

Introduction
The microbes that persistently colonize their vertebrate hosts are not accidental (1). Although highly numerous and diverse, there is specificity by site and substantial conservation between individuals. The genus Helicobacter includes spiral, highly motile, urease-positive, gram-negative bacteria that colonize the stomach in many mammals. Each mammal has one or more dominant Helicobacter species and they are highly, if not exclusively, host species-specific (2). Such observations are consistent with the hypothesis that when ancestral mammals diverged from reptiles about 150 million years ago, they contained ancestral helicobacters, which then diverged as their hosts changed. According to this hypothesis, helicobacters represent ancestral biota (flora) in the mammalian stomach. The human-adapted strain is H. pylori (3), which has not been reproducibly observed in any animals other than humans and other primates (3).
Although we can not reliably estimate how long H. pylori has been in the human stomach, its ancestors may have been present when our humanoid ancestors diverged from other primates about four million years ago. Consistent with this view are results from phylogeographic studies; strong and consistent evidence indicates that our ancestors already were carrying gastric H. pylori when a group that ultimately populated much of the world last left Africa, more than 58,000 years ago (4). In any case, H. pylori has been colonizing the stomach of humans since at least Paleolithic times.
In this paper, we examine the evidence concerning the relationship of this ancient member of the human microbiota, and particularly its absence, with the recent and on-going epidemic of asthma and related allergic disorders. We discuss the possibility that gastric H. pylori colonization protects against these disorders and that its disappearance has fueled their rise.
H. pylori acquisition and persistence
H. pylori is acquired, and may be detected, in early childhood usually after the first year of life (5). Transmission is fecal-oral, oral-oral, and vomitus-oral (6). Once acquired, in the absence of antibiotic use, H. pylori persists at least for decades, and most often for the full life of its host (7). H. pylori strains are highly variable, and several loci affect H. pylori-host interactions. In particular, strains with an intact cag island inject the CagA protein into host gastric epithelial cells (8); this heightened interaction in relation to cag-negative strains affects disease risk (9,10).
For most of human existence, we have lived in small, intimate groups (11), in which our microbiota mingled extensively with that of other group members (12). Under the conditions of poor hygiene that have predominated for most of human existence, transmission of gastro-intestinal microbes has been easily accomplished. In present-day developing countries in which such enteric transmission occurs, H. pylori is ubiquitous, with estimates for its prevalence in adults exceeding 80%; its presence is possibly nearly universal, when multiple detection methods are used (7, 13). In populations in which H. pylori is highly prevalent, gastric colonization with several distinct strains appears common (14).
H. pylori is disappearing
Despite the substantial evidence for the antiquity and ubiquity of H. pylori colonization in humans, it now has become clear that the prevalence of H. pylori is rapidly decreasing! This is a birth cohort effect, that began in the early 20th century in many developed countries, and accelerated after World War II (15–17). The effect has been so profound that fewer than 10% of children under 10 in the United States and in other industrialized countries now are H. pylori-positive, compared to the historic 70–90% levels (15–17). As a result of this change, occurring around the developed world to variable extents, risk factors for H. pylori acquisition can be determined. These include large family size, having parents, (especially mothers) carrying H. pylori, H. pylori-positive older siblings, and household crowding during childhood (18,19). Thus, as disappearance begins, the effects compound with each generation, especially as water has become more clean, family sizes have shrunk, mothers pre-masticate food less, and nutrition has improved (20).
Another phenomenon that may contribute to H. pylori disappearance is the widespread usage of antibiotics, especially during childhood (21). To reliably eradicate H. pylori requires combinations of two to four antimicrobial agents, but early studies with monotherapies including beta-lactam and macrolide antibiotics showed eradication rates from 10–50% (22, 23). If comparable effects occur every time a child is treated with antibiotics for an upper respiratory or skin infection or for otitis media, then the rapid (and compounding) decline in H. pylori prevalence in childhood in developed countries in recent decades is not difficult to understand.
Consequences of the presence or absence of H. pylori
By comparing persons with and without the organism, medical scientists have studied the costs and benefits of carrying H. pylori. First, came the observation that the presence of H. pylori in the gastric lumen is associated with the presence in the gastric lamina propria of phagocytic and immune cells (24). Warren and Marshall recognized the association of H. pylori with these histological findings, which pathologists call ”chronic gastritis” (24, 25); the presence of inflammatory cells leads to use of terms implying pathological processes. However, as biologists, we believe that the collection of immune and inflammatory cells in the tissue should be considered as “the physiologic response to the indigenous microbiota” (or PRIM). Similarly, the lamina proprias of the mammalian mouth, vagina, and colon are populated by phagocytic and immunologic cells that respond to the local indigenous microbiota. In contrast, germ-free animals have essentially no phagocytic and immune cells in their lamina propria, but “conventionalizing” these animals, by restoring their normal biota restores these cells, which is considered as the normal histopathology (reviewed in 26).
One difficulty with terming the host response to H. pylori as “chronic gastritis” is not in the observation, which is correct, but in interpreting the finding as pathological, and not as physiological. However, in at least one context PRIM also is pathological, since it is associated with increased risk for development of peptic ulceration (27, 28), and gastric adenocarcinoma and lymphoma (10, 29). Further, the highly interactive CagA-positive strains induce the strongest PRIM and confer the greatest risk of ulceration and carcinoma (10, 28, 30). Thus, H. pylori and the PRIM it induces are clearly associated with risk of disease, and even fatality. The decline in the incidence of these diseases in the 20th century is consistent with the decline in H. pylori prevalence.
However, it now has become clear that there is an inverse association between H. pylori with reflux esophagitis (GERD), and its consequences, including Barrett’s esophagus, and esophageal adenocarcinoma (10). Although the gastric PRIM is a risk factor for the development of peptic ulceration and gastric adenocarcinoma, it is inversely associated with the development of these esophageal diseases, and the more interactive CagA-positive strains are associated with the strongest inverse effects (10). Thus, a paradigm exists of a host-microbial interaction that in some cases may promote pathological conditions, whereas in other cases may be protective from pathology. This is not a simple concept for most physicians, but in fact fits well with Rosebury’s definition of an “amphibiont” as a microbe that could be pathogen or symbiont, depending on context (31). The phenomenon of “amphibiosis” can be used to characterize our indigenous microbiota (32), in which, for example, residential oral alpha-hemolytic streptococci protect against oral invaders, but also can cause endocarditis.
Effects of H. pylori on human physiology
The stomach is an organ with multiple functions; H. pylori and its induced PRIM affects at least three of these. First, the stomach secretes acid, which is under complex regulatory control, involving autonomic innervation, and the neurendocrine peptides, gastrin and somatostatin. It is evident that H. pylori status affects this homeostasis (9). Second, the stomach has adaptive immunologic activity in terms of both T- and B-cell function (33–37). The H. pylori-positive and the H. pylori-free stomachs are immunologically different, not only in terms of H. pylori-specific responses (36), but in more general responses as well (37), and including a far greater population of regulatory T-cells (33–35). Third, the stomach produces leptin, and is the major site for production of ghrelin (9). These neurendocrine peptides play important roles in mammalian energy homeostasis, and emerging evidence indicates that H. pylori status is relevant to their regulation (38, 39).
It thus becomes clear that a generation or more of children in developed countries have been growing up without H. pylori to guide or influence these physiological functions, and others not yet described (Figure 1). It is predictable that such altered acid secretion, immunologic activation, and neurendocrine regulation have a variety of consequences.
Figure 1.
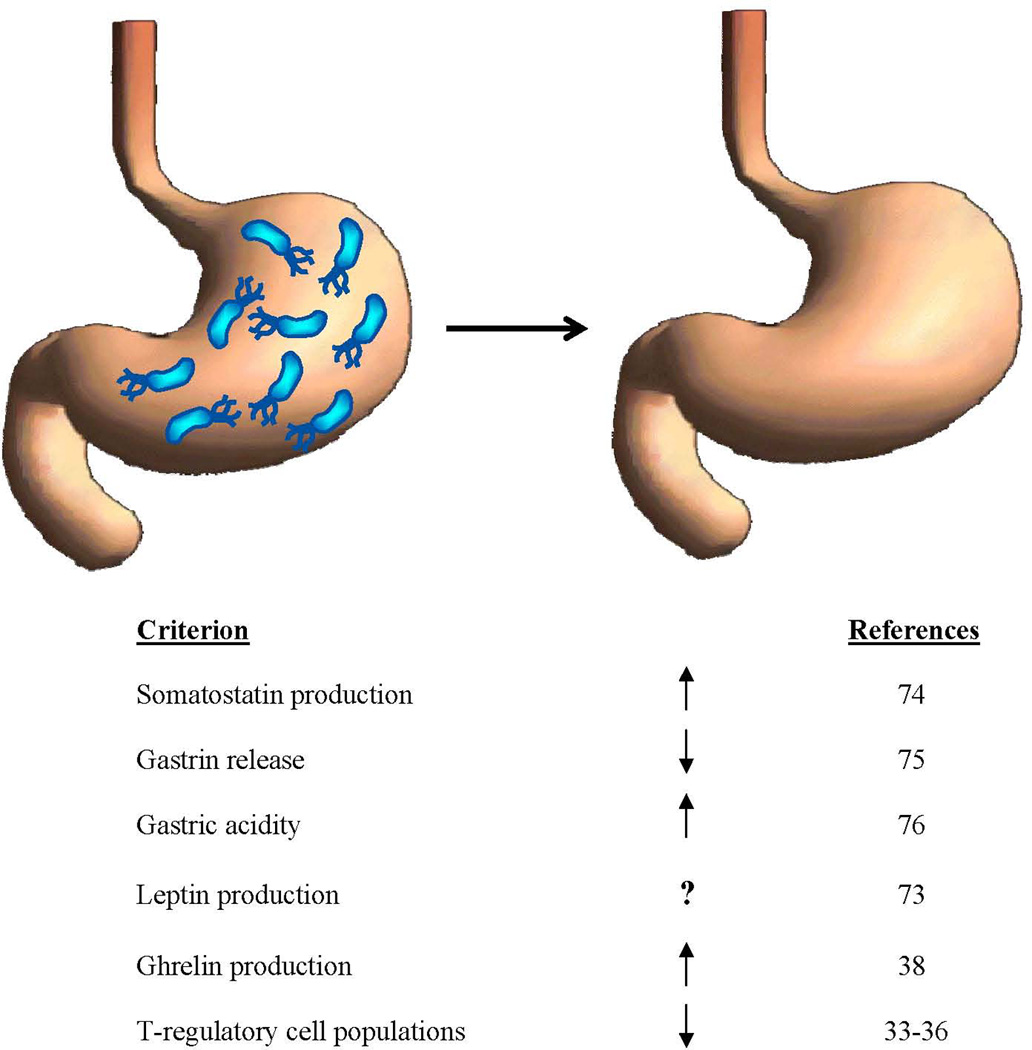
Changes in gastric physiology as the ancient (H. pylori–colonized) stomach is becoming the post-modern (H. pylori–free) stomach. Representative references are cited.
Association with asthma and allergic conditions?
In recent years, there has been a rise in the prevalence of asthma, hay fever (allergic rhinitis) and atopy (including eczema) in developed countries (40). This change, which begins in early childhood, is present across many populations in the world, and is considerable in its extent. A perturbation of such magnitude must be environmentally caused, and some of the leading candidates include exposure to tobacco smoke, air pollution, allergens, exogenous infections and microbial substances in the environment, as well as obesity (40–42)
In addition to these exogenous causes, an alternative hypothesis could relate to a change in our indigenous microbiota (1). As such, it is plausible to consider H. pylori, since its well-documented disappearance is extensive and involves developed country populations (15–17). Further, the disappearance of H. pylori has preceded the rise in asthma, but are they related?
Table 1 summarizes 12 recent cross-sectional and 4 case-control studies in which the relationships of H. pylori with asthma, atopy, allergic rhinitis, and/or eczema were examined (43–57). In general, the cross-sectional studies, involving a variety of populations and somewhat differing definitions of atopy and asthma, show significant inverse relationships of these conditions with H. pylori. The published case-control studies, in general much smaller in scale, do not show any significant direct or inverse relationships (Table 1). However, a case-control study we conducted in New York showed an inverse relationship between H. pylori, especially cagA+ strains, with asthma and atopy (58).
Table 1.
Association of H. pylori and asthma, allergic rhinitis, and atopic disease from prior literature
| CROSS-SECTIONAL STUDIES | |||||||
|---|---|---|---|---|---|---|---|
| Author, year (reference) |
Location | Study population | Age (years) |
H. pylori measure |
Definition of outcome | Major findings: Condition and OR (95% CI) in relation to H pylori+ |
|
| Matricardi, 2000 (43) | Caserta, Italy | 1659 Italian male military cadets | 17–24 | IgG ELISA | Total IgE Atopy: logRU > 1.2 Non-atopic: logRU < 0 |
Atopy For H pylori, T gondii, Hep A 1 vs. 0 2 or 3 vs. 0 |
0.70 (0.52–0.94)* 0.37 (0.22–0.63)* |
| Kosunen, 2002 (44) | Vammala, Finland | 326 and 319 healthy subjects in 1973 and 1994, respectively | 15–54 | IgA & IgG ELISA | Atopy: any IgE >0.35 IU/mL |
Atopy In 1973: In 1994: |
0.97 (0.46–2.05) 0.20 (0.05–0.71)* |
| McCune, 2003 (45) | Bristol, England | 3244 healthy subjects | 20–59 | 13C-urea breath test | Current medications for the disorders: asthma (inhalers), allergic rhinitis (antihistamines), and eczema (topical corticosteroids) |
Asthma: Allergic rhinitis: Eczema: Any of the three: |
0.78 (0.59–1.05) 0.60 (0.36–1.00)* 0.29 (0.06–1.26) 0.70 (0.54–0.91)* |
| Linneberg, 2003 (46) | Denmark | 1101 subjects | 15–69 | IgG ELISA | Self-reported allergic rhinitis Specific IgE to 6 allergens Atopy: any IgE >0.35 kU/L |
Atopy Allergic rhinitis: |
0.78 (0.57–1.08) 0.74 (0.51–1.07) |
| Jarvis, 2004 (47) | East Anglia, England | 907 randomly invited from 15,000 young adults | 20–44 | IgG ELISA | Self-reported symptoms in the prior year suggestive of hay fever and asthma Total IgE and specific IgE to house dust mite, cat, grass, Cladosporium, and birch |
Hay fever/nasal allergies: Wheeze with no cold: Allergy to grass: Allergy to >1 allergens: |
1.01 (0.70–1.52) 0.80 (0.51–1.24) 0.65 (0.43–0.99)* 1.13 (0.81–1.59) |
| Radon, 2004 (48) | Northern Germany | 321 with blood samples from 930 randomly selected from 3112 inhabitants | 18–44 | IgG ELISA, IgG CagA | Specific IgE against a panel of aeroallergens Atopy: any IgE>0.70 kU/L |
Atopy: | 0.70 (0.39–1.28) |
| von Hertzen, 2006 (49) | Eastern Finland, Western Russia | Healthy adults; 790 from Finland, 387 from Russia |
25–54 | IgG ELISA | Skin prick testing with a panel of 11 common airborne allergens Atopy: any wheal diameter ≥ 3 mm |
Atopy, in Russians: In Finns: |
0.55, p < 0.01*1 0.72, p = 0.53 1 |
| Janson, 2007 (50) | Iceland, Sweden, Estonia | 1249 healthy adults | Mean 42 | IgG ELISA | Detection of specific Atopy: any IgE >0.35 kU/L Self-reported hay fever, asthma in the prior year |
Atopy: For IgG antibodies to ≤3 specified infectious Atopy: Allergic asthma: Allergic rhinitis: |
0.57 (0.43–0.77)* 0.70 (0.52–0.94)* 0.55 (0.34–0.89)* 0.59 (0.42–0.83)* |
| Chen, 2007 (51) | USA | 7663 adults | 20–90; Mean, 43 | IgG ELISA, IgG CagA | Self-reported asthma and hay fever (current and lifetime) Skin sensitization tests |
OR in relation to CagA+ Ever asthma: Onset age ≤ 15: |
0.79 (0.63–0.99)* 0.63 (0.43–0.93)* |
| Herbarth, 2007 (52) | Germany | 2487 children | Mean 6 | 13C-urea breath test | Lifetime physician-diagnosed eczema | Eczema: | 0.37, p < 0.01*1 |
| Shiotani, 2007 (53) | Japan | 777 university students | Mean 19 | IgG ELISA | Self-reported atopic dermatitis, bronchial asthma, allergic rhinoconjunctivitis, urticaria | Any allergic disease: Ever asthma: (in ≤19 years) |
0.60 (0.40–0.90)* 0.65 (0.45–1.06) |
| Chen, 2007 (54) | USA | 7412 adults | 3–85, Mean 25 | IgG ELISA | Self-reported asthma and hay fever (current and lifetime) | Current asthma: (in ≤ 13 years) Early childhood: (onset < 5 years) |
0.41 (0.24–0.69)* 0.58 (0.38–0.88)* |
| CASE-CONTROL STUDIES | |||||||
|---|---|---|---|---|---|---|---|
| Author, year (reference) |
Location | Study population | Age (years) |
H. pylori measure |
Definition of outcome | Major findings: Condition and OR (95% CI) in relation to H pylori+ |
|
| Matricardi, 2000 (43) | Caserta, Italy | 240 atopic cases and 240 non-atopic controls | 17–24 | IgG ELISA | Total IgE Atopy: logRU > 1.2 Non-atopic: logRU < 0 |
Atopy: | 0.76 (0.47–1.24) |
| Bodner, 2000 (55) | Grampion, Scotland | 97 cases and 208 controls | 39–45 | IgG ELISA | Skin & specific IgE tests Atopy: weal ≥ 3 mm, or any IgE > 0.35 IU/ml Self-reported adult-onset wheeze and asthma |
Wheeze: Wheeze and asthma: Atopy: |
1.20 (0.70–2.20) 0.50 (0.20–1.50) 0.90 (0.60–1.60) |
| Tseng, 2000 (56) | Hong Kong | 90 cases with stable asthma and 97 controls | Mean 43 | IgG ELISA | Current asthma diagnosed by ATS guidelines | Asthma: | 1.55 (0.83–2.90) |
| Jun, 2005 (57) | Japan | 46 cases with asthma, and 48 healthy controls | Mean 52 | IgG ELISA IgG CagA | Current asthma diagnosed by ATS guidelines | Compared with healthy controls Asthma: For CagA+ Asthma: |
1.10 (0.45–2.69) 1.20 (0.39–3.69) |
P < 0.05
CI was not estimated because information on covariates is not available; the study reported a p-value adjusted for covariates only.
P < 0.05
To consider the findings of the cross-sectional analyses, we focus on two other studies that we conducted (51, 54). We first examined a large, publicly available database from the National Health and Nutrition Survey (NHANES) III, conducted between 1988 and 1994 (59). In the mid-1990’s, H. pylori and CagA serology were performed on stored specimens from more than 10,000 NHANES III subjects, with the laboratory workers and statisticians blinded to asthma or atopy status. In 2006, we were able to link 7,663 records that contained information on both asthma and H. pylori status (51). For all subjects, there was an inverse association of ever having had asthma with having a cagA+ H. pylori strain (OR (95% CI) = 0.79(0.63–099), with a stronger inverse association in those less than the median (43 years) age (0.63(0.43–0.93), and no association in the older persons. Similarly, the inverse association was strongest in those who had asthma onset before the age of 15 years (0.63(0.43–0.93), with no association with those with older-onset asthma. Highly similar trends were observed in relation to allergic rhinitis and allergy symptoms, with some inverse relationships also occuring in persons with cagA-negative H. pylori strains. We also linked records for 2,386 persons who had skin tests performed for pollens and molds, and who had H. pylori status ascertained (51). For 4 of the 6 antigens tested, there were inverse associations in persons with cagA+ strains, especially those below the median age. Thus, we found inverse associations between H. pylori, especially cagA+ strains, with asthma and related allergic disorders, especially involving younger individuals, and with early life disease onset.
Because of these findings, we sought independent assessment of the relationships. We then examined the subsequent survey, NHANES 1999–2000 (60). For that study, we found 7,412 subjects who had data on asthma and related conditions as well as on H. pylori status; no testing for cagA status had been performed. The median age in this study was 25, and because our prior results highlighted asthma with early age of onset, we focused on children less than 20 years old. We found significant inverse associations of H. pylori positivity with early onset of asthma and allergic rhinitis in children and teens under 20, as well as ever having had asthma and current asthma in children 3–13 years old(54). H. pylori also was inversely related to having recently had wheezing, allergic rhinitis, and dermatitis, eczema, or rash. These two large, cross-sectional, independent studies show highly consistent results across asthma and related allergic disorders, and extend the prior studies which were more limited in sample size, age range of study populations, as well as data on potential confounders, H. pylori strains, and age of onset of asthma (Table 1).
Biological plausibility for H. pylori to play a protective role against asthma
H. pylori status could be causally related to asthma and its related disorders, with colonized persons having a partial protection. Considering the Bradford Hill criteria (61) provides evidence that supports such a causal role. First, the secular trend is consistent and reverse causation in not likely; H. pylori is disappearing while asthma incidence is rising. Importantly, the decline in H. pylori acquisition, beginning early in the 20th century, precedes the increase in asthma. However, all of the epidemiological studies to date are cross-sectional or case-control studies, and not prospective. Nevertheless, it is not likely that asthma and related disorders could themselves be leading to the disappearance of H. pylori. Once acquired early in life, if not treated with antibiotics, H. pylori persists at least for decades, if not for life. The cross-sectional studies could measure an effect of asthma, or of its treatment. For example, if asthmatics receive more antibiotics than non-asthmatics, that could reduce H. pylori prevalence. However, the specificity of the inverse association with early life asthma and not with long-standing asthma seen in adults is one argument against that proposition.
Second, a dose-response relationship between exposure and disease is present. Studies of differences among H. pylori strains show the strongest effects for cagA+ strains, in terms of risk of disease (ulcers, gastric cancer) or protection from disease (GERD and esophageal adenocarcinoma). A similar dose-response to that related to GERD is present with asthma, with cagA+ strains having the strongest inverse association (54, 58). Third, as shown in Table 1, a variety of cross-sectional studies show protective effects, suggesting consistency of the data. The increasing number of these studies, especially our two large, independent, and population-based studies, point toward a correct association. Nevertheless, that not all studies, especially the case-control studies, show this inverse association could indicate that there is population-based specificity for the observation, and/or differences in study design.
Fourth, is the role of specificity; asthma is considered as predominantly allergic or not. The strong inverse associations with H. pylori are present for asthma and other allergic disorders consistent with the allergic (atopic) spectrum. In addition, the inverse association with H. pylori appears stronger for childhood-onset asthma. There may be etiologic differences between childhood-onset and adulthood-onset asthma. Childhood asthma often remits during adolescence, although many of these patients in remission have relapses during young adulthood (62). Consistently, the case-control studies of H. pylori and current asthma in adults did not find any association (Table 1). The effect of H. pylori may be less important in adult-onset asthma, since the risk factors may be much more heterogeneous than in childhood asthma. In addition, asthma in adults may be new onset, persistent from childhood, or exacerbated from childhood asthma. Although commonly associated with atopy, adult asthma is more complex and onset may be complicated by environmental exposures (e.g. tobacco, occupation) (63, 64). Finally, the misclassification of current status of asthma and H. pylori could be more serious in adults. Since the misclassifications of asthma and H. pylori status do not depend on one another, it is non-differential, which would lead to a bias toward the null.
Fifth, the inverse association is coherent with our knowledge, and there is no evidence of plausible competing theories or rival hypotheses. One possibility is that H. pylori status, while related to asthma risk, is merely a marker for other phenomena. For example, early life antibiotic use (65, 66) that eliminates H. pylori carriage also could eliminate one or more other microbes that actually are the protective agents. There are insufficient data at present to rule out this possibility. Several studies that have evaluated multiple infections suggest their additive effect in the etiology of asthma (43, 50) (Table 1). In addition, the inverse association between H. pylori and asthma is independent of indicators of socioeconomic status, age, gender, ethnic background, smoking status, and hepatitis A infection (51). An independent phenomenon that makes asthma more likely and H. pylori carriage less likely could be underlying the inverse association. Such a phenomenon could be due to enhancement of Th-2 immunity due to another microbe, for example, and a consequent effect on H. pylori status could provide a maker of risk. Sixth, mechanisms exist (see below) that could explain a protective effect. In total, there is considerable biological plausibility for a protective role of H. pylori (especially cagA+) strains toward asthma and related disorders.
Mechanisms by which gastric H. pylori colonization might affect asthma risk
In the simplest statement, it is increasingly clear that the gastric physiology of the H. pylori-positive and negative subjects differs (9, 20, 67). Several non-exclusive mechanisms could be playing a role. First, if H. pylori, is actually protecting against GERD (10), it also could protect against asthma, since some proportion of asthma is due to GERD (68); this component may actually be underestimated (69). However, this mechanism is unlikely to be sufficient to explain protective H. pylori effects in hay fever and atopic dermatitis. Second, the constellation of asthma, atopy, hay fever, and skin sensitization suggests immunologic mediation. H. pylori-positive persons have a gastric population of immunocytes, including regulatory T-cells (33–36), that is largely or completely absent from H. pylori-negative subjects. Such cells may have systemic immunomodulatory activities. Recent studies indicate an interaction of H. pylori colonization with Mycobacterium tuberculosis, with colonization associated with the maintenance of tuberculosis latency (70), again pointing to a global immunomodulatory role. A third mechanism may relate to the effects of H. pylori-induced inflammation on gastric hormonal levels (9). Both leptin and gastrin have immunomodulatory activities as well as intermediary effects on energy homeostasis (71, 72). There is increasing evidence that H. pylori gastric colonization affects both ghrelin and leptin production (38, 73) which thus would affect the immunoregulatory environment. Finally, H. pylori’s effects on the autonomic nervous system might play a role. Individual differences in the host-microbial interaction could account for differential risk and disease expression. Prospective studies that evaluate the influence of H. pylori on both indicators of causal intermediates and asthma risk will help delineate the mechanisms.
Conclusions
For probably the first time in human history, generations of children are growing, without H. pylori in their stomachs, guiding the development of their immunologic capabilities, their hormonal regulation of energy homeostasis, and their regulation of gastric acidity (Figure 1). The loss of this ancient, dominant, and persistent member of the normal biota of humans would be predicted to have consequences, and there now is much information about the beneficial and deleterious aspects of this change on gastrointestinal tract health and disease (1, 10, 77, 78). However, increasing evidence is pointing to extra-intestinal manifestations of the disappearance of H. pylori, including disorders of energy homeostasis (38, 39) and asthma. An inverse association of H. pylori and childhood asthma, allergic rhinitis, and atopy is becoming increasing obvious. Although this may represent an epiphenomenon as part of a more general change in human microecology (1), there is substantial biological plausibility for a role of the disappearance of H. pylori and the rise of these allergic disorders of children. Nevertheless, if H. pylori, and especially cagA status, only is a marker for asthma risk, it could become useful for clinical and epidemiological studies. These questions are of sufficient importance that confirmatory and prospective studies in different populations should be done.
Clearly, the interactions of H. pylori are complex, somewhat host-specific, and certainly incompletely understood. Ten years ago, one of us predicted that doctors of the future will have the tools to perform relevant phenotyping and genotyping of young children and then take the appropriate stocks of H. pylori from their pharmacy and deliberately colonize that child with that strain (or combination of strains) most likely to optimize their life-long health (79). The continuing beneficial associations of H. pylori with reduction of risk for esophageal diseases (including malignancy), now with asthma and atopy, and possibly with obesity and diabetes (9, 38, 39), should be considered in H. pylori treatment and intervention plans, and move that earlier prediction closer to reality.
It is possible that for most individuals, H. pylori is beneficial in childhood and more deleterious later in life. Within such a paradigm, a public health framework for H. pylori introduction and eradication can be envisioned.
Acknowledgements
The authors thank Michael Marmor, Maria-Elena Fernandez-Beros, Linda Rogers, and Guillermo Perez-Perez for their participation in the initial studies at Bellevue Hospital that stimulated this work.
Funders
This research was supported by grant ES000260 from the National Institute of Environmental Health Sciences, grant CA016087 from the National Cancer Institute, grant RO1GM63270 from the National Institutes of Health, the Diane Belfer Program in Human Microbial Ecology, the Senior Scholar Award of the Ellison Medical Foundation, Ellison Medical Foundation, and Colten Family Foundation.
Footnotes
The Corresponding Author has the right to grant on behalf of all authors and does grant on behalf of all authors, an exclusive license on a worldwide basis to the BMJ Publishing Group Ltd and its Licensees to permit this article to be published in Gut editions and any other BMJPGL products to exploit all subsidiary rights, as set out in the Gut license (http://gut.bmjjournals.com/ifora/licence.dtl).
Competing Interests
Disclosure: Dr. Blaser, as a co-discoverer of cagA at Vanderbilt University, can receive royalties from the commercial exploitation of cagA. No diagnostic tests for cagA are currently licensed.
References
- 1.Blaser MJ. Who are we? Indigenous microbes and the ecology of human diseases. EMBO Rep. 2006;7:956–960. doi: 10.1038/sj.embor.7400812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kusters JG, van Vliet AHM, Kuipers EJ. Pathogenesis of Helicobacter pylori Infection. Clin Microbiol Rev. 2006;19:449–490. doi: 10.1128/CMR.00054-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bik EM, Eckburg PB, Gill SR, Nelson KE, Purdom EA, Francois F et al. Molecular analysis of the bacterial microbiota in the human stomach. Proc Natl Acad Sci U S A. 2006;103:732–737. doi: 10.1073/pnas.0506655103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Linz B, Balloux F, Moodley Y, Manica A, Liu H, Roumagnac P et al. An African origin for the intimate association between humans and Helicobacter pylori. Nature. 2007;445:915–918. doi: 10.1038/nature05562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Malaty HM, El Kasabany A, Graham DY, Miller CC, Reddy SG, Srinivasan SR et al. Age at acquisition of Helicobacter pylori infection: a follow-up study from infancy to adulthood. Lancet. 2002;359:931–935. doi: 10.1016/S0140-6736(02)08025-X. [DOI] [PubMed] [Google Scholar]
- 6.Parsonnet J, Shmuely H, Haggerty TD. Fecal and oral shedding of Helicobacter pylori from healthy, infected adults. JAMA. 1999;282:2240–2245. doi: 10.1001/jama.282.23.2240. [DOI] [PubMed] [Google Scholar]
- 7.Taylor DN, Blaser MJ. The epidemiology of Helicobacter pylori infection. Epidemiol Rev. 1991;13:42–59. doi: 10.1093/oxfordjournals.epirev.a036078. [DOI] [PubMed] [Google Scholar]
- 8.Odenbreit S, Puls J, Sedlmaier B, Gerland E, Fischer W, Haas R. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science. 2000;287:1497–1500. doi: 10.1126/science.287.5457.1497. [DOI] [PubMed] [Google Scholar]
- 9.Blaser MJ, Atherton JC. Helicobacter pylori persistence: biology and disease. J Clin Invest. 2004;113:321–333. doi: 10.1172/JCI20925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peek RM, Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer. 2002;2:28–37. doi: 10.1038/nrc703. [DOI] [PubMed] [Google Scholar]
- 11.Barnard AJ, editor. Hunter-gatherers in history, archeology and anthropology. Oxford: Berg; 2004. p. 278. [Google Scholar]
- 12.Blaser MJ, Kirschner D. The equilibria that permit bacterial persistence in human hosts. Nature. 2007;449:843–849. doi: 10.1038/nature06198. [DOI] [PubMed] [Google Scholar]
- 13.Romero-Gallo J, Perez-Perez GI, Novick RP, Kamath K, Norboo T, Blaser MJ. Responses to Helicobacter pylori whole cell and CagA antigens amongst Ladakh patients undergoing endoscopy. Clin Diag Lab Immunol. 2002;9:1313–1317. doi: 10.1128/CDLI.9.6.1313-1317.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghose C, Perez-Perez GI, van Doorn LJ, Dominguez-Bello MG, Blaser MJ. High frequency of gastric colonization with multiple Helicobacter pylori strains in Venezuelan subjects. J Clin Microbiol. 2005;43:2635–2641. doi: 10.1128/JCM.43.6.2635-2641.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Banatvala N, Mayo K, Megraud F, Jennings R, Deeks JJ, Feldman RA. The cohort effect and Helicobacter pylori. J Infect Dis. 1993;168:219–221. doi: 10.1093/infdis/168.1.219. [DOI] [PubMed] [Google Scholar]
- 16.Kosunen TU, Aromaa A, Knekt P, et al. Helicobacter antibodies in 1973 and 1994 in the adult population of Vammala, Finland. Epidemiol Infect. 1997;119:29–34. doi: 10.1017/s0950268897007565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perez-Perez GI, Salomaa A, Kosunen TU, Daverman B, Rautelin H, Aromaa A et al. Evidence that cagA(+) Helicobacter pylori strains are disappearing more rapidly than cagA(-) strains. Gut. 2002;50:295–298. doi: 10.1136/gut.50.3.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goodman K, Correa P, Tenganá Aux HJ, Ramirez H, DeLany JP, Pepinosa OG, Quiñones ML, Parra TC. Helicobacter pylori infection in the Colombian Andes: a population-based study of transmission pathways. Am J Epidemiol. 1996;144:290–299. doi: 10.1093/oxfordjournals.aje.a008924. [DOI] [PubMed] [Google Scholar]
- 19.Goodman K, Correa P. Transmission of Helicobacter pylori among siblings. Lancet. 2000;355:358–362. doi: 10.1016/S0140-6736(99)05273-3. [DOI] [PubMed] [Google Scholar]
- 20.Wunder C, Churin Y, Winau F, et al. Cholesterol glucosylation promotes immune evasion by Helicobacter pylori. Nat Med. 2006;12:1030–1038. doi: 10.1038/nm1480. [DOI] [PubMed] [Google Scholar]
- 21.McCaig LF, Besser RE, Hughes JM. Trends in antimicrobial prescribing rates for children and adolescents. JAMA. 2002;287:3096–3102. doi: 10.1001/jama.287.23.3096. [DOI] [PubMed] [Google Scholar]
- 22.Peterson WL, Graham DY, Marshall B, Blaser MJ, Genta RM, Klein PD et al. Clarithromycin as monotherapy for eradication of Helicobacter pylori: a randomized, double-blind trial. Am J Gastroenterol. 1993;88:1860–1864. [PubMed] [Google Scholar]
- 23.Rauws EAJ, Langenberg W, Houthoff HJ, Zanen HC, Tytgat GNJ. Campylobacter pyloridis-associated chronic active antral gastritis. Gastroenterology. 1988;94:33–40. [PubMed] [Google Scholar]
- 24.Warren JR. Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet. 1983;1:1273. [PubMed] [Google Scholar]
- 25.Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet. 1984;1:1311–1315. doi: 10.1016/s0140-6736(84)91816-6. [DOI] [PubMed] [Google Scholar]
- 26.Blaser MJ. Helicobacters are indigenous to the human stomach: duodenal ulceration is due to changes in gastric microecology in the modern era. Gut. 1998;43:721–727. doi: 10.1136/gut.43.5.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nomura A, Stemmerman GN, Chyou P-H, Pérez-Pérez GI, Blaser MJ. Helicobacter pylori infection and the risk for duodenal and gastric ulceration. Ann Intern Med. 1994;120:977–981. doi: 10.7326/0003-4819-120-12-199406150-00001. [DOI] [PubMed] [Google Scholar]
- 28.Nomura AMY, Perez-Perez GI, Lee J, Stemmerman G, Blaser MJ. Relationship between H. pylori cagA status and risk of peptic ulcer disease. Am J Epidemiol. 2002;155:1054–1059. doi: 10.1093/aje/155.11.1054. [DOI] [PubMed] [Google Scholar]
- 29.Parsonnet J, Hansen S, Rodriguez L, Gelb AB, Warnke RA, Jellum E, et al. Helicobacter pylori infection and gastric lymphoma. N Engl J Med. 1994;330:1267–1271. doi: 10.1056/NEJM199405053301803. [DOI] [PubMed] [Google Scholar]
- 30.Blaser MJ, Pérez-Pérez GI, Kleanthous H, Cover TL, Peek RM, Chyou PH, Stemmermann GN, Nomura A. Infection with Helicobacter pylori strains possessing cagA associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Research. 1995;55:2111–2115. [PubMed] [Google Scholar]
- 31.Rosebury T. Microorganisms indigenous to man. New York: McGraw Hill; 1962. pp. 1–8. [Google Scholar]
- 32.Blaser MJ. Ending the war metaphor: the changing agenda for unraveling the host-microbe relationship. Washington, DC: Institute of Medicine. National Academies Press; 2006. Pathogenesis and symbiosis: Human gastric colonization by Helicobacter pylori as a model system of amphibiosis; pp. 115–130. [Google Scholar]
- 33.Lundgren A, Suri-Payer E, Enarsson K, Svennerholm AM, Lundin BS. Helicobacter pylori-specific CD4+CD25high regulatory T cells suppress memory T-cell responses to H. pylori in infected individuals. Infect Immun. 2003;71:1755–1762. doi: 10.1128/IAI.71.4.1755-1762.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rad R, Brenner L, Bauer S, Schwendy S, Layland L, da Costa CP, et al. CD25+/Foxp3+ T cells regulate gastric inflammation and Helicobacter pylori colonization in vivo. Gastroenterology. 2006;131:525–537. doi: 10.1053/j.gastro.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 35.Lundgren A, Stromberg E, Sjoling A, Lindholm C, Enarsson K, Edebo A et al. Mucosal FOXP3-expressing CD4+ CD25high regulatory T cells in Helicobacter pylori-infected patients. Infect Immun. 2005;73:523–531. doi: 10.1128/IAI.73.1.523-531.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goll R, Gruber F, Olsen T, Cui G, Raschpichler G, Buset M et al. Helicobacter pylori stimulates a mixed adaptive immune response with a strong T-regulatory component in human gastric mucosa. Helicobacter. 2007;12:185–192. doi: 10.1111/j.1523-5378.2007.00495.x. [DOI] [PubMed] [Google Scholar]
- 37.Mattson A, Lönroth H, Qiuding-Järbrink M, Svennerholm AM. Induction of B cell responses in the stomach of Helicobacter pylori-infected subjects after oral cholera vaccination. J Clin Invest. 1998;102:51–56. doi: 10.1172/JCI22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nwokolo CU, Freshwater DA, O'Hare P, Randeva HS. Plasma ghrelin following cure of Helicobacter pylori. Gut. 2003;52:637–640. doi: 10.1136/gut.52.5.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marini E, Maldinado A, Cabras S, Hidalgo G, Buffa R, Marin A, Flores G, Racugno W, Pericchi L, Castellanos M, Groschl M, Blaser MJ, Dominguez M. Helicobacter pylori and intestinal parasites are not detrimental to the nutritional status of Amerindians. Am J Trop Med Hyg. 2007;76:534–540. [PubMed] [Google Scholar]
- 40.Eder W, Ege MJ, von Mutius E. The asthma epidemic. N Engl J Med. 2006;355:2226–2235. doi: 10.1056/NEJMra054308. [DOI] [PubMed] [Google Scholar]
- 41.Strachan DP. Hay fever, hygiene, and household size. BMJ. 1989;299:1259–1260. doi: 10.1136/bmj.299.6710.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Matricardi PM, Rosmini F, Panetta V, Ferrigno L, Bonini S. Hay fever and asthma in relation to markers of infection in the United States. J Allergy Clin Immunol. 2002;110:381–387. doi: 10.1067/mai.2002.126658. [DOI] [PubMed] [Google Scholar]
- 43.Matricardi PM, Rosmini F, Riondino S, Fortini M, Ferrigno L, Rapicetta M et al. Exposure to foodborne and orofecal microbes versus airborne viruses in relation to atopy and allergic asthma: epidemiological study. BMJ. 2000;320:412–417. doi: 10.1136/bmj.320.7232.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kosunen TU. Increase of allergen-specific immunoglobulin E antibodies from 1973 to 1994 in a Finnish population and a possible relationship to Helicobacter pylori infections. Clin Exp Allergy. 2002;32:373–378. doi: 10.1046/j.1365-2222.2002.01330.x. [DOI] [PubMed] [Google Scholar]
- 45.McCune A, Lane A, Murray L, Harvey I, Nair P, Donovan J et al. Reduced risk of atopic disorders in adults with Helicobacter pylori infection. Eur J Gastroenterol Hepatol. 2003;15:637–640. doi: 10.1097/00042737-200306000-00010. [DOI] [PubMed] [Google Scholar]
- 46.Linneberg A. IgG antibodies against microorganisms and atopic disease in Danish adults: the Copenhagen Allergy Study. J Allergy Clin Immunol. 2003;111:847–853. doi: 10.1067/mai.2003.1335. [DOI] [PubMed] [Google Scholar]
- 47.Jarvis D, Luczynska C, Chinn S, Burney P. The association of hepatitis A and Helicobacter pylori with sensitization to common allergens, asthma and hay fever in a population of young British adults. Allergy. 2004;59:1063–1067. doi: 10.1111/j.1398-9995.2004.00539.x. [DOI] [PubMed] [Google Scholar]
- 48.Radon K. Farming exposure in childhood, exposure to markers of infections and the development of atopy in rural subjects. Clin Exp Allergy. 2004;34:1178–1183. doi: 10.1111/j.1365-2222.2004.02005.x. [DOI] [PubMed] [Google Scholar]
- 49.von Hertzen LC, Laatikainen T, Makela MJ, Jousilahti P, Kosunen TU, Petays T et al. Infectious burden as a determinant of atopy-- a comparison between adults in Finnish and Russian Karelia. Int Arch Allergy Immunol. 2006;140:89–95. doi: 10.1159/000092251. [DOI] [PubMed] [Google Scholar]
- 50.Janson C. The effect of infectious burden on the prevalence of atopy and respiratory allergies in Iceland, Estonia, and Sweden. J Allergy Clin Immunol. 2007;120:673–679. doi: 10.1016/j.jaci.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 51.Chen Y, Blaser MJ. Inverse associations of Helicobacter pylori with asthma and allergy. Arch Intern Med. 2007;167:821–827. doi: 10.1001/archinte.167.8.821. [DOI] [PubMed] [Google Scholar]
- 52.Herbarth O et al. Helicobacter pylori colonisation and eczema. J Epidemiol Community Health. 2007;61:638–640. doi: 10.1136/jech.2006.046706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shiotani A, Miyanishi T, Kamada T, Haruma K. Helicobacter pylori infection and allergic diseases: Epidemiological study in Japanese university students. J Gastroenterol Hepatol. 2007 doi: 10.1111/j.1440-1746.2007.05107.x. [DOI] [PubMed] [Google Scholar]
- 54.Chen Y, Blaser M. Helicobacter pylori colonization is inversely associated with childhood asthma. Abstract presented at IDSA; October 2007; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bodner C, Anderson WJ, Reid TS, Godden DJ. Childhood exposure to infection and risk of adult onset wheeze and atopy. Thorax. 2000;55:383–387. doi: 10.1136/thorax.55.5.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tsang KW, Lam WK, Chan KN, Hu W, Wu A, Kwok E et al. Helicobacter pylori sero-prevalence in asthma. Respir Med. 2000;94:756–759. doi: 10.1053/rmed.2000.0817. [DOI] [PubMed] [Google Scholar]
- 57.Jun ZJ, Lei Y, Shimizu Y, Dobashi K, Mori M. Helicobacter pylori seroprevalence in patients with mild asthma. Tohoku J Exp Med. 2005;207:287–291. doi: 10.1620/tjem.207.287. [DOI] [PubMed] [Google Scholar]
- 58.Reibman J, Marmor M, Fernandez-Beros M, Rogers L, Perez-Perez GI, Blaser MJ. Asthma in an urban population is inversely associated with Helicobacter pylori status. Abstract presented at the American Thoracic Society; May 2005; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.National Center for Health Statistics. Plan and operation of the Third National Health and Nutrition Examination Survey. 1988–94. Vital and Health Statistics, series 1, no. 32. Hyattsville, MD: National Center for Health Statistics; 1994. [PubMed] [Google Scholar]
- 60.National Center for Health Statistics. NHANES 1999–2000 data files-data, docs, codebooks, SAS code. Hyattsville, MD: National Center for Health Statistics; 2005. [Google Scholar]
- 61.Hill AB. The environment and disease: association or causation? Proc Soc Med. 1965;58:295–300. [PMC free article] [PubMed] [Google Scholar]
- 62.Vonk JM, Postma DS, Boezen HM, Grol MH, Schouten JP, Koëter GH, Gerritsen J. Childhood factors associated with asthma remission after 30 year follow up. Thorax. 2004;59:925–929. doi: 10.1136/thx.2003.016246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Arif AA, Whitehead LW, Delclos GL, Tortolero SR, Lee ES. Prevalence and risk factors of work related asthma by industry among United States workers: data from the third national health and nutrition examination survey (1988–94) Occup Environ Med. 2002;59:505–511. doi: 10.1136/oem.59.8.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Butland BK, Strachan DP. Asthma onset and relapse in adult life: the British 1958 birth cohort study. Ann Allergy Asthma Immunol. 2007;98:337–343. doi: 10.1016/S1081-1206(10)60879-4. [DOI] [PubMed] [Google Scholar]
- 65.Kozyrskyj AL, Ernst P, Becker AB. Increased risk of childhood asthma from antibiotic use in early life. Chest. 2007;131:1753–1759. doi: 10.1378/chest.06-3008. [DOI] [PubMed] [Google Scholar]
- 66.Spiro DM, Arnold DH, Barbone F. Association between antibiotic use and primary idiopathic intussusception. Arch Pediatr Adolesc Med. 2003;157:54–59. doi: 10.1001/archpedi.157.1.54. [DOI] [PubMed] [Google Scholar]
- 67.Pillinger MH, Marjanovic N, Kim S-Y, Lee YC, Scher JV, Roper J, Abeles AM, Izmirly P, Fischer M, Pillinger MY, Tolani S, Dinsell V, Abramson SB. Helicobacter pylori stimulates gastric epithelial cell MMP-1 secretion via CagA-dependent and independent ERK activation. J Biol Chem. 2007;282:18722–18731. doi: 10.1074/jbc.M703022200. [DOI] [PubMed] [Google Scholar]
- 68.Simpson WG. Gastroesophageal reflux disease and asthma: diagnosis and management. Archives of Internal Medicine. 1995;155;8:798–803. [PubMed] [Google Scholar]
- 69.Field SK, Underwood M, Brant R, Cowie RL. Prevalence of gastroesophageal reflux symptoms in asthma. Chest. 1996;109;2:316–322. doi: 10.1378/chest.109.2.316. [DOI] [PubMed] [Google Scholar]
- 70.Perry S, de Jong BC, Hill P, Adegbola B, Parsonnet J. Helicobacter pylori and the outcome of M. tuberculosis infection. Abstract LB-22 presented at the 45th Infectious Diseases Society of America. Annual Meeting; October 4–7, 2007; San Diego, CA. (page 256 Program and Abstracts). [Google Scholar]
- 71.Matarese G, Moschos S, Mantzoros CS. Leptin in immunology. J Immunol. 2005;174:3137–3142. doi: 10.4049/jimmunol.174.6.3137. [DOI] [PubMed] [Google Scholar]
- 72.Matsuda Y, Okamatsu H, Tani K, Kimura T. Matsuishi. Correlation of plasma ghrelin and serum immunoglobulin levels: a hormonal link between immunity and obesity? J Allergy Clin Immunol. 2007;119:S174. [Google Scholar]
- 73.Azuma T, Suto H, Ito Y, Ohtani M, Dojo M, Kuriyama M, Kato T. Gastric leptin and Helicobacter pylori infection. Gut. 2001;49:324–329. doi: 10.1136/gut.49.3.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Moss SF, Legon S, Bishop AE, Polak JM, Calam J. Effect of Helicobacter pylori on gastric somatostatin in duodenal ulcer disease. Lancet. 1992;340:930–932. doi: 10.1016/0140-6736(92)92816-x. [DOI] [PubMed] [Google Scholar]
- 75.Levi S, Beardshall K, Haddad G, Playford R, Ghosh P, Calam J. Campylobacter pylori and duodenal ulcers: the gastrin link. Lancet. 1989;1:1167–1168. doi: 10.1016/s0140-6736(89)92752-9. [DOI] [PubMed] [Google Scholar]
- 76.El-Omar EM, Penman ID, Ardill JE, Chittajallu RS, Howie C, McColl KE. Helicobacter pylori infection and abnormalities of acid secretion in patients with duodenal ulcer disease. Gastroenterology. 1995;109:681–691. doi: 10.1016/0016-5085(95)90374-7. [DOI] [PubMed] [Google Scholar]
- 77.Kuipers EJ, Uyterlinde AM, Pena AS, Roosendaal R, Pals G, Nelis GF, Festen HP, Meuwissen SG. Long-term sequelae of Helicobacter pylori gastritis. Lancet. 1995;345:1525–1528. doi: 10.1016/s0140-6736(95)91084-0. [DOI] [PubMed] [Google Scholar]
- 78.Blaser MJ, Nomura A, Lee J, Stemmerman GN, Perez-Perez GI. Early life family structure and microbially-induced cancer risk. PLOS Medicine. 2007;4(1):e7. doi: 10.1371/journal.pmed.0040007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Blaser MJ. Science, medicine, and the future: Helicobacter pylori and gastric diseases. BMJ. 1998;316:1507–1510. doi: 10.1136/bmj.316.7143.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]