

Acute lymphoblastic leukemia (ALL) is a neoplasm of lymphocytic progenitors and represents the most common malignancy in children. The disease is frequently characterized by large-scale chromosomal alterations including hyper- or hypodiploidy, translocations, deletions and insertions. Genomic profiling of ALL patient samples identified frequent alterations in pathways regulating B-cell development, differentiation and cell cycle progression.1
Mutations in genes encoding epigenetic regulators are common in hematologic malignancies, including ALL. These mutations can occur in genes encoding writers, readers or erasers of the epigenetic marks and can result in a gain or loss of function. Methylation of lysine 27 on histone H3 (H3K27) is commonly dysregulated in both myeloid and lymphoid malignancies. Mutations of EZH2, SUZ12 and EED, members of the Polycomb repressive complex 2 that methylates H3K27, have recently been described in T-cell precursor ALL.2 In addition, loss-of-function mutations of UTX, which functions as a H3K27 demethylase, are found in a number of solid and hematological tumors, including multiple myeloma (MM) and ALL.3 These data suggest that precise regulation of H3K27 methylation is important for normal hematopoiesis.
Epigenetic regulators are also misregulated through chromosomal translocations that can effectively alter their normal biological roles. In 20% of multiple myeloma patients, translocation 4;14 leads to overexpression of histone methyltransferase MMSET/NSD2/WHSC1, which is believed to be the driving lesion of the disease. MMSET is a member of the NSD family of proteins, which includes NSD1 and NSD3 also implicated in oncogenesis. Multiple myeloma patients with t(4;14) have inferior survival rates and do not respond well to cytotoxic chemotherapy. MMSET dimethylates lysine 36 on histone H3 (H3K36me2) and its overexpression leads to a global increase in this modification, accompanied by a concomitant global decrease in H3K27me3 methylation, a mark associated with gene repression.4 These chromatin changes are associated with major changes in gene expression, enhanced proliferation and survival of myeloma cells. Additionally, MMSET overexpression has been found in a number of advanced stage solid tumors, including prostate, colon and skin cancers.5
Recent studies have attempted to reveal novel cancer-associated mutations through genomic sequencing of patient specimens and established cancer cell lines. In particular, the Cancer Cell Line Encyclopedia (CCLE) has cataloged mutations and gene expression analysis for a large set of genes in more than 1000 different cell lines and allows for identification of recurrent mutations in various tumors.6 Examination of this database identified a recurrent glutamic acid to lysine mutation at amino acid 1099 (E1099K) of MMSET in eight different cell lines. The mutation is located within the catalytic SET domain of the protein, suggesting that it could affect the ability of MMSET to methylate H3K36 (Figure 1a).
Figure 1.
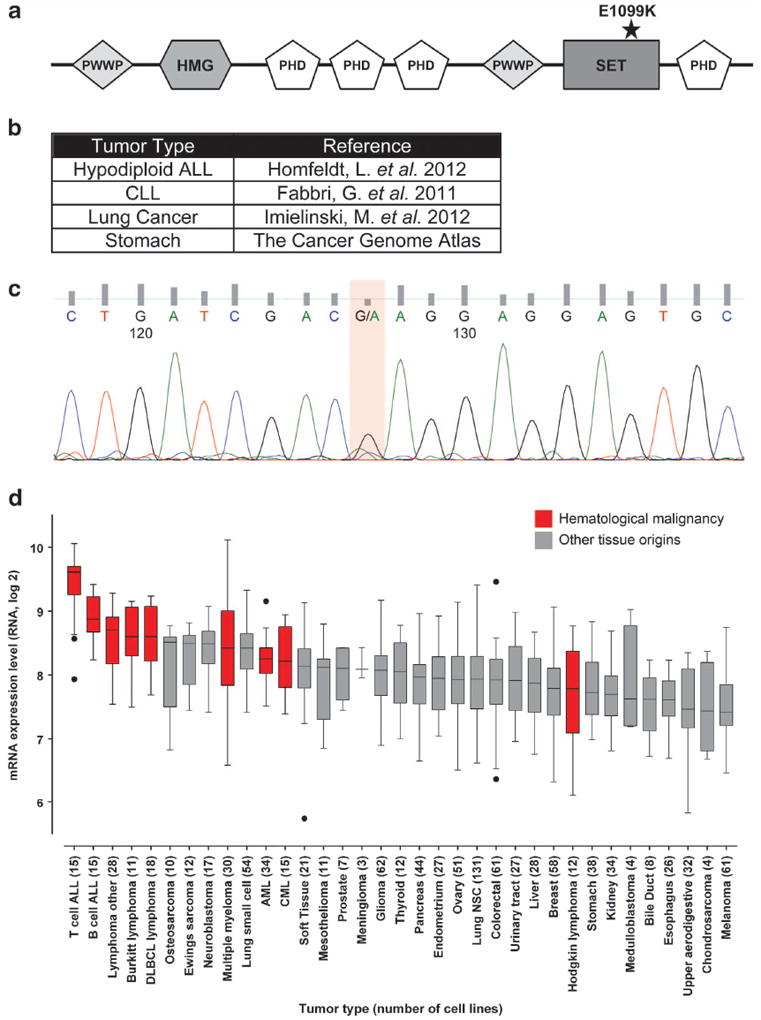
Recurrent E1099K mutation of MMSET in cancer. (a) Diagram of the MMSET protein. E1099K mutation within the SET domain is denoted by a star. (b) Table summarizing previously published studies identifying MMSET E1099K mutation in various tumors. (c) Sequence chromatogram for the B-precursor ALL patient carrying an E1099K MMSET mutation. The presence of both A and G peaks (orange highlight) indicates heterozygous mutation at this position. (d) Expression of MMSET mRNA across various cancer cell lines. The highest level of expression can be seen in lymphoid cells. The figure has been adapted from the Broad Cancer Cell Line Encyclopedia6.
Examination of previously sequenced patient data identified the E1099K mutation of MMSET in several different tumors, including 1/40 (2.5%) cases of hypodiploid ALLs, one patient with chronic lymphocytic leukemia (CLL), one case of lung adenocarcinoma and one patient with adenocarcinoma of the stomach (Figure 1b).7-9 In an independent cohort that included 11 pediatric B-precursor ALLs and 21 pediatric T-ALLs, we identified one patient with B-precursor ALL carrying the same heterozygous E1099K mutation (Figure 1c). Furthermore, seven out of eight cell lines containing MMSET E1099K mutation and sequenced by the CCLE are of lymphoid origin (1 MM and 6 ALL), suggesting that this mutation may predominantly be associated with lymphoid malignancies.6 Particularly, this mutation may be more predominant in pediatric ALL cases because most of the established MMSET mutant cell lines originated from younger patients. In agreement with this, our analysis of 200 cases of adult B-ALL from the ECOG E2993 study did not identify a single instance of the MMSET E1099K mutation. Additionally, analysis of CCLE data showed MMSET to be most highly expressed in T- and B-ALLs, Burkitt’s lymphoma and diffuse large B cell lymphomas (Figure 1d),6 further implicating MMSET in the development of lymphoid malignancies.
The recurrence of the heterozygous E1099K mutation, as well as its overrepresentation in lymphoid cell lines, suggests that it acts as a novel ‘driver’ of oncogenesis through altered MMSET function. Previously, similar mutations were described for EZH2, the enzyme responsible for di- and trimethylation of lysine 27 on histone H3 (H3K27me2/3). Activating mutations in the EZH2 SET domain are present in ~30% of germinal center diffuse large B-cell lymphomas and ~10% of follicular lymphomas. These mutations alter the activity of the enzyme, enhancing its ability to trimethylate lysine 27 and repress genes critical for the exit of the B cell from the germinal center.10 Similarly, we hypothesized that E1099K mutation within the SET domain of MMSET may enhance its ability to dimethylate lysine 36. In support of this idea, we compared histone methylation in cell lines with and without the E1099K mutation by immunoblot analysis. MMSET mRNA and protein levels in cells harboring the E1099K mutation were comparable to those with wild-type MMSET (Figures 2a and b). Nevertheless, RS411, SEM and RCH-AV cells, all containing the mutant MMSET, exhibit a global increase in H3K36me2 levels, strongly suggesting that MMSET E1099K has enhanced histone methyltransferase activity (Figure 2a). MM.M1S, a myeloma cell line, also harbors the E1099K mutation and has an enhanced level of H3K36me2, mimicking the MMSET overexpression found in t(4:14)+ cells. This suggests that, as in t(4;14)+ MM cases, ALL and other lymphoid malignancies with MMSET E1099K, global chromatin dysfunction may drive aberrant gene expression and contribute to disease pathogenesis.
Figure 2.
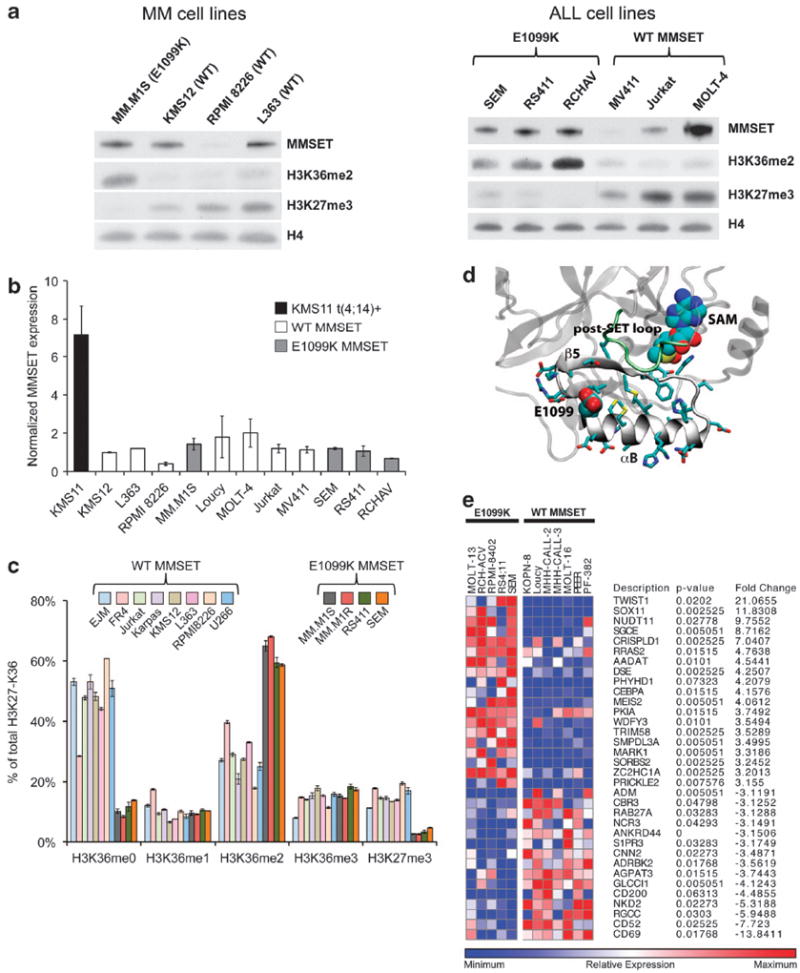
E1099K mutation of MMSET is associated with altered global histone methylation and gene expression. (a) Western blots using nuclear extracts from MM (left) and ALL (right) cell lines expressing the wild-type or E1099K mutant MMSET. The presence of E1099K mutation correlates with enhanced H3K36 dimethylation and concomitant global decrease in H3K27 trimethylation. Total H4 is used as a loading control. (b) Quantitative RT-PCR for MMSET in MM and ALL cell lines. KMS11 cells contain t(4;14) translocation and thus express elevated levels of MMSET, whereas the rest of the cell lines express lower levels of MMSET, independently of the presence of the mutation. (c) Mass spectrometry analysis of histones from MM and ALL cell lines expressing the wild type or E1099K mutant MMSET. The analysis was performed as described previously.12 Mutant cells have increased methylation of lysine 36 and decreased methylation of lysine 27. (d) Proposed substrate-binding site of MMSET. The αB and β-sheet 5 of SET domain are represented in gray and post-SET loop in green. Side chains of residues in αB and β-sheet 5 are shown in licorice representation and E1099 and S-adenosyl-L-methionine (SAM) are shown in atom colored vdW representation. A homology model of MMSET (NSD2) was developed based on NSD1 crystal structure (3OOI.pdb)15 using the program Modeller. The SET domain of NSD1 shares 75.9% sequence identity and 88.8% positive conservation with the MMSET SET domain without any gaps. As in the 3D structure of NSD1, the site that binds the C-terminal side of the H3 peptide in the MMSET model is in the cleft between SET and post-SET loops and contains E1099. (e) Heat map representing the most highly differentially expressed genes in MMSET wild-type (n = 7) and E1099K mutant (n = 5) ALL cell lines. The data and the figure were extracted from the Broad Cancer Cell Line Encyclopedia6.
Histone modifications show distinct patterns of interplay, and certain chromatin marks are dependent on each other whereas others are mutually exclusive. Our previous comparison of myeloma cell lines with and without the t(4;14) translocation showed that overexpression of MMSET and increased H3K36 methylation induce a dramatic decrease in the levels of H3K27me3.4 This inhibition is thought to occur through prevention of Polycomb recruitment to histones methylated on lysine 36.11 Similarly, E1099K mutant ALL cell lines demonstrate a lower abundance of H3K27me3 compared with wild-type ALL cells (Figure 2a). The differences in H3K36 and H3K27 methylation were confirmed independently by quantitative mass spectrometry (Figure 2c). Aberrant H3K27 methylation, through enhanced MMSET enzymatic activity, mimics the previously described Polycomb loss-of-function mutations in ALL and could lead to misregulation of genes and pathways that regulate normal lymphoid differentiation and cell survival.
In t(4;14)+ myelomas, overexpression of MMSET leads to aberrant dimethylation of normally unmethylated K36.12 Similarly, E1099K mutation enhances H3K36 dimethylation at the expense of unmethylated K36 (Figure 2c), suggesting that the mutation does not alter the enzyme’s substrate specificity. Thus, in comparison with the wild-type protein, E1099K mutation likely enhances the kinetic properties and processivity of the enzyme. Because the crystal structure of MMSET has not been solved, we used the structure of NSD1, a closely related family member of MMSET, to predict the position of the mutated glutamic acid residue within the SET domain. Analysis of a homology model of MMSET derived from the NSD1 catalytic domain structure shows E1099 to lie within the histone tail-binding groove (Figure 2d). Notably, the region on the C-terminal side of the K36 contains a lysine (K37) and two arginine residues (R40 and R42); such basic residues have been shown to play a role in histone–enzyme ionic interactions in other SET domain-containing proteins.13 Accordingly, introduction of a positively charged lysine by E1099K mutation may disrupt the favorable histone–enzyme interactions leading to the gain of function.
Collectively, these results establish epigenetic changes correlated with the presence of the MMSET E1099K mutation that are likely to affect transcriptional regulation (H3K27 hypomethylation and H3K36 hypermethylation). To identify specific gene targets that are dysregulated by these global changes in histone methylation, we utilized genome-wide gene expression data from the CCLE. Specifically, we compared two classes of female ALL cell lines either with the E1099K mutation (n = 5) or with the WT MMSET (n = 7). The mean expression calculated for individual genes within each class was used to discern the top 200 genes that are differentially expressed. All protein-coding genes with a greater than threefold difference in mean expression between E1099K and WT cell lines are shown in Figure 2e. Remarkably, this analysis identified TWIST1, a target of MMSET in both prostate cancer and MM, as one of the most upregulated genes in cells with the E1099K mutation.14
In conclusion, we report on a novel recurrent mutation affecting the catalytic domain of MMSET protein. This mutation is most predominant in pediatric lymphoid malignancies, although its involvement in other tumors cannot be ruled out. Our data suggest that, similar to t(4;14) translocations in multiple myeloma, E1099K mutation within MMSET enhances global H3K36 dimethylation in lymphoid cells. This is likely due to a charge alteration within the histone-binding pocket of MMSET and putative disruption of histone–enzyme ionic interactions. In addition, E1099K mutation and aberrantly high H3K36 methylation lead to a genome-wide decrease in H3K27me3, possibly affecting expression of a number of genes important for normal lymphoid development. As the number of small-molecule inhibitors of epigenetic regulators grows, it will be of interest to determine whether inhibition of MMSET activity can be beneficial not only for t(4;14)+ MM patients but also for patients who express a mutant allele of this protein.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Contributor Information
JD Licht, Email: j-licht@northwestern.edu.
R Popovic, Email: r-popovic@northwestern.edu.
References
- 1.Inaba H, Greaves M, Mullighan CG. Acute lymphoblastic leukaemia. Lancet. 2013;381:1943–1955. doi: 10.1016/S0140-6736(12)62187-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–163. doi: 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van Haaften G, Dalgliesh GL, Davies H, Chen L, Bignell G, Greenman C, et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nature Genet. 2009;41:521–523. doi: 10.1038/ng.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martinez-Garcia E, Popovic R, Min DJ, Sweet SM, Thomas PM, Zamdborg L, et al. The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood. 2011;117:211–220. doi: 10.1182/blood-2010-07-298349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hudlebusch HR, Santoni-Rugiu E, Simon R, Ralfkiaer E, Rossing HH, Johansen JV, et al. The histone methyltransferase and putative oncoprotein MMSET is overexpressed in a large variety of human tumors. Clin Cancer Res. 2011;17:2919–2933. doi: 10.1158/1078-0432.CCR-10-1302. [DOI] [PubMed] [Google Scholar]
- 6.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150:1107–1120. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fabbri G, Rasi S, Rossi D, Trifonov V, Khiabanian H, Ma J, et al. Analysis of the chronic lymphocytic leukemia coding genome: role of NOTCH1 mutational activation. J Exp Med. 2011;208:1389–1401. doi: 10.1084/jem.20110921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holmfeldt L, Wei L, Diaz-Flores E, Walsh M, Zhang J, Ding L, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nature Gen. 2013;45:242–252. doi: 10.1038/ng.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, et al. EZH2 is required for germinal center formation and somatic ezh2 mutations promote lymphoid transformation. Cancer Cell. 2013;23:677–692. doi: 10.1016/j.ccr.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yuan W, Xu M, Huang C, Liu N, Chen S, Zhu B. H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. J Biol Chemistry. 2011;286:7983–7989. doi: 10.1074/jbc.M110.194027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng Y, Sweet SM, Popovic R, Martinez-Garcia E, Tipton JD, Thomas PM, et al. Total kinetic analysis reveals how combinatorial methylation patterns are established on lysines 27 and 36 of histone H3. Proc Natl Acad Sci USA. 2012;109:13549–13554. doi: 10.1073/pnas.1205707109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xiao B, Jing C, Wilson JR, Walker PA, Vasisht N, Kelly G, et al. Structure and catalytic mechanism of the human histone methyltransferase SET7/9. Nature. 2003;421:652–656. doi: 10.1038/nature01378. [DOI] [PubMed] [Google Scholar]
- 14.Ezponda T, Popovic R, Shah MY, Martinez-Garcia E, Zheng Y, Min DJ, et al. The histone methyltransferase MMSET/WHSC1 activates TWIST1 to promote an epithelial-mesenchymal transition and invasive properties of prostate cancer. Oncogene. 2013;32:2882–2890. doi: 10.1038/onc.2012.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qiao Q, Li Y, Chen Z, Wang M, Reinberg D, Xu RM. The structure of NSD1 reveals an autoregulatory mechanism underlying histone H3K36 methylation. J Biol Chem. 2011;286:8361–8368. doi: 10.1074/jbc.M110.204115. [DOI] [PMC free article] [PubMed] [Google Scholar]