING
Supported by grants from the Research & Development Office of the Department of Veterans Affairs (Career Development Award and Merit Review Grant) and the National Institute on Aging, NIH, AG08017.
DISCLOSURE
D. Erten-Lyons receives research support from the Department of Veterans Affairs (Career Development Award grant). She also receives reimbursement through Medicare or commercial insurance plans for providing clinical assessment and care for patients and is salaried to see patients at the Portland VA Medical Center. R. Woltjer reports no disclosures. J. Kaye received research support from the Department of Veterans Affairs and the NIH. Individuals that work in the research centers he directs received research support from Eisai, Roche, Phylogeny, and Merck; Dr. Kaye was compensated for consulting with Grifols. Dr. Kaye received reimbursement through Medicare or commercial insurance plans for providing clinical assessment and care for patients; he has been salaried to see patients at the Portland VA Medical Center; he served as an unpaid Vice-Chair for the International Professional Interest Area Work Group of the International Society to Advance Alzheimer's Research and Treatments and as an unpaid Commissioner for the Center for Aging Services and Technologies. He serves on the editorial advisory board of the journals Alzheimer's & Dementia and Frontiers of Aging Neuroscience. N. Mattek reports no disclosures. H. Dodge receives research support from the NIH (P30 AG008017, R01 AG033581), is a member of the Data Core Steering Committee at the National Alzheimer's Coordinating Center and serves on the Statistical Review Board for the International Psychogeriatrics and as a statistical editor for the journal Alzheimer's & Dementia. S. Green and H. Tran report no disclosures. D. Howieson receives book royalties from Oxford University Press. She received an honorarium to speak to the Pacific Northwest Neuropsychological Society in 2013. K. Wild reports no disclosures. L. Silbert receives research support from the NIH (1R01AG036772, P30 AG008017, P50 NS062684). She also receives reimbursement through Medicare or commercial insurance plans for providing clinical assessment and care for patients and for intraoperative neurophysiologic monitoring, and is salaried to see patients at the Portland VA Medical Center. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Poggesi A, Pantoni L, Inzitari D, et al. 2001-2011: a decade of the LADIS (Leukoaraiosis and DISability) Study: what have we learned about white matter changes and small-vessel disease? Cerebrovasc Dis 2011;32:577–588 [DOI] [PubMed] [Google Scholar]
- 2.Silbert LC, Howieson DB, Dodge H, Kaye JA. Cognitive impairment risk: white matter hyperintensity progression matters. Neurology 2009;73:120–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Silbert LC, Nelson C, Howieson DB, Moore MM, Kaye JA. Impact of white matter hyperintensity volume progression on rate of cognitive and motor decline. Neurology 2008;71:108–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Debette S, Markus HS. The clinical importance of white matter hyperintensities on brain magnetic resonance imaging: systematic review and meta-analysis. BMJ 2010;341:c3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carmichael O, Schwarz C, Drucker D, et al. Longitudinal changes in white matter disease and cognition in the first year of the Alzheimer disease neuroimaging initiative. Arch Neurol 2010;67:1370–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Longstreth WT, Jr, Arnold AM, Beauchamp NJ, Jr, et al. Incidence, manifestations, and predictors of worsening white matter on serial cranial magnetic resonance imaging in the elderly: the Cardiovascular Health Study. Stroke 2005;36:56–61 [DOI] [PubMed] [Google Scholar]
- 7.van Dijk EJ, Prins ND, Vrooman HA, Hofman A, Koudstaal PJ, Breteler MM. Progression of cerebral small vessel disease in relation to risk factors and cognitive consequences: Rotterdam Scan Study. Stroke 2008;39:2712–2719 [DOI] [PubMed] [Google Scholar]
- 8.Black S, Gao F, Bilbao J. Understanding white matter disease: imaging-pathological correlations in vascular cognitive impairment. Stroke 2009;40:S48–S52 [DOI] [PubMed] [Google Scholar]
- 9.Brown WR, Moody DM, Thore CR, Challa VR, Anstrom JA. Vascular dementia in leukoaraiosis may be a consequence of capillary loss not only in the lesions, but in normal-appearing white matter and cortex as well. J Neurol Sci 2007;257:62–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fernando MS, Simpson JE, Matthews F, et al. White matter lesions in an unselected cohort of the elderly: molecular pathology suggests origin from chronic hypoperfusion injury. Stroke 2006;37:1391–1398 [DOI] [PubMed] [Google Scholar]
- 11.Gouw AA, Seewann A, van der Flier WM, et al. Heterogeneity of small vessel disease: a systematic review of MRI and histopathology correlations. J Neurol Neurosurg Psychiatry 2011;82:126–135 [DOI] [PubMed] [Google Scholar]
- 12.Thomas AJ, O'Brien JT, Davis S, et al. Ischemic basis for deep white matter hyperintensities in major depression: a neuropathological study. Arch Gen Psychiatry 2002;59:785–792 [DOI] [PubMed] [Google Scholar]
- 13.Young VG, Halliday GM, Kril JJ. Neuropathologic correlates of white matter hyperintensities. Neurology 2008;71:804–811 [DOI] [PubMed] [Google Scholar]
- 14.de la Torre JC. Alzheimer disease as a vascular disorder: nosological evidence. Stroke 2002;33:1152–1162 [DOI] [PubMed] [Google Scholar]
- 15.Howieson DB, Camicioli R, Quinn J, et al. Natural history of cognitive decline in the old old. Neurology 2003;60:1489–1494 [DOI] [PubMed] [Google Scholar]
- 16.Silbert LC, Dodge HH, Perkins LG, et al. Trajectory of white matter hyperintensity burden preceding mild cognitive impairment. Neurology 2012;79:741–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Folstein MF, Robins LN, Helzer JE. The Mini-Mental State Examination. Arch Gen Psychiatry 1983;40:812. [DOI] [PubMed] [Google Scholar]
- 18.Morris JC. Clinical Dementia Rating: a reliable and valid diagnostic and staging measure for dementia of the Alzheimer type. Int Psychogeriatr 1997;9(suppl 1):173–176 [DOI] [PubMed] [Google Scholar]
- 19.Kiernan RJ, Mueller J, Langston JW, Van Dyke C. The Neurobehavioral Cognitive Status Examination: a brief but quantitative approach to cognitive assessment. Ann Intern Med 1987;107:481–485 [DOI] [PubMed] [Google Scholar]
- 20.Mueller EA, Moore MM, Kerr DC, et al. Brain volume preserved in healthy elderly through the eleventh decade. Neurology 1998;51:1555–1562 [DOI] [PubMed] [Google Scholar]
- 21.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 1984;34:939–944 [DOI] [PubMed] [Google Scholar]
- 22.Chui HC, Victoroff JI, Margolin D, Jagust W, Shankle R, Katzman R. Criteria for the diagnosis of ischemic vascular dementia proposed by the State of California Alzheimer's Disease Diagnostic and Treatment Centers. Neurology 1992;42:473–480 [DOI] [PubMed] [Google Scholar]
- 23.Braak H, Braak E. Staging of Alzheimer's disease-related neurofibrillary changes. Neurobiol Aging 1995;16:271–278 [DOI] [PubMed] [Google Scholar]
- 24.Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 1991;41:479–486 [DOI] [PubMed] [Google Scholar]
- 25.Green MS, Kaye JA, Ball MJ. The Oregon Brain Aging Study: neuropathology accompanying healthy aging in the oldest old. Neurology 2000;54:105–113 [DOI] [PubMed] [Google Scholar]
- 26.Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging–Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol 2012;123:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sachdev P, Wen W, Chen X, Brodaty H. Progression of white matter hyperintensities in elderly individuals over 3 years. Neurology 2007;68:214–222 [DOI] [PubMed] [Google Scholar]
- 28.Jagust WJ, Zheng L, Harvey DJ, et al. Neuropathological basis of magnetic resonance images in aging and dementia. Ann Neurol 2008;63:72–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bozzali M, Falini A, Franceschi M, et al. White matter damage in Alzheimer's disease assessed in vivo using diffusion tensor magnetic resonance imaging. J Neurol Neurosurg Psychiatry 2002;72:742–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gavett BE, Stern RA, McKee AC. Chronic traumatic encephalopathy: a potential late effect of sport-related concussive and subconcussive head trauma. Clin Sports Med 2011;30:179–188, xi [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci 2011;12:723–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gordon-Krajcer W, Kozniewska E, Lazarewicz JW, Ksiezak-Reding H. Differential changes in phosphorylation of tau at PHF-1 and 12E8 epitopes during brain ischemia and reperfusion in gerbils. Neurochem Res 2007;32:729–737 [DOI] [PubMed] [Google Scholar]
- 33.Koike MA, Green KN, Blurton-Jones M, Laferla FM. Oligemic hypoperfusion differentially affects tau and amyloid-beta. Am J Pathol 2010;177:300–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang X, Xing A, Xu C, Cai Q, Liu H, Li L. Cerebrovascular hypoperfusion induces spatial memory impairment, synaptic changes, and amyloid-beta oligomerization in rats. J Alzheimers Dis 2010;21:813–822 [DOI] [PubMed] [Google Scholar]
- 35.Knopman DS, Roberts R. Vascular risk factors: imaging and neuropathologic correlates. J Alzheimers Dis 2010;20:699–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yarchoan M, Xie SX, Kling MA, et al. Cerebrovascular atherosclerosis correlates with Alzheimer pathology in neurodegenerative dementias. Brain 2012;135:3749–3756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Honig LS, Kukull W, Mayeux R. Atherosclerosis and AD: analysis of data from the US National Alzheimer's Coordinating Center. Neurology 2005;64:494–500 [DOI] [PubMed] [Google Scholar]