

Abstract
We studied the mechanisms and sites of activator actions of 2-[2-(3,4-dichloro-phenyl)-2,3-dihydro-1H-isoindol-5-ylamino]-nicotinic acid [PD307243 (PD)] and 1,3-bis-(2-hydroxy-5-trifluoromethyl-phenyl)-urea [NS1643 (NS)] on the human ether-ago-go-related gene (hERG) channel expressed in oocytes and COS-7 cells. PD and NS affected hERG in a concentration-dependent manner, reaching a maximal increase in current amplitude by 100% and ≥300% (1-s test pulse to 0 mV), with apparent Kd values of 3 and 20 μM, respectively. Both drugs slowed hERG inactivation. NS additionally shifted the activation curve in the negative direction, accelerated activation, and slowed deactivation. Kinetic model simulations suggested that the activator effects of PD and NS could be largely accounted for by their effects on the hERG gating kinetics. Both drugs worked from outside the cell membrane but their binding sites seemed to be distinctly different. Perturbing the conformation of outer vestibule/external pore entrance (by cysteine substitution at high-impact positions or cysteine side chain modification at intermediate-impact positions) prevented the activator effect of NS but not that of PD. Furthermore, the peptide toxin BeKm-1, which bound to the outer mouth of the hERG channel, suppressed NS effect but potentiated PD effect. We propose that NS is a “gating-modifier”: it binds to the outer vestibule/ pore entrance of hERG and increases current amplitudes by promoting channel activation while retarding inactivation. The activator effect of PD was prevented by external quaternary ammonium cations or dofetilide, which approached the hERG selectivity filter from opposite sides of the membrane and depleted K+ ions in the selectivity filter. We suggest that PD may work as a “pore-modifier” of the hERG channel.
The human ether-a-go-go-related gene encodes the pore-forming component of the rapid delayed rectifier (IKr) channel (Sanguinetti et al., 1995). The hERG/IKr channel is important for action potential repolarization in the heart (Tseng, 2001). Mutations in hERG/IKr have been linked to congenital long QT (LQT2) or short QT syndrome (Brugada et al., 2004). A more common problem is the sensitivity of hERG/IKr channel to a wide range of drugs (Recanatini et al., 2005). Unintentional drug blockade of hERG/IKr is the major cause for the acquired LQT syndrome (aLQT) (Roden, 2004). This problem has prompted the development of “hERG activators” to combat aLQT and perhaps LQT2. To achieve this goal, the hERG activators should be able to increase currents through the channel under a wide range of conditions: channel blockade by different chemicals, as seen in aLQT, and channel malfunction as a result of various mutations, as seen in LQT2. To design such robust hERG activators, knowledge about the mechanisms and sites of actions of available hERG activators is important.
Three major classes of hERG channel activators are available, represented by the following prototypes: RPR260243 (Kang et al., 2005), NS1643 (Casis et al., 2006; Hansen et al., 2006), and PD118057 (Zhou et al., 2005). The mechanism and site of action of RPR260243 have been elucidated recently. RPR260243 slows hERG channel deactivation (Kang et al., 2005). Mutations of specific residues in the S4-S5 linker, cytoplasmic halves of S5 and S6 could attenuate or abolish the activator effect of RPR260243 (Perry et al., 2007). Mapping these positions to the Kv1.2 crystal structure showed that they are close to each other in three-dimensional space, in a region critically involved in the voltage-dependent activation/deactivation gating processes (Long et al., 2005). These observations provided a satisfying explanation for the pharmacological action of RPR260243.
A previous study on NS1643 suggested that this hERG activator mainly increased outward but not inward currents through the channels (Casis et al., 2006). Furthermore, mutations at the external pore entrance and in the pore loop that perturb the inactivation process of the hERG channel can attenuate the activator effect of NS1643. On the other hand, mutations of an aromatic residue (Phe656) on S6 that is critical for hERG blocker binding within the inner cavity (Kamiya et al., 2006) potentiate the activator effect of NS1643 (Casis et al., 2006). It was suggested that NS1643 works mainly by hindering channel inactivation. However, detailed analysis of the effects of NS1643 on hERG gating kinetics has been limited, so that the mechanism and site of its activator action are not entirely clear.
A previous study on PD118057 showed that this drug increased outward currents through the hERG channels with little effect on the voltage-dependence or kinetics of activation and deactivation (Zhou et al., 2005). PD118057 also slowed the rate of channel inactivation at 36°C. A recent study on PD307243, a structure analog of PD118057, showed that this hERG activator could dramatically slow the rate of channel deactivation, to the extent that some of the channels became constitutively active at hyperpolarized voltages (Gordon et al., 2008). The authors also reported that PD307243 greatly slowed the rate of hERG inactivation at depolarized voltages. A combination of these two effects created a transient outward current (Ito)-like channel phenotype for the hERG channel (Gordon et al., 2008). There have been no mutagenesis experiments to probe for the binding site of PD118057 or PD307243 in the hERG channel. The mechanism for the peculiar pharmacological actions of PD307243 is not clear.
We undertook the present study to investigate the mechanisms and sites of activator actions of PD307243 and NS1643 [chemical structures shown in Zhou et al. (2005) and Casis et al. (2006)]. Experiments were designed to systematically address the following questions:
How do these drugs modify the gating kinetics of the hERG channel, and can the drug effects on channel gating account for their activator actions?
Is there a membrane “sidedness” in the activators’ actions? This information is needed for the design of the following experiments to probe for the drug binding sites.
How do perturbations of the outer vestibule and external pore entrance of the hERG channel affect the activator actions of PD307243 and NS1643?
How do pore blockers from outside or inside the cell membrane affect the activator actions of PD307243 and NS1643?
Materials and Methods
Molecular Biology
The hERG1 WT cDNA was a kind gift from Dr. Gail A. Robertson (University of Wisconsin, Madison, WI). The hERG cDNA was subcloned into a vector, pAlterMax, and site-specific cysteine (Cys) substitutions were made using the oligonucleotide-directed method and a commercial kit (Altered Site Mammalian Mutagenesis System; Promega, Madison, WI) as described in our previous publications (Liu et al., 2002). Mutations were confirmed by DNA sequencing. Transcription of cRNAs was performed using the mMessage mMachine kit from Ambion (Austin, TX). The concentrations and quality of cRNA products were examined using cRNA gel densitometry (ChemiImager model 4000; Alpha Innotech, San Leandro, CA) before oocyte injection.
Oocyte Experiments
Oocytes were isolated as described previously (Liu et al., 2002). Each oocyte was microinjected with 40 nl of cRNA solution (total cRNA, 10–18 ng). After cRNA injection, oocytes were incubated in ND96 medium (96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 5 mM HEPES, and 2.5 sodium pyruvate, pH 7.5) supplemented with horse serum (4%) and antibiotics (50 U/ml penicillin and 50 U/ml streptomycin) for 3 to 5 days before experiments. Voltage clamp was performed using the “2-cushion pipette” method (Schreibmayer et al., 1994). Oocytes were superfused or bathed in a low-[Cl] ND96 solution (Cl− ions in ND96 replaced by methanesulfonate to minimize interference from oocyte endogenous chloride currents), and membrane currents were recorded using an oocyte clamp amplifier (model 725B or 725C; Warner Instruments, Hamden, CT). Voltage clamp protocol generation and data acquisition were controlled by pClamp 5.5 via computer and a 12-bit D/A and A/D converter (Molecular Devices, Sunnyvale, CA).
COS-7 Experiments
COS-7 cells were maintained in Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal calf serum (Hyclone, Logan, UT) and penicillin/streptomycin, in a moist 5% CO2 chamber at 36°C. Cells were plated at a subconfluence level the day before transfection. To each 35-mm dish were added cDNAs for hERG (1 μg) and CD8 (0.2 μg), reaching a cDNA molar ratio of 1:0.25. Currents were recorded 24 to 48 h after transfection. Cells were exposed to CD8 monoclonal antibody-coated beads (Dynal Biotech, Lake Success, NY) at room temperature for 10 min and treated with trypsin-EDTA (Invitrogen) at room temperature for 5 min; cell suspension was added to a poly-L-lysine–coated coverslip placed in the cell chamber. Cells decorated with CD8 monoclonal antibody beads were used for patch clamping. The pipette solution contained 115 mM potassium aspartate, 20 mM KCl, 10 mM EGTA, 10 mM ATP (potassium salt), 1 mM MgCl2, and 10 mM HEPES, pH 7.3. Pipette tip resistance was 1.9 to 2.1 MΩ, and 95% of series resistance was compensated. The bath solution was normal Tyrode’s solution: 147 mM NaCl, 4 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 5 mM HEPES, and 5.5 mM dextrose, pH 7.3. Whole-cell patch clamp was performed using an AxoPatch 200B controlled by pClamp10 via DigiData1440A. Methods of data analysis were similar to those described for oocyte experiments.
Guinea Pig Ventricular Myocyte Experiments
Myocytes were isolated from a young adult male guinea pig (375 g) using collagenase digestion followed by gentle mechanical trituration as described previously (Yao et al., 1999). Whole-cell current recording conditions were similar to those described for COS-7 experiments, except (1) during current recording, myocytes were superfused with a nominally sodium- and calcium-free Tyrode’s solution (NaCl and CaCl2 replaced by equimolar choline chloride and MgCl2), and (2) recording temperature was 33–34°C. Guinea pig ventricular myocytes express a sizable IKr as well as a robust slow delayed rectifier current (IKs) (Sanguinetti and Jurkiewicz, 1990). We could not identify reagents or conditions that could selectively suppress IKs without affecting IKr, to allow a direct detection of agonist effects on IKr. Therefore, we relied on the difference in the voltage range of activation between IKr and IKs (Sanguinetti and Jurkiewicz 1990). At 33–34°C, the most suitable voltage range for recording IKr (as a time-dependent outward current largely free of IKs) was −20 to −10 mV. Removing extracellular sodium and calcium ions was necessary because this voltage range overlapped with those of sodium and calcium channel currents.
Data Analysis and Kinetic Model Simulations
The voltage clamp protocols and methods of data analysis used to characterize the effects of PD307243 and NS1643 on voltage-dependence and rate of activation, deactivation, and inactivation of the hERG channel are described in Fig. 1 and legend. Simulations of hERG gating kinetics and drug effects were based on a hERG gating model described previously (Wang et al., 1997) using a computer program, Model-Maker4 (ModelKinetix, Oxfordshire, UK).
Fig. 1.

Characterization of the effects of PD on the hERG channel expressed in oocytes. A, top, superimposed hERG current traces before (black) and after PD application (1, 2, 5, 10, 20, and 50 μM, traces shown in the same colors as those of symbols in the lower panel). The currents were elicited by the voltage clamp protocol diagrammed on top applied once every min. Bottom, time course of changes in hERG current amplitude from the same experiment. Current amplitudes were measured from the time-dependent current during the 0 mV step and normalized by the control current right before the start of PD application. B, concentration-response relationship. Numbers of measurements are shown in parentheses. The superimposed curve was calculated from the equation Id/Ic = 1+ (A − 1)/(1 + Kd/[D]), with A (-fold increase in current amplitude) = 2 and apparent dissociation constant (Kd) =3 μM. In the following panels, data points and current traces recorded under the control conditions are shown in black, and those recorded in the presence of PD (10 μM) are shown in red. C, PD did not shift the 1-s isochronal activation curve of hERG. Inset, voltage-clamp protocol and representative current traces. The relationship between Vt and peak amplitude of tail current (Itail) was fit with a Boltzmann function: Itail = Imax/[1 + exp(V0.5 − Vt)/k], to estimate the maximal peak tail current amplitude (Imax), half-maximal activation voltage (V0.5) and slope factor (k). Itail was normalized by Imax (Itail/Imax = fraction activated) and plotted against Vt. D, PD increased hERG test pulse currents in a voltage-dependent manner. The voltage clamp protocol was the same as that described for C. For each oocyte, amplitudes of time-dependent currents during 1-s test pulses to Vt were normalized by the control current at Vt −10 mV. The current ratios, Id/Ic, are also plotted based on the right coordinate. E, PD did not alter the time course of hERG activation. Inset, “envelope” voltage clamp protocol and representative current traces. The rate of hERG activation was slower than inactivation. Thus the time course of activation could not be directly measured from the test pulse currents. Because the outward tail currents signified channels recovered from inactivation, the time course of growth of peak tail current amplitude after increasing durations of depolarizing pulses could be used to infer the time course of channel activation. The membrane voltage was stepped to Vt for durations varying from 5 to 1000 ms in 50-ms increments. The time course of growth of peak tail current amplitude was fit with a single exponential function to generate τactivation (plotted on a logarithmic scale so that the slope indicates the voltage-sensitivity of the underlying gating process). F, PD did not alter the time course of hERG deactivation. Inset, voltage-clamp protocol. Tail currents during 2- to 5-s step to Vr were fit with a double exponential function to generate the fast and slow time constants of deactivation (τfast and τslow, right and left coordinates, respectively). In Vr range ≤ − 100 mV, channel deactivation was almost exclusively the fast component; thus τslow values are not shown. G, PD modestly slowed the rate of hERG inactivation. Inset, triple-pulse voltage-clamp protocol and representative current traces during the third step (indicated by gray shade in the protocol diagram). The blue trace represents control current trace scaled to match the amplitude of current in PD. hERG inactivation was faster than activation. To bypass the slow activation process, the hERG channels were first fully activated and inactivated by a 1-s step to +60 mV. This was followed by a brief 15-ms step to −80 mV, during which the channels recovered from inactivation without appreciable deactivation. The third depolarizing step to various Vt caused channel transition from open to inactivated states. The decay of current traces during the third pulse was fit with a single exponential function to generate time constant of inactivation (τinactivation). The rate of inactivation (reciprocal of τinactivation, left coordinate, logarithmic scale) was plotted against Vt. The relationship between the rate of inactivation and Vt in the range of 0 to + 60 mV was fit with the equation K(Vt) = K(0)exp(zinaVtF/RT), where K(Vt) and K(0) are rates of inactivation at Vt and 0 mV, respectively; zina is the equivalent gating charge of inactivation; and F, R, and T are the Faraday constant, the gas constant, and absolute temperature, respectively. The lines superimposed on the data points were calculated from this equation, with K(0) and zina values of 0.23 ms −1and 0.72 for control, 0.12 ms−1 and 0.71 for PD. * p < 0.05, PD versus control. H, PD increased both outward and inward currents through the hERG channel. The voltage clamp protocol was the same as that diagrammed in F. For each cell, current amplitudes were measured from the peak or plateau level of tail currents (defined as fully activated current, or If-a) and normalized by the control If-a at −50 mV.
Drug Solutions and Application
PD307243 and NS1643 were generous gifts from Dr. Xiaoping Xu (GlaxoSmithKline, King of Prussia, PA). Their stock solutions (10 mM in DMSO) were divided into 100-μl aliquots and stored at −20°C until experiments. On the day of experiments, an aliquot was thawed, kept on ice, and protected from light during the course of the experiments. In early experiments, we applied these drugs using the superfusion (flow-bath) method. The stock solutions were added to the low-[Cl] ND96 to reach desired final concentrations and the oocytes were super-fused with the drug-containing solutions. In most of the experiments reported here, we applied the drugs using a “nonflow” bath method. The oocytes were placed in 0.8 ml of low-[Cl] ND96 without super-fusion and impaled with two electrodes. After obtaining the control data, PD307243 or NS1643 stock solution was mixed with 0.2 ml of low-[Cl] ND96 (to reach the desired final concentration when diluted 5-fold) and added to the bath. The solution was mixed by gently pipetting the bath solution up and down. When using the nonflow bath method, the agar salt bridges connecting the 3 M KCl/silver/ AgCl grounding electrodes to the bath solution were made in 2 mM potassium to avoid leakage of K+ ions into the bath solution. There were no discernible differences in results from these two methods of drug application and the data were pooled.
Other drugs used in the experiments included: BeKm-1 [recombinant form; Alomone Labs (Jerusalem, Israel), 200 μM stock in ND96 containing 0.5% bovine serum albumin, diluted to 10 nM final concentration by adding 50 nl of the stock to 1 ml of bath solution containing 0.5% bovine serum albumin, which prevented peptide from sticking to plastic surface], tetrapentylammonium chloride (1 M stock in water; final concentration, 50 μM; Sigma-Aldrich, St. Louis, MO) and dofetilide (1 mM stock in water, pH 3; final concentration, 0.5 μM; a gift from Pfizer, New York, NY).
In the experiments shown in Fig. 5, PD307243, NS1643, and TEA were injected into oocytes during the course of recording. The micro-dispenser injecting device was the same as that used for cRNA injection. Assuming an oocyte volume of 0.5 μl (sphere with 1-mm diameter), the following stock solutions and injection volumes were used to reach the estimated final cytoplasmic concentrations: PD307243, 15 nl of 10 mM stock to reach 300 μM; NS1643, 20 nl of 25 mM stock to reach 1000 μM; and TEA, 20 nl of 50 mM stock to reach 2 mM.
Fig. 5.

Both PD307243 and NS1643 worked from outside the cell membrane, yet their effects were independent of each other. A, left, time course of changes in hERG current amplitude expressed in a COS-7 cell M), and total before, during, and after bath application of PD (PDo, 10 μ current suppression by dofetilide (DOF). Currents were elicited by the voltage-clamp protocol diagrammed on top of the right panel. Current amplitudes were measured from the peak of tail current at −60 mV and normalized by the control right before PD application. The time course of onset of PD effect was fit with a single exponential function with τ value of 6 min. Right, superimposed current traces from the same experiment at time points a, b, and c. B and C, left, time course of changes in hERG current amplitude expressed in oocytes before and after intraoocyte drug injection (estimated cytoplasmic concentrations denoted: PDi, ~ 300 μM; and NSi, ~1000 μM), during exposure to drugs applied to the bath solution (PDo, 10 μM; NSo, 30 μM) and after intraoocyte injection of TEA (TEAi, ~ 2 mM). In each experiment, the same injecting pipette was used first for PD or NS injection and then for TEA injection. Right, superimposed current traces from the same experiments as in left at the specified time points. Inset of right in B, percentage increase in hERG current amplitude caused by 10 μM PD tested in COS-7 cells and in oocytes measured from the peak amplitudes of outward tail currents at −60 mV (66 ± 10 and 61 ± 8, n = 4 and 20, p > 0.05). The same conclusion could be reached by measuring time-dependent test pulse currents at +40 mV: PD increased the current by 187 ± 67% and 185 ± 16% in COS-7 cells and in oocytes, respectively. D, left, time course of changes in hERG current amplitude before and during bath application of 20 μM PD and after adding NS 30 μM in the continuous presence of PD. Right, percentage increase in current amplitude caused by NS in the absence (−PD) or presence of PD (+PD).
Chemical Analysis of PD307243
Because the effects of PD307243 on hERG reported here differed markedly from those reported previously (Gordon et al., 2008), we analyzed the chemical structure of PD307243 used in our experiments. 1H-NMR and 13C-NMR spectra were recorded at 300 and 75 MHz, respectively. Electronspray ionization mass spectrum was performed on a Waters MZQ Micromass ZQ 4000 (Waters, Milford, MA). High-resolution mass spectrum (HRMS) was recorded on a Mat 95XP (Thermo Fisher Scientific, Waltham, MA). Results are presented as Supplemental Data.
Results
PD307243 and NS1643 Exerted “Activator” Effects on the hERG Channel and Altered Channel Gating Kinetics
Figs. 1 and 2 illustrate detailed characterization of the effects of PD307243 and NS1643 (abbreviated as PD and NS in the following text) on the hERG channel expressed in oocytes. Both drugs increased the hERG current amplitude in a concentration-dependent manner (Figs. 1A and 2A). Such activator effects were seen in both outward and inward currents (Figs. 1H and 2H). In most experiments, we used the test pulse current amplitude at 0 mV to quantify the activator effects (except data in Figs. 5–7, noted in the figures or legends). We wished to compare the apparent binding affinity and potency of the activator effect between PD and NS. Assuming that both activators bound to the hERG channel in a 1:1 stoichiometry and activator binding increased the current amplitude to “A” fold of control, the drug-channel interaction could be described by Scheme 1.
Fig. 2.

Characterization of the effects of NS on the hERG channel expressed in oocytes. Voltage clamp protocols and methods of data analysis are the same as those described for corresponding panels in Fig. 1. [NS] for experiments shown in C to H was 30 μM. A, time course of changes in hERG current amplitude before and during exposure to NS at the specified concentrations. B, concentration-response relationship. The superimposed curve was calculated from the equation Id/Ic =1+ (A − 1)/(1 + Kd/[D]) with A (-fold increase in current amplitude) = 3.24 and Kd = 20 μM. C, NS shifted the 1-s isochronal activation curve of hERG in the negative direction by −9.8 ± 1.4 mV (n = 7). D, NS increased hERG test pulse current in a voltage-dependent manner. E, NS accelerated hERG activation (shortened τactivation). F, NS slowed hERG deactivation, manifested as a prolongation of τfast and τslow. G, NS slowed hERG inactivation. The lines superimposed on the data points were calculated from K(Vt) = K(0)exp(zinaVtF/RT) (described for Fig. 1G), with values of K(0) and zina of 0.02 ms−1and 0.6 for control and 0.01 ms−1 and 0.5 for PD, respectively. H, NS increased both outward and inward currents through the hERG channel. * p < 0.05, NS versus control.
Fig. 7.

Modifications of Cys side chains introduced into 2 pore entrance positions differentially affected the “activator” effects of PD307243 and NS1643. Currents were elicited by different voltage clamp protocols to 1) illustrate their gating behavior as WT-like or mutant and 2) probe for drug effects on both outward and inward currents through the channels. For the sake of simplicity, individual voltage-clamp protocols are not shown; only the step voltages are noted (holding voltage was −80 mV in all cases). A, T613C manifested WT-like gating behavior under the control conditions (gray trace), and switched to the mutant behavior after MTSET modification (black trace). B, S631C manifested WT-like behavior (gray trace) when DTT treated, but switched to mutant behavior (black trace) after H2O2 treatment with pulsing. C and D, PD was effective in increasing currents through the T613C and S631C channels in both WT-like and mutant behavior. On the other hand, NS was effective in increasing currents through the T613C and S631C channels in the WT-like mode but incapable of increasing currents when the channels were in the mutant mode.
Scheme 1.
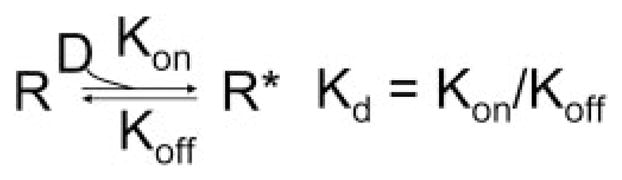
R and R* represent fractions of channels in activator-free and activator-bound states, D is activator drug, Kon and Koff are binding and unbinding rate constants, and Kd is dissociation constant.
At equilibrium in the presence of activator concentration [D], R = Kd/([D] + Kd), R* = [D]/([D] + Kd). The ratio of current amplitude in the presence of activator (Id) to control (Ic) will be: Id/Ic = R + (R*A). This leads to the following equation describing the relationship between Id/Ic and [D]: Id/Ic = 1+ (A − 1)/(1 + Kd/[D]).
Because PD had a secondary current-suppressing effect on the hERG channel (that became more apparent at concentrations ≥20 μM, or in the presence of pore blockers; see Fig. 9), we fit the concentration-response relationship for 1 to 10 μM PD. The A value was 2, and the Kd value was 3 μM (Fig. 1B). In some oocytes, NS at concentrations ≥60 μM induced a membrane leak conductance (thus the large standard error bars at 60 and 90 μM in Fig. 2B). Nevertheless, we fit the complete concentration-response relationship of NS (3–90 μM) with the equation above, and obtained A and Kd values of 3.24 and 20 μM, respectively. In the following experiments, we used 10 or 20 μM PD and 30 μM NS (i.e., concentrations that could induce significant effects on the hERG current amplitude without appreciable secondary current-suppressing or leak-inducing effects).
Fig. 9.

An outer pore blocker, TPeA+, or an inner cavity blocker, dofetilide (DOF) prevented the activator effect of PD307243 on hERG. A, cartoon of a TPeA+ ion binding to the outer pore entrance of hERG. B, time course of changes in hERG current amplitude before and after application of 50 μM TPeA+ and after application of increasing concentrations of PD in the continuous presence of TPeA+. Current amplitudes were measured from time-dependent currents during 1-s step to 0 mV and normalized by control. Inset: summary of PD (20 μM)-induced changes in hERG current amplitude in the absence or presence of TPeA+. C and D, the format is the same as that in A and B. DOF binds from the intracellular side of the membrane to the inner cavity.
Both activators slowed the inactivation rate of the hERG channel to ~50% of control without altering its voltage sensitivity (Figs. 1G and 2G). However, the two differed in how they influenced the activation/deactivation gating processes of the hERG channel. PD did not shift the voltage-dependence of activation (measured by the 1-s isochronal activation curve; Fig. 1C). It had no effect on the rate of hERG activation (measured by the “envelope” voltage-clamp protocol shown in Fig. 1E), or the time course of deactivation (quantified by double-exponential curve fitting; Fig. 1F). On the other hand, NS consistently shifted the 1-s isochronal activation curve in the negative direction (by ~10 mV) and accelerated hERG activation (Fig. 2, C and E). NS also slowed the rate of hERG deactivation, which manifested as a prolongation of the fast and slow time constants (Fig. 2F), although the distribution of the 2 components at different voltages was not affected (data not shown). PD and NS also differed in the voltage-dependence of their activator effects on the test pulse current (measured by current ratio or Id/Ic). Although the value of Id/Ic for PD showed an increase with stronger depolarization (Fig. 1D), the opposite was seen with NS (Fig. 2D).
Could the Activator Effects of PD307243 and NS1643 Be Accounted for by Their Effects on hERG Channel Gating Kinetics?
We used a kinetic model for hERG channel gating described previously (Wang et al., 1997) to test whether the activator effect of PD on the hERG channel could be accounted for by the drug-induced slowing of inactivation. The kinetic model and parameter values used in the simulations are described in Scheme 2 and Table 1, respectively. The only change we made for PD was a 50% decrease in the forward rate of channel inactivation, in accordance with the experimental data shown in Fig. 1: PD reduced the rate of hERG inactivation by 50% without changing its voltage sensitivity or parameter values related to the activation/deactivation gating processes. The simulation results are shown in Fig. 3A. The model could qualitatively mimic the effects of PD on hERG test pulse currents (a and c), tail currents (a) and rate of inactivation (d). There was no shift in the voltage-dependence of activation (b).
Scheme 2.
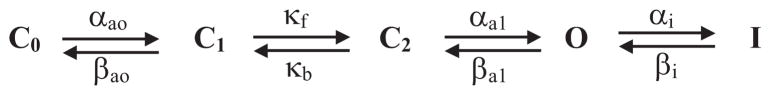
The kinetic model of hERG gating was based on that of Wang et al (1997). Cx, O, and I represent closed states, open state, and inactivated state, respectively. The transitions between Co and C1, C2 and O, and O and I are voltage-dependent, with rate constants calculated as shown: αa0(V) = αao(0)*exp(zaoVF/RT); βa0(V) = βao(0)*exp(zboVF/RT); αa1(V) = αa1(0)*exp(za1VF/RT); βa1(V) = βa1(0)*exp(zb1VF/RT); αi(V) = αi(0)*exp(zaiVF/RT); and βi(V) = βi(0)*exp(zbiVF/RT). αx(V) and αx(0) are forward (or rightward according to the model) rate constants at voltages of V and 0 mV, respectively, and zax is the equivalent gating charge involved in the gating transition. βx(V), βx(0), and zbx are the corresponding parameters for the backward or leftward transitions. The transition between C1 and C2 is voltage-independent, with forward and backward rate constants of κf and κb, respectively. F, R, and T, are the Faraday constant, the gas constant, and absolute temperature, respectively.
TABLE 1.
Parameter values used in kinetic model simulation of the effects of PD(307243) and NS(1643) on the hERG channel
| Parameter | Control | PD | NS |
|---|---|---|---|
| αao(0) (s −1) | 22.3 | 22.3 | 22.3 |
| zao | 0.294 | 0.294 | 0.294 |
| βoa(0) (s −1) | 0.047 | 0.047 | 0.047 |
| zbo | −1.577 | −1.577 | −1.577 |
| κf (s−1) | 23.8 | 23.8 | 23.8 |
| κb (s−1) | 36.8 | 36.8 | 36.8 |
| αa1(0) (s−1) | 13.7 | 13.7 | 54.9 |
| za1 | 0.955 | 0.955 | 0.955 |
| βa1(0) (s−1) | 0.069 | 0.069 | 0.017 |
| zb1 | −1.045 | −1.045 | −1.045 |
| αi(0) (s−1) | 90.8 | 45.4 | 45.4 |
| zai | 0.585 | 0.585 | 0.585 |
| βi(0) (s−1) | 6.5 | 6.5 | 6.5 |
| zbi | −0.817 | −0.817 | −0.817 |
See Scheme 2 for kinetic model. The only change in parameter values for PD simulation was a 50% reduction in the “O” to “I” transition rate (highlighted in bold). The changes in parameter values for NS simulation included: 50% reduction in the “O” to “I” transition rate, a 4-fold increase in the “C2” to “O” transition rate, and a 75% reduction in “O” to “C2” transition rate (highlighted in bold).
Fig. 3.

Model simulation of the effects of PD307243 (A) and NS1643 (B) on the hERG channel. The model and parameter values are listed in Table1. Simulated current traces and data points for control, PD and NS are shown in black, red and blue, respectively. For each column, a and d show superimposed simulated current traces elicited by the voltage clamp protocols diagrammed in the insets (time scale was the same as protocols shown in Fig. 1), b depicts simulated 1-s isochronal activation curves, and c illustrates simulated test pulse current-voltage relationship (open symbols, left coordinate) and current ratio (Id/Ic, right coordinate). Inset of A, a, shows horizontally shifted inward tail currents at −120 mV, to illustrate the difference in the peak current amplitude.
To check whether we could simulate the NS effects on the hERG channel, we made the following changes in the model parameters: 1) forward rate constant of inactivation reduced by 50%, based on the slowing effect of NS on the rate of inactivation without altering the voltage-sensitivity; 2) forward rate constant from C2 to O states increased to 4-fold; and 3) backward rate constant from O to C2 decreased by 75%. We chose to modify the transition rates between the O state and the immediate C2 state based on the experimental data shown in Fig. 2F. The time course of hERG channel deactivation could be described by a double exponential function in the Vr range > −100 mV, but mainly a single (fast) exponential function in more negative Vr range. The probable explanation for this kinetic behavior is that the fast deactivation component is mainly due to the backward transition from the O state to the immediate closed state, whereas the slow deactivation component is due to the residual forward transitions between closed states in the less negative voltage range. Therefore, to be consistent with the finding that NS significantly slowed the fast component of deactivation even at −120 mV, we altered the transition between O and C2 states. The amount of changes described above was needed to reproduce the negative shift in the activation curve by −10 mV (Fig. 2C). The results of simulation of the NS effects are shown in Fig. 3B. The model qualitatively mimicked the effects of NS on the test pulse current (a and c), tail current (a), rate of inactivation (d), and voltage-dependence of activation (b).
Figure 4 shows the concentration-response relationships of PD and NS on the hERG channel. The data support the above notion that the agonist effect of PD was accompanied mainly by a slowing of hERG inactivation, whereas that of NS had contribution from both a slowing of hERG inactivation and a stabilization of the channel in the activated versus closed states (denoted by the negative shift in V0.5 of activation). Model prediction of the agonist effects of PD and NS seemed less than those observed experimentally (Figs. 1 and 2). Prolonged whole-cell recording was accompanied by a gradual run-up of the hERG current amplitudes (Supplemental Fig. S1) that could cause an overestimation of the agonist effects.
Fig. 4.

Concentration-response relationships of PD307243 (A) and NS1643 (B) on hERG current amplitude and gating kinetics. ΔAMP, percentage increase in current amplitude measured as time-dependent current during 1-s step to 0 mV; Δ V0.5, shift in V0.5 of activation; ΔRateINAC, percentage decrease in rate of channel inactivation at 0 mV. Data were averaged from four to six measurements each.
The model-predicted relationship between Vt and Id/Ic provided an explanation for the experimental findings of opposite trends for PD versus NS (Figs. 1D and 2D). For the PD simulation, the values of Id/Ic gradually increased as the Vt became more depolarized, and reached a plateau at Vt ≥ +40 mV, because the only change we introduced for PD in the hERG gating model was a reduction in the forward transition rate from O to I states, which was voltage-sensitive. As Vt became more depolarized, the rate of inactivation became a more dominating factor in determining the current amplitude, and thus slowing it would cause a higher percentage of increase in the current amplitude. For the NS simulation, the Id/Ic value was highest at the threshold depolarization voltage and declined steadily as Vt became more depolarized, approaching a steady level at Vt ≥ +20 mV. This was because NS caused a more prominent increase in channel open probability at more negative Vt (but above the threshold of activation, b). This trend contributed to the higher Id/Ic values from −60 to +20 mV. At more positive Vt, slowing of inactivation was the main factor for the increase in current amplitude, as in PD simulation, and Id/Ic approached a similar value of ~2.
Both PD307243 and NS1643 Acted on the hERG Channel from the Extracellular Side of the Cell Membrane but Their Effects Were Additive
A prerequisite for the search of a drug’s binding site in a membrane channel is to know whether there is “membrane sidedness” in the drug’s actions (i.e., whether the drug is effective only when applied from inside or outside the cell membrane). Once this information is available, proper strategies can be designed (e.g., site-specific mutations, pharmacological or ionic perturbations) to gain further insights into the channel domain(s) where the drug may bind. Figures 1A and 2A show that the effects of PD and NS [applied by directly adding the drug solution to the bath solution followed by pipetting up and down to mix the two (i.e., the “nonflow” bath); see Materials and Methods] had very rapid onset, and the activator effects could be seen in the low micromolar concentration range. It is noteworthy that the potency of PD tested in oocytes was not different from that of PD tested in mammalian COS-7 cells (Figs. 5, A versus B). If these activators worked on the hERG channel from the intracellular side of the cell membrane, the yolk inside oocytes would provide a huge sink for the lipophilic drug molecules, leading to a decrease in the apparent drug potency and a delay in the onset of drug effects, as is the case for quinidine (Yeola et al., 1996; Zhang et al., 1998). Because COS-7 cells do not have yolk, we expected to see much higher drug potency when tested in the mammalian cells versus in oocytes. To check whether PD and NS worked on the hERG channel from the extracellular side of the cell membrane, we directly compared the potency of these drugs applied by intraoocyte injection versus by bath application. Figure 5, B and C, shows that intraoocyte injection of a high concentration of PD (300 μM) or NS (1000 μM) had no discernible effect on the hERG current amplitude or gating kinetics. However, subsequent bath application of a 30-fold lower concentration of PD (10 μM) or NS (30 μM) effectively increased the hERG current amplitude. To confirm the effectiveness of our intraoocyte injection technique, in both experiments we pulled out the injecting pipettes, expelled the remaining drug solutions in the pipette tips, refilled the pipette tips with a TEA solution, reimpaled the oocytes and injected ~20 nl of TEA solution into the oocytes (comparable with the volume of drug solution injected). In both cases, intraoocyte injection of TEA to reach an estimated cytoplasmic concentration of 2 mM effectively suppressed the hERG current. These observations confirmed that both PD and NS worked on the hERG channel from the extracellular side of the cell membrane. The apparent slow onset of drug effects shown in Fig. 5, A–C, was due to the slow superfusion rate in these experiments to conserve drugs.
We further tested whether the effects of PD and NS were related to each other in binding site or mechanism of action. In these experiments, PD 20 μM was applied to the hERG channel expressed in oocytes using direct pipetting. After PD effect stabilized, NS 30 μM was applied in the continuous presence of PD. NS effects were compared with those measured in the absence of PD. Figure 5D shows that NS was equally effective in increasing hERG current without or with PD, suggesting that the binding sites and mechanisms of action of these two hERG activators are independent of each other.
Cys Substitutions Introduced into the Outer Vestibule or the External Pore Entrance of the hERG Channel, or Modifications of the Engineered Cys Side Chains, Differentially Affected the Activator Actions of PD307243 and NS1643
Because both PD and NS worked from the extracellular side of the cell membrane and both slowed the rate of hERG inactivation, our search for the drug binding sites targeted the outer vestibule region and the pore entrance. It has been proposed that the hERG channel’s inactivation process is similar to the P-type inactivation described for the Shaker or Shaker-like channels (López-Barneo et al., 1993; Smith et al., 1996; Fan et al., 1999). The P-type inactivation is believed to arise from conformational changes around the outer mouth and in the pore-loop that alter ion selectivity or conductance of the selectivity filter and shut down K+ ion flux through the pore. We have shown that the unusually long (~43 amino acids) extracellular S5-P linker in the hERG channel also plays a critical role in the channel’s inactivation process (Liu et al., 2002). Mutations introduced into specific “high-impact” positions along the S5-P linker can disrupt the inactivation process (Dun et al., 1999; Liu et al., 2002). The four S5-P linkers in a tetrameric hERG channel, along with the P-S6 linkers, line the outer vestibule of the channel. Because mutations along the S5-P linker that disrupted inactivation also impaired the hERG channel’s ability to select for K+ ions (Dun et al., 1999; Liu et al., 2002), the linker must interact with the pore loop in 3-D space. Indeed, our recent 3-D model for hERG’s pore domain, based on peptide toxin foot-printing experiments and homology modeling, suggested that side chains at several “high-impact” positions could make contact with pore-loop residues (Tseng et al., 2007). We have performed a detailed Cys-scanning mutagenesis study of the S5-P linker in the hERG channel, and we identified not only high-impact positions but also “intermediate-impact” positions (Liu et al., 2002). Cys substitution per se at the intermediate-impact positions does not disrupt the inactivation process or the pore’s K+ selectivity. However, modification of Cys side chains at these positions markedly affected channel function. Furthermore, our data suggested that the middle segment of the S5-P linker (positions 583–597) might adopt an α-helical structure. This was confirmed by subsequent experiments using NMR spectroscopy (Torres et al., 2003; Jiang et al., 2005). It is noteworthy that Cys side chains engineered into most positions along this “S5-P helix” could form intersubunit disulfide bonds (Jiang et al., 2005). We further showed that Cys side chain introduced into position 584, 585, 588, and 589 on the S5-P helix could form intersubunit disulfide bond with counterparts from adjacent subunits (Jiang et al., 2005). This may also be the case for the Cys side chain introduced into position 631 at the external pore entrance. In all cases, di-sulfide bond formation disrupted the inactivation process and the K+ selectivity.
We took advantage of the available information and mutant channels to address the following question: how do Cys side chains introduced into the S5-P linker and the pore entrance positions, or a modification of these engineered Cys side chains, affect the activator effects of PD and NS? We selected eight Cys substituted mutants that covered the region of interest (diagrammed in Fig. 6A). In all cases, oocytes were treated with DTT to reduce spontaneously formed di-sulfide bonds. In cases when disulfide bonds were needed to create the mutant phenotype (G584C, H587C, and S631C), oocytes were treated with H2O2 (0.3%) in conjunction with depolarizing pulses (or incubated in elevated [K+]o, 20 mM) to facilitate intersubunit disulfide bond formation. The mutant phenotype was identified by a lack of bell-shaped current-voltage relationships (as shown in Fig. 1, D and H) as a result of a disruption of the inactivation process. Furthermore, tail currents recorded at −60 or −80 mV turned in the inward direction as a result of a loss of K+ selectivity (so that extracellular Na+ ions carried inward tail currents at these voltages). To guard against artifacts generated by interference from oocyte endogenous currents, we confirmed that 1) currents in DTT-treated oocytes that had been injected with cRNAs for the Cys-substituted mutants were significantly larger than currents in uninjected oocytes, and (2) PD at 20 μM had no noticeable effects on currents in uninjected oocytes (data not shown).
Fig. 6.

Summary of effects of Cys substitution or Cys side chain modification introduced into the outer vestibule or pore entrance of the hERG channel on the activator action of PD307243 and NS1643. A, top, two-dimensional diagram of a hERG subunit, marking S1 to S6 transmembrane helices, pore loop, and the extracellular S5-P helix. Bottom, amino acid sequence of the S5-P linker, marking the positions that participate in forming the S5-P helix. High- and intermediate-impact positions are highlighted by white lettering on black shade and by gray shade, respectively. The positions examined in Figs. 6 and 7 are noted below the sequence. Thr613 and Ser631 are marked in the two-dimensional diagram. B and C, percentage change in current amplitude measured at 0 mV (WT or WT-like) or +60 mV (mutant) induced by 20 μM PD or 30 μM NS plotted against the channel types along the abscissa in C, where Cys side chains modified by di-sulfide bonding (-S-S-) or MTSET modification (+MTS) are noted. Hatched histogram bars for WT or WT-like channel phenotype are shown in white background, whereas those for mutant phenotype are shown in black background (C, inset). D, superimposed current traces recorded from oocytes expressing the specified hERG mutants. Currents were elicited by the voltage clamp protocol diagrammed on top under three conditions: control (light gray traces), after adding 30 μM NS (dark gray traces), and after adding 20 μM PD in the continuous presence of NS (black traces).
We quantified drug effects on current amplitudes through channels with the mutant phenotype from test pulse currents measured at +60 mV, the highest amounts of outward current in the voltage range tested. PD (20 μM) or 30 μM NS was applied to the bath solution by direct pipetting. Drug effects were evaluated in a yes-or-no fashion. We did not attempt to quantitatively compare the degrees of drugs’ activator effects among the channels. Figure 6B shows that PD was effective in increasing currents through all the channels in either the WT-like or the mutant mode. Therefore, conformational changes in the outer vestibule or the pore entrance did not prevent the activator effect of PD. This is the case even with intersubunit disulfide bond formation in G584C, H587C, or S631C, which was expected to alter the peptide backbone conformation and flexibility. On the other hand, although NS was effective in increasing currents through the channels when they were in the WT-like gating mode, the drug could not induce the activator effect when the channels were in the mutant mode (Fig. 6C).
Original data are shown in Figs. 6D and 7 to support the above conclusions. Figure 6D illustrates representative current traces recorded from G572C, G584C (disulfide bonded), W585C, H587C (disulfide bonded), and N588C. NS (30 μM) did not induce the activator effect (and a modest current-suppressing effect on W585C was revealed). In the continuous presence of NS, 20 μM PD effectively increased outward current at +60 mV as well as inward current at −120 mV. Figure 7 illustrates the impact of modifying Cys side chains introduced into the external pore entrance, T613C and S631C. Cys substitution per se (in the free thiol state) at these 2 positions did not affect the channel function: they were WT-like in showing strong inactivation and K+ selectivity. However, MTSET modification of T613C impaired the channel’s ability to inactivate or to select for K+ ions (Fig. 7A). Treating S631C with 0.3% H2O2 while depolarizing the membrane to cause channel activation and inactivation effectively converted the S631C channel to the mutant phenotype: impaired inactivation and K+ selectivity (Fig. 7B) (Fan et al., 1999). PD (20 μM) effectively increased both outward currents and inward currents through the T613C and S631C channels in both WT-like and mutant modes (Fig. 7, C and D, left columns). On the other hand, although 30 μM NS was effective in increasing currents through the two channels when they were in the WT-like mode, it could not induce any activator effect when the channels were in the mutant mode (Fig. 7, C and D, right columns). Note that (2-triethylammonium) ethylmethane thiosulfonate, DTT, or H2O2 had no effect on the gating kinetics or K+ selectivity of WT hERG (Fan et al., 1999). DTT or H2O2 treatment did not impair the agonist effect of NS on WT hERG (data not shown).
Using External and Internal hERG Pore Blockers to Further Probe the Sites for the Activator Actions of PD307243 and NS1643
We have shown that a scorpion peptide toxin, BeKm-1, binds to the outer vestibule of the hERG channel to suppress its currents (Fig. 8A) (Zhang et al., 2003). We further identified the following hERG residues that are likely to make direct contacts with the bound BeKm-1 peptide: Arg582, Ile583, Tyr597 (at the two ends of the S5-P helix), and Ser631 (at the pore entrance) (Tseng et al., 2007). We hypothesized that BeKm-1 binding might differentially affect the binding and/or action of PD and NS. Indeed, Fig. 8, B to D, shows that BeKm-1 potentiated PD’s activator effect but suppressed NS’s activator effect.
Fig. 8.

BeKm-1 potentiated the activator effect of PD307243 but suppressed the activator effect of NS1643. A, cartoon of hERG pore domain with an extracellular BeKm-1 structure. Three hERG subunits are depicted, with the fourth one closest to viewers removed to reveal the narrow selectivity filter and wide inner cavity of the pore. Cylinders represent the S5-P helices. B, main plot, two time courses of changes in hERG current amplitude before and after exposure to 20 μM PD. Current amplitudes were measured from time-dependent currents during 1-s steps to 0 mV and normalized by the current right before PD application. One experiment (open symbols) was conducted in the absence of BeKm-1. The other (black symbols) was conducted in the presence of 10 μM BeKm-1. Inset, the complete time course of the second experiment. BeKm-1 suppressed the current by 80% and subsequent application of PD in the continuous presence of BeKm-1 effectively increased the current amplitude. C, NS (30 μM) effect on hERG in the absence or presence of BeKm-1. The format is the same as that of B. D, data summary. *, p < 0.05, data obtained in the presence of BeKm-1 versus those without BeKm-1.
Positively charged quaternary ammonium cations (QA+) have been used as a tool to probe the ion conduction pathway of potassium channels (Ahern et al., 2006). In the Shaker or Shaker-like channels, QA+ applied from outside the cell membrane binds to the external pore entrance to occlude currents through the pore. Although extracellular TEA has a low binding affinity to hERG (50% reduction at ~50 mM) (Pardo-Lopez et al., 2002), lengthening the hydrocarbon chain length greatly increased the binding affinity, so that 50 μM tetrapentyl ammonium cation (TPeA+) could suppress the current by ≥80% (Fig. 9A). TPeA+ effectively prevented the activator effect of PD. Instead, TPeA+ unmasked a concentration-dependent current suppressing effect of PD (Fig. 9B).
Dofetilide binds to the hERG channel from the intracellular side of the cell membrane (Kamiya et al., 2006). An Ala-scanning mutagenesis approach has been used to probe the dofetilide binding site in the hERG channel (Kamiya et al., 2006): Y652A, F656A on the S6 helix, T623A, S624A, and V625A along the selectivity filter of the pore-loop, as well as Gly648 (on S6, juxtaposed to the pore-loop) greatly attenuated the dofetilide binding affinity. Therefore the dofetilide binding site is likely formed with S6 helices and roofed by the pore-loop (cartoon in Fig. 9C). Applying dofetilide 0.5 μM to hERG expressed in oocytes in conjunction with constant pulsing to cause channel activation led to a gradual decrease in current amplitude, reaching a suppression of ≥80% in 15 to 20 min (Fig. 9D). Under these conditions, dofetilide effectively prevented the activator effect of PD, and unmasked its current suppressing effect. Thus, although PD’s activator effect on the hERG channel could not be disrupted by interventions applied to the extracellular S5-P linker or the external pore entrance, it was prevented by blockers that could reduce K+ ion occupancy in the selectivity filter of the hERG channel (more in Discussion).
Discussion
The major findings in this study can be summarized as follows. The concentration-response relationships of PD and NS on the hERG channel could be described by a 1:1 binding stoichiometry, the maximal effect being 100% and ≥300% increases, respectively, in the current amplitude (measured as time-dependent current during 1-s step to 0 mV), with apparent Kd values of 3 and 20 μM, respectively. The activator effect of PD was accompanied by a slowing of hERG inactivation. On the other hand, the activator effect of NS involved not only a slowing of hERG inactivation but also an acceleration of channel activation as well as retardation of channel deactivation. Both drugs worked on the hERG channel from the extracellular side of the cell membrane. Inactivation-disrupting mutations or side chain modifications introduced into the outer vestibule or pore entrance of the hERG channel abolished the activator effect of NS but did not prevent the activator effect of PD. BeKm-1, a peptide toxin that bound to the outer vestibule of the hERG channel (Tseng et al., 2007) greatly attenuated the activator effect of NS but potentiated the activator effect of PD. Finally, the activator effect of PD was prevented by an external pore blocker, tetrapentylammonium (Ahern et al., 2006), or by dofetilide, which is known to bind to the inner cavity of the hERG channel (Kamiya et al., 2006).
Binding Site and Mechanism of Action of NS1643
Our data support the notion that the NS binding site is formed by the outer vestibule and pore entrance of the hERG channel. The positions at which Cys substitution or side-chain modification abolished NS’s activator effects (e.g., 572 at the extracellular end of S5, positions along the S5-P helix between 584 and 593, 613 and 631 at the external pore entrance) may be close to each other in three-dimensional space (Supplemental Fig. S2). Our previous studies suggested that the S5-P helices in the hERG channel must be able to adopt dynamic conformational changes during membrane depolarization, so that a considerable portion of their surfaces can come close to each other to allow intersubunit disulfide bond formation between Cys side chains at equivalent positions (Jiang et al., 2005). Furthermore, our three-dimensional model of the hERG channel pore domain suggested that Trp585 and Leu589 side chains on the S5-P helices can make contacts with pore-loop side chains (Tseng et al., 2007), providing an explanation for why mutations along the S5-P linker can affect the channel’s K+ selectivity (Tseng et al., 2007).
It is not surprising that putative NS binding to a domain formed by the outer vestibule/external pore entrance can influence the rates of activation and deactivation. These gating processes are believed to occur at the cytoplasmic halves of the S6 segments, controlled by the voltage-sensor S4 via the S4-S5 linkers (Long et al., 2005). The extracellular end of S4 in the activated state can come close to the extracellular end of S5 from an adjacent subunit (to allow intersubunit disulfide bond formation) (Gandhi et al., 2003; Lainé et al., 2003). Therefore, drug binding to the outer vestibule/external pore entrance may allosterically affect the stability of S4 segment in the activated state. Indeed, we have shown that Cys substitutions at high-impact positions in the outer vestibule of the hERG channel could affect not only inactivation/K+ selectivity, but also the voltage-dependence of activation (Liu et al., 2002).
Our data are subject to alternative explanations. Data in Figs. 5 and 6 can be alternatively explained by a removal of the mechanism for NS action. Data in Fig. 8 can be alternatively explained by a reorientation of the channel upon BeKm-1 binding, preventing access to an NS binding site in other area(s) of the channel.
Binding Site and Mechanism of Action of PD307243
The activator effect of PD was very robust. PD remained effective even when intersubunit disulfide bonds were formed between S5-P helices. Such intersubunit disulfide bonds were expected to perturb the peptide backbone conformation and flexibility. However, extracellularly applied TPeA+ and intracellularly acting dofetilide prevented the activator effect of PD. What are the common features shared by these two pore blockers that acted from the opposite sides of the cell membrane? An X-crystallographic study has revealed the binding sites for external TEA and internal tetrabutylammonium (TBA) and their effects on Tl+ (a K+ analog) ion occupancy in the selectivity filter of the KcsA channel (Lenaeus et al., 2005). It is reasonable to apply information learned from the crystallographic work on KcsA to our study for the following reasons. First, it has been shown that the binding site and probably the mechanism of action of extracellular QA+ cations are similar among different K channels (Ahern et al., 2006). Second, although TBA and dofetilide are distinctly different molecules, TBA binding to the KcsA channel and dofetilide binding to the hERG channel share two important features: 1) they both bind deep in the channel inner cavity, right beneath and likely making contact with the pore-loop, and 2) binding is stabilized by hydrophobic interactions between the blockers and aromatic residues facing toward the inner cavity (Phe103 in KcsA and the equivalent Tyr652 in hERG) (Lenaeus et al., 2005; Kamiya et al., 2006). External TEA binding and internal TBA binding to the KcsA channel share two features: 1) the selectivity filter becomes largely devoid of Tl+ ions, and 2) the peptide backbone of the selectivity filter exhibits subtle conformational changes (Lenaeus et al., 2005). Although these observations explain why external TEA and internal TBA (or external TPeA+ and internal dofetilide, in the case of hERG) can totally occlude K+ ion flux through the pore, they also suggest that PD may be a “pore modifier.”
To explain why PD is sensitive to the degree of K+ ion occupancy in the selectivity filter, we hypothesize that PD acts directly on the pore-loop to increase channel conductance and secondarily/additionally to retard hERG inactivation. Although we were able to qualitatively simulate PD effects on the WT hERG channel by a 50% reduction in the inactivation rate, introducing an increase in channel conductance in the parameter set might improve model simulation quantitatively. Interventions that disrupted hERG inactivation did not prevent PD’s agonist effect. In these cases, PD might work by primarily increasing channel conductance, although the interventions might have also affected PD binding affinity and/or degree of increase in the channel conductance. To provide direct experimental evidence for the hypothesis, we need to perform outside-out patch recordings of single channel currents through WT and inactivation-disrupted channels under the control conditions and after the application of increasing concentrations of PD. Such extensive experiments are beyond the scope of the current study.
BeKm-1 potentiated the activator effect of PD. What are the differences between TPeA+ and BeKm-1, both of which bound to the outer mouth of the hERG channel? BeKm-1 differs from TPeA+ in two aspects. First, the peptide toxin binds above the pore entrance and does not totally occlude K+ ion flux through the pore (Zhang et al., 2003; Tseng et al., 2007). Second, BeKm-1 binds to the hERG outer vestibule in the resting and activated states but unbinds in the inactivated state, probably because of the dynamic conformational changes in this region discussed above (Tseng et al., 2007). Therefore, the extracellular BeKm-1 peptides may go through binding and unbinding cycles under our voltage clamp conditions, and the presence of PD through its pore-modifier effect may retard BeKm-1 binding or facilitate BeKm-1 unbinding, producing an apparent potentiation of its activator effect.
Comparison with Previous Studies
Our results on NS1643 are consistent with a previous study (Casis et al., 2006): pore mutations that impair hERG inactivation (S631A, G628C/S631C, and S620T) attenuate the activator effect of NS. Our results on PD307243 are similar to a previous study on its structure analog, PD118057 (Zhou et al., 2005); PD118057 did not affect the voltage dependence or kinetics of activation and deactivation, and it slowed the rate of hERG inactivation at 35°C. A recent report on PD307243 (1–10 μM, applied to the hERG channel stably expressed in CHO cells) had some very novel findings (Gordon et al., 2008). The authors showed that PD307243 not only increased the hERG current amplitude but also created a prominent constitutive component and greatly slowed the rate of channel inactivation. The combined effects of constitutive channel activation and markedly slowed inactivation created a peculiar transient outward (Ito)-like current. We have not observed such a PD-induced Ito-like current or constitutive channel activation in oocytes or COS-7 cells. We have confirmed the chemical structure of PD307243 used in our experiments (Supplemental Data). It is noteworthy that we have confirmed the agonist effect of PD on native IKr in guinea pig ventricular myocytes (Fig. 10). In three separate experiments, 10 μM PD increased the time-dependent outward current recorded at −10 or −20 mV (mainly reflecting IKr) by 41, 95, and 193% after exposure times of 1.5, 4, and 7 min, respectively. PD did not induce any Ito-like current in guinea pig ventricular myocytes.
Fig. 10.

Agonist effect of PD307243 on native IKr in a guinea pig ventricular myocyte. Shown are superimposed current traces recorded from the same myocyte before (thin gray trace) and 5 min after application of PD 10 μM (thick black trace). Currents were elicited by the pulse protocol diagrammed. Based on the difference in voltage-dependence of activation (Sanguinetti and Jurkiewicz, 1990), time-dependent current during the −10 mV step and the following tail current at −50 mV mainly represented IKr. Time-dependent current during the +50 mV step mainly represented IKs, although the following tail current resulted from both IKr and IKs.
Supplementary Material
Acknowledgments
We thank Dr. Xiaoping Xu (Cardiovascular and Urogenital Center of Excellence in Drug Discovery, GlaxoSmithKline, King of Prussia, PA) for providing PD307243 and NS1643.
This study was supported by R01-HL46451 and HL67840 (to G.N.T.) from the National Heart, Lung and Blood Institute of the National Institutes of Health.
ABBREVIATIONS
- hERG
human ether-a-go-go-related gene
- LQT2
long QT
- aLQT
acquired LQT
- RPR260243
(3R,4R)-4-(3-(6-methoxyquinolin-4-yl)-3-oxo-propyl)-1-(3-(2,3,5-trifluoro-phenyl)-prop-2-ynyl)-piperidine-3-carboxylic acid
- NS
NS1643 (1,3-bis-(2-hydroxy-5-trifluoromethyl-phenyl)-urea)
- PD118057
2-(4-(2-(3,4-dichlorophenyl)ethyl)phenylamino)benzoic acid
- PD
PD307243 (2-[2-(3,4-dichloro-phenyl)-2,3-dihydro-1H-isoindol-5-ylamino]-nicotinic acid)
- TEA
tetraethylammonium
- DTT
dithiothreitol
- BeKm-1
toxin from Buthus eupeus venom
- TPeA
tetrapentylammonium
- TBA
tetrabutylammonium
- QA
quaternary ammonium
- WT
wild type
- Vt
test pulse voltage
- Vr
repolarization voltage
Footnotes
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
References
- Ahern CA, Eastwood AL, Lester HA, Dougherty DA, Horn R. A cation-π interaction between extracellular TEA and an aromatic residue in potassium channels. J Gen Physiol. 2006;128:649–657. doi: 10.1085/jgp.200609654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugada R, Hong K, Dumaine R, Cordeiro JM, Gaita F, Borggrefe M, Menendez TM, Brugada J, Pollevick GD, Wolpert C, et al. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation. 2004;109:30–35. doi: 10.1161/01.CIR.0000109482.92774.3A. [DOI] [PubMed] [Google Scholar]
- Casis O, Olesen SP, Sanguinetti MC. Mechanism of action of a novel human ether-a-go-go-related gene channel activator. Mol Pharmacol. 2006;69:658–665. doi: 10.1124/mol.105.019943. [DOI] [PubMed] [Google Scholar]
- Dun W, Jiang M, Tseng GN. Allosteric effects of mutations in the extracellular S5-P loop on the gating and ion permeation properties of hERG. Pflugers Arch. 1999;439:141–149. doi: 10.1007/s004249900101. [DOI] [PubMed] [Google Scholar]
- Fan JS, Jiang M, Dun W, McDonald TV, Tseng GN. Effects of outer mouth mutations on hERG channel function: a comparison with similar mutations in Shaker. Biophys J. 1999;76:3128–3140. doi: 10.1016/S0006-3495(99)77464-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi CS, Clark E, Loots E, Pralle A, Isacoff EY. The orientation and molecular movement of a K+ channel voltage-sensing domain. Neuron. 2003;40:515–525. doi: 10.1016/s0896-6273(03)00646-9. [DOI] [PubMed] [Google Scholar]
- Gordon E, Lozinskaya IM, Lin Z, Seus SF, Blaney FE, Willette RN, Xu X. 2-[2-(3,4-Dichloro-phenyl)-2,3-dihydro-1H-isoindol-5-ylamino]-nicotinic acid (PD-307243) causes instantaneous current through human ether-a-go-go-related gene potassium channels. Mol Pharmacol. 2008;73:639–651. doi: 10.1124/mol.107.041152. [DOI] [PubMed] [Google Scholar]
- Hansen RS, Diness TG, Christ T, Demnitz J, Ravens U, Olesen SP, Grunnet M. Activation of human ether-a-go-go-related gene potassium channels by the diphenylurea 1,3-bis-(2-hydroxy-5-trifluoromethy-phenyl)-urea (NS1643) Mol Pharmacol. 2006;69:266–277. doi: 10.1124/mol.105.015859. [DOI] [PubMed] [Google Scholar]
- Jiang M, Zhang M, Maslennikov IV, Liu J, Wu DM, Korolkova YV, Arseniev AS, Grishin EV, Tseng GN. Dynamic conformational changes of extracellular S5-P linkers in the hERG channel. J Physiol. 2005;569:75–89. doi: 10.1113/jphysiol.2005.093682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya K, Niwa R, Mitcheson JS, Sanguinetti MC. Molecular determinants of hERG channel block. Mol Pharmacol. 2006;69:1709–1716. doi: 10.1124/mol.105.020990. [DOI] [PubMed] [Google Scholar]
- Kang J, Chen XL, Wang H, Ji J, Cheng H, Incardona J, Reynolds W, Viviani F, Tabart M, Rampe D. Discovery of a small molecule activator of the human ether-a-go-go-related gene (HERG) cardiac K+ channel. Mol Pharmacol. 2005;67:827–836. doi: 10.1124/mol.104.006577. [DOI] [PubMed] [Google Scholar]
- Lainé M, Lin MCA, Bannister JPA, Silverman WR, Mock AF, Roux B, Papazian DM. Atomic proximity between S4 segment and pore domain in Shaker potassium channels. Neuron. 2003;39:467–481. doi: 10.1016/s0896-6273(03)00468-9. [DOI] [PubMed] [Google Scholar]
- Lenaeus MJ, Vamvouka M, Focia PJ, Gross A. Structural basis of TEA blockade in a model potassium channel. Nat Struct Mol Biol. 2005;12:454–459. doi: 10.1038/nsmb929. [DOI] [PubMed] [Google Scholar]
- Liu J, Zhang M, Jiang M, Tseng GN. Structural and functional role of the extracellular S5-P linker in the HERG potassium channel. J Gen Physiol. 2002;120:723–737. doi: 10.1085/jgp.20028687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long SB, Campbell EB, MacKinnon R. Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science. 2005;309:903–908. doi: 10.1126/science.1116270. [DOI] [PubMed] [Google Scholar]
- López-Barneo J, Hoshi T, Heinemann SH, Aldrich RW. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors Channels. 1993;1:61–71. [PubMed] [Google Scholar]
- Pardo-Lopez L, Zhang M, Liu J, Jiang M, Possani LD, Tseng GN. Mapping the binding site of a HERG-specific peptide toxin (ErgTx) to the channel’s outer vestibule. J Biol Chem. 2002;277:16403–16411. doi: 10.1074/jbc.M200460200. [DOI] [PubMed] [Google Scholar]
- Perry M, Saches FB, Sanguinetti MC. Structural basis of action of a human ether-a-go-go-related gene 1 potassium channel activator. Proc Natl Acad Sci U S A. 2007;104:13827–13832. doi: 10.1073/pnas.0703934104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recanatini M, Poluzzi E, Masetti M, Cavalli A, De Ponti F. QT prolongation through hERG K+ channel blockade: current knowledge and strategies for the early prediction during drug development. Med Res Rev. 2005;25:133–166. doi: 10.1002/med.20019. [DOI] [PubMed] [Google Scholar]
- Roden DM. Drug-induced prolongation of the QT interval. N Engl J Med. 2004;350:1013–1022. doi: 10.1056/NEJMra032426. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Jurkiewicz NK. Two components of cardiac delayed rectifier K+ current. Differential sensitivity to block by class III antiarrhythmic agents. J Gen Physiol. 1990;96:195–215. doi: 10.1085/jgp.96.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreibmayer W, Lester HA, Dascal N. Voltage clamping of Xenopus laevis oocytes utilizing agarose-cushion electrodes. Pflugers Arch. 1994;426:453–458. doi: 10.1007/BF00388310. [DOI] [PubMed] [Google Scholar]
- Smith PL, Baukrowitz T, Yellen G. The inward rectification mechanism of the HERG cardiac potassium channel. Nature. 1996;379:833–836. doi: 10.1038/379833a0. [DOI] [PubMed] [Google Scholar]
- Torres AM, Bansal P, Sunde M, Clarke CE, Bursill JA, Smith DJ, Bauskin A, Breit SN, Campbell TJ, Alewood PF, et al. Structure of the HERG K+ channel S5P extracellular linker: role of an amphipathic α-helix in C-type inactivation. J Biol Chem. 2003;278:42136–42148. doi: 10.1074/jbc.M212824200. [DOI] [PubMed] [Google Scholar]
- Tseng GN. IKr: the hERG channel. J Mol Cell Cardiol. 2001;33:835–849. doi: 10.1006/jmcc.2000.1317. [DOI] [PubMed] [Google Scholar]
- Tseng GN, Sonawane KD, Korolkova YV, Zhang M, Liu J, Grishin E, Guy HR. Probing outer mouth structure of the hERG channel with peptide toxin foot printing and molecular modeling. Biophys J. 2007;92:3524–3540. doi: 10.1529/biophysj.106.097360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Liu S, Morales MJ, Strauss HC, Rasmusson RL. A quantitative analysis of the activation and inactivation kinetics of HERG expressed in Xenopus oocytes. J Physiol. 1997;502:45–60. doi: 10.1111/j.1469-7793.1997.045bl.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao JA, Jiang M, Fan JS, Zhou YY, Tseng GN. Heterogeneous changes in K currents in rat ventricles three days after myocardial infarction. Cardiovasc Res. 1999;44:132–145. doi: 10.1016/s0008-6363(99)00154-6. [DOI] [PubMed] [Google Scholar]
- Yeola SW, Rich TC, Uebele VN, Tamkun MM, Snyders DJ. Molecular analysis of a binding site for quinidine in a human cardiac delayed rectifier K+ channel. Role of S6 in antiarrhythmic drug binding. Circ Res. 1996;78:1105–1114. doi: 10.1161/01.res.78.6.1105. [DOI] [PubMed] [Google Scholar]
- Zhang H, Zhu B, Yao JA, Tseng GN. Differential effects of S6 mutations on binding of quinidine and 4-aminopyridine to rKv1.4: common site but different factors in determining blockers’ binding affinity. J Pharmacol Exp Ther. 1998;287:332–343. [PubMed] [Google Scholar]
- Zhang M, Korolkova YV, Liu J, Jiang M, Grishin EV, Tseng GN. BeKm-1 is a hERG-specific toxin that shares the structure with ChTx but the mechanism of action with ErgTx1. Biophys J. 2003;84:3022–3036. doi: 10.1016/S0006-3495(03)70028-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Augelli-Szafran CE, Bradley JA, Chen X, Koci BJ, Volberg WA, Sun Z, Cordes JS. Novel potent human ether-a-go-go-related gene (hERG) potassium channel enhancers and their in vitro antiarrhythmic activity. Mol Pharmacol. 2005;68:876–884. doi: 10.1124/mol.105.014035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.