

Abstract
Manipulation of gene expression has been used to elucidate gene function, explore fundamental biological processes and to identify potential drug targets in Trypanosoma brucei. We show in bloodstream forms that CDC2-related kinase CRK12 (Tb11.01.4130) is essential since transcriptional inactivation in conditional null mutants is lethal but 19 other protein kinases are not essential since null mutants are viable. We did so using efficient methods for the generation of null and conditional null cell lines of T. brucei by approaches that generate transfection constructs with large targeting sequences and which use reliable transfection and selection conditions. These methods, which are described in detail in the supplementary material, employ multiple oligonucleotides and PCR reactions and several transfections but are cost effective and can simultaneously generate 24 transfectants thus shifting the rate limiting experimental steps from the production of cell lines to their analysis.
Keywords: Trypanosoma brucei, Protein kinases, Gene knockout, Conditional expression, Fusion PCR Method
Manipulation of gene expression is a powerful tool for elucidating the functions of genes in the context of a biological system and for assessing the potential of the gene products as drug targets. The functions of numerous genes have been examined in Trypanosoma brucei in order to explore fundamental biological processes and to search for drug targets. In addition, this diploid protozoan extracellular pathogen has served as an experimental surrogate for the related intracellular kinetoplastid pathogens T. cruzi and Leishmania because of their highly homologous and syntenic genomes and because experimental genetic manipulation is more developed in T. brucei [1].
The robust homologous recombination system that is naturally present in T. brucei has been exploited to make gene knockouts (KOs) and other genome modifications. For gene KOs, cells are transfected by electroporation with a linear DNA construct that contains an antibiotic resistance gene, i.e. selectable marker, with flanking sequences that target recombination which results in replacement of the endogenous gene with the selectable marker. The creation of a null mutant, i.e. a cell in which both alleles eliminated, is a definitive way to identify non-essential genes. Conditional RNA interference (RNAi) [1], by which expression of a target gene is repressed via mRNA destruction in a tetracycline(tet)-dependant manner, has been commonly used in T. brucei to assess gene function and essentiality. RNAi cell line production entails the cloning of single RNAi construct and single transfection step into the appropriate cell line. The ability to efficiently generate RNAi cell lines has led to its systematic use to assess many genes on one chromosome [2] and for genome-wide analysis of genes using deep sequencing of a population of RNAi cell lines [3]. An additional advantage to the RNAi approach is that mRNAs from nearly identical paralogs can be simultaneously targeted with a single construct. However, RNAi has the disadvantages that it may not sufficiently reduce the mRNA levels to generate a definitive phenotype and can have off-target effects, i.e. degradation of mRNA from genes that were not intentionally targeted. Little is known about off-target RNAi effects in T. brucei, however, and genetic approaches can be used to verify that RNAi is acting on-target [4]. We prepared null cell lines by knocking out both endogenous alleles in cells and a “conditional null” cell line by knocking out both endogenous alleles in cells that have a tet-regulatable copy of the target gene’s coding DNA sequence (CDS) to determine the essentiality of several protein kinases. Expression knockdown is very robust in conditional nulls cell lines but these cell lines have not been widely used because their preparation requires multiple transfections and cloning steps. However, we improved the efficiency of their production by minimizing the need to prepare plasmid constructs and improving transfection and selection conditions.
We employed an efficient PCR approach to make gene KO DNA constructs instead of a sequential PCR/cloning approach (e.g. [5–9]) since the latter is time consuming and has pitfalls that reflect the limitations of restriction enzyme digestion, DNA fragment purification, and ligation efficiency. A PCR approach has been used to directly produce the DNA constructs for transfection by a single PCR reaction of drug resistance markers with long oligos (~100 nucleotides) that contain targeting sequences for gene-specific KOs (e.g. [10] and http://tryps.rockefeller.edu/trypsru2_genetics.html). We instead employed a “fusion PCR” approach to generate KO constructs with sizeable homologous targeting sequences (Fig. 1) since the use of long oligos for PCR adds significant cost and effectively limits the length of homologous targeting region. This targeting region limit can affect the frequency of homologous recombination since it has been shown that shorter targeting sequences result in lower recombination frequency in T. brucei [11] as has been seen for other organisms. Hence, we used long (~500 nt) targeting sequences to potentially provide for a greater recombination frequency, and thus a greater frequency of transfection. These larger targeting regions were used with a fusion PCR approach to enhance the efficiency of transfection construct production.
Figure 1. Schema for generation of transfection constructs.
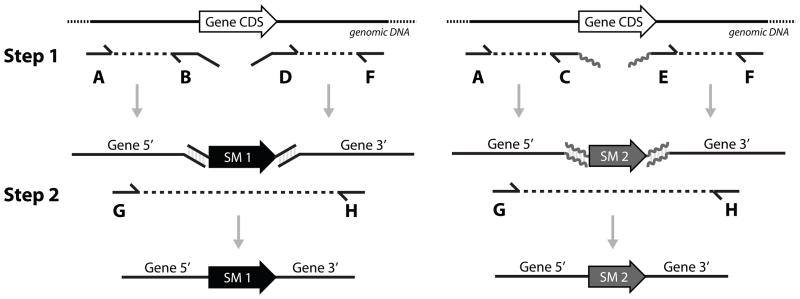
Fusion PCR method that generates gene knockout (KO) constructs. Step 1: gene-specific 5′ and 3′ targeting sequences of both alleles are amplified by separate PCRs using six different oligonucleotides (oligos). Step 2: The 5′ and 3′ targeting fragments of each allele are fused by PCR with the coding DNA sequence (CDS) of their respective selectable marker (SM) that is amplified from a plasmid stock. Complementary sequences are designed into the termini of the oligos as illustrated and nested oligos G and H provide for specific amplification of the fusion PCR product. Approximately 25–50ng each of 5′ and 3′ targeting sequences and a drug resistance CDS PCR products were combined and fused by PCR amplification. The fusion PCR products were verified and purified on an agarose gel and recovered with a Qiagen gel extraction kit. Fusion PCR protocols and oligo sequences are detailed in supplementary material.
The fusion PCR method that we used (Fig. 1) was adapted from that used in other systems (e.g. [12, 13]). Multiple PCR reactions were used to rapidly, inexpensively, and effectively generate DNA for direct use in transfections. Two separate PCR reactions were used to amplify 5′ and 3′ targeting sequences from T. brucei genomic DNA for each gene. The inner oligos each contain a sequence that is complementary to the outer sequences of a selectable marker that has been PCR amplified in bulk from plasmid DNA and gel purified. The 5′, 3′ and selectable marker PCR products were combined after removal of the PCR primers using a commercial spin column (Qiagen) and amplified by another PCR reaction which produces the fusion PCR product for transfection. Six unique oligos are needed to create the fusion product for KO of each allele (Fig 1). For the 1st allele the two inner oligos (oligos B, D) contain sequences that complement those of the 1st selectable marker, the two outer oligos (oligos A, F) provide for amplification of ~500 nts of targeting sequence flanking the gene CDS, and two oligos (G, H) that are nested (i.e. internal to oligos A and F) for specificity and to create the fusion PCR product that will be used for transfection. Two additional oligos (oligos C, E) are needed which contain sequences that are complementary to the 2nd selectable marker to generate the PCR product that will be used in the fusion PCR to generate the construct to KO the 2nd allele. The original two outer oligos and those for the nested PCR can be the same (i.e. oligos A, F, G, H).
The fusion PCR transfection constructs for two alleles made in this manner will have the same targeting sequences. This can result in the construct for the 2nd allele KO recombining with the site of 1st allele KO. However the use of two drugs in combination will select against such cells making this approach suitable for high throughput studies. Nevertheless, recombination of the 2nd allele KO construct with the site of 1st allele KO can (and does) occur. This will not eliminate the 2nd endogenous allele but will insert at the site of the 2nd selectable marker and either retain or replace of the 1st selectable marker. This can be averted as detailed in the supplementary material by using oligo pairs that are internal to A, B and D, F to PCR amplify the targeting sequences for the 2nd selectable marker. This results in the need for two additional oligos and for awareness of possible consequences to adjacent genes (e.g. deletion of important regulatory or coding sequences). Thus, minimally eight, and maximally ten, oligos are needed to KO two alleles, which is more than the number used for the long oligo KO approach. However, the total cost of these eight to ten oligos is a fraction of the cost of the longer ones due to their smaller size. Currently the total reagents costs for the fusion PCR is 3–4 times less than for the single PCR method using long oligos and is due to the substantially greater cost of the long oligos. In addition, the larger flanking gene targeting regions of the fusion PCR products provide for higher recombination and transfection efficiencies. The oligos are designed to have similar annealing temperatures so that all of the PCR products can be generated under the same conditions. This PCR protocol has been scaled up to a 96-well format which enables simultaneous production of PCR products for the KO of 12–24 genes (see supplementary materials for details). More PCR products can be produced at a time (e.g. for 48–96 genes) but the subsequent steps of transfection, cell maintenance and confirmation of the desired transfections effectively limit production to 24 gene KOs at a time by one person.
Fusion PCR products were prepared and used to KO target genes in T. brucei to attempt to create null mutants, i.e. eliminate both endogenous alleles, for 20 predicted protein kinases. The creation of null mutants would definitively demonstrate that the eliminated gene is not essential for viability. A conditional null mutant was produced when null mutants could not be obtained. The protein kinases chosen are primarily members of the Sterile (STE) and Other protein kinase groups which had not been thoroughly studied genetically in T. brucei. Null mutants were generated by the higher-throughput fusion PCR transfection method using two different selectable markers to sequentially KO both endogenous copies of target genes (see supplementary materials for KO schematic). These constructs were designed to precisely delete the target gene from the start to the stop codon. One endogenous allele was eliminated by transfecting purified fusion PCR products by electroporation into bloodform (BF) T. brucei single marker 427 cells (SM427) [14]. The SM427 cell line expresses T7 polymerase and Tet repressor proteins in the β tubulin locus (maintained with NEO selectable marker) to control tet conditional expression of transgenes [14]. The electroporation of each PCR fusion product was done using approximately 2×107 mid log phase SM427 cells as outlined in the supplementary materials. Several conditions can affect transfection efficiency and hence the generation of KO cell lines. These include the amount and quality of the DNA used, the length of its targeting sequences, the number, growth phase and strain of cells, the transfection device and the settings used, the selectable markers, and the concentrations and plating conditions used during selection for transfectants. Many of these variables have been examined previously ([11, 15]) and a few are addressed herein. We found that the choice of transfection device used greatly affects transfection frequency as have others ([16], http://tryps.rockefeller.edu/trypsru2_protocols_index.html). More importantly, we found that the drug resistance markers used and the concentrations of drugs used for selection were critical for reliably obtaining KO, especially null, cell lines.
An average of 58 independent clones (determined by limiting dilution) with a 1st allele knocked out were obtained from ~2×107 cells using ~200–600ng of PCR fusion product. For the 20 genes analyzed, the range was over an order of magnitude. No correlation was observed between the number of clones obtained and the amount of PCR product used above 200ng, or the length of target gene’s ORF between one and five kb (data not shown). Transfections under similar conditions using blasticidin, puromycin, and phleomycin selectable markers (BSD, PAC, and BLE) resulted in 1st allele KOs although the numbers of clones obtained with different genes and with different drug concentrations were not analyzed. Success rates for 2nd allele KOs under similar conditions were 15–140 fold lower. Success rates for 2nd allele KOs for other genes are also lower even when a tet-regulatable copy is expressed (data not shown). Thus lower success rates for 2nd allele KOs cannot be explained solely by a response to the elimination of gene expression. Electroporation using the Amaxa Nucleofection device, which has a higher transfection efficiency than the BTX device ([16], http://tryps.rockefeller.edu/trypsru2_protocols_index.html), resulted in a greater number of 2nd allele KOs (7–30 fold more). The lower number of transfectants of 2nd versus 1st allele KOs may be due to increased selection as a consequence of the absence of one allele, potential effects on expression of other genes (e.g. those in the same cluster) of the other gene KO, competition for recombination with the 1st allele KO locus, and/or the effect of using multiple antibiotics. Competition for recombination can be reduced if not eliminated by using sequences in the fusion PCR product for the 2nd allele that have been eliminated in the 1st allele KO as outlined above (see supplementary material).
The choice of selectable marker, drug concentration, and conditions for selection were significant variables affecting the frequency with which allele KO clones were obtained, especially for the 2nd allele KO (see supplementary material). Reducing drug concentrations below those generally used for selection in T. brucei (5 μg/ml hygromycin, 5 μg/ml blasticidin, 0.1 μg/ml puromycin, 2.5 μg/ml phelomycin) (http://tryps.rockefeller.edu/trypsru2_genetics.html) and omitting one drug during part of the selection time resulted in a greater frequency of obtaining KO cell clones (see supplementary material). This suggests that the expression levels of drug resistance markers that are governed by the target genes endogenous UTRs may not be ideal for selection depending on the selectable marker. In support of this, we found that providing the selectable marker gene with UTRs that support higher levels of expression provided a wider window for drug selection when the level of expression with the endogenous gene UTRs is limiting, perhaps those with lower levels of expression (see supplementary material).
To verify the desired gene KOs, genomic DNA of clones obtained after the 1st and 2nd allele transfection were genotyped using diagnostic oligos by qRT-PCR and, in some cases, by PCR and gel analysis. All clones obtained after attempting to KO the 1st allele (with HYG) of each of 20 protein kinases showed the loss of one endogenous coding sequence (CDS) copy by qRT-PCR (Fig. 2). Attempts to KO the 2nd allele (with BSD or PAC) in these 20 1st allele KO cell lines resulted in 46/47 clones (multiple clones for most genes) in which both endogenous CDSs were absent (Fig. 2, data not shown). However, we were unable to knockout the second CRK12 allele (see below). We did not analyze in detail the one cell line that was resistant to both selectable markers but it still had one copy of the target gene CDS. Several KO cell lines were also examined by PCR using oligos outside of targeting region and subsequent gel analysis showed presence of bands consistent with the replacement of the target CDS with the selectable markers (Fig. 2B, data not shown). These data suggest that the approach described herein may only infrequently generate cell lines that are resistant to the selectable marker but do not contain the desired 2nd allele KO due to events such as mis-targeting or gene duplication. This minimizes the time needed to confirm the identity of the desired clones after transfection.
Figure 2. Genotyping of putative KO cell lines.

A. qRT-PCR analysis of genomic DNA isolated from putative 1st allele KO and 2nd allele KO (putative null) cell lines. For each cell line, genomic DNA was isolated from approximately 1×107 cells with DNeasy Blood & Tissue Kit (Qiagen). QRT-PCR was performed in triplicate with SsoFast EvaGreen Supermix With Low ROX (BioRad) with a 7500 Real-Time PCR System (Applied Biosystems). Oligos for qRT-PCR were designed using Primer3Plus (sequences given in supplementary material). Tert was used as an internal control. All 1st allele KO cell lines (n=19) showed loss of one copy by qRT-PCR. Most 2nd allele KO cell lines (putative nulls) showed loss of both copies by qRT-PCR. Genotypically null cell lines were obtained for all of these 19 genes, but one of two lines of Tb11.02.4860 (CAMKK) retained a copy of the target CDS (* RT < 0.0001, ** RT < 0.005, ¥ no amplification, # off target amplification). B. Example of genotyping of a Tb927.10.14780 null cell line by PCR using oligos outside of the targeting regions and subsequent gel analysis showed presence of novel bands consistent with the replacement of both target CDS’s with selectable marker CDSs.
Identification of genes that are essential for viability is of interest in order to determine their function and their potential as drug targets. Stable null cell lines cannot be obtained for such genes without genetic or other compensation. Attempts to knock out the 2nd endogenous allele of an essential gene will either not result in viable stable cell lines even after multiple attempts, as we and other have repeatedly observed (data not shown), or will result in unintended genetic changes. Stable cell lines obtained from such attempts that are resistant to both drugs encoded in the 1st and 2nd KO selectable markers can retain a copy of the gene CDS by mis-integrating a selectable marker at an off-target locus or by integrating at the targeted locus without the loss of the resident allele (e.g. as a result of gene duplication event). For example, CK1.2 (Tb927.5.800) has a strong growth defect by RNAi in BFs and attempts by others to create null cell lines were unsuccessful suggesting that it is essential [8]. Our multiple attempts to create null cell lines for CK1.2 only resulted in cell lines that contained both resistance markers but still contained a copy of the CK1.2 CDS, consistent with the gene being essential for growth in BF T. brucei (data not shown).
Conditional null cell lines can be generated to study genes that are essential for viability. By this approach, a tet-regulatable copy of the gene CDS is inserted into a transcriptionally silent rRNA intergenic locus of SM427 T. brucei cells prior to the KO of both endogenous alleles. For efficiency, this tet-regulatable copy can be introduced in cells in which the 1st allele has been knocked out simultaneously and in parallel with attempts to generate a null mutant. The 2nd endogenous allele is then knocked out with a fusion PCR product for the 2nd allele KO with selection performed in media that contains tet to ensure expression of the regulatable copy in the rRNA intergenic region. Proper gene insertion is assayed by PCR as indicated above and tet-regulation is assayed by monitoring cell growth in the presence (gene expressed) and absence of tet (gene repressed) by monitoring mRNA knockdown with qPCR or northern blot. Expression of the regulatable copy can also be monitored by western analysis if an antibody is available against the protein of interest or if an epitope tag has been fused to the regulatable copy. In addition, the level of mRNA and protein knockdown can be controlled within a moderate range by adjusting the level of tet in the cell grown media.
The tet-regulatable copy of the gene CDS can be cloned with any of the many vectors that are widely available (see http://www.zbh.uni-heidelberg.de/clayton/vectors.html, http://tryps.rockefeller.edu/trypsru2_plasmids.html and references in supplementary materials). Insertion of the CDS into the appropriate tet-regulatable vector can be performed by traditional ligation cloning with restriction enzyme sites designed into CDS PCR oligos or more efficiently by in vitro recombination using the Gateway system (Invitrogen) with the recombinase-specific sequence tails added to the oligos that amplify the CDS. To expedite cloning of several regulatable transgenes simultaneously, we have modified a commonly used regulatable expression vector (pLEW100v5) so that the gene CDS can be inserted via in vitro recombination (see supplementary materials).
Using the scheme outlined in the supplementary materials, we created several conditional null cell lines in BF and procyclic form (PF) T. brucei including one for CDC2-related kinase CRK12 (Tb11.01.4130) in BF cells (Fig. 3) which had previously been shown to have a growth defect upon RNAi [17]. This cell line was created in SM427 cells which express the tet repressor protein that is necessary for regulation of the CDS allele in the rRNA locus. For this cell line, fusion PCR products constructed as described above were used to KO the two endogenous alleles and a Gateway version of pLEW100v5 was used to produce the tet-regulatable copy (Fig. 3A). The genotype of this cell line was verified by qRT-PCR with genomic DNA as a template (Fig. 3B). Growth of cells monitored over several days in media containing tet (regulatable CRK12 on) or lacking tet (regulatable CRK12 off) and showed a strong growth phenotype in cells in which the regulatable copy was no longer expressed (Fig. 3C). Growth resumed after four days presumably due to loss of repression of the regulatable allele in the rRNA locus although other means could have resulted in this growth recovery. However, similar loss of repression has been seen for other conditional null cell lines [18–20] and has been shown to correspond with mutations that result in the loss of tet repressor coding sequences [19].
Figure 3. CRK12 conditional null cell line created by the fusion PCR and Gateway approach.

A. Fusion PCR products were used to KO the two CRK12 endogenous alleles with HYG and BSD. The Gateway cloning approach was used to generate the tet-regulatable copy of CRK12 for transfection. To clone this tet-regulatable copy of CRK12, the CRK12 CDS (Tb11.01.4130) was PCR amplified from SM427 genomic DNA using Phusion polymerase (NEB) and oligos KO_078(CRK12)GWF and KO_078(CRK12)GWR, which contain attB recombination sequences for Gateway cloning. The product was gel purified and then cloned into pDONR221 using BP Clonase II (Invitrogen). The proper clone was verified by restriction enzyme digestion and DNA sequencing. The resultant Entry Clone (pCM013) was then recombined into a Gateway compatible version of the pLEW100v5 vector (pCM038) and a proper clone was verified by restriction enzyme digestion and sequencing. The resultant plasmid (pCM092) was linearized with NotI prior to transfection of T. brucei to generate a cell line with tet-dependent CRK12 expression from the rRNA intergenic region. B. QRT-PCR verification of CRK12 conditional null cell line. Oligos amplifying a region of the CRK12 CDS show one copy of the CDS is present (tet-regulatable copy) and two sets of oligos to amplify 5′/CDS and 3′/CDS regions specific to endogenous allele of CRK12 show absence of endogenous CRK12 CDS (* RT < 0.0005). The tet-regulatable CRK12 has different 5′ and 3′ UTRs from those of the endogenous alleles. C. Growth curve of CRK12 conditional null cell line shows growth rate inhibition upon withdraw of tet. CRK12 (Tb11.01.4130) conditional null cells were maintained in sequential dilutions of media containing tet to maintain cells in mid-log phase growth. CRK12 conditional null cell cultures at densities of 7×105 – 1×106 parasites/ml were used as inocula for growth curves. The cells were spun down and washed twice in media lacking tet to wash out tet and 5×104 parasites/ml were diluted into 20 ml and split into two 10 ml cultures (+tet and −tet). Cultures with a conditional null copy were maintained in media with 1 μg/ml tet to maintain expression. Cells were Incubated at 37°C, 5% CO2 and counted every 24hrs using the Z2 Cell Counter (Beckman Coulter Inc.). Cells were then diluted to 5×104 parasites/ml and incubated for 24 hrs and this was repeated over seven consecutive days.
The method described herein and in the supplementary materials enable efficient analysis of gene essentiality and gene function in T. brucei. It does so by substituting PCR for gene cloning to a substantial extent and by preparing numerous transfection constructs and performing multiple transfections simultaneously. In this way numerous gene KO cell lines and conditional cell lines are rapidly generated. In addition, variations are described that further increase the utility of the conditional null cell lines. We also show that the selectable drug markers used and selection conditions are important factors for obtaining KO cells lines (see supplementary materials). The number of selectable drug markers that are used for cell line construction can be reduced when using this KO method. One approach is to employ loss-of-heterozygosity by increasing drug pressure to select for recombinants in which the inserted drug marker in a single allele gene KO converts the wild type allele through mitotic recombination thus eliminating it. This approach has been used infrequently in T. brucei although gene conversion occurs frequently in association with antigenic variation. Selectable markers can also be excised after integration to allow reuse in subsequent transfectants. This can be accomplished by generating KO cell lines from PCR products that contain the positive selectable marker (i.e. HYG, PAC, BSD, BLE) fused to a negative selectable marker (HSVTK CDS) and flanked by loxP sites. Excision can be accomplished by transient transfection of Cre recombinase and selection against the HSVTK protein by addition of Ganciclovir to the media (see references and details in supplementary materials).
Overall, we used a fusion PCR-based KO technique to stringently assess gene essentiality in BF T. brucei by producing null or conditional null cell lines. Obtaining 19 null cell lines for protein kinases illustrates its efficiency since only 6 nulls for protein kinases in BF T. brucei have been reported to date [5–9]. The methods used provide an alternative to and greater stringency than conditional RNAi for the analysis of essential genes. Additional methodological details are provided in the supplementary material.
Supplementary Material
Highlights.
CRK12 is an essential protein kinase in BF T. brucei.
19 protein kinases are not essential in bloodstream form T. brucei.
Efficient methods for producing transgenic T. brucei cell lines.
Use of fusion PCR, the Gateway system, and improved selection conditions.
Supplementary material with methodological details and protocols.
Acknowledgments
We thank Ngoc Tran for technical assistance and Jef Boeke for oligo ordering advice. This work was supported by the National Institutes of Health grant AI078962 with some studies initiated with support from National Institutes of Health grant AI075641.
References
- 1.Kolev NG, Tschudi C, Ullu E. RNA interference in protozoan parasites: achievements and challenges. Eukaryot Cell. 2011;10:1156–63. doi: 10.1128/EC.05114-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Subramaniam C, Veazey P, Redmond S, Hayes-Sinclair J, Chambers E, Carrington M, et al. Chromosome-wide analysis of gene function by RNA interference in the african trypanosome. Eukaryot Cell. 2006;5:1539–49. doi: 10.1128/EC.00141-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alsford S, Turner DJ, Obado SO, Sanchez-Flores A, Glover L, Berriman M, et al. High-throughput phenotyping using parallel sequencing of RNA interference targets in the African trypanosome. Genome Res. 2011;21:915–24. doi: 10.1101/gr.115089.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ringpis GE, Lathrop RH, Aphasizhev R. iCODA: RNAi-based inducible knock-in system in Trypanosoma brucei. Methods Mol Biol. 2011;718:23–37. doi: 10.1007/978-1-61779-018-8_2. [DOI] [PubMed] [Google Scholar]
- 5.Domenicali Pfister D, Burkard G, Morand S, Renggli CK, Roditi I, Vassella E. A Mitogen-activated protein kinase controls differentiation of bloodstream forms of Trypanosoma brucei. Eukaryot Cell. 2006;5:1126–35. doi: 10.1128/EC.00094-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jensen BC, Kifer CT, Parsons M. Trypanosoma brucei: Two mitogen activated protein kinase kinases are dispensable for growth and virulence of the bloodstream form. Exp Parasitol. 2011;128:250–5. doi: 10.1016/j.exppara.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muller IB, Domenicali-Pfister D, Roditi I, Vassella E. Stage-specific requirement of a mitogen-activated protein kinase by Trypanosoma brucei. Mol Biol Cell. 2002;13:3787–99. doi: 10.1091/mbc.E02-02-0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Urbaniak MD. Casein kinase 1 isoform 2 is essential for bloodstream form Trypanosoma brucei. Mol Biochem Parasitol. 2009;166:183–5. doi: 10.1016/j.molbiopara.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vassella E, Kramer R, Turner CM, Wankell M, Modes C, van den Bogaard M, et al. Deletion of a novel protein kinase with PX and FYVE-related domains increases the rate of differentiation of Trypanosoma brucei. Mol Microbiol. 2001;41:33–46. doi: 10.1046/j.1365-2958.2001.02471.x. [DOI] [PubMed] [Google Scholar]
- 10.Gaud A, Carrington M, Deshusses J, Schaller DR. Polymerase chain reaction-based gene disruption in Trypanosoma brucei. Mol Biochem Parasitol. 1997;87:113–5. doi: 10.1016/s0166-6851(97)00048-0. [DOI] [PubMed] [Google Scholar]
- 11.Barnes RL, McCulloch R. Trypanosoma brucei homologous recombination is dependent on substrate length and homology, though displays a differential dependence on mismatch repair as substrate length decreases. Nucleic Acids Res. 2007;35:3478–93. doi: 10.1093/nar/gkm249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Szewczyk E, Nayak T, Oakley CE, Edgerton H, Xiong Y, Taheri-Talesh N, et al. Fusion PCR and gene targeting in Aspergillus nidulans. Nat Protoc. 2006;1:3111–20. doi: 10.1038/nprot.2006.405. [DOI] [PubMed] [Google Scholar]
- 13.Noble SM, Johnson AD. Strains and strategies for large-scale gene deletion studies of the diploid human fungal pathogen Candida albicans. Eukaryot Cell. 2005;4:298–309. doi: 10.1128/EC.4.2.298-309.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wirtz E, Leal S, Ochatt C, Cross GA. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol Biochem Parasitol. 1999;99:89–101. doi: 10.1016/s0166-6851(99)00002-x. [DOI] [PubMed] [Google Scholar]
- 15.Wirtz E, Hartmann C, Clayton C. Gene expression mediated by bacteriophage T3 and T7 RNA polymerases in transgenic trypanosomes. Nucleic Acids Res. 1994;22:3887–94. doi: 10.1093/nar/22.19.3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burkard G, Fragoso CM, Roditi I. Highly efficient stable transformation of bloodstream forms of Trypanosoma brucei. Mol Biochem Parasitol. 2007;153:220–3. doi: 10.1016/j.molbiopara.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 17.Mackey ZB, Koupparis K, Nishino M, McKerrow JH. High-throughput analysis of an RNAi library identifies novel kinase targets in Trypanosoma brucei. Chem Biol Drug Des. 2011;78:454–63. doi: 10.1111/j.1747-0285.2011.01156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martin KL, Smith TK. The glycosylphosphatidylinositol (GPI) biosynthetic pathway of bloodstream-form Trypanosoma brucei is dependent on the de novo synthesis of inositol. Mol Microbiol. 2006;61:89–105. doi: 10.1111/j.1365-2958.2006.05216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roper JR, Guther ML, Milne KG, Ferguson MA. Galactose metabolism is essential for the African sleeping sickness parasite Trypanosoma brucei. Proc Natl Acad Sci U S A. 2002;99:5884–9. doi: 10.1073/pnas.092669999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Young SA, Smith TK. The essential neutral sphingomyelinase is involved in the trafficking of the variant surface glycoprotein in the bloodstream form of Trypanosoma brucei. Mol Microbiol. 2010;76:1461–82. doi: 10.1111/j.1365-2958.2010.07151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.