

Abstract
Amyotrophic lateral sclerosis (ALS) causes motor neuron degeneration and paralysis. No treatment can significantly slow or arrest the disease progression. Mutations in the SOD1 gene cause a subset of familial ALS by a gain of toxicity. In principle, these cases could be treated with RNAi that destroys the mutant mRNA, thereby abolishing the toxic protein. However, no system is available to efficiently deliver the RNAi therapy. Recombinant adenoassociated virus (rAAV) is a promising vehicle due to its long-lasting gene expression and low toxicity. However, ALS afflicts broad areas of the central nervous system (CNS). A lack of practical means to spread rAAV broadly has hindered its application in treatment of ALS. To overcome this barrier, we injected several rAAV serotypes into the cerebrospinal fluid. We found that some rAAV serotypes such as rAAVrh10 and rAAV9 transduced cells throughout the length of the spinal cord following a single intrathecal injection and in the broad forebrain following a single injection into the third ventricle. Furthermore, a single intrathecal injection of rAAVrh10 robustly transduced motor neurons throughout the spinal cord in a non-human primate. These results suggested a therapeutic potential of this vector for ALS. To test this, we injected a rAAVrh10 vector that expressed an artificial miRNA targeting SOD1 into the SOD1G93A mice. This treatment knocked down the mutant SOD1 expression and slowed the disease progression. Our results demonstrate the potential of rAAVs for delivering gene therapy to treat ALS and other diseases that afflict broad areas of the CNS.
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) causes progressive motor neuron degeneration, leading to skeletal muscle atrophy, paralysis and death. Approximately 90% of cases are sporadic and 10% are inherited. Among the inherited cases, multiple genetic mutations have been identified but the mechanisms whereby these mutations cause disease remain unclear (1,2). Due to limited understanding of the disease mechanisms, it is not yet clear what a rational therapeutic strategy should be. Nevertheless, strong evidence suggests that mutations in one of the disease genes, SOD1, cause motor neuron degeneration by a gain of toxicity (3). Based on this understanding, it has been postulated that by lowering the mutant protein expression, the toxicity can be reduced and motor neuron degeneration slowed (4). Various strategies to reduce the expression of the toxic mutant protein have been tested, including silencing SOD1 expression using RNAi or antisense oligonucleotides and neutralizing mutant protein toxicity by antibodies targeting the misfolded protein. These strategies were efficacious in animal models (5–10) but remain to be evaluated in humans.
The rAAV-based gene delivery system holds promise for delivering functional genes to treat diseases caused by loss-of-function gene mutations as well as therapeutic RNAi to treat disorders caused by gain-of-toxicity gene mutations (11). Some of the most attractive features of the rAAV system for gene therapy include its low toxicity and the capacity to mediate long-term stable transgene expression (12,13). Recent expansion of known rAAV serotypes with varied cell tropism has also broadened the range of diseases to which rAAV may be applied (14). Particularly, encouraging is the recent clinical trials in human central nervous system (CNS) diseases, where rAAV is shown to be safe and demonstrates therapeutic efficacy (15,16).
Various rAAV serotypes have been tested for gene transduction in the CNS by different routes of administration. A long-practiced method is locally injecting rAAV into a region of the CNS afflicted in a human disease, such as substantia nigra for Parkinson's disease, striatum for Huntington's disease and cerebellum for spinocerebellar ataxia, resulting in transgene expression in the vicinity of the injected site (17–20). However, ALS causes degeneration of motor neurons that are distributed throughout the CNS including the cortex, brainstem and the entire length of the spinal cord. Effective delivery of gene therapy to these broad areas remains elusive. Indeed, injection of rAAV-based RNAi therapeutics into the spinal cord parenchyma transduced limited numbers of cells. While the treatment protected the transduced cells, it produced no overall clinical benefit in an animal model for ALS (6).
In order to transduce broadly distributed motor neurons by rAAVs, several strategies have been explored. One method was injecting the vectors into skeletal muscle and allowing the nerve terminals to internalize the vector genomes, which were then transported in retrograde direction to the spinal motor neurons. This approach has shown some success in mouse models (5,21,22). However, applying this method in adult humans would be difficult if not impossible. The transduction efficiency of motor neurons by muscle injection was relatively low, resulting in disappointing outcomes for treating ALS (23). A much larger muscle mass in humans than in mice also poses additional challenges including manufacturing and delivering large doses of rAAV and the risk for potential adverse effects ranging from immune reaction to transduction of non-targeting cells.
An alternative strategy for delivery of transgenes into motor neurons is to inject the virus into large nerves, which maximizes the exposure of motor axons to the virus, allowing the motor neurons to internalize the viral genome and transport it back to the cell bodies (9,24). This method has been shown to be more efficient in transducing motor neurons than muscle injection (9). However, translation of this method to human applications will also be challenging because many nerves will need to be injected in order to cover all the various motor neuron groups. Additionally, the degenerating motor axons may be inefficient in transporting the vector genomes to the cell body, resulting in poor transduction efficiency.
Another approach is to administer rAAV by intravenous injection. It has been demonstrated that systemically delivered rAAV9 and several other AAV serotypes can cross the blood–brain barrier and transduce cells in broad areas of the CNS (25–29). However, the relatively low levels of transgene expression in the CNS, the high levels of peripheral tissue transduction and potential immunotoxicity as well as the requirement of large doses for human applications may compromise the balance between therapeutic benefit and untoward side effects.
Recently, several groups have tested injection of rAAV9 into cerebrospinal fluid (CSF) (27, 30–32) and have generally reported wider transduction of cells. However, widespread strong transgene expression and therapeutic efficacy for ALS remain to be demonstrated. To this end, we tested five rAAV serotypes (rAAV1, 2, 8, 9 and rh.10) for their capacity to spread and transduce broad CNS areas following injections into the CSF. Of these five serotypes, rAAV9 and rh.10 displayed a remarkable capacity for spreading transduction broadly. A single intrathecal injection in the lumbar region achieved an unprecedented high level of transgene expression along the full length of the spinal cord and even to the brainstem. Likewise, a single injection into the third ventricle also delivered similar high levels of transgene expression to broad areas of the forebrain. To test whether this strategy can be vertically translated into adult primates, we performed the same experiment in a non-human primate species and observed similar spreading of transgene expression along the full length of the monkey spinal cord. To test this approach for therapeutic RNAi delivery to treat ALS, we constructed a rAAVrh10 vector that co-expressed GFP and an artificial miRNA (amiRNA) targeting human Sod1 mRNA (amiR-Sod1). Following the intrathecal injection of this vector into a mouse ALS model that expresses mutant SOD1G93A (33), this rAAVrh10 vector mediated broad transduction along the spinal cord, knocked down the mutant SOD1 expression and significantly slowed the disease progression. These results suggest that CSF injection of rAAVrh10, rAAV9 and possibly other serotypes can be effective in delivering therapeutic genes to treat human diseases that afflict broad areas of the CNS.
RESULTS
rAAVrh10 and rAAV9 mediate widespread and sustained transgene expression in the CNS after a single injection into the CSF
To improve the prospect of gene therapy for diseases that afflict broad areas of the CNS such as ALS, we took advantage of a collection of various rAAV serotypes (14) and screened a subset of those rAAVs (rAAV1, 2, 8, 9 and rh.10) for EGFP reporter gene delivery to the spinal cord. We directly injected each of the rAAVs into the CSF in the area of the lumbar spine. Three to four weeks later, we dissected out the entire CNS and examined GFP directly under a fluorescence microscope. We observed that three serotypes, rAAV1, 2 and 8 transduced relatively poorly along the spinal cord. Among these, AAV1 generated localized EGFP expression in the cells around the injection site (Fig. 1A). In contrast, two other rAAVs, rAAVrh10 and rAAV9, achieved widespread EGFP transduction along the entire spinal cord and as far as the brainstem (Fig. 1A and B; Supplementary Material, Fig. S1). The CNS structures rostral to the brainstem were generally insignificantly transduced, although scattered astrocytes and ascending fibers tracks could be observed in midbrain and cerebral cortex (Supplementary Material, Fig. S1). In the spinal cord, the transgene expression was strong, widespread, asymmetric and uneven (Fig. 1B, Supplementary Material, Fig. S1). Based on cellular morphology, most of the transduced cells were astrocytes (see confirmation below) with a few scattered motor neurons (Fig. 1C).
Figure 1.

Widespread transduction of rAAVrh10 and rAAV9 compared with AAV serotype 1, 2 and 8 after a single intrathecal injection into the lumbar CSF. (A) GFP fluorescence in whole mount brains and spinal cords. rAAV (4.8 × 1010 vg) were injected into each mouse. Whole brains and spinal cords were dissected out at 28 days after the injection. The GFP fluorescence was photographed directly from the whole mount tissue. The panels in the third column on the right are false color heatmap of the second panel showing GFP intensity in various regions. The heatmap was generated by taking an 8bit monochrome image and applying a ‘heatmap’ palette on NIS-elements software (Nikon). Each color represents a different fluorescence intensity: pink corresponds to maximum intensity of 255 and violet corresponds to minimum of 1. (B) GFP fluorescence in longitudinal sections throughout the brainstem and the spinal cord in a mouse injected with AAVrh10. From left to right: brainstem (BS), cervical (CSC), thoracic (TSC) and lumbar spinal cord (LSC) segments. Scale bars represent 250 µm. (C) Panel a shows an enlarged segment of cervical spinal cord, which is representative to all levels of the spinal cord. The white arrow points to a motor neuron among numerous astrocytes (red arrows). The scale bar = 20 µm. Panel b shows an enlarged view of a cell with astrocyte morphology. The scale bar = 5 µm. Panel c shows an enlarged view of a cell with motor neuron morphology. The scale bar = 10 µm. In all images in this and other figures, the GFP fluorescence was captured directly from unstained sections.
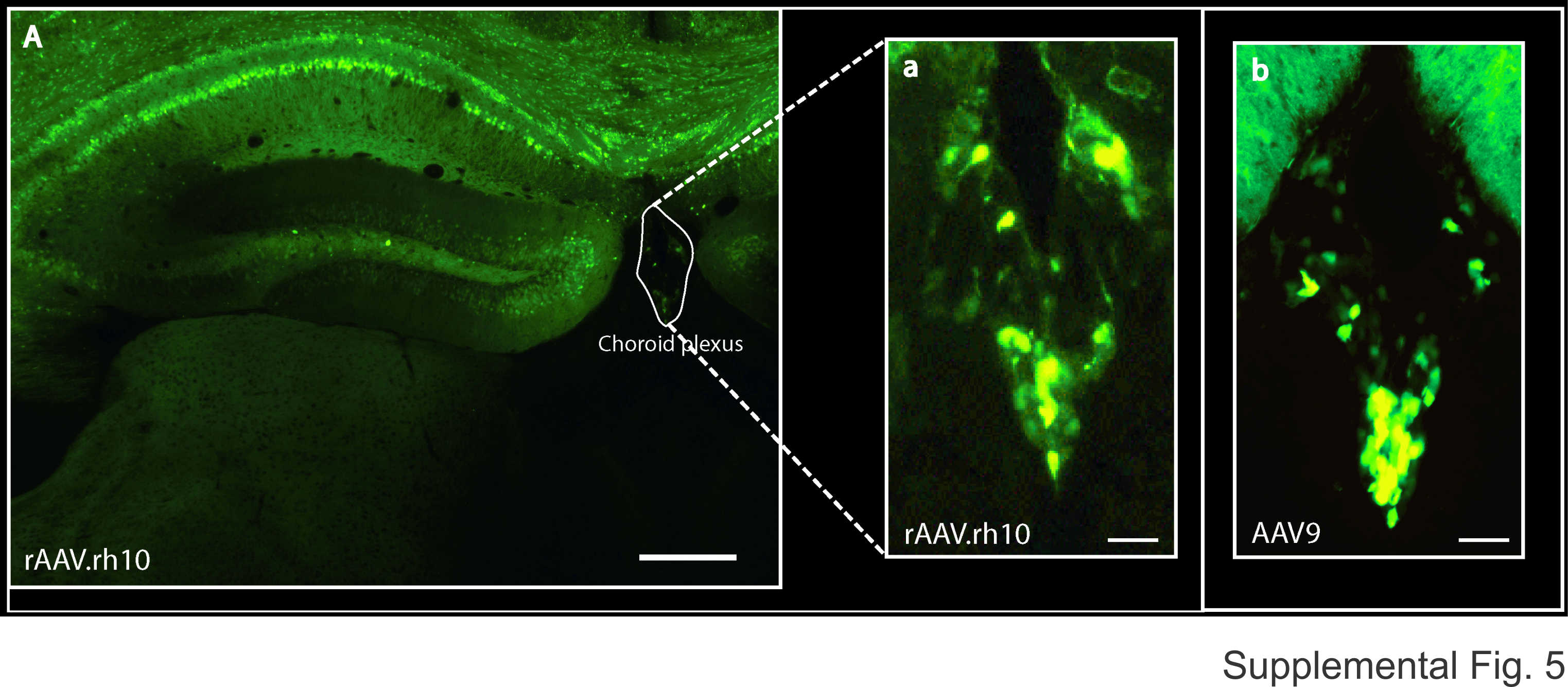
Consistent with these findings, when injected into the CSF in the third ventricle, rAAV1, 2 and 8 generated relatively poor transduction, whereas rAAVrh10 and rAAV9 transduced well throughout wide areas of the CNS, spreading from olfactory bulb to sacral spinal cord (Fig. 2A and B; Supplementary Material, Fig. S2). The most prominent transductions were in the areas of the cerebral cortex, hippocampus, striatum, corpus callosum, amygdala, cerebellum and other areas (Fig. 2B; Supplementary Material, Figs S2–S4). Additionally, cells in choroid plexus were transduced (Supplementary Material, Fig. S5). In general, the transduction levels were highly variable among the individual animals and in different regions of the same animal. For example, in the best transduced animals that were injected into the 3rd ventricle, we estimate that ∼50–80% of the DAPI-stained cells were GFP positive in the best transduced regions, which included the layer 5/6 of the cerebral cortex, the CA1 region of hippocampus, corpus callosum and upper part of the striatum. Even in some relatively weak regions, including thalamus, amygdala, the lateral sides of the cortex, ∼30–50% cells were transduced. However, the regions posterior to the forebrain, only the cerebellum showed a modest level of transduction (∼10–30%). In other regions including midbrain, brainstem and spinal cord, only small clusters and scattered cells were transduced (Fig. 2B, Supplementary Material, Figs S2–S4).
Figure 2.

Broad and robust transduction of forebrain cells after a single injection of 4.8 × 1010 rAAV9 and rAAVrh10 particles into the third ventricle. (A) A comparison of transduction efficiency demonstrates that rAAV9 and AAVrh10 are the best serotypes. GFP fluorescence in whole mount brains was photographed at 28 days after the injection. The panels in the third column on the right are false color heatmap of the second panels showing GFP intensity in the brains, which was generated as described in the legend of Fig. 1. (B) Coronal sections of the CNS from a rAAV9-injected mouse. The section plains are marked in the bright field image at the bottom. OB, olfactory bulb; FB, forebrain; CB, cerebellum; BS, brainstem; CSC, TSC, LSC and SSC represent cervical, thoracic, lumbar and sacral spinal cords, respectively.
The major cell types transduced in the forebrain included neurons, astrocytes and oligodendrocytes based on cellular morphology (Supplementary Material, Figs S3 and S4). In cerebellum, some Purkinje cells were transduced (Fig. 2B; Supplementary Material, Fig. S2). In the lower brain regions, the major GFP-fluorescent structures were the corticospinal track and scattered or clustered astrocytes (Fig. 2B; Supplementary Material, Fig. S2). To confirm the transduced cell types, we stained for various cellular markers. The results confirmed EGFP transduction of neurons, astrocytes and oligodendrocytes, but not microglia (Fig. 3; Supplementary Material, Fig. S6).
Figure 3.
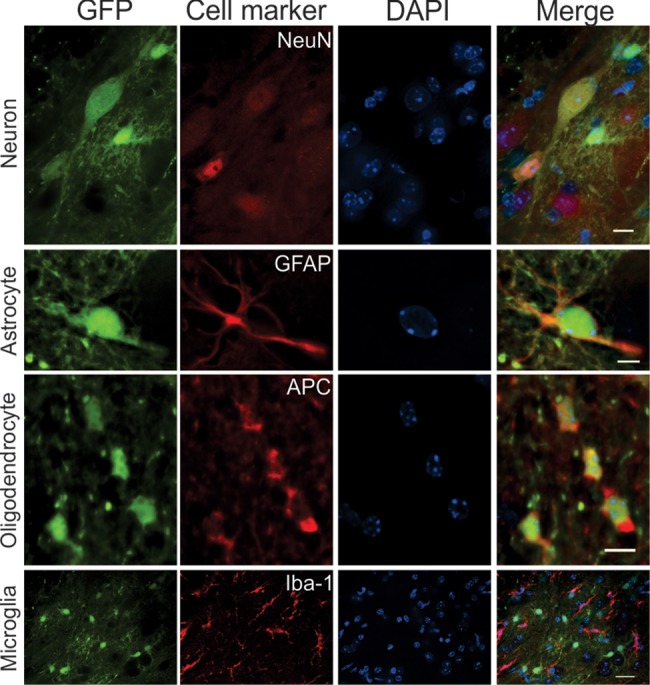
rAAVrh10-GFP transduced neurons, astrocytes and oligodendrocytes, but not microglia after its intrathecal injection into the CSF. Spinal cord sections were stained for neurons (NeuN), astrocytes (GFAP), oligodendrocytes (APC) and microglia (Iba-1). Scale bar is 10 µm in the first row, 5 µm in the second and third row and 20 µm in the fourth row.
Intrathecal injection of rAAVrh10-GFP transduces motor neurons along the whole length of the spinal cord in a primate species
The experimental data in mice demonstrated that rAAVrh10 could spread broadly in the mouse CNS. To determine its performance in primates, we chose to test in marmoset monkey (Callithrix jacchus), which is thought to be phyletic dwarfs that have evolved from a larger ancestor. Its small size (∼300–600 g) and many genetic and physiologic similarities to humans afford many advantages as an experimental model system (34). Indeed, marmoset has been used in modeling neurodegenerative diseases such as Parkinson's disease and Huntington's disease (35–40). Recently, technology for transgenic marmoset has been developed (41), further enhancing the promise of marmoset as a model system for neurodegenerative diseases in the future. We intrathecally injected a marmoset monkey with the rAAVrh10 vector. A single intrathecal injection of rAAVrh10-GFP into the CSF in the subarachnoid space of the lumbar spine resulted in transduction of cells along the entire length of the spinal cord. Robust GFP signal was detected in motor neurons and their axons at the cervical (Fig. 4A, D and G), thoracic (Fig. 4B, E and H) and lumbar (Fig. 4C, F and I) levels. Similarly, strong GFP signal was observed in the dorsal root axons at the cervical (Fig. 4J) and thoracic (Fig. 4K) levels, and to a lesser degree, lumbar (Fig. 4L) levels, thus indicating that the neurons in the dorsal root ganglia (DRG) at cervical and thoracic, and to a lesser degree at lumbar levels, were well transduced. Consistent with this pattern of transduction in the DRG neurons, axon fibers in the cuneate fasciculus expressed GFP strongly (Fig. 4M and N) while fibers in the gracile fasciculus expressed GFP relatively weakly (Fig. 4O). The cells in the intermediate zone of the spinal cord were generally weakly transduced at all levels (Fig. 4P –R). Thus, although further study will be needed to determine the consistency of this transduction pattern, our results demonstrate that intrathecal injection of AAVrh10 into the CSF can spread transgene expression along the full length of the spinal cord in adult primates, just as in mice.
Figure 4.

Intrathecal injection of rAAVrh10-GFP into the lumbar CSF of a marmoset led to a robust expression of GFP in motor neurons along the full length of the spinal cord as revealed by immunostaining in (A) cervical, (B) thoracic and (C) lumbar spinal cord. Scale bars = 200 µm. (D–F) Enlarged views of ventral horn motor neurons (red boxes) in (A)–(C), respectively. Scale bars are 100 µm in (D) and (F), and 50 µm in (E). (G–I) GFP expression in motor axons in ventral white matter (blue boxes) in (A)–(C), respectively. Scale bars = 100 µm. (J–L) GFP expression in dorsal root axons (yellow boxes) in (A)–(C), respectively. Scale bars = 50 μm. (M–O) Dorsal funiculus (purple boxes) in (A)–(C), respectively. Scale bars = 100 µm (M and O) and 50 µm (N). Notice the strong GFP signals in the cuneate fasciculus (cu) in the cervical (M) and thoracic (N) spinal cord, but only modest GFP signal in gracile fasciculus (gr) in the lumbar (O) spinal cord. GM means gray matter. (P–R) The intermediate zone of the gray matter (green boxes) in (A)–(C), respectively. Scale bars in (J) to R = 50 µm. All sections were counterstained with haematoxylin.
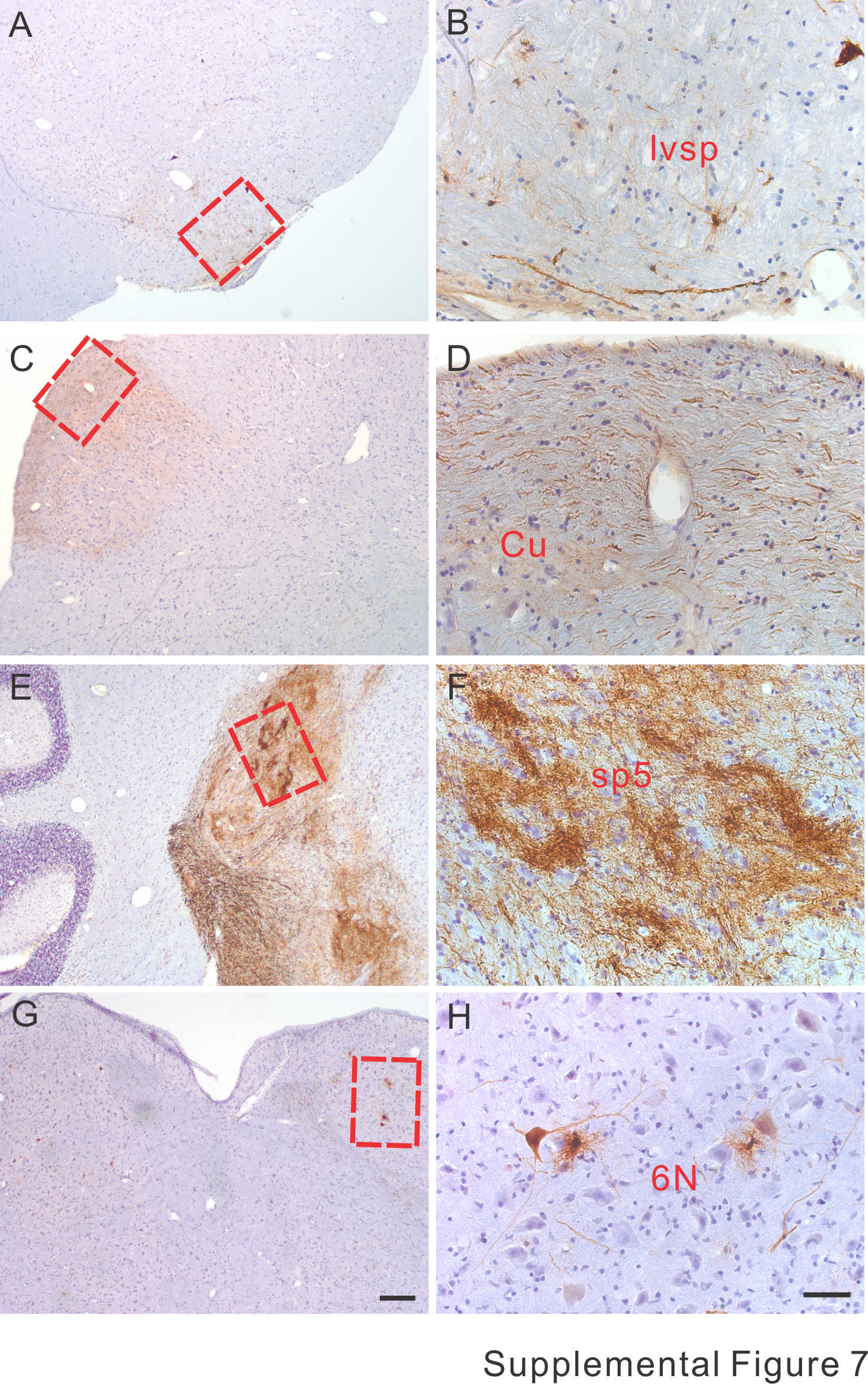
Intrathecal injection of AAVrh10 also led to transduction in some areas of the brainstem, although the transduction levels were generally lower than in the spinal cord. For example, in the medulla oblongata, GFP signal was undetectable in most cells. Only a few cells and some fibers expressed GFP in lateral vestibulospinal tract (Supplementary Material, Fig. S7A and B). However, robust GFP signal was observed in the cuneate funiculus (Supplementary Material, Fig. S7C and D), which confirms the transduction of the DRG neurons along the sides of the cervical and thoracic spinal cord (Fig. 7J and K). In the pons, strong GFP expression was observed in the spinal trigeminal tract (Supplementary Material, Fig. S7E and F). Additionally, a few motor neurons and glial cells in the abducens nucleus expressed GFP (Supplementary Material, Fig. S7G and H). In areas further rostral to the brainstem, the transduction was generally weak. Only sporadic single or clustered cells were transduced in cerebellum, hippocampus and cerebral cortex (Supplementary Material, Fig. S8). Therefore, the overall transduction area following intrathecal injection of AAVrh.10 was similar between the marmoset and the mice.
Figure 7.

Therapeutic benefit of AAVrh10-GFP-amiR-SOD1. Female SOD1G93A mice were injected intrathecally with the amiR-SOD1 (red curves) and amiR-Scr vectors (green curves) between 55 and 60 days of age, and were followed by weekly body weight measurements. (A) The disease progression as monitored by body weight, which peaked at the same time in the two groups (arrow). However, the decline was slowed in the amiR-SOD1-treated group compared with the amiR-Scr-treated group. (B) The disease onset, which is defined as the day of peak body weight, was not significantly different between the two groups (see also Table 1). (C) The early disease stage, which is defined as a 10% weight loss from the peak body weight, was delayed in the amiR-SOD1-treated group (see also Table 1). (D) The end disease stage, which is defined as the onset of paralysis in any two limbs, was significantly delayed in the amiR-SOD1-treated group (see also Table 1). (E) The lifespan was correlated with the transduction level in AAVrh10-GFP-amiR-SOD1-injected mice. (F) There was not a correlation between the lifespan and transduction level in AAVrh10-GFP-amiR-Scr-injected mice.
rAAVrh10 delivers widespread expression of amiR-Sod1 in the CNS and knocks down the expression of mutant SOD1 in vivo
The widespread reporter gene transduction by rAAVrh10 and rAAV9 following a single CSF injection suggested that these vectors would be useful to deliver therapeutic genes to treat ALS. To test this, we constructed nine plasmids that co-expressed GFP and a panel of amiRNAs designed for silencing human SOD1 expression (amiR-Sod1; Supplementary Material, Table S1). These amiRNAs matched the SOD1 mRNA perfectly so that they would mediate silencing through the RNAi mechanism. The amiR-Sod1 was embedded in an intron in the 3′-UTR (Fig. 5A). We chose a miRNA construct instead of a shRNA construct based on studies that documented lower side effects of miRNA constructs as compared with the shRNA constructs (18). Additionally, the miRNA and GFP co-expression were driven by the same Pol II promoter. Therefore, the GFP expression should provide an unambiguous indication of the spatial and temporal expression pattern of the amiRNA. As a control, we also constructed a plasmid that expresses GFP and a scrambled artificial miRNA (amiR-Scr; Supplementary Material, Table S1) that does not target any known genes. We screened the nine constructs that expressed different amiR-Sod1 for their efficiency in knocking down SOD1 expression in cells in culture (Fig. 5B and C). We found that construct #5 most consistently and potently inhibited human SOD1 expression, whereas the amiR-Scr construct had no effect (Fig. 5B and C). Therefore, we used construct #5 to produce the rAAVrh10-GFP-amiR-Sod1. As a control, we also produced rAAVrh10-GFP-Scr. The rAAVrh10-GFP-amiR-Sod1 could reproducibly knockdown human SOD1 expression in vitro (Fig. 5D).
Figure 5.

In vitro test of SOD1 knockdown by amiR-SOD1 constructs. (A) A schematic illustration of the construct, which uses the chicken β-actin (CBA) promoter to drive transcription of GFP and amiR-Sod1 expression. pA = poly A signal. (B) Testing SOD1 knockdown activity by different amiR-SOD1s using Northern blot. Human HEK293 cells were transfected with the plasmid constructs. The total RNA was used in Northern blot for SOD1 mRNA. (C) Western blot for SOD1 protein from the transfected HEK293 cells. Notice that amiR-SOD1#1, 2, 3, 5, 6 and 8 knocked down SOD1 significantly, whereas amiR-SOD1#4, 7 and 9 showed poor knockdown activity. (D) Testing AAVrh10-amiR-SOD1#5 by transducing HEK293 cells. SOD1 was detected by western blot of the total protein extracted from cells transduced for 48 h. Scr represents AAVrh10-amiR-Scr that expressed a scrambled miRNA. M represents mock transfection. β-Actin was used as a loading control for all the blots.
To determine therapeutic efficacy of this viral construct in vivo, we intrathecally injected this vector into the SOD1G93A transgenic mice. We experienced some variable success rates in those injections as monitored by EGFP fluorescence at the end point of our study. Briefly, ∼25% of the injections produced high levels of transduction of the spinal cord, ∼50% generated medium levels and ∼25% resulted in low levels of transduction. This variation was likely caused by various degrees of leakage during intrathecal injection, as we observed an inverse correlation between the EGFP transduction efficiency of the spinal cord and the liver (Supplementary Material, Fig. S9). Interestingly, the expression level of amiR-Sod1 and the degree of SOD1 knockdown were well correlated with the levels of EGFP transduction (Fig. 6A–C). By double immunofluorescence, we observed reduced SOD1 staining intensity in the GFP-positive cells but not in the GFP-negative cells in the rAAVrh10-GFP-amiR-Sod1-treated animals (Fig. 6D). rAAVrh10-GFP-amiR-Scr-treated mice did not show evidence of SOD1 knockdown (data not shown). Therefore, the rAAVrh10-GFP-amiR-Sod1 vector could silence mutant SOD1 expression in vivo.
Figure 6.
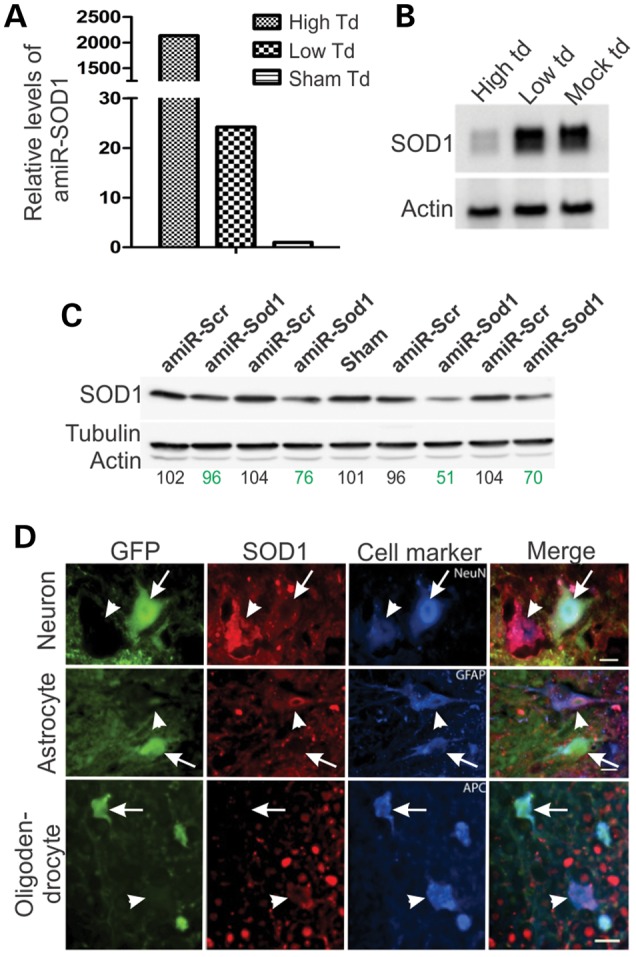
AAVrh10-GFP-amiR-SOD1 expresses amiR-SOD1 and knocks down SOD1 expression in vivo. (A) Real-time PCR for amiR-SOD1 from mouse spinal cords that were transduced (td) at high and low levels. The levels were compared with a sham transduced spinal cord, which was from a mouse injected with PBS. (B) Northern blot for SOD1 mRNA from spinal cord with high, low and sham vector transduction. (C) Western blot for SOD1 from spinal cords of individual animals injected with amiR-Scr or amiR-SOD1. Tubulin and actin were detected as loading controls. The numbers below the blot panels are the relative SOD1 band density normalized to tubulin band density and then the average SOD1 density in the uninjected mice, which is set at 100. Notice the various degrees of knockdown among the four AAV-amiR-SOD1-injected animals. (D) Immunofluorescence staining of spinal cord sections from a mouse injected with AAVrh10-GFP-miR-SOD1. Neurons were marked with NeuN, astrocytes with GFAP and oligodendrocytes with APC. The scale bars are 10, 5 and 10 µm in top, middle and low panels, respectively. Notice that GFP-positive cells (arrows) were stained weak for hSOD1 compared with the GFP-negative cells (arrowheads).
rAAVrh10 delivered amiR-SOD1 expression in the CNS slows disease progression in the SOD1G93A mice
To determine whether the rAAVrh10-GFP-amiR-Sod1 vector was therapeutically efficacious in the high expresser line of SOD1G93A mice, we administered the vector to the animals by intrathecal injection and compared the disease progression with a group of rAAVrh10-GFP-amiR-Scr-treated control mice. The amiR-Sod1 vector did not significantly alter the disease onset based on the age of peak body weight (Fig. 7A and B). However, the vector significantly slowed the disease progression and extended survival (Fig. 7C and D, Table 1). Notably, there was a larger variation of survival in the treated group than the control group (Fig. 7D). To determine whether the variability in survival was associated with variability in transduction, we evaluated the GFP fluorescence in each of the freshly dissected spinal cords from the end stage animals using a semiquantitative scale and then correlated this result with the survival time. We found a positive correlation in the group that was treated with the amiR-Sod1 vector, but not in the group that was treated with the amiR-Scr vector (Table 2, Fig. 7E and F), thus suggesting that the transduction of amiR-Sod1 led to the therapeutic benefit.
Table 1.
AAVrh10-amiR-Sod1 slows ALS progression
| Treatment | Onset | ErS | EdS | Onset to ErS | Onset to EdS | n |
|---|---|---|---|---|---|---|
| AAVrh10-amiR-SOD1 | 97 ± 11 | 123 ± 12 | 136 ± 14 | 26 ± 11 | 38 ± 14 | 23 |
| AAVrh10-amiR-Scr | 95 ± 8 | 114 ± 9 | 123 ± 7 | 19 ± 7 | 29 ± 9 | 18 |
| P (Gehan–Breslow–Wilcoxon test) | 0.8026 | 0.011 | 0.0011 | 0.0277 | 0.0135 |
The numbers are average days ± standard deviation. Onset, the day each mouse reached maximum body weight. ErS, early stage of disease, the day a mouse lost 10% or more of its maximum body weight. EdS, end stage of disease, the day a mouse was completely paralyzed in two or more limbs.
Table 2.
Correlation between the transduction level and survival
| Mouse group | GFP intensity in spinal cord |
Correlation | |||||
|---|---|---|---|---|---|---|---|
| Virus injected | Survival | Cervical | Thoracic | Lumbar | Total score | Spearman's rank CC | n |
| AAVrh10-amiR-SOD1 | 166 | +++ | ++ | ++++ | 9 | 0.813 | 15 |
| 151 | ++ | + | +++ | 6 | |||
| 148 | ++ | ++ | ++ | 6 | |||
| 147 | ++ | ++ | ++ | 6 | |||
| 141 | + | + | ++ | 4 | |||
| 140 | ++ | ++ | ++ | 6 | |||
| 139 | + | ++ | +++ | 6 | |||
| 135 | + | ++ | ++++ | 7 | |||
| 135 | + | + | ++ | 4 | |||
| 134 | + | + | ++ | 4 | |||
| 131 | + | + | − | 2 | |||
| 128 | − | − | + | 1 | |||
| 123 | − | + | + | 2 | |||
| 122 | ++ | − | − | 2 | |||
| 111 | + | + | ++ | 4 | |||
| AAVrh10-amiR-Scr | 135 | ++ | + | ++ | 5 | −0.182 | 11 |
| 123 | ++++ | ++++ | ++ | 10 | |||
| 122 | ++++ | ++++ | +++ | 11 | |||
| 122 | ++ | + | − | 3 | |||
| 119 | ++++ | ++++ | +++ | 11 | |||
| 119 | ++ | +++ | ++++ | 9 | |||
| 119 | ++ | ++ | − | 4 | |||
| 118 | ++ | +++ | ++ | 7 | |||
| 117 | ++++ | ++++ | ++++ | 12 | |||
| 115 | ++ | ++++ | ++++ | 10 | |||
| 111 | ++ | ++ | ++ | 6 | |||
Mice that reached end disease stage were sacrificed. Spinal cords were dissected and visually assessed for GFP intensity level in different segments. The semiquantitative assessment was conducted as described in Materials and Methods and converted to a numeric score. The correlation between the score and the survival time was calculated. There was a positive correlation between AAVrh10-amiRr-SodOD1-injected mice, but no correlation between AAVrh10-amiRr-Scr injected mice.
DISCUSSION
Our study demonstrates that widespread, strong CNS transduction is achievable by single bolus CSF injections of rAAVrh10 and rAAV9, and in addition, therapeutic genes delivered by this strategy can be efficacious. These results have built on previously established methods and have brought gene therapy closer to clinical application in ALS and other diseases that afflict broad areas of the CNS.
Previous reports have shown that CSF injection can improve the spread of transduction by rAAV9 but key obstacles for its application in ALS remained. Federici and colleagues compared intraparenchymal and intra-CSF injections of rAAV6 and rAAV9 into the lumbar spine in mice (20). They found that the intraparenchymal injection transduced cells in the vicinity of the injection site, whereas the CSF injection resulted in a wide transduction, covering predominantly lumbar and lower thoracic spinal cord, which, nevertheless, was less extensive than our results (Fig. 1). In another rodent study (32), Gray and colleagues found that injection of rAAV9 into the third ventricle only transduced limited areas around the ventricle, significantly less widespread than what we have observed (Fig. 2). Additionally, their intrathecal injection led to widespread scattered EGFP-positive cells throughout the entire CNS. However, the overall fluorescence intensities in their study appeared lower than what we have observed in our study (Fig. 1).
Several recent studies have tested CSF injection in large animals. Kaspar and colleagues reported that intrathecal or intracisternal injection of rAAV9 into the CSF of neonatal pigs generated widespread transduction in motor neurons along the spinal cord (27), similar to our observations in an adult monkey in the current study. Bankiewicz and colleagues reported that injection of rAAV9 into the CSF in cisterna magna of adult monkeys generated relatively stronger transduction in the CNS than intravascular injection. They found a predominance of transduction in the cerebellum but the level of spinal cord transduction was not reported (31). Boulis and colleagues reported that intrathecal injection of rAAV9 resulted in robust transduction of motor neurons in adult pigs. However, transduction of the full length of the spinal cord was only achieved after bolus injection at three spinal cord levels (lumbar, thoracic and cervical) by maneuvering an intrathecal catheter, which was implanted through a surgical lumbar laminectomy procedure (30). Most recently, Gray and colleagues reported that CSF injection of rAAV9 resulted in widespread but relatively scattered transduction throughout the CNS in monkeys (32). They included sorbitol in the injection mix to enhance the osmolality, but it was not clear whether such an inclusion was necessary.
Our experiments demonstrate that rAAV9 and rAAVrh10 are capable of spreading along the full length of the spinal cord and mediate strong transgene expression following a single intrathecal injection into the CSF in mice (Fig. 1) and in monkeys (Fig. 3) without addition of chemical enhancers. Additionally, these rAAV serotypes are capable of transducing cells in broad forebrain areas following a single injection into the third ventricle (Fig. 2). Furthermore, our preclinical therapeutic trial in the SOD1G93A model using intrathecal injection of rAAVrh10-GFP-amiR-Sod1 vector into the CSF is unprecedented and demonstrates that this approach can effectively deliver RNAi therapy and elicit therapeutic benefit (Fig. 7). These data suggest that intrathecal and intraventricular injection of rAAV9 and rAAVrh10 can be an effective strategy to deliver therapeutic genes to treat ALS and other adult onset diseases that afflict broad areas of the CNS.
Our results also point out areas where further preclinical investigation is needed in order to bring this therapeutic strategy to clinical application. First, differences among various studies need to be reconciled. Because GFP expression following rAAV administration has only been detected by immunostaining using GFP antibodies in previous studies (20,27,30–32), our detection of GFP fluorescence without the aid of GFP immunostaining suggests that we have achieved an unprecedented high level of transgene expression in broad areas of the CNS following single intrathecal or intraventricular injections in rodents (Figs 1 and 2). Many differences in the experimental procedure, including rAAV constructs, preparations and doses, surgery, animal species and injection methods might have led to the differences. Reconciling these differences will be required to establish more consistent methods for delivering rAAV transduction in broad CNS areas.
Second, we noticed that in our intrathecal experiments there were significant variations in the levels of transduction between individual animals (Table 2). The reasons for the variation are not clear but they could explain the different results that have been reported in the rodent experiments from different groups (20,32). The inverse correlation between the CNS transduction levels and the peripheral leaky transduction levels (Supplementary Material, Fig. S3) suggests that various degrees of leakiness during the injection in individual animals may be a major cause for the variation. If this is the case, this problem may be solved when the injection is carried out in large animals by a standard lumbar puncture procedure, where a seal around the needle can be reliably attained. The successful spreading of transduction along the full length of the monkey spinal cord injected using the standard lumbar puncture (Fig. 3) suggests that this is achievable.
Third, in our mouse experiments we have observed a predominance of transgene expression in astrocytes in the spinal cord but poor expression in motor neurons (Fig. 1). This is consistent with previous reports and may be attributable to the CBA promoter that we used in our rAAV vectors (20,29). Given the understanding that mutant gene expression in both neurons and glia contribute to the pathogenesis in ALS (42–46), it will be desirable to deliver the RNAi therapy to both neurons and glia. The lack of the transgene expression in motor neurons may have limited the therapeutic efficacy to slowing the disease progression but not delaying the disease onset in our experiments (Fig. 7). Solutions to this problem, however, may have already been developed. Samulski and colleagues have reported the construction of a new promoter called CBh, which they demonstrated to be capable of driving transgene expression in both glia and motor neurons in the spinal cord (32,47).
Fourth, compared with the peripheral rAAV administration methods (25–27,29), the CSF-injection strategy is more CNS specific. We observed an inverse correlation between the levels of transduction in the spinal cord and in the liver (Supplementary Material, Fig. S3), thus suggesting that a tight seal and minimizing leakage will be the key to producing consistent, high-level CNS transduction with minimal peripheral leakage transduction. Our observation is consistent with reports from other groups, which have generally observed robust CNS transduction with low peripheral transduction levels (20,27,30–32). As pointed out generally in these studies, an additional benefit is the dramatically lower doses of rAAV that will be required to deliver therapeutic genes.
Fifth, while our results demonstrate in principle that rAAVrh10 and rAAV9 can spread widely in the CNS after injection into the CSF, it is likely that additional serotypes possess this property since we have only screened five among more than a hundred AAV serotypes now known (48). It will be highly worthwhile screening additional serotypes because being able to choose from multiple AAV serotypes for therapeutic purposes will enhance the flexibility in gene therapy and provide means to skirt potential immunity developed against one serotype if multiple gene delivery is needed.
Lastly, although our study demonstrates that robust widespread transduction and therapeutic efficacy can be achieved in small animal species, it remains to be demonstrated whether such levels of transduction is achievable in large species with dimensions of the CNS closer to humans. For this reason, future studies in large animal species, particularly large primate species will be necessary to determine whether this approach will be feasible to treat human diseases.
An interesting question arisen from our observations is why rAAV9 and rAAVrh.10, but not other serotypes spread widely after injection into the CSF. The mechanism that determines the cell tropism of AAVs is currently incompletely understood. rAAV9 binds to terminal N-linked galactose and laminin receptor to gain cell entry (49,50). The receptor for rAAVrh.10 has not been determined but the similarity to the transduction pattern of rAAV9 suggests that both rAAVs may bind to the same type of receptors. A surprising and puzzling finding in our study is that the strongest transduced areas after injection into the third ventricle were largely above the third and lateral ventricles. Thus, the direction of viral diffusion appeared to be opposite to the direction of CSF circulation. One possibility was that this was an artifact from the viral injection, i.e. the virus simply diffused back through the needle track to the subarachnoid space where it might penetrate the brain parenchyma from the above through the paravascular glymphatic system (51). However, this did not explain why it was the deep layers of the cortex that received the strongest transduction. Another possibility was that the virus was actively transported from the injection site against the flow of the CSF to the deep layers of the cortex and hippocampus. However, there is not a known system that provides such a mechanism. Therefore, further studies will be necessary to answer the question why the transduction by AAV9 and AAVrh.10 spreads in a direction opposite to the CSF flow.
In summary, our experiments demonstrate that CSF injection of rAAV can be used to deliver therapeutic genes to treat diseases that afflict broad areas of the CNS and that such treatment is practical and efficacious in a mouse model for ALS. Therefore, our study suggests that CSF injection of rAAV may be further developed to treat human diseases. Furthermore, our study indicates that further studies will be needed to discover more AAV serotypes that are capable of spreading throughout the CNS, to understand the mechanism whereby rAAV9 and rAAVrh.10 spread after CSF injection, to develop a reliable method for CSF injection and to experiment with this new strategy in large primate species and in various animal models for diseases that afflict broad areas of the CNS, including ALS, lysosome storage diseases, spinal muscular atrophy and Alzheimer's disease.
MATERIALS AND METHODS
AAV vector construction and production
A previously described pCAG-RFP-miRNAint-KX plasmid was used to construct nine amiRNA constructs targeting the human SOD1 gene (52). The miRNAs were designed based on the human mir-30a folding structure and constructed by PCR using three overlapping oligonucleotides. The PCR products were digested with Kpn1 and Xho1 and inserted into the pCAG-RFP-miRNAint-KX backbone. All the amiRNA sequences were verified by sequencing and are shown in Supplementary Material, Table S1. The constructs, which express RFP and an amiR-Sod1, were tested for their knockdown efficiency by transfection into HEK293 cells as described below. The amiR-Sod1#5, which had the highest SOD1 knockdown efficiency, and the scrambled control construct amiR-Scr were PCR cloned into the 3′-UTR of pAAVCB6EGFP, a self-complementary AAV2 vector (28), between the 5′ XbaI and 3′ BamH1 restriction sites. These AAV vectors were designed to express GFP and the amiRNAs. They were verified by sequencing and tested for GFP expression and knockdown of SOD1 by transfection into HEK293 cells. They were then packaged into the AAVrh.10 or AAV9 capsid by the Viral Vector Core of the Gene Therapy Center at University of Massachusetts Medical School, as described previously (28).
Cell culture and transfection
HEK293 cells were maintained on DMEM media supplemented with 10% fetal bovine serum, 100 units/ml of penicillin and 100 µg/ml of streptomycin. Lipofectamine 2000™ (Invitrogen) was used to transfect the plasmid DNA into the cells according to a reverse transfection protocol from the manufacturer. Briefly, 1.6 µg of plasmid DNA were used for each well of a 12-well plate. After incubation of DNA with Lipofectamine 2000™, 25 000 cells in 1 ml of complete growth medium without antibiotics were added to each well and incubated for 48 h. For testing rAAV virus, 2.4 × 1010 viral genomes (vg) were added to 25 000 cells in a well of a 12-well plate and incubated for 48 h. Under this condition, all cells (100%) were transduced based on GFP expression. Cells were harvested for RNA or protein extraction as described below.
Northern blot and quantitative RT–PCR
Spinal cords and brains from mice were quickly harvested, snap frozen in liquid nitrogen and stored at −80°C. To extract RNA, frozen tissues were homogenized in cold TRIzol reagent (Invitrogen) and processed according to the manufacturer's protocol. For northern blot detection of SOD1 mRNA, 3 µg of total RNA was electrophoresed on a 1% denaturing agarose gel, transferred to a nylon membrane (Roche) and probed with a Digoxigenin-labeled probe synthesized using a PCR DIG Probe Synthesis Kit (Roche) against the human SOD1 coding region. The primers were forward CAGTGCAGGGCATCATCAATTTC and reverse CCCAATTACACCACAAGCCAAAC. After probing for SOD1, the blot was stripped and re-used to detect β-actin with probes synthesized as described above. The primers were forward GGATGACGATATCGCTGCGCTGGT and reverse TCACGCACGATTTCCCTCTCAGCT. For quantitative RT–PCR to detect the amiR-Sod1 expression, 5 µg of total RNA for each sample was used to synthesize the cDNA using the QuantiMir™ RT Kit and the Small RNA Quantitation System (System Biosciences). The quantitative PCR was carried out using IQ SYBR Green Supermix (Bio-Rad Laboratories) on a Bio-Rad CFX96 Real Time System. The primers were forward CTTTGTCAGCAGTCACATTGC for antisense miRNA and forward GCAATGTGACTTCGCTGACAAAG for sense miRNA. The reverse primer was the universal reverse primer provided by the manufacturer.
Western blot
Total protein was extracted from frozen tissue or cells by homogenization in a solution containing 1% SDS, 25 mm phosphate pH 7.6, 1 mm EGTA and Halt Protease Inhibitor Cocktails (Thermo Scientific). The protein concentration was determined by BCA assay (Thermo Scientific) according to the manufacturer's protocol. To detect SOD1 protein, 15 µg of total protein were separated on 15% SDS–polyacrylamide gels (Bio-Rad) and wet transferred to a Protran® (Whatman GmbH) nitrocellulose transfer membrane. The membrane was cut in half between 25 and 37 kDa. The membrane <25 kDa was probed with sheep anti-SOD1 (Biodesign, #K90077C, now Meridian Life Science) primary antibody and rabbit anti-sheep IgG (Calbiochem, #402100) secondary antibody. The upper half of the membrane was probed with mouse anti-actin primary antibody JLA20 (Developmental Studies Hybridoma Bank) and goat anti-mouse IgM (Bethyl #A90-101P) secondary antibody. The protein bands were visualized using SuperSignal West Pico kit (Thermo Scientific).
Histological processing and immunostaining
Mice were transcardially perfused with ice-chilled PBS followed by cold fixation buffer containing 4% paraformaldehyde and 0.1% of glutaraldehyde in PBS. Tissues were post-fixed by soaking in the same fixative for 48 h. Brain and spinal cord and other tissues from fixed mice were cryoprotected in 30% sucrose for 24–48 h. Tissues were then embedded in OCT compound and frozen sectioned using a cryostat. For immunofluorescent staining, the sections were washed in 1% Triton X-100 in PBS for 30 min and with PBS for 2 min, and then blocked in 10% serum with 0.05% Triton X-100 and 0.05% Tween 20 for 30 min at room temperature. Primary antibodies were added to the sections and incubated at 4°C overnight. The sources of primary antibodies and dilutions are as follows: SOD1 (BioDesign K90077C, now Meridian Life Science, 1:1000), NeuN (Millipore MAB377, 1:200), GFAP (Abcam Ab7260, 1:1000), IBA1 (BioCare Medical CP290A, B, 1:200), APC (EMD Bioscience OP80-100UG, 1:200), GFP (Abcam AB13970, 1: 500) and ChAT (Millipore AB1044P, 1:200). Following the incubation, sections were washed three times for 5 min each, incubated in the appropriate secondary antibody at room temperature for 90 min, washed three times in PBS for 5 min each, mounted with Vectashield Mounting solution and sealed with nail polish. Images of the brain and spinal cord sections were taken with a Nikon Eclipse Ti Widefield fluorescence microscope equipped with a Retiga-2000RV cooled-CCD camera.
For immunohistochemistry, sections were washed three times in PBS containing 0.25% Tween 20 and then stained following the manufacturer's instructions for Vectastain ABC kit, Elite PK-6100 standard ImmPact tm DAB peroxidase Substrate kit SK-4105 (Vector Lab). The sections were then mounted on slides and dried overnight at 55°C. After soaking in Xylene two times for 2 min each, the slides were sealed with Permount (Vector Lab).
Mice and viral injections
Wild-type and transgenic mice expressing SOD1G93A (B6.Cg-Tg(SOD1*G93A)1Gur/J) were purchased from The Jackson Laboratory. They were maintained on a FVB/NJ background for >10 generations in the animal facility of the University of Massachusetts Medical School. All mouse experiments were approved by IACUC and conducted according to UMMS policies and procedures regulating the use of animals in research and the provisions of the PHS/NIH Guide for the Care and Use of Laboratory Animals.
Intracerebroventricular injection was carried out to test the transduction efficiency of various AAV serotypes. For each serotype, six or more animals were injected. The injection was conducted as follows: the animal was immobilized on a Stoeling stereotaxis instrument. The scalp was shaved and cleaned with betadine and alcohol. A 1-cm longitudinal skin incision was made in the midline on the top of the head. The periosteum was scraped with a sterile cotton tip. A hole ∼1 mm in diameter was drilled through the cranium using a sterile ophthalmic burr. A 30-gauge Hamilton syringe connected to a Micro4 injection pump system (World Precision Instruments) containing rAAV was placed into the brain following brain coordinates for the third ventricle: caudal to Bregma 0.9–1 mm, midline 0 mm and ventral to skull surface 2.3–2.6 mm. The AAV was injected at 1-µl/min. After injection, the needle was maintained in place for an additional 2 min before withdrawal and the skin was closed with stainless steel wound clips.
Intrathecal injection of rAAV virus into mouse lumbar subarachnoid space was carried out to test the transduction efficiency of various serotypes. Each serotype was tested in six or more animals between 60 and 150 days old. Additionally, the intrathecal injection was carried out to deliver AAVrh10-GFP-amiR-Sod1 into the SOD1G93A mice at ∼65 days of age to test RNAi therapy. The injection was conducted using a method as described previously (10). In brief, a thin catheter was made by stretching PE10 tubing to an inner diameter of ∼0.12 mm. The stretched section was cut to 1.9 mm and two beads (1 mm apart) were made at the junction of the stretched and the thick sections by heating and pressing with flat pointed forceps. The thick section of the tube was 2.5 cm long. To implant the catheter, the mouse was anesthetized by intraperitoneal injection of Avertin (1.2% 2,2,2-tribromoethanol in 2% tert-amyl alcohol and PBS) at 0.23 ml/10 g body weight. The catheter was implanted between L5 and L6 vertebrae and stitched to the surface muscle. Viruses in 8 µl PBS were injected into each mouse by a Hamilton syringe at ∼1 µl/in freehand.
For serotype screening, 4.8 × 1010 vg in PBS were injected for each mouse, which was equivalent to 2.4 × 1012 vg/kg. For RNAi therapy, 2.4 × 1010 vg were injected into each mouse, which was equivalent to 1.2 × 1012 vg/kg. The catheters were removed 1 day after injection. For determining knockdown, the mice were sacrificed and their tissues dissected and processed as described above.
For testing therapy, female mice were used. All surgically operated mice were housed normally after catheter removal and checked for weight twice weekly. The end stage of SOD1G93A mice was defined as the day when complete paralysis of two limbs occurred, which was judged by an independent observer who was blind to the treatment that the mouse received. Whenever possible, the whole spinal cord was dissected out and evaluated for GFP fluorescence. The spinal cord was laid in a plastic dish with PBS. The GFP fluorescence was visualized with the fluorescence microscope and the scores were assigned to each segment of the spinal cord. − = fluorescence that was indistinguishable from the background in the uninjected mice. + = weak fluorescence that nevertheless was distinguishable from the background in the uninjected mice. ++ = moderate fluorescence that was readily observable under the microscope. +++ = bright fluorescence under the microscope and faint yellowish-green taint without the microscope. ++++ = very bright fluorescence that could be observed without the microscope.
Intrathecal injection of rAAVrh10 in a marmoset monkey
The animal (0.602 kg) was housed at the New England Primate Research Center and maintained in accordance with the ‘Guide for Care and Use of Laboratory Animals’ of the Institute of Laboratory Resources, National Research Council. The facility is accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care International and all work was approved by Harvard Medical School's Standing Committee on Animals. The marmoset was screened for AAV antibodies as described previously (53) and was negative. For intrathecal injection, the animal was sedated with ketamine and the area over the lumbar spine was shaved and prepped with betadine and alcohol. The space between L4 and L5 was palpated and a 30-gauge needle inserted into the space until a small flash of CSF was obtained in the hub of the needle. At this time, a 1 ml syringe containing ∼2.7 × 1012 vg in 250 µl of PBS was attached to the needle and this volume was injected into the CSF slowly. The needle was then removed and the animal was allowed to recover. The marmoset was returned to its home cage and had blood samples taken at 1 week. At 2 weeks, the animal was sedated with ketamine, a blood sample was obtained, a 24-gauge catheter placed in the saphenous vein, and ∼1000 units of heparin administered intravenously. The animal was deeply anesthetized with Nembutal and perfused with 4% paraformaldehyde in PBS. Further histological analysis was conducted as described above.
Statistics
Disease onset, early stage, end stage, onset to early stage duration and onset to end stage duration were obtained from individual animals. The averages and standard deviations were calculated for each treatment group. Statistical comparisons were done using Gehan–Breslow–Wilcoxon test.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by grants from NIH/NINDS (RO1NS059708), the ALS Association, ALS Therapeutic Alliance, the Packard Center for ALS Research at Johns Hopkins to Z.X.; from NIH/NINDS (1R01NS076991) and a gift grant from Jacob's Cure to G.G.; from NINDS (5RO1-NS050557-05), NINDS American Recovery and Reinvestment Act Award (RC2-NS070-342), the Angel Fund, the ALS Association, P2ALS, Project ALS, the Pierre L. de Bourgknecht ALS Research Foundation and the ALS Therapy Alliance to R.H.B.; and from University of Massachusetts Center for Clinical Translational Science (UL1RR 031982) to Z.X. G.G. and R.H.B.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Dr Jemeen Sreedharan for critically reading the manuscript, Dr Ronald Desrosiers for his help in obtaining the monkeys and animal use approvals, Drs Miguel Esteves and Phillip D. Zamore for sharing equipment, Dr Hong Cao for assisting with confocal imaging, Ms Weijia Tan for technical assistance and Ms Sili Zhou for animal care.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Andersen P.M., Al-Chalabi A. Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat. Rev. Neurol. 2011;7:603–615. doi: 10.1038/nrneurol.2011.150. [DOI] [PubMed] [Google Scholar]
- 2.Ludolph A.C., Brettschneider J., Weishaupt J.H. Amyotrophic lateral sclerosis. Curr. Opin. Neurol. 2012;25:530–535. doi: 10.1097/WCO.0b013e328356d328. [DOI] [PubMed] [Google Scholar]
- 3.Xu Z. Mechanism and treatment of motoneuron degeneration in ALS: what have SOD1 mutants told us? ALS Other Mot. Neu. Disord. 2000;1:225–234. doi: 10.1080/14660820050515052. [DOI] [PubMed] [Google Scholar]
- 4.Ding H., Schwarz D.S., Keene A., Affarel B., Fenton L., Xia X., Shi Y., Zamore P.D., Xu Z. Selective silencing by RNAi of a dominant allele that causes amyotrophic lateral sclerosis. Aging Cell. 2003;2:209–217. doi: 10.1046/j.1474-9728.2003.00054.x. [DOI] [PubMed] [Google Scholar]
- 5.Ralph G.S., Radcliffe P.A., Day D.M., Carthy J.M., Leroux M.A., Lee D.C.P., Wong L.-F., Bilsland L.G., Greensmith L., Kingsman S.M., et al. Silencing mutant SOD1 using RNAi protects against neurodegeneration and extends survival in an ALS model. Nat. Med. 2005;11:429–433. doi: 10.1038/nm1205. [DOI] [PubMed] [Google Scholar]
- 6.Raoul C., Abbas-Terki T., Bensadoun J.-C., Guillot S., Haase G., Szulc J., Henderson C.E., Aebischer P. Lentiviral-mediated silencing of SOD1 through RNA interference retards disease onset and progression in a mouse model of ALS. Nat. Med. 2005;11:423–428. doi: 10.1038/nm1207. [DOI] [PubMed] [Google Scholar]
- 7.Smith R.A., Miller T.M., Yamanaka K., Monia B.P., Condon T.P., Hung G., Lobsiger C.S., Ward C.M., McAlonis-Downes M., Wei H., et al. Antisense oligonucleotide therapy for neurodegenerative disease. J. Clin. Investig. 2006;116:2290–2296. doi: 10.1172/JCI25424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xia X., Zhou H., Huang Y., Xu Z. Allele-specific RNAi selectively silences mutant SOD1 and achieves significant therapeutic benefit in vivo. Neurobiol. Dis. 2006;23:578–586. doi: 10.1016/j.nbd.2006.04.019. [DOI] [PubMed] [Google Scholar]
- 9.Wu R., Wang H., Xia X., Zhou H., Liu C., Castro M., Xu Z. Nerve injection of viral vectors efficiently transfers transgenes into motor neurons and delivers RNAi therapy against ALS. Antioxid. Redox Signal. 2009;11:1523–1534. doi: 10.1089/ars.2009.2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang H., Ghosh A., Baigude H., Yang C.S., Qiu L., Xia X., Zhou H., Rana T.M., Xu Z. Therapeutic gene silencing delivered by a chemically modified small interfering RNA against mutant SOD1 slows amyotrophic lateral sclerosis progression. J. Biol. Chem. 2008;283:15845–15852. doi: 10.1074/jbc.M800834200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boudreau R.L., Rodriguez-Lebron E., Davidson B.L. RNAi medicine for the brain: progresses and challenges. Hum. Mol. Genet. 2011;20:R21–R27. doi: 10.1093/hmg/ddr137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grieger J.C., Samulski R.J. Adeno-associated virus vectorology, manufacturing, and clinical applications. Methods Enzymol. 2012;507:229–254. doi: 10.1016/B978-0-12-386509-0.00012-0. [DOI] [PubMed] [Google Scholar]
- 13.Manfredsson F.P., Mandel R.J. Development of gene therapy for neurological disorders. Discov. Med. 2010;9:204–211. [PubMed] [Google Scholar]
- 14.Vandenberghe L.H., Wilson J.M., Gao G. Tailoring the AAV vector capsid for gene therapy. Gene Ther. 2009;16:311–319. doi: 10.1038/gt.2008.170. [DOI] [PubMed] [Google Scholar]
- 15.Mittermeyer G., Christine C.W., Rosenbluth K.H., Baker S.L., Starr P., Larson P., Kaplan P.L., Forsayeth J., Aminoff M.J., Bankiewicz K.S. Long-term evaluation of a phase 1 study of AADC gene therapy for Parkinson's disease. Hum. Gene Ther. 2012;23:377–381. doi: 10.1089/hum.2011.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bennett J., Ashtari M., Wellman J., Marshall K.A., Cyckowski L.L., Chung D.C., McCague S., Pierce E.A., Chen Y., Bennicelli J.L., et al. AAV2 gene therapy readministration in three adults with congenital blindness. Sci. Transl. Med. 2012;4:120ra115. doi: 10.1126/scitranslmed.3002865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim S.R., Kareva T., Yarygina O., Kholodilov N., Burke R.E. AAV transduction of dopamine neurons with constitutively active Rheb protects from neurodegeneration and mediates axon regrowth. Mol. Ther. 2012;20:275–286. doi: 10.1038/mt.2011.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McBride J.L., Boudreau R.L., Harper S.Q., Staber P.D., Monteys A.M., Martins I., Gilmore B.L., Burstein H., Peluso R.W., Polisky B., et al. Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: Implications for the therapeutic development of RNAi. Proc. Natl. Acad. Sci. USA. 2008;105:5868–5873. doi: 10.1073/pnas.0801775105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xia H., Mao Q., Eliason S.L., Harper S.Q., Martins I.H., Orr H.T., Paulson H.L., Yang L., Kotin R.M., Davidson B.L. RNAi suppresses polyglutamine-induced neurodegeneration in a model of spinocerebellar ataxia. Nat. Med. 2004;10:816–820. doi: 10.1038/nm1076. [DOI] [PubMed] [Google Scholar]
- 20.Snyder B.R., Gray S.J., Quach E.T., Huang J.W., Leung C.H., Samulski R.J., Boulis N.M., Federici T. Comparison of adeno-associated viral vector serotypes for spinal cord and motor neuron gene delivery. Hum. Gene. Ther. 2011;22:1129–1135. doi: 10.1089/hum.2011.008. [DOI] [PubMed] [Google Scholar]
- 21.Kaspar B.K., Llado J., Sherkat N., Rothstein J.D., Gage F.H. Retrograde Viral Delivery of IGF-1 Prolongs Survival in a Mouse ALS Model. Science. 2003;301:839–842. doi: 10.1126/science.1086137. [DOI] [PubMed] [Google Scholar]
- 22.Azzouz M., Ralph G.S., Storkebaum E., Walmsley L.E., Mitrophanous K.A., Kingsman S.M., Carmeliet P., Mazarakis N.D. VEGF delivery with retrogradely transported lentivector prolongs survival in a mouse ALS model. Nature. 2004;429:413–417. doi: 10.1038/nature02544. [DOI] [PubMed] [Google Scholar]
- 23.Towne C., Setola V., Schneider B.L., Aebischer P. Neuroprotection by gene therapy targeting mutant SOD1 in individual pools of motor neurons does not translate into therapeutic benefit in fALS mice. Mol. Ther. 2010;19:274–283. doi: 10.1038/mt.2010.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boulis N.M., Turner D.E., Dice J.A., Bhatia V., Feldman E.L. Characterization of adenoviral gene expression in spinal cord after remote vector delivery. Neurosurgery. 1999;45:131–138. doi: 10.1097/00006123-199907000-00029. [DOI] [PubMed] [Google Scholar]
- 25.Foust K.D., Nurre E., Montgomery C.L., Hernandez A., Chan C.M., Kaspar B.K. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 2009;27:59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duque S., Joussemet B., Riviere C., Marais T., Dubreil L., Douar A.M., Fyfe J., Moullier P., Colle M.A., Barkats M. Intravenous administration of self-complementary AAV9 enables transgene delivery to adult motor neurons. Mol. Ther. 2009;17:1187–1196. doi: 10.1038/mt.2009.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bevan A.K., Duque S., Foust K.D., Morales P.R., Braun L., Schmelzer L., Chan C.M., McCrate M., Chicoine L.G., Coley B.D., et al. Systemic gene delivery in large species for targeting spinal cord, brain, and peripheral tissues for pediatric disorders. Mol. Ther. 2011;19:1971–1980. doi: 10.1038/mt.2011.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang H., Yang B., Mu X., Ahmed S.S., Su Q., He R., Wang H., Mueller C., Sena-Esteves M., Brown R., et al. Several rAAV vectors efficiently cross the blood-brain barrier and transduce neurons and astrocytes in the neonatal mouse central nervous system. Mol. Ther. 2011;19:1440–1448. doi: 10.1038/mt.2011.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gray S.J., Matagne V., Bachaboina L., Yadav S., Ojeda S.R., Samulski R.J. Preclinical differences of intravascular AAV9 delivery to neurons and glia: a comparative study of adult mice and nonhuman primates. Mol. Ther. 2011;19:1058–1069. doi: 10.1038/mt.2011.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Federici T., Taub J.S., Baum G.R., Gray S.J., Grieger J.C., Matthews K.A., Handy C.R., Passini M.A., Samulski R.J., Boulis N.M. Robust spinal motor neuron transduction following intrathecal delivery of AAV9 in pigs. Gene Ther. 2012;19:852–859. doi: 10.1038/gt.2011.130. [DOI] [PubMed] [Google Scholar]
- 31.Samaranch L., Salegio E.A., San Sebastian W., Kells A.P., Foust K.D., Bringas J.R., Lamarre C., Forsayeth J., Kaspar B.K., Bankiewicz K.S. Adeno-associated virus serotype 9 transduction in the central nervous system of nonhuman primates. Hum. Gene Ther. 2012;23:382–389. doi: 10.1089/hum.2011.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gray S.J., Nagabhushan Kalburgi S., McCown T.J., Jude Samulski R. Global CNS gene delivery and evasion of anti-AAV-neutralizing antibodies by intrathecal AAV administration in non-human primates. Gene Ther. 2013;20:465. doi: 10.1038/gt.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gurney M.E., Pu H., Chiu A.Y., Dal Canto M.C., Polchow C.Y., Alexander D.D., Caliendo J., Hentati A., Kwon Y.W., Deng H.-X., et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 34.Baker H.F., Ridley R.M. Use of common marmoset (Callithrix jacchus) in psychopharmacological research. In: Waddington S., Joseph M.H., editors. Working Methods in Multidisciplinary Psychopharmacology. Manchester: Manchester University Press; 1987. pp. 41–73. [Google Scholar]
- 35.Annett L.E., Torres E.M., Clarke D.J., Ishida Y., Barker R.A., Ridley R.M., Baker H.F., Dunnett S.B. Survival of nigral grafts within the striatum of marmosets with 6-OHDA lesions depends critically on donor embryo age. Cell Transplant. 1997;6:557–569. doi: 10.1177/096368979700600606. [DOI] [PubMed] [Google Scholar]
- 36.Kendall A.L., Rayment F.D., Torres E.M., Baker H.F., Ridley R.M., Dunnett S.B. Functional integration of striatal allografts in a primate model of Huntington's disease. Nat. Med. 1998;4:727–729. doi: 10.1038/nm0698-727. [DOI] [PubMed] [Google Scholar]
- 37.Besret L., Kendall A.L., Dunnett S.B. Aspects of PET imaging relevant to the assessment of striatal transplantation in Huntington's disease. J. Anat. 2000;196:597–607. doi: 10.1046/j.1469-7580.2000.19640597.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Costa S., Iravani M.M., Pearce R.K.B., Jenner P. Glial cell line-derived neurotrophic factor concentration dependently improves disability and motor activity in MPTP-treated common marmosets. Eur. J. Pharmacol. 2001;412:45–50. doi: 10.1016/s0014-2999(00)00933-x. [DOI] [PubMed] [Google Scholar]
- 39.Eslamboli A., Georgievska B., Ridley R.M., Baker H.F., Muzyczka N., Burger C., Mandel R.J., Annett L., Kirik D. Continuous low-level glial cell line-derived neurotrophic factor delivery using recombinant adeno-associated viral vectors provides neuroprotection and induces behavioral recovery in a primate model of Parkinson's disease. J. Neurosci. 2005;25:769–777. doi: 10.1523/JNEUROSCI.4421-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maratos E.C., Jackson M.J., Pearce R.K., Cannizzaro C., Jenner P. Both short- and long-acting D-1/D-2 dopamine agonists induce less dyskinesia than L-DOPA in the MPTP-lesioned common marmoset (Callithrix jacchus) Exp. Neurol. 2003;179:90–102. doi: 10.1006/exnr.2002.8055. [DOI] [PubMed] [Google Scholar]
- 41.Okano H., Hikishima K., Iriki A., Sasaki E. The common marmoset as a novel animal model system for biomedical and neuroscience research applications. Semin. Fetal Neonatal. Med. 2012;17:336–340. doi: 10.1016/j.siny.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 42.Ilieva H., Polymenidou M., Cleveland D.W. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 2009;187:761–772. doi: 10.1083/jcb.200908164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Papadeas S.T., Kraig S.E., O'Banion C., Lepore A.C., Maragakis N.J. Astrocytes carrying the superoxide dismutase 1 (SOD1G93A) mutation induce wild-type motor neuron degeneration in vivo. Proc. Natl. Acad. Sci. USA. 2011;108:17803–17808. doi: 10.1073/pnas.1103141108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nagai M., Re D.B., Nagata T., Chalazonitis A., Jessell T.M., Wichterle H., Przedborski S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 2007;10:615–622. doi: 10.1038/nn1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Di Giorgio F.P., Carrasco M.A., Siao M.C., Maniatis T., Eggan K. Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat. Neurosci. 2007;10:608–614. doi: 10.1038/nn1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee Y., Morrison B.M., Li Y., Lengacher S., Farah M.H., Hoffman P.N., Liu Y., Tsingalia A., Jin L., Zhang P.W., et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature. 2012;487:443–448. doi: 10.1038/nature11314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gray S.J., Foti S.B., Schwartz J.W., Bachaboina L., Taylor-Blake B., Coleman J., Ehlers M.D., Zylka M.J., McCown T.J., Samulski R.J. Optimizing promoters for recombinant adeno-associated virus-mediated gene expression in the peripheral and central nervous system using self-complementary vectors. Hum. Gene Ther. 2011;22:1143–1153. doi: 10.1089/hum.2010.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gao G., Vandenberghe L.H., Wilson J.M. New recombinant serotypes of AAV vectors. Curr. Gene Ther. 2005;5:285–297. doi: 10.2174/1566523054065057. [DOI] [PubMed] [Google Scholar]
- 49.Shen S., Bryant K.D., Brown S.M., Randell S.H., Asokan A. Terminal N-linked galactose is the primary receptor for adeno-associated virus 9. J. Biol. Chem. 2011;286:13532–13540. doi: 10.1074/jbc.M110.210922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Akache B., Grimm D., Pandey K., Yant S.R., Xu H., Kay M.A. The 37/67-kilodalton laminin receptor is a receptor for adeno-associated virus serotypes 8, 2, 3, and 9. J. Virol. 2006;80:9831–9836. doi: 10.1128/JVI.00878-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Iliff J.J., Wang M., Liao Y., Plogg B.A., Peng W., Gundersen G.A., Benveniste H., Vates G.E., Deane R., Goldman S.A., et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci. Transl. Med. 2012;4:147ra111. doi: 10.1126/scitranslmed.3003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang C., Qiu L., Xu Z. Specific gene silencing using RNAi in cell culture. Methods Mol. Biol. 2011;793:457–477. doi: 10.1007/978-1-61779-328-8_30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gao G., Lu Y., Calcedo R., Grant R.L., Bell P., Wang L., Figueredo J., Lock M., Wilson J.M. Biology of AAV serotype vectors in liver-directed gene transfer to nonhuman primates. Mol. Ther. 2006;13:77–87. doi: 10.1016/j.ymthe.2005.08.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}