

Abstract
Cell-mediated regenerative approaches using muscle progenitor cells hold promises for the treatment of many forms of muscle disorders. Their applicability in the clinic, however, is hindered by the low levels of regeneration obtained after transplantation and the large number of cells required to achieve an effect. To better understand the mechanisms that regulate the temporal switch of replicating muscle progenitor cells into terminally differentiated cells and to develop new strategies that could enhance muscle regeneration, we have developed and performed a high-throughput screening (HTS) capable of identifying genes that play active roles during myogenesis. Secondary and tertiary screens were used to confirm the effects of RNAi in vitro and in vivo and to select for candidate hits that significantly increase regeneration into skeletal muscles. Downregulation of cyclin D2 (CCND2) was shown to dramatically enhance myogenic differentiation of muscle progenitor cells and to induce a robust regeneration after cell transplantation into skeletal muscles of dystrophin-deficient mice. Protein interaction network and pathway analysis revealed that CCND2 directly interacts with the cyclin-dependent kinase Cdk4 to inhibit phosphorylation of the retinoblastoma protein (pRb), thus blocking the activation of the myogenic switch during fusion. These studies identify CCND2 as a new key regulator of terminal differentiation in muscle progenitor cells and open new possibilities for the treatment of many forms of muscle disorders characterized by impaired regeneration and loss of muscle mass.
INTRODUCTION
Many of the diseases affecting muscle are distinguished by the impaired ability of muscle progenitor cells to fuse and form new fibers and the inability of the existing fibers to maintain normal homeostasis. Among those, Duchenne muscular dystrophy (DMD) is one of the most severe diseases. It is caused by mutations in the dystrophin gene that abolish dystrophin expression in muscle of DMD patients (1). The disease is characterized by a progressive loss of muscle mass which has been attributed primarily to the progressive atrophy and loss of individual muscle fibers. By the age of ten, the majority of Duchenne boys have lost a significant amount of muscle tissue. Beyond that point, the lack of mobility and physical inactivity accelerate muscle loss, causing a rapid and irreversible decline.
A number of different approaches have been explored, aimed at ameliorating the disease by preventing loss of muscle mass (2–10). Among those, transplantation of healthy or normal muscle stem cells holds promise due to its potential of promoting formation of de-novo myofibers lost as a result of the disease. Muscle stem cells are located beneath the basal lamina of muscle fibers and are responsible for the homeostasis and tissue repair of skeletal muscles. Upon activation, they undergo replicative expansion to produce daughter cells that will either enter the myogenic lineage progression to ultimately become myoblasts (Mbs) or withdraw from the cell cycle to reenter the quiescent stage (11–14). Mbs can be easily propagated in vitro over a prolonged period of time without compromising their ability to differentiate and form myotubes (Mts) (13,15).
Most of the regenerative approaches to DMD have been focused on isolating stem cells that can efficiently reconstitute muscle after intramuscular injection or after vascular delivery. These studies have focused primarily on identifying a population of cells capable of committing almost exclusively to the myogenic program. Ultimately however, each cell type will have to enter the final stages of cell commitment and treatments to DMD and their application into humans will require for the transplanted cell to efficiently differentiate into Mbs and form new fibers.
To date, clinical trials for DMD using Mb transplantation have been disappointing (16–18). The large number of cells required to achieve an effect and the inability of the transplanted cells to efficiently fuse and reconstitute muscles severely hamper the applicability of cell-mediated regenerative applications to DMD. Effective treatments to DMD using transplantation procedures are likely to require the use of complementary approaches aimed at enhancing terminal differentiation of Mbs after engraftment.
Encouraging results have been obtained using myostatin blockades. Myostatin is a member of the transforming growth factor-β (TGF-β) superfamily of secreted growth and differentiation factors, which is essential for proper regulation of skeletal muscle mass. A dramatic increase in muscle mass has been observed in animal models (5,19,20), as well as in humans (21), lacking myostatin. When combined with the cell transplantation procedure, downregulation of myostatin in Mbs has been shown to significantly increase the ability of transplanted cells to fuse into pre-existing or nascent myofibers (22). Altogether, these results have demonstrated that activation of muscle progenitor cells and terminal differentiation of Mbs can be influenced by controlling the expression of a single gene. To date, however, the identification of genetic targets that could be used to potentiate regeneration of skeletal muscles after transplantation has been limited.
We have implemented high-throughput screening (HTS) technologies in the study of terminal differentiation of muscle progenitor cells, and we have developed an HTS assay capable of identifying genes that inhibit Mb fusion and control Mt growth. The screen of 16 000 individual small-interfering RNAs (siRNAs) has led to the identification of 635 genes involved in myogenesis. Among those, downregulation of CCND2 in Mbs showed to significantly promote muscle regeneration and muscle repair in vivo. The number of dystrophin-positive (DYS+) fibers was much higher in muscles that received Mbs downregulating CCND2 compared with those transplanted with Mbs downregulating myostatin, suggesting that CCND2 is a potent regulator of myofibrillogenesis. We also show that CCND2 acts by inhibiting the activation of the myogenic switch and by influencing the phosphorylation of pRb. Our work identifies a new genetic target for DMD therapeutics which could ultimately lead to the development of an effective treatment for the disease.
RESULTS
Assay development and primary screen
Optimization of the screening procedure was performed using a high-content imager (HCI) and Mbs isolated from a transgenic mouse expressing the green fluorescent protein (GFP) under the control of a ubiquitous promoter. As a positive control, we used an siRNA specifically designed to target and disrupt expression of the myostatin gene, previously shown to promote Mb differentiation and to enhance muscle growth (23). Differentiation was followed for up to 48 h in live cells treated with the siRNA downregulating myostatin expression (MSTN-siRNA). Analysis was performed using the MetaXpress analysis imaging software (Molecular Device) adapted to discriminate multinucleated cells (Mts) from mononucleated (non-fused) cells (Fig. 1). A statistically significant difference in the percentage of non-fused cells was detected in Mts downregulating myostatin compared with sham-transfected Mts but only at densities comprised between 1 × 103 and 5 × 103 cells per well. The screen was carried out using a density of 2.5 × 103 cells per well (Fig. 1D). At this density, the percentage of non-fused cells treated with MSTN-siRNA was ∼10-fold lower than that of sham-transfected cells and the signal to background ratio (S/B) between the two groups was greater than 3.
Figure 1.

Optimization of the screening protocol. (A) The MetaXpress imaging software allows us to determine the number of non-fused cells in each imaged field. Viable hoechst (33342) is used to visualize nuclei in the 355/360 nm wavelength spectrum and appears blue. GFP is imaged in the 485/520 nm spectrum to identify green fluorescent cytoplasmic content. Initially, the software analyzes each image separately to determine the total number of nuclei (hoechst staining). Green fluorescent cells are discriminated based on the size and shape. The imaging software distinguishes mononucleated (non-fused) cells from Mts containing two or more nuclei. (B) An siRNA targeting myostatin mRNA (MSTN-siRNA) was used as a positive control and to optimize the screening protocol. Note the difference in Mt size when compared with untransfected cells. (C) Mbs were seeded in 384-well plates at different concentrations and transfected with the MSTN-siRNA or transfection reagent alone (Sham) for 24 h. Cells were allowed to differentiate for 48 h in differentiation medium before acquiring the images. Each bar represents the average count of total number of nuclei per well (N = 16 wells). (D) Changes in the fusion index were measured 48 h after induction of differentiation in sham-transfected cells or cells transfected with the MSTN-siRNA. Fusion index was calculated as a percentage of cells containing two or more nuclei over total number of cells. (*P < 0.05; **P < 0.005).
Screens were conducted using siRNA libraries designed to target the Mouse Kinases (MK) library (767 genes), the Mouse Genome Wide (MGW) library (2700 genes) and the Mouse G Protein-Coupled Receptor (GPCR) library (518 genes), targeting specific genes of known and unknown function across the genome. Images were acquired from four different fields per well and the cells were imaged 48 h after induction of differentiation. Screens were performed in duplicates. Data were analyzed using analytic metrics to assess and rank the extent of siRNA inhibition on differentiation and by visual inspection of each of the images acquired. To avoid off-target effects, each siRNA was transfected separately and only genes with at least three siRNA hits were selected for further analysis. Positive hits were originally ranked based on the individual signals obtained from each of the wells. Then, the rank distribution of all siRNAs (wells) targeting the same gene and showing activity was examined based on the P-value. Of nearly 4000 genes analyzed, downregulation of 635 genes showed a significant effect compared with control (sham-transfected) cells (Fig. 2A). Of those, inhibition of 155 genes was shown to promote Mb fusion significantly better than the MSTN-siRNA (Supplementary Material, Table S1).
Figure 2.
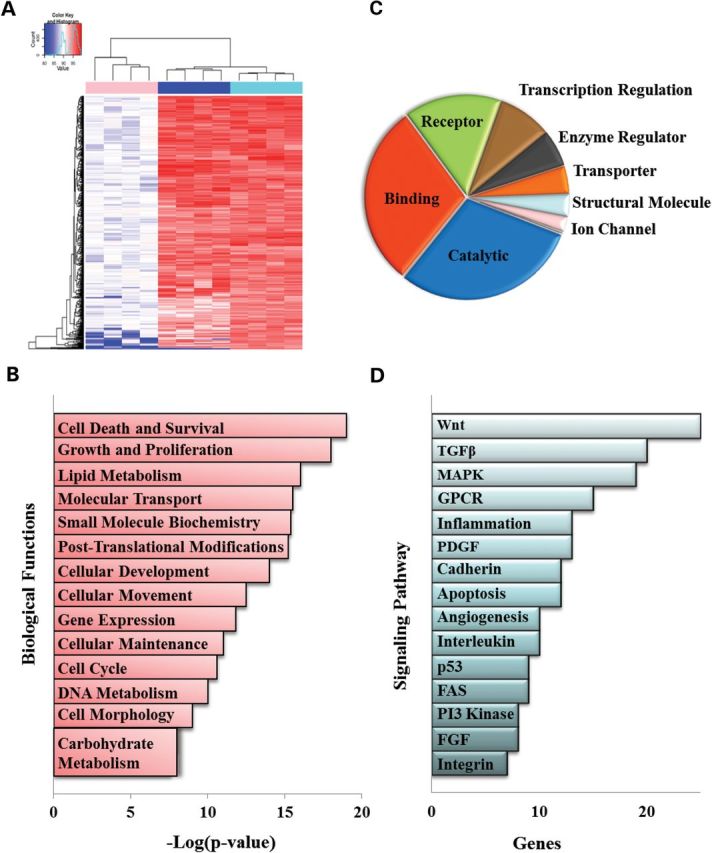
Heat map and pathway analysis of relevant genes. (A) Heat map showing differences in fusion of the genes identified by HTS (columns 5 through 8) compared with the fusion index of sham-transfected muscle cells (columns 1 through 4) or cells transfected with the siRNA targeting myostatin (columns 9 through 12). Genes are clustered by similarity. Changes in fusion are depicted in shades of red. Complete gene lists and analysis tools are available online (Supplementary Material, Table S1). (B) Top 14 biological functions performed using IPA software of the complete gene list of positive hits calculated as the negative base-10 logarithm of the P-value [−log(P-value)] and indicate the probability of finding the input proteins in a given network as a result of random chance. (C) Genes were classified into molecular function according to the PANTHER classification system. (D) Signaling pathways identified by the PANTHER classification system plotted based on the number of genes identified within each pathway. Categories with at least eight genes are displayed in the chart. (TGF-β, transforming growth factor β; MAPK, mitogen activator protein kinase; GPCR, G protein-coupled receptors; PDGF, platelet derived growth factor; FGF, fibroblast growth factor).
Functional characterization of the genes identified by the imaging software was conducted using ingenuity pathway analysis (IPA) and Protein ANalysis THrough Evolutionary Relationships (PANTHER) by comparing the data set in the context of known biological response and functional databases (Fig. 2B–D). Among the biological functions identified, the predominant processes involved were cell survival, growth and differentiation (Fig. 2B) demonstrating that inhibition of proliferation is a critical step in the activation of the myogenic program. Interestingly, the majority of the genes were hydrolases, peptidases or receptors suggesting that differentiation in Mbs is mediated primarily through the activation of signaling pathways (Fig. 2C). Among those pathways, the Wnt, the TGFβ and the mitogen activator protein (MAP) kinase pathways were shown to play a primary role (Fig. 2D).
For therapeutic purposes, genes whose downregulation can efficiently promote muscle regeneration and muscle repair are likely to have a significant impact on treating muscle wasting disorders such as DMD. The clinical relevance of this approach should be further enhanced if the regeneration induced is greater than that detected following myostatin inhibition. We therefore focused our analysis on genes that, when downregulated, showed to promote fusion better than muscle cells downregulating myostatin. The fusion index of the 155 positive hits was determined using the images acquired by HCI and was expressed as the percentage of nuclei within Mts containing 2-to-5, 6-to-10, or more than 10 nuclei per Mt. Downregulation of six of the genes identified showed to significantly increase the percentage of Mts containing 6-to-10 and more than 10 nuclei compared with Mts transfected with vehicle alone (Fig. 3B and C). However, only the downregulation of CCND2 and CDC42EP1 was shown to significantly enhance fusion in Mts containing 6-to-10 and greater than 10 nuclei, respectively, when compared with cells treated with the MSTN-siRNA (Fig. 3B and C). The results were confirmed in primary Mb cultures isolated from C57BL/10 and mdx mice and treated with the siRNAs duplexes used for the screen. Altogether, these results demonstrate that the effects observed in the HTS were consistent in different cell lines and different strains. A representative image of Mts obtained following downregulation of each of the eight hits is presented in Supplementary Material, Figure S1.
Figure 3.
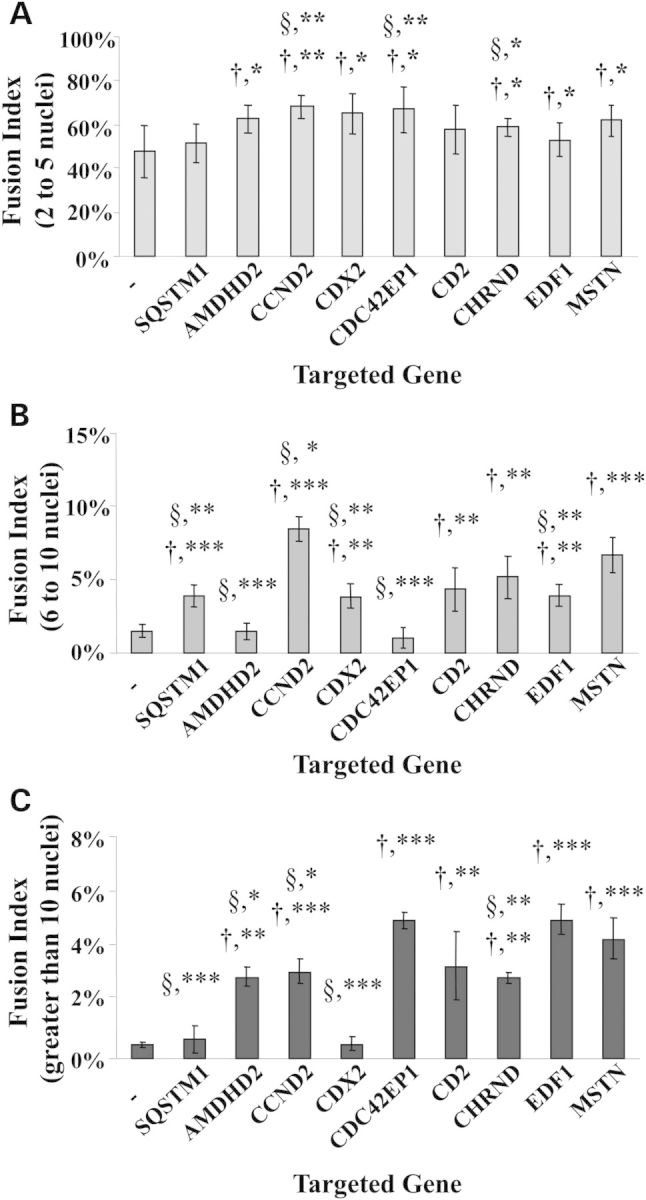
Fusion indexes of major hits identified in the screen. The mean number of nuclei in multinucleated muscle cells containing either 2-to-5 (A), 6-to-10 (B), or more than 10 nuclei per Mt (C) was determined 48 h after induction of differentiation and was expressed as a percentage of the total number of nuclei. Statistical significance of each group transfected with the siRNA targeting the gene of interest over fusion indexes of cultures treated with vehicle alone is depicted using the † symbol. Groups that showed a statistically significant difference in the percentage of fusion than cultures treated with the myostatin siRNA are indicated by the § symbol. (N = 8; *P < 0.03; **P < 0.005; ***P < 0.0005).
Functional activity of RNAi in vivo after transplantation
To determine whether silencing of the eight selected genes would have biological activity in skeletal muscle, we examined the regenerative potential of Mbs treated with siRNAs downregulating their expression. Lower limb muscles of mdx/nude mice were irradiated 3 days prior to transplantation to ablate all satellite cells and tibialis anterior (TA) muscles were injured with cardiotoxin 1 day prior to transplantation to induce regeneration and enhance cell engraftment. Mbs isolated from GFP transgenic mice were transfected with the siRNAs duplex showing activity in vitro. Mbs were then transplanted into recipient muscles 10 h after initiation of transfection. As a negative control, we used GFP Mbs treated with transfection reagent alone or Mbs transfected with a control, non-targeting siRNA (CTL-siRNA). GFP Mbs downregulating myostatin were used as a positive control. TAs were isolated 1 week after transplantation and analyzed for expression of GFP and dystrophin, which were used as an index of regeneration and to determine the efficacy of the transplantation procedure (Fig. 4 and Supplementary Material, Fig. S2). Bright fluorescent GFP-positive (GFP+) fibers were readily recognized in all injected muscles. The results were confirmed by immunostaining analysis using an antibody specific to dystrophin. No significant differences were detected in the number of GFP+ and DYS+ fibers. Muscle transplanted with Mbs downregulating myostatin showed a significantly higher number of GFP+ and DYS+ myofibers than those that received sham-transfected Mbs or Mbs transfected with the CTL-siRNA (Fig. 4). Downregulation of six of the eight genes identified by HTS showed a significant increase in the number of GFP+ and DYS+ myofibers when compared with those detected in TAs that were transplanted with Mbs treated with the CTL-siRNA (Fig. 4). However, only muscles that received Mbs transfected with an siRNA specific to CCND2 showed a significant higher number of positive fibers than those transplanted with Mbs downregulating myostatin (Fig. 4). These results clearly demonstrated that CCND2 is a strong inhibitor of differentiation and a key target for further therapeutic development.
Figure 4.

Regeneration of skeletal muscles after transplantation of Mbs transfected with siRNAs. The number of DYS+ fibers correlated to that of GFP+ fibers as assessed by immunostaining analysis of consecutive sections of muscles isolated from treated and untreated mice. The differences between DYS+ fibers detected in muscles that received sham-transfected Mbs were not statically different from those detected in muscles that received Mbs transfected with a CTL-siRNA or untreated muscles. Muscles injected with Mbs downregulating CCND2, CDX2, CDC42EPI, CD2, CHRND and EDF1 showed a statistically significant difference in the number of GFP+ and DYS+ fibers compared with muscles that received sham-transfected Mbs or Mbs treated with a control siRNA. Only Mbs transfected with a CCND2-siRNA showed a significant increase in their ability to fuse and regenerate muscles compared with those downregulating myostatin. (N = 5; *P < 0.03; **P ≤ 0.0003)
Primary hit selection
We next focused our studies on better understanding the function of CCND2 during myogenesis.
The specificity of CCND2 was demonstrated using siRNAs different from the four duplexes present in the library (Supplementary Material, Table S2). Fusion indexes were calculated for all cultures treated with siRNAs targeting the CCND2 mRNA for up to 3 days after induction of differentiation. Each siRNA tested showed to significantly increase fusion in muscle culture of mdx and wild-type cells. Real-time PCR analysis revealed that CCND2 mRNA is highly expressed in proliferating Mbs as well as cultures induced to differentiate and maintained in differentiation medium for 24 h (Fig. 5A). Transcript levels significantly decreased at 48 h in differentiation medium and were almost undetectable 72 h after induction of differentiation. The results were confirmed by western blot analysis (Fig. 5B).
Figure 5.

CCND2 expressions in vitro during differentiation and in response to siRNA treatment. (A) CCND2 mRNA profiles in C57 Mbs after switching to differentiation medium calculated as fold changes to the level of mRNA in untransfected Mbs. Transcript levels were normalized to those of glyceraldehyde 3-phosphate dehydrogenase (GAPDH). (B) Western blot analysis of total proteins isolated from Mbs and from differentiating Mts. Protein expression was clearly detected in cultures induced to differentiate for up to 72 h. Expression decreased as Mts matured. GAPDH was used as loading control. (C) Real-time PCR analysis of CCND2 mRNA in Mbs transfected with a control siRNA(CTL-siRNA) or an siRNA designed to downregulate CCND2 expression (CCND2-siRNA) and then induced to differentiate. A significant reduction in the level of expression of CCND2 was evident as early as 24 h after differentiation. Maximal effects were achieved at 48 h. No significant differences were detected in CCND2 expression in cultures treated with the CTL-siRNA compared with untreated cells. (*P ≤ 0.004; **P ≤ 0.0003). (D) Protein analysis in Mts maintained in differentiation medium for 48 h after siRNA transfection confirmed a reduction in the level of CCND2 in those cultures. All analyses were confirmed in multiple experiments and using different primary cell lines.
Downregulation of CCND2 mediated by an siRNA targeting the coding sequence of the mRNA resulted in a significant decrease of CCND2 transcript levels compared with cells treated with a non-targeting siRNA as early as 24 h after induction of differentiation (48 h after siRNA transfection). Transcripts remained lower throughout all time points analyzed (Fig. 5C) and returned to levels similar to those of untransfected cells or cells transfected with the control siRNA by 96 h after induction of fusion. CCND2 protein was markedly decreased in Mts isolated from cultures transfected with the CCND2-siRNA and maintained in differentiation medium for 48 h (Fig. 5D). Immunostaining analysis demonstrated that CCND2 was localized predominantly in the nuclear compartment of Mts in cells treated with the CTL-siRNA, but was absent in cultures transfected with CCND2-siRNA (Supplementary Material, Fig S3). No effects on cell cycle progression were detected in Mbs undergoing RNAi and then induced to differentiate or Mbs maintained in growth medium for up to 4 days after transfection, suggesting that CCND2 is not required to regulate cell division (data not shown). Furthermore, Mb cultures transfected with targeting siRNAs for 4 days and then induced to differentiate showed no significant differences in the fusion index when compared with untransfected cells or cells transfected with the CTL-siRNA under similar experimental conditions.
To further confirm the specificity of CCND2 in regulating differentiation and to determine whether constitutive downregulation of CCND2 would affect regeneration following Mb transplantation, we generated cell lines expressing different lentiviral vectors which were transduced in C57BL/10 primary Mbs, as described in greater detail in the Materials and Methods section. Real-time RT–PCR analysis confirmed the expected decrease in CCND2 mRNA levels in all clones isolated. On average, all cell lines analyzed were shown to have a 50–70% reduction in CCND2 expression compared with Mbs infected with a non-targeting shRNA (CTL-shRNA). No changes in the cell cycle distribution were found in cells treated with the targeting lentiviral vectors and that were maintained in selection medium two weeks after infection (Supplementary Material, Table S4), suggesting that CCND2 is dispensable in actively dividing cells. The rate of cell division in those cells remained unchanged at all of the time points analyzed and no differences in the number of apoptotic cells were detected in Mbs as determined by Tunnel staining.
Mt formation was monitored in those cells for up to 4 days after induction of differentiation (Supplementary Material, Fig. S4). Multinucleated Mts containing more than six nuclei per Mt were evident in culture as early as 24 h after switching to differentiation medium and fusion continued through all time points analyzed. By 72 h in differentiation medium, the majority of the culture contained large (>50) nuclei per Mt. These results confirmed the specificity of CCND2 and provided strong evidence for a role of CCND2 during differentiation.
In vivo analysis of mdx muscles following Mb transplantation
The regenerative capacity of Mbs constitutively downregulating CCND2 was assessed in vivo following transplantation into TA muscles of dystrophin-deficient mice. Muscles were irradiated and regeneration was induced by cardiotoxin injury as described above. Mbs infected with a CCND2-shRNA or the CTL-shRNA were transplanted at the dosages indicated in Figure 6 and regeneration was followed over time. The number of eMHC-positive fibers (eMHC+) detected in TAs isolated 4 days after transplantation increased in a dose-dependent manner. Muscle that received CCND2-shRNA Mbs showed a significantly higher number of positive fibers than those injected with CTL-shRNA Mbs, demonstrating that transplanted cells were all capable of forming newly regenerated fibers and that the downregulation of CCND2 enhanced regeneration (Fig. 6A and Supplementary Material, Fig. S5).
Figure 6.
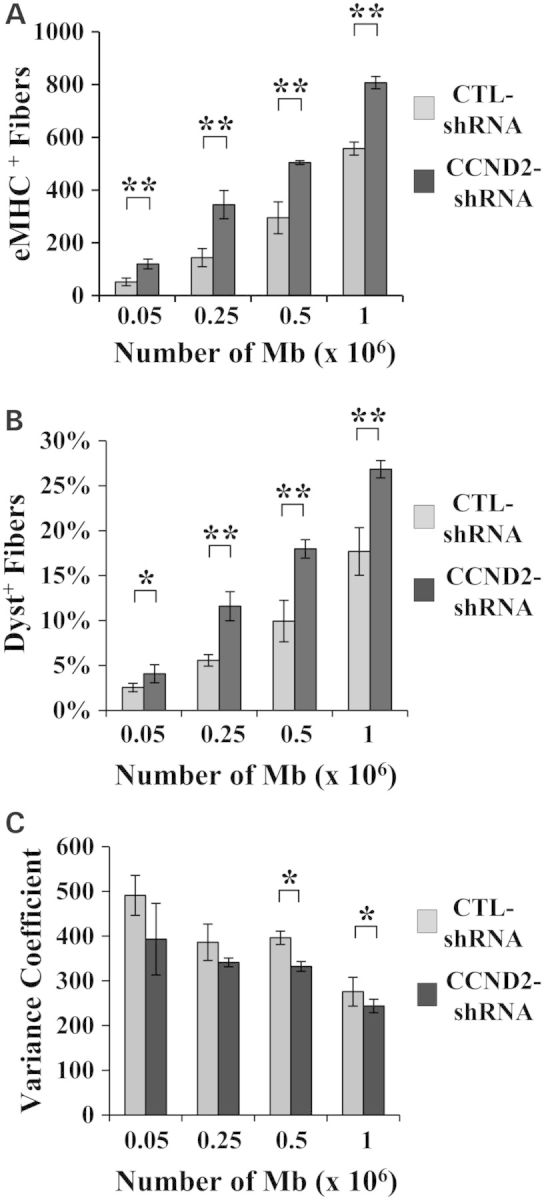
Transplantation of CCND2-shRNA Mbs increases regeneration. (A) The number of eMHC+ fibers was significantly higher in muscles engrafted with CCND2-shRNA Mbs compared with muscles that received CTL-shRNA Mbs. (**P ≤ 0.004). (B) A significant increase in the percentage of DYS+ fibers was evident in all muscles that received CCND2-shRNA Mbs compared with muscles transplanted with CTL-shRNA Mbs. (N = 4 muscles per treatment group; *P = 0.02; **P ≤ 0.001). (C) A gradual decline in variance coefficient was seen with increasing Mb dosage. A significant difference was detected in muscles that received CCND2-shRNA Mbs compared with muscle transplanted with CTL-shRNA Mbs but only at the doses of 2.5 × 105 and 1 × 106 Mbs per injected TA. (**P ≤ 0.04). Analyses were confirmed in duplicate experiments (N = 4 muscles per treatment group).
Similar results were obtained in muscles immunoassayed for dystrophin and analyzed 1 month after transplantation. On average, the number of DYS+ fibers was consistently 1.5- to 2-fold higher in muscles that received CCND2-shRNA Mbs compared with muscles transplanted with CTL-shRNA Mbs (Fig. 6B; Supplementary Material, Fig. S6). Furthermore, a reduction in the diameter of the fibers that expressed dystrophin was evident in all muscles that received Mbs at doses ≥0.5 × 106 (Fig. 6C).
Mbs infected with a CCND2-shRNA or the CTL-shRNA were transplanted at a dosage of 5 × 105 Mbs per injected muscle and regeneration was followed for up to 1 month following engraftment (Supplementary Material, Fig. S7). The highest number of eMHC-positive (eMHC+) fibers was detected at 4 days following transplantation in both the treatment groups. Muscles analyzed 15 days and 1 month after transplantation showed few although clearly detectable clusters of eMHC+ fibers scattered throughout the muscle (data not shown). Dystrophin expression was evident even at the earliest time point analyzed and was shown to progressively increase over time (Supplementary Material, Fig. S7B). The percentage of DYS+ fibers was significantly higher in muscles that received CCND2-shRNA compared with muscles transplanted with CTL-shRNA at all the time points analyzed. Altogether, these data suggest that constitutive downregulation of CCND2 prior to transplantation promotes myofibrillogenesis further supporting future studies aimed at investigating the therapeutic potential of targeting CCND2 gene expression for the treatment of muscle disorders like DMD.
Knockdown of CCND2 influences the expression of desmin, Myf5 and myogenin in primary muscle cultures
To better characterize the downstream effects of CCND2 downregulation during myogenesis, we have examined the expression pattern of key markers of Mb differentiation, desmin and the myogenic regulator factors (MRFs) MyoD, Myf5 and myogenin in cells downregulating CCND2 (Supplementary Material, Fig. S8 and S9). Experiments were conducted in primary cell lines isolated from C57BL/10 muscles transfected with one of the siRNAs showing to efficiently inhibit CCND2 expression (Supplementary Material, Table S2). The results were compared with those obtained from untransfected cells or cells transfected with the CTL-siRNA. A significant increase in desmin mRNA expression was evident as early as 48 h after induction of differentiation (Supplementary Material, Fig. S8A). No changes were detected in the levels of MyoD at any of the time points analyzed (Supplementary Material, Fig. S8B). Interestingly, expression of Myf5 was significantly increased as early as 24 h after induction of differentiation and remained higher throughout all time points analyzed (Supplementary Material, Fig. S8C). A significant increase in myogenin expression was also detected 48 h but not 24 or 72 h after induction of differentiation (Supplementary Material, Fig. S8D).
The results were confirmed at the protein level using western blot analysis. Cells were transfected with the CTL-siRNA or the CCND2-siRNA and were induced to differentiate 10 h later. Proteins were analyzed for expression of desmin, MyoD, Myf5 and myogenin 48 h later (Supplementary Material, Fig. S9). Taken together, the results suggest that CCND2 acts through mechanisms that are independent of MyoD activation and that regulation of differentiation mediated by CCND2 occurs predominantly within the first 48 h after induction of differentiation.
Protein interaction network and protein linkage analyses of CCND2
To better elucidate the role of CCND2 and the mechanisms of action that allow this gene to influence myogenesis, we have performed a comprehensive search of genes and proteins shown to interact directly or indirectly with CCND2 using primarily the Entrez Gene and the String-functional protein interaction network (String 8.2: http://string-db.org). We have identified a number of proteins that interact with CCND2 and constructed a protein interaction tree (Supplementary Material, Fig. S10). To test for functional interconnectedness between the tree and other biological processes, we have used the NIH DAVID pathway analysis tool (http://david.abcc.ncifcrf.gov) (24,25). The analysis revealed that CCND2 is a member of a dozen different cellular pathways all of which require the interaction of CCND2 with a cyclin-dependent kinase (Cdk).
Cdks are serine/threonine kinases that phosphorylate proteins on serine and threonine amino acid residues. Direct interaction of the D-type cyclins with Cdks has been previously shown to control terminal differentiation of Mbs (26–29). To determine whether the inhibitory effect of CCND2 observed during differentiation is mediated through the direct interaction of CCND2 with members of the Cdk family, we generated plasmid constructs encoding full-length CCND2 cDNA (pCCND2-Δ3′UTR) or encoding a truncated version of the vector lacking the N-terminal region of the gene encoding for the binding region of the Cdks (pCCND2- Δ3′UTR-ΔN) (Fig. 7A). All constructs lacked the 3′-UTR region. Plasmids were co-transfected with an siRNA targeting the 3′UTR of the CCND2 mRNA (Supplementary Material, Table S2). Cells were induced to differentiate and analyzed using the MetaXpress fusion software (Fig. 7B and 7C). Overexpression of full-length CCND2 inhibited fusion while expression of the pCCND2- Δ3′UTR- ΔN construct had no significant effects on differentiation when compared with cells treated with the CCND2-siRNA targeting the 3′-UTR region of the CCND2 mRNA (Fig. 7C).
Figure 7.

CCND2 regulates fusion through the interaction with Cdks. (A) Structure of the plasmids used to study the interaction of CCND2 with Cdk. The pCCND2-Δ3′UTR construct encodes a cDNA expressing full-length CCND2 but lacking the 3′-UTR region while the pCCND2- Δ3′UTR-ΔN vector encodes a shorter version of the vector sequence predicted to express an in frame, although truncated version of the CCND2 lacking the N-terminal region of the protein which contains the binding motif for the Cdk. (B) Plasmids were co-transfected with an siRNA targeting the 3′UTR of the CCND2 mRNA and were allowed to recover for 24 h before switching to differentiation medium. Images were acquired 48 h after induction of differentiation using a HCI. Mts were much smaller in cultures that over-expressed full-length CCND2 when compared with cells transfected with the CTL-siRNA or the CCND2-siRNA. Fusion was not impaired in cells co-transfected with the pCCND2-Δ3′UTR-ΔN construct and the CCND2-siRNA (scale bar: 50 μm). (C) Fusion was assessed using the MetaXpress fusion analysis software 48 h after induction of differentiation. The average number of fused cells detected in cultures treated with CCND2-siRNA was significantly different from that of cultures co-transfected with pCCND2-Δ3′UTR and CCND2-siRNA but not in those treated with pCCND2- Δ3′UTR-ΔN and CCND2-siRNA. (N = 16; * P ≤ 0.04). (D) Western blot analysis was performed 48 h after induction of differentiation (72 h after siRNA transfection). Phosphorylated pRb (ppRb) was markedly reduced in culture treated with CCND2-siRNA but not in sham-transfected cells or cells treated with CTL-siRNA and maintained in differentiation medium for the same period of time. Results were consistent across triplicate experiments.
To identify the specific Cdk interacting with CCND2 and responsible for inhibiting Mb differentiation, we analyzed all the images acquired by HTS in cells treated with the siRNA duplexes targeting each member of the Cdk family. Downregulation of Cdk4, but not CdkN1A, CdkN1B, Cdk2, Cdk6 and Cdk5, significantly increased fusion, suggesting that inhibition of Mb differentiation is likely mediated by the CCND2/Cdk4 complex (data not shown).
A key component of the pCCND2-Δ3′UTR- ΔN vector is the lack of the LxCxE motif required for the CCND2/Cdk complex to bind and phosphorylate the retinoblastoma protein (pRb) (Fig. 7A). To further clarify the role of the CCND2/Cdk complex during differentiation, we have analyzed the effects of CCND2 downregulation on pRb. Mbs were transfected with the CTL- or the CCND2-siRNA for 10 h and were then induced to differentiate for 48 h before protein analysis. A decrease in phosphorylated pRb (ppRb) was detected in all cultures transfected with the siRNA targeting CCND2 expression, but not in cultures treated with the CTL-siRNA (Fig. 7D). Similar results were obtained in cultures maintained in differentiation media for 24 h following transfection with the targeting and control siRNAs (data not shown).
Altogether, these results demonstrate that the effects observed on fusion and Mt formation are dependent upon the inactivation of CCND2 during the stages of differentiation downstream of cell cycle withdrawal of Mbs. Furthermore, the results suggest that CCND2 acts primarily during early stages of myocyte formation and activation of the myogenic program by maintaining pRb in an unphosphorylated state. Thus, inhibition of pRb phosphorylation is likely to be responsible for the dramatic increase in regeneration observed after transplantation.
DISCUSSION
We have developed an HTS system capable of performing a genome-wide functional genetic screen in primary muscle progenitor cells and able to identify regulators of Mb differentiation. The screening platform utilized primary, secondary and tertiary screenings based on independent endpoint measurements to maximize its specificity. Several key features made the assay particularly desirable and highly reliable. First, it was designed to study the effects of downregulation of a single gene at the time using individual siRNA sequences, thus minimizing off-target effects obtained as a result of pooled siRNAs. Second, the system was completely void of reporter vectors and relayed exclusively on determining changes in differentiating cells under physiological conditions following transfection with siRNAs. Third, it utilized freshly isolated primary cells as opposed to immortalized cell lines such as C2C12. Although the latter have been employed extensively to study muscle regeneration and muscle repair, they do not mimic perfectly the system and differences have been reported in their response to environmental cues and their level of differentiation and maturation (30). Finally, genes to be taken into secondary screenings were selected on the bases of their morphology and fusion index analyses using the images acquired by HCI, narrowing down the number of positives to few genes, which showed consistent and reproducible effects in at least two independent runs and using more than one duplex targeting the same gene.
In our screen, we selected as positives only those genes that showed a response in at least three distinct duplexes and individual siRNAs that showed poor or no effects were excluded from subsequent mathematical and statistical analyses. Since the P-value was associated with a gene, all the wells of the same gene that were included in the analysis were assigned identical P-values. Using this probability-based approach, a gene with multiple moderately active wells was weighed less heavily than a gene with more active wells and therefore, should be ranked more heavily given their higher activity. Thus, we are confident that the criteria used to select the genes were unbiased. Nonetheless, it is possible that poor target affinity of some of the siRNAs contained in the library may have caused genes important for regulating Mb differentiation to be missed. However, the stringency of the parameters used to select positive hits enabled us to ensure that all the genes identified have biological significance.
Confirmation of the activity of positive hits in vivo was performed in muscles depleted of endogenous stem cells. Variability between each individual experiment and within experimental groups should be greatly reduced by the lack of possible competitive effects between transplanted cells and endogenous stem cells present in the microenvironment of recipient muscles. Therefore, an enhancement in regeneration following transplantation of Mbs undergoing silencing of each specific hit could only be attributed to ability of the transplanted cells to fuse more efficiently compared with untransfected cells and ensured consistent and reproducible results among replicates.
Many of the several hundred genes identified were shown to cluster into specific pathways known to play key roles during terminal differentiation of Mbs (Fig. 2D). Among the pathways identified through gene ontology analyses, genes appertaining to the Wnt, the TGF-β and the MAP kinase signaling pathways were over-represented. These pathways have been studied extensively in muscles both in vitro and in vivo and the role of many of the genes identified in our screen has been clearly established during myogenesis. In addition, we found enrichment in genes involved directly or indirectly in cell cycle regulation, in particular Cdks consistent with the observation that withdrawal of Mbs from cell cycle mediated by Cdk inhibitors promotes their differentiation (31).
Importantly, many of the genes that were identified by the screen have been linked directly or indirectly to the GPCR signaling pathway (Fig. 2D) whose role in muscle is still largely unknown. Those genes are particularly attractive therapeutic targets since many of the pharmaceutical drugs currently on the market are known to inhibit these receptors. Active studies in our laboratory are currently aimed at better characterizing the role of those receptors in muscles and at testing some of those inhibitors. The use of drugs that are already approved for use in humans is likely to expedite the preclinical developmental stages required for their testing in DMD and may prove beneficial in the treatment of various forms of muscular dystrophies.
Our study clearly identifies CCND2 as the lead candidate of the screen. The specificity of the gene was confirmed using different siRNA sequences targeting various regions of the CCND2 mRNA. Similar effects were achieved using lentiviral-mediated shRNA knockdown of CCND2 (Fig. 5). Gain- and loss-of-function studies suggested that CCND2 acts by interacting with Cdk4 and by maintaining pRb in the hypophosphorylated (active) state. The role of pRb during myogenesis has been studied in detail and its activity has been linked to both cell cycle exit and differentiation (32–36). Our studies further expand the role of pRb in muscle repair and demonstrate that regulation of its phosphorylated state is mediated, at least in part, by CCND2. When transplanted into skeletal muscle of recipient mice, Mbs downregulating CCND2 through siRNA or shRNA all showed to significantly improve the ability of Mbs to fuse and form new myofibers compared with untreated or control Mbs (Supplementary Material, Fig. S5 and S6). Altogether, these studies suggest that downregulation of CCND2 may be an important target to enhance regeneration in diseases like DMD when combined with cell-mediated regenerative approaches.
Further studies will be required before this approach can be brought into the clinic and the clinical potential of downregulating CCND2 expression will have to be studied in detail using different types of muscle stem cells to ensure its broad applicability for the treatment of muscle disorders. Finally, the role of CCND2 in mature myofibers will have to be clearly determined to guarantee that the beneficial effects achieved through transplantation are maintained over time and that this approach is safe to use in patients.
In conclusion, our study identified known and unknown genes that function during myogenesis and many new targets that could be used to enhance muscle regeneration and muscle repair. The clinical applicability of these new targets is not restricted to diseases like DMD, but has potential for the treatment of many other disorders characterized by loss of muscle mass including aging and many other forms of muscular dystrophy. Characterizing the role of these genes during myogenesis will be crucial at better understanding their functions in muscles during regeneration and repair and is likely to broaden our knowledge on the tightly regulated and complex processes that control muscle function. Furthermore, the identification of CCND2 as the lead gene in our HTS may open new possibilities for the treatment of DMD using cell-mediated regenerative approaches.
MATERIALS AND METHODS
Mice
Wild-type (C57BL/10SnJ), the GFP transgenic mice (GFPC57BL/6-Tg(ACTB-EGFP)1Osb/J), the nude strain (C57Bl/6 Foxn1nu) and mdx mice (C57BL/10ScSn-Dmdmdx/J) offspring from breeder colonies were originally obtained from Jackson Laboratory (Bar Harbor, ME, USA). The mdx/nude strain was derived as previously described (37). Mice were housed and maintained in the Veterinary Medical Unit at the Division of the Laboratory Animal Medicine (DLAM) in accordance with guidelines of the Administrative Panel on Laboratory Animal Care of the University of California, Los Angeles.
Cell culture and transfection
Cells were derived from limb muscle of neonatal GFP, mdx and C57 mice as previously described (38). For growth, cells were plated on dishes coated with 5 µg/ml laminin (Life Technologies, Inc.) and maintained in growth medium consisting of Ham's F10 nutrient mixture (Mediatech, Herndon, VA, USA) supplemented with 20% fetal bovine serum, 4 ng/ml of human fibroblast growth factor (FGF) basic (Peprotech Inc., Rocky Hill, NJ, USA), penicillin and streptomycin. Cell differentiation was induced by maintaining the cells in low serum medium (differentiation medium) consisting of Dulbecco's modified Eagle's medium (DMEM) supplemented with 2% horse serum, penicillin and streptomycin.
The siRNA libraries used in the primary and secondary screens and the non-targeting control were purchased from Dharmacon (Thermo Fisher Scientific, Fremont, CA, USA). The siRNA designed to target myostatin, and the additional siRNAs used in target validation experiments were obtained from Invitrogen (Invitrogen Corporation, Carlsbad, CA, USA) and are listed in Supplementary Material, Table S2. No homologous sequences were found in mouse genome. The double-stranded siRNAs were synthesized chemically and modified into stealth siRNA (Invitrogen Corp) to enhance the stability in vitro.
For primary siRNA screens, tgGFP Mbs were reverse transfected with siRNAs in 384-well plates. In brief, cells were trypsinized and resuspended in growth medium containing 20% FBS and 1:250 w/v extracellular matrix (ECM, Sigma, St Louis, MO, USA), at a concentration of 6 × 104 cells/ml. 10 µl of a transfection reagent mixture containing 10 µl/ml LipofectamineTM 2000 in Opti-MEM (Invitrogen Corporation) was added to each well prespotted with 1 pmol of siRNA and incubated at room temperature for 20 min. Cells (2500 cells/well) were added to wells using a robotic multichannel pipette. Approximately 10 h after transfection, the medium was replaced with prewarmed differentiation medium containing DMEM, 2% HS and antibiotics. Two days after transfection, 10 µl of fresh differentiation medium containing 2 µm Hoechst 33342 (Invitrogen Corporation) was added to each well and Mts were imaged using a high-content imager (Molecular Device, Carlsbad, CA, USA).
Secondary and tertiary screenings were performed in 12-well or 6-well plates. Cells (1.5 × 104 cells/well) were seeded 12 h prior to transfection. For each transfection, 5 μl of siRNA (100 mm) was complexed with 5 µl of Lipofectamine™ 2000 for 30 min at room temperature following the manufacturer's instructions. The complex was then added to the wells containing growth medium without FGF. Transfection was stopped after 10 h by replacing the transfection solution with fresh differentiation medium.
Lentiviral vectors and clones selection
Lentiviral particles were purchased from Sigma (Supplementary Material, Table S3). Infection conditions in tgGFP Mbs were optimized with MISSON TurboGFP Control Transduction particles (SHC003V) and non-targeting shRNA Control Transduction particles (SHC002V; Sigma) in 24-well plates for optimal growth conditions, viral dosage and puromycin selection concentration. TgGFP cells were seeded at 1.6 × 104 per well in triplicate on 12-well plates and for 12 h. The lentiviral vectors encoding shRNA (50 particles/cell) were added onto cells in the presence of 8 μg/ml hexadimethrine bromide and incubated for 24 h at 37°C. A total of five different shRNA sequences were tested (Supplementary Material, Table S3). Clones were selected using puromycin at a final concentration of 2 µg/ml for 6 days. Cells were evaluated from days 7 to 10 for cell proliferation and cytotoxicity under the microscope. On day 10, the number of viable cells was determined via CellTiter 96 AQueous One Solution Cell Cytotoxicity Assay (Promega).
Intramuscular injections
Transplantation of muscle progenitor cells was performed as described previously (39–41). Mice were anesthetized using either methoxyfluorane followed by intraperitoneal ketamine:xylazine:water (1:1:2) at 0.9 µl/g or isofluorane through a nose cone. All mice were injected at the age of 3 months. Lower limb muscles of mdx/nude mice were exposed to 18 Gy γ-irradiation 3 days before grafting to ablate all circulating muscle progenitor cells (42). Regeneration into recipient TA muscles was stimulated by intramuscular injection of 50 µl of notexin (10 µg/ml) (43) 24 h before Mb transplantation (39,41,44,45). On the day of transplantation, cells were detached from the flask with 0.1% trypsin in Hank's balanced salt solution (HBSS) containing 0.02% EDTA followed by three suspensions in HBSS and centrifugations at 1800 rpm for 5 min. The last centrifugation was at 6500 rpm for 5 min. The volume of injection was maintained constant at 50 μl for all the engraftment procedures. Cells were injected percutaneously into TA muscles using a 0.3 cc insulin syringe with a 30-gauge needle (Becton Dickinson). Cell viability was assessed using trypan blue staining. Transplantation of Mbs transfected with siRNAs was performed using cells resuspended at a final concentration of 1 × 104 cells/µl. This concentration was chosen based on pilot studies that were performed to determine the minimal number of cells needed to detect a significant difference in the number of GFP-positive and DYS+ fibers in recipient muscles injected with sham-transfected Mbs or Mbs treated with the CTL-siRNA when compared with those that received Mbs downregulating myostatin. Transplantation of Mbs infected with the lentiviral vectors was carried out on clones isolated from cells transduced with the CTL-shRNA or the CCND2a-shRNA and was performed as described above. Cells were resuspended at different concentrations (Fig. 6) and injections were performed using a final volume of 50 μl.
Plasmids
The pCCND2-Δ3′UTR construct expressing wild-type cyclin D2 and the pCCND2-Δ3′UTR-ΔN lacking the N-terminal region of the protein were generated by cloning the open reading frame of the two proteins into a pcDNA3.3 vector (Invitrogen Corporation). The mouse complementary DNA (cDNA) encoding CCND2 (GenBank accession number NM_009829) was obtained by direct amplification of a cDNA library using a forward primer (CCND2-forw: 5′-CAGTCGGGACCGAGTGGTGG-3′) homologous to the region of the gene located in position −30 and a reverse primer (CCND2-rev: 5′-GCCGCCCGAATGGCTTCC-3′) annealing to the first 18 bp immediately downstream of the stop codon. The open reading fame for the pCCND2-Δ3′UTR-ΔN was amplified with the forward primer (CCND2-forwΔN: 5′-GTGCGTGCAGAAGGACATCC-3′) located in position +130 of the CCND2 cDNA paired with the CCND2-rev primer. Amplification was carried out for 30 cycles in the presence of Pfu DNA polymerase (Stratagene) at an annealing temperature of 65°C for 1 min and an extension temperature of 68°C for 2 min. Amplification was terminated by an additional extension of 30 min at 72°C. PCR products were run on a 1.2% agarose gel and purified using the gel extraction kit (Qiagen, Valencia, CA, USA) as previously described (46). Vector sequences were confirmed using an ABI377 automated sequencer.
Western blot analysis
Assessment of CCND2, desmin and MRF expression was performed using standard procedures and was carried out on proteins isolated from untransfected cells or cells transfected with the CTL-siRNA or the siRNAC10 oligomer targeting the CCND2 mRNA (Supplementary Material, Table S2). Briefly, cells were lysed in RIPA buffer (50 mm Tris–HCl, pH 7.4, 150 mm NaCl, 0.5% deoxycholate and 1% Nonidet P-40) containing aprotinin (20 µg/ml), leupeptin (20 µg/ml), phenylmethylsulfonyl fluoride (10 µg/ml) and sodium orthovanadate (1 mm). Total protein in the extract was determined by the Bio-Rad protein assay (Bio-Rad, Hercules, CA, USA). Immunoblot analysis was performed on 100 μg of total protein from samples separated by electrophoresis using 12% SDS-polyacrylamide gels and then transferred onto nitrocellulose membranes. The membranes were blocked with 5% milk in Phosphate buffered saline (PBS) for 1 h at room temperature and were then incubated with primary antibody overnight at 4°C. The following antibodies were used: mouse monoclonal anti-CCND2 (Thermo Fisher Scientific; 1:200), anti-MyoD (BD Biosciences Pharmingen, San Diego, CA; 1:200), anti-myogenin (Abcam, Cambridge, MA, USA; 1:500) and anti-desmin (D33; Sigma-Aldrich, St Louis, MO; 1:500), and rabbit polyclonal anti-Myf5 (C-20; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA; 1:800). Blots were washed with 0.05% Tween-20 in PBS and then incubated with a horseradish peroxidase-coupled anti-mouse or anti-rabbit secondary antibody (Amersham Pharmacia Biotech, Piscataway, NJ, USA). Specific antibody binding was detected using an enhanced chemiluminescent system (Amersham).
Extraction of pRb was achieved using a high-salt buffer containing 50 mm Tris–HCl, 250 mm NaCl, 5 mm EDTA, 0.1% Triton X-100, 50 mm NaF, 0.1 mm sodium orthovanadate, 5 μg/ml leupeptin, 5 μg/ml aprotinin and 5 μg/ml pepstatin as previously described (47). Protein extracts were separated on a 6% SDS-polyacrylamide gel and electroblotted onto nitrocellulose. Membrane was blocked for 1 h with 5% milk, 0.05% Tween-20 in PBS followed by overnight incubation with a mouse monoclonal anti-human pRb antibody (BD Pharmingen; 1:200). Protein expression was detected with a peroxidase-conjugated secondary antibody and a chemiluminescent substrate as described above.
Flow cytometry
Fluorescence was measured using a Becton Dickinson FACScalibur flow cytometry (Becton Dickinson, Franklin Lakes, NJ, USA). Cells were trypsinized, harvested, resuspended in 0.5% BSA, 2 mm EDTA in PBS and processed immediately. Cells were fixed in 70% ice-cold ethanol, washed in PBS and resuspended in 0.5 ml of PBS containing 50 mg/ml RNase A, 1% FBS and 2.5 mg/ ml propidium iodide. A total of 2 × 104 cells were used for each analysis. Experiments were repeated in triplicate for each time point analyzed and a total of three independent experiments were performed.
RT–PCR analysis of transcript levels
Total RNA was extracted from cultured cells using TRI-REAGENT (Sigma). For each reaction, 5 µg of RNA was treated with 1 U of DNase I (GIBCO/BRL) at room temperature and reverse transcribed using the first-strand cDNA synthesis kit (Invitrogen Corporation) in the presence of SuperscriptIII (Invitrogen Corporation) to a final volume of 20 µl. Amplification was carried out using the combination of primers described in Supplementary Material, Table S5.
Real-time PCR was performed using a My iQ single-color detection system under the following thermal cycler conditions: 95°C for 10 min followed by 40 cycles of a three-step reaction consisting of denaturation at 95°C for 30 s, annealing at 55°C for 1 min and extension and data collection at 72°C for 30 s. The relative expression ratio of mRNA transcripts was calculated based on real-time PCR efficiencies using a standard ΔΔCT method (48). Glyceraldehyde 3-phosphate dehydrogenase was used as an internal standard control for all samples and to normalize transcript levels.
Immunofluorescence analysis
Dystrophin immunostaining of cultured cells was performed as previously described (38,49). Briefly, cells cultured on coverslips were fixed in ice-cold ethanol for 10 min, rinsed in PBS and permeabilized in 0.3% Triton X-100 for 10 min at room temperature. A solution containing 5% heat-inactivated normal goat serum and 1 mg/ml BSA in PBS was used as blocking solution for 30 min. Cells were then incubated with a primary rabbit polyclonal antibody to CCND2 (M-20, Santa Cruz Biotechnology, Inc.; 1:300) overnight at 4°C. Cells were washed in PBS before incubation for 1 h at room temperature with an Alexa 546-coupled goat-anti-mouse (H + L) secondary antibody (Invitrogen Corporation; 1:250). Coverslips were mounted using Vectashield (Vector Laboratories, Inc., Burlingame, CA, USA) for fluorescence microscopy.
Dystrophin staining of muscles was done using serial 10 μm sections as previously described (50). TA muscles were cut for at least two-thirds of the muscle length at intervals of 400 µm and examined for dystrophin expression. Air-dried sections were rehydrated in physiological solutions and blocked using a solution containing normal goat serum diluted 1:20 in PBS. Consecutive sections isolated from injected muscles were immunoassayed using a polyclonal antibody (Thermo Fisher Scientific; 1:200) specific for the C-terminal region of the dystrophin protein. Specific antibody binding was detected with the Alexa 546-coupled goat-anti-rabbit secondary antibodies (1:500). Quantitative determination of the number of DYS+ fibers was performed on the entire cross-sectional area for each section analyzed.
Quantitative measurements of muscle fibers
Myofiber minimal Feret's diameter was calculated as previously described (51) by measuring 200–400 fibers in muscles that received the lower dosage of Mbs (1 × 104) and at least 500 randomly selected fibers per muscle for all the remaining treatment groups. Quantitative analysis was performed using the Image J 1.37c software (National Institute of Health, USA). The variance coefficient of the muscle fiber size was calculated by dividing standard deviation of the muscle fiber size with the mean muscle fiber size and by multiplying the value obtained by 1000 (51).
Statistical analysis
Data are presented as means and standard deviations. Comparisons between groups were done using Student's t-test assuming two-tailed distribution and equal variances.
SUPPLEMENTARY MATERIAL
AUTHOR'S CONTRIBUTION
M.V.K. and J.Y. performed experiments, analyzed data and wrote the manuscript. R.K. and T.C. assisted with data acquisition and analysis. C.B. designed and directed the study, performed experiments, analyzed and integrated the data and wrote the manuscript.
Conflict of Interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Dr Robert D. Damoiseaux for suggestions, discussion and assistance with the screening of the siRNA libraries and Dr Giovanni Coppala and Fuying Gao for generating the heatmap. This work was supported in part by the End Duchenne Parent Project and by the Berkeley endowment startup funds to CB. The UCLA vector Core Facility used for the lentiviral vector production is supported by JCCC/P30 CA01042 and Cure/P30 DK041301. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
REFERENCES
- 1.Monaco A.P., Neve R.L., Colletti-Feener C., Bertelson C.J., Kurnit D.M., Kunkel L.M. Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature. 1986;323:646–650. doi: 10.1038/323646a0. doi:10.1038/323646a0. [DOI] [PubMed] [Google Scholar]
- 2.Wehling-Henricks M., Lee J.J., Tidball J.G. Prednisolone decreases cellular adhesion molecules required for inflammatory cell infiltration in dystrophin-deficient skeletal muscle. Neuromuscul. Disord. 2004;14:483–490. doi: 10.1016/j.nmd.2004.04.008. doi:10.1016/j.nmd.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 3.Shavlakadze T., White J., Hoh J.F., Rosenthal N., Grounds M.D. Targeted expression of insulin-like growth factor-I reduces early myofiber necrosis in dystrophic mdx mice. Mol. Ther. 2004;10:829–843. doi: 10.1016/j.ymthe.2004.07.026. doi:10.1016/j.ymthe.2004.07.026. [DOI] [PubMed] [Google Scholar]
- 4.Quinlan J.G., Wong B.L., Niemeier R.T., McCullough A.S., Levin L., Emanuele M. Poloxamer 188 failed to prevent exercise-induced membrane breakdown in mdx skeletal muscle fibers. Neuromuscul. Disord. 2006;16:855–864. doi: 10.1016/j.nmd.2006.09.016. doi:10.1016/j.nmd.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 5.Bogdanovich S., Krag T.O., Barton E.R., Morris L.D., Whittemore L.A., Ahima R.S., Khurana T.S. Functional improvement of dystrophic muscle by myostatin blockade. Nature. 2002;420:418–421. doi: 10.1038/nature01154. doi:10.1038/nature01154. [DOI] [PubMed] [Google Scholar]
- 6.Yasuda S., Townsend D., Michele D.E., Favre E.G., Day S.M., Metzger J.M. Dystrophic heart failure blocked by membrane sealant poloxamer. Nature. 2005;436:1025–1029. doi: 10.1038/nature03844. doi:10.1038/nature03844. [DOI] [PubMed] [Google Scholar]
- 7.Ng R., Metzger J.M., Claflin D.R., Faulkner J.A. Poloxamer 188 reduces the contraction-induced force decline in lumbrical muscles from mdx mice. Am. J. Physiol. Cell Physiol. 2008;295:C146–C150. doi: 10.1152/ajpcell.00017.2008. doi:10.1152/ajpcell.00017.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Granchelli J.A., Pollina C., Hudecki M.S. Pre-clinical screening of drugs using the mdx mouse. Neuromuscul. Disord. 2000;10:235–239. doi: 10.1016/s0960-8966(99)00126-1. doi:10.1016/S0960-8966(99)00126-1. [DOI] [PubMed] [Google Scholar]
- 9.Rolland J.F., De L.A., Burdi R., Andreetta F., Confalonieri P., Conte C.D. Overactivity of exercise-sensitive cation channels and their impaired modulation by IGF-1 in mdx native muscle fibers: beneficial effect of pentoxifylline. Neurobiol. Dis. 2006;24:466–474. doi: 10.1016/j.nbd.2006.08.010. doi:10.1016/j.nbd.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 10.Gosselin L.E., Williams J.E. Pentoxifylline fails to attenuate fibrosis in dystrophic (mdx) diaphragm muscle. Muscle Nerve. 2006;33:820–823. doi: 10.1002/mus.20523. doi:10.1002/mus.20523. [DOI] [PubMed] [Google Scholar]
- 11.Hawke T.J., Garry D.J. Myogenic satellite cells: physiology to molecular biology. J. Appl. Physiol. 2001;91:534–551. doi: 10.1152/jappl.2001.91.2.534. [DOI] [PubMed] [Google Scholar]
- 12.Conboy M.J., Karasov A.O., Rando T.A. High incidence of non-random template strand segregation and asymmetric fate determination in dividing stem cells and their progeny. PLoS. Biol. 2007;5:e102. doi: 10.1371/journal.pbio.0050102. doi:10.1371/journal.pbio.0050102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Charge S.B., Rudnicki M.A. Cellular and molecular regulation of muscle regeneration. Physiol. Rev. 2004;84:209–238. doi: 10.1152/physrev.00019.2003. doi:10.1152/physrev.00019.2003. [DOI] [PubMed] [Google Scholar]
- 14.Gopinath S.D., Rando T.A. Stem cell review series: aging of the skeletal muscle stem cell niche. Aging Cell. 2008;7:590–598. doi: 10.1111/j.1474-9726.2008.00399.x. doi:10.1111/j.1474-9726.2008.00399.x. [DOI] [PubMed] [Google Scholar]
- 15.Dhawan J., Rando T.A. Stem cells in postnatal myogenesis: molecular mechanisms of satellite cell quiescence, activation and replenishment. Trends Cell Biol. 2005;15:666–673. doi: 10.1016/j.tcb.2005.10.007. doi:10.1016/j.tcb.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 16.Moisset P.A., Skuk D., Asselin I., Goulet M., Roy B., Karpati G., Tremblay J.P. Successful transplantation of genetically corrected DMD myoblasts following ex vivo transduction with the dystrophin minigene. Biochem. Biophys. Res. Commun. 1998;247:94–99. doi: 10.1006/bbrc.1998.8739. doi:10.1006/bbrc.1998.8739. [DOI] [PubMed] [Google Scholar]
- 17.Gussoni E., Blau H.M., Kunkel L.M. The fate of individual myoblasts after transplantation into muscles of DMD patients. Nat. Med. 1997;3:970–977. doi: 10.1038/nm0997-970. doi:10.1038/nm0997-970. [DOI] [PubMed] [Google Scholar]
- 18.Miller R.G., Sharma K.R., Pavlath G.K., Gussoni E., Mynhier M., Lanctot A.M., Greco C.M., Steinman L., Blau H.M. Myoblast implantation in Duchenne muscular dystrophy: the San Francisco study. Muscle Nerve. 1997;20:469–478. doi: 10.1002/(sici)1097-4598(199704)20:4<469::aid-mus10>3.0.co;2-u. doi:10.1002/(SICI)1097-4598(199704)20:4<469::AID-MUS10>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 19.Wagner K.R., McPherron A.C., Winik N., Lee S.J. Loss of myostatin attenuates severity of muscular dystrophy in mdx mice. Ann. Neurol. 2002;52:832–836. doi: 10.1002/ana.10385. doi:10.1002/ana.10385. [DOI] [PubMed] [Google Scholar]
- 20.McPherron A.C., Lee S.J. Double muscling in cattle due to mutations in the myostatin gene. Proc. Natl Acad. Sci. USA. 1997;94:12457–12461. doi: 10.1073/pnas.94.23.12457. doi:10.1073/pnas.94.23.12457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schuelke M., Wagner K.R., Stolz L.E., Hubner C., Riebel T., Komen W., Braun T., Tobin J.F., Lee S.J. Myostatin mutation associated with gross muscle hypertrophy in a child. N. Engl. J. Med. 2004;350:2682–2688. doi: 10.1056/NEJMoa040933. doi:10.1056/NEJMoa040933. [DOI] [PubMed] [Google Scholar]
- 22.Benabdallah B.F., Bouchentouf M., Tremblay J.P. Improved success of myoblast transplantation in mdx mice by blocking the myostatin signal. Transplantation. 2005;79:1696–1702. doi: 10.1097/01.tp.0000167379.27872.2b. doi:10.1097/01.TP.0000167379.27872.2B. [DOI] [PubMed] [Google Scholar]
- 23.Magee T.R., Artaza J.N., Ferrini M.G., Vernet D., Zuniga F.I., Cantini L., Reisz-Porszasz S., Rajfer J., Gonzalez-Cadavid N.F. Myostatin short interfering hairpin RNA gene transfer increases skeletal muscle mass. J. Gene Med. 2006;8:1171–1181. doi: 10.1002/jgm.946. doi:10.1002/jgm.946. [DOI] [PubMed] [Google Scholar]
- 24.Dennis G., Sherman B., Hosack D., Yang J., Gao W., Lane H.C., Lempicki R. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4:3. doi:10.1186/gb-2003-4-5-p3. [PubMed] [Google Scholar]
- 25.Huang D.W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2008;4:44–57. doi: 10.1038/nprot.2008.211. doi:10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 26.Rao S.S., Kohtz D.S. Positive and negative regulation of D-type cyclin expression in skeletal myoblasts by basic fibroblast growth factor and transforming growth factor. J. Biol. Chem. 1995;270:4093–4100. doi: 10.1074/jbc.270.8.4093. doi:10.1074/jbc.270.8.4093. [DOI] [PubMed] [Google Scholar]
- 27.Rao S.S., Chu C., Kohtz D.S. Ectopic expression of cyclin D1 prevents activation of gene transcription by myogenic basic helix–loop–helix regulators. Mol. Cell. Biol. 1994;14:5259–5267. doi: 10.1128/mcb.14.8.5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Skapek S.X., Rhee J., Kim P.S., Novitch B.G., Lassar A.B. Cyclin-mediated inhibition of muscle gene expression via a mechanism that is independent of pRB hyperphosphorylation. Mol. Cell Biol. 1996;16:7043–7053. doi: 10.1128/mcb.16.12.7043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Skapek S.X., Rhee J., Spicer D.B., Lassar A.B. Inhibition of myogenic differentiation in proliferating myoblasts by cyclin D1-dependent kinase. Science. 1995;267:1022–1024. doi: 10.1126/science.7863328. doi:10.1126/science.7863328. [DOI] [PubMed] [Google Scholar]
- 30.Maley M.A., Davies M.J., Grounds M.D. Extracellular matrix, growth factors, genetics: their influence on cell proliferation and myotube formation in primary cultures of adult mouse skeletal muscle. Exp. Cell Res. 1995;219:169–179. doi: 10.1006/excr.1995.1217. doi:10.1006/excr.1995.1217. [DOI] [PubMed] [Google Scholar]
- 31.Puri P.L., Sartorelli V. Regulation of muscle regulatory factors by DNA-binding, interacting proteins, and post-transcriptional modifications. J. Cell Physiol. 2000;185:155–173. doi: 10.1002/1097-4652(200011)185:2<155::AID-JCP1>3.0.CO;2-Z. doi:10.1002/1097-4652(200011)185:2<155::AID-JCP1>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 32.Gu W., Schneider J.W., Condorelli G., Kaushal S., Mahdavi V., Nadal-Ginard B. Interaction of myogenic factors and the retinoblastoma protein mediates muscle cell commitment and differentiation. Cell. 1993;72:309–324. doi: 10.1016/0092-8674(93)90110-c. doi:10.1016/0092-8674(93)90110-C. [DOI] [PubMed] [Google Scholar]
- 33.Zacksenhaus E., Jiang Z., Chung D., Marth J.D., Phillips R.A., Gallie B.L. pRb controls proliferation, differentiation, and death of skeletal muscle cells and other lineages during embryogenesis. Genes Dev. 1996;10:3051–3064. doi: 10.1101/gad.10.23.3051. doi:10.1101/gad.10.23.3051. [DOI] [PubMed] [Google Scholar]
- 34.Puri P.L., Iezzi S., Stiegler P., Chen T.T., Schiltz R.L., Muscat G.E., Giordano A., Kedes L., Wang J.Y., Sartorelli V. Class I histone deacetylases sequentially interact with MyoD and pRb during skeletal myogenesis. Mol. Cell. 2001;8:885–897. doi: 10.1016/s1097-2765(01)00373-2. doi:10.1016/S1097-2765(01)00373-2. [DOI] [PubMed] [Google Scholar]
- 35.Blais A., van Oevelen C.J., Margueron R., Acosta-Alvear D., Dynlacht B.D. Retinoblastoma tumor suppressor protein-dependent methylation of histone H3 lysine 27 is associated with irreversible cell cycle exit. J. Cell Biol. 2007;179:1399–1412. doi: 10.1083/jcb.200705051. doi:10.1083/jcb.200705051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huh M.S., Parker M.H., Scime A., Parks R., Rudnicki M.A. Rb is required for progression through myogenic differentiation but not maintenance of terminal differentiation. J. Cell Biol. 2004;166:865–876. doi: 10.1083/jcb.200403004. doi:10.1083/jcb.200403004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morgan J.E., Hoffman E.P., Partridge T.A. Normal myogenic cells from newborn mice restore normal histology to degenerating muscles of the mdx mouse. J. Cell Biol. 1990;111:2437–2449. doi: 10.1083/jcb.111.6.2437. doi:10.1083/jcb.111.6.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bertoni C., Morris G.E., Rando T.A. Strand bias in oligonucleotide-mediated dystrophin gene editing. Hum. Mol. Genet. 2005;14:221–233. doi: 10.1093/hmg/ddi020. doi:10.1093/hmg/ddi020. [DOI] [PubMed] [Google Scholar]
- 39.Partridge T.A., Morgan J.E., Coulton G.R., Hoffman E.P., Kunkel L.M. Conversion of mdx myofibres from dystrophin-negative to - positive by injection of normal myoblasts. Nature. 1989;337:176–179. doi: 10.1038/337176a0. doi:10.1038/337176a0. [DOI] [PubMed] [Google Scholar]
- 40.Huard J., Tremblay G., Verreault S., Labrecque C., Tremblay J.P. Utilization of an antibody specific for human dystrophin to follow myoblast transplantation in nude mice. Cell Transplant. 1993;2:113–118. doi: 10.1177/096368979300200204. [DOI] [PubMed] [Google Scholar]
- 41.Karpati G., Pouliot Y., Zubrzycka-Gaarn E., Carpenter S., Ray P.N., Worton R.G., Holland P. Dystrophin is expressed in mdx skeletal muscle fibers after normal myoblast implantation. Am. J. Pathol. 1989;135:27–32. [PMC free article] [PubMed] [Google Scholar]
- 42.Gross J.G., Bou-Gharios G., Morgan J.E. Potentiation of myoblast transplantation by host muscle irradiation is dependent on the rate of radiation delivery. Cell Tissue Res. 1999;298:371–375. doi: 10.1007/s004419900062. doi:10.1007/s004419900062. [DOI] [PubMed] [Google Scholar]
- 43.Harris J.B., Johnson M.A., Karlsson E. Proceedings: histological and histochemical aspects of the effect of notexin on rat skeletal muscle. Br. J. Pharmacol. 1974;52:152P. [PMC free article] [PubMed] [Google Scholar]
- 44.Huard J., Acsadi G., Jani A., Massie B., Karpati G. Gene transfer into skeletal muscles by isogenic myoblasts. Hum. Gene Ther. 1994;5:949–958. doi: 10.1089/hum.1994.5.8-949. doi:10.1089/hum.1994.5.8-949. [DOI] [PubMed] [Google Scholar]
- 45.Satoh A., Huard J., Labrecque C., Tremblay J.P. Use of fluorescent latex microspheres (FLMs) to follow the fate of transplanted myoblasts. J. Histochem. Cytochem. 1993;41:1579–1582. doi: 10.1177/41.10.8245416. doi:10.1177/41.10.8245416. [DOI] [PubMed] [Google Scholar]
- 46.Bertoni C., Rustagi A., Rando T.A. Enhanced gene repair mediated by methyl-CpG-modified single-stranded oligonucleotides. Nucleic Acids Res. 2009;37:7468–7482. doi: 10.1093/nar/gkp757. doi:10.1093/nar/gkp757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sacco A., Siepi F., Crescenzi M. HPV E7 expression in skeletal muscle cells distinguishes initiation of the postmitotic state from its maintenance. Oncogene. 2003;22:4027–4034. doi: 10.1038/sj.onc.1206353. doi:10.1038/sj.onc.1206353. [DOI] [PubMed] [Google Scholar]
- 48.Pfaffl M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. doi:10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bertoni C., Rando T.A. Dystrophin gene repair in mdx muscle precursor cells in vitro and in vivo mediated by RNA-DNA chimeric oligonucleotides. Hum. Gene Ther. 2002;13:707–718. doi: 10.1089/104303402317322276. doi:10.1089/104303402317322276. [DOI] [PubMed] [Google Scholar]
- 50.Kayali R., Bury F., Ballard M., Bertoni C. Site directed gene repair of the dystrophin gene mediated by PNA-ssODNs. Hum. Mol. Genet. 2010;19:3266–3281. doi: 10.1093/hmg/ddq235. doi:10.1093/hmg/ddq235. [DOI] [PubMed] [Google Scholar]
- 51.Briguet A., Courdier-Fruh I., Foster M., Meier T., Magyar J.P. Histological parameters for the quantitative assessment of muscular dystrophy in the mdx-mouse. Neuromuscul. Disord. 2004;14:675–682. doi: 10.1016/j.nmd.2004.06.008. doi:10.1016/j.nmd.2004.06.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.