

Abstract
This survey was conducted in two protected areas in Nigeria to genetically identify individual lions and to determine the genetic variation within and between the populations. We used faecal sample DNA, a non-invasive alternative to the risky and laborious task of taking samples directly from the animals, often preceded by catching and immobilization. Data collection in Yankari Game Reserve (YGR) spanned through a period of five years (2008 –2012), whereas data in Kainji Lake National Park (KLNP) was gathered for a period of three years (2009, 2010 and 2012). We identified a minimum of eight individuals (2 males, 3 females, 3 unknown) from YGR and a minimum of ten individuals (7 males, 3 females) from KLNP. The two populations were found to be genetically distinct as shown by the relatively high fixation index (FST = 0.17) with each population exhibiting signs of inbreeding (YGR FIS = 0.49, KLNP FIS = 0.38). The genetic differentiation between the Yankari and Kainji lions is assumed to result from large spatial geographic distance and physical barriers reducing gene flow between these two remaining wild lion populations in Nigeria. To mitigate the probable inbreeding depression in the lion populations within Nigeria it might be important to transfer lions between parks or reserves or to reintroduce lions from the zoos back to the wild.
Introduction
Human pressure, agricultural developments and industrialization are leading increasingly to the destruction, fragmentation and isolation of natural populations [1]. A consequence of these changes is loss of genetic variability [2] and increasing risk of extinction [3]–[5]. Their requirements for large home ranges, low fecundity and low numbers have made mammalian carnivores vulnerable to local extinction in fragmented habitats [6]. Mammals are often the dominant carnivores where they occupy the top position in the food chain, thereby serving as ecologically fundamental species for the stability of the ecosystem. Their decline or extinction may disrupt the food chains and alter the structure of ecological communities [7]. Sadly over the years, the activities of man have put on verge the future prospects of existence of many wild mammal species, especially large carnivores [8].
Lions (Panthera leo) once roamed most parts of Africa, Southern Europe, the Middle East and Asia [9]. Today they are only found in sub-Saharan Africa and at one locality in India where they are being increasingly restricted to protected areas and often in declining numbers [10]–[11]. In West Africa, lions are found only in protected areas such as national parks, game reserves and zoos. In Nigeria, the only protected areas known to still have wild lions are Yankari Game Reserve and Kainji-Lake National Park. The number of lions in these two isolates in West Africa has been poorly investigated. Population size estimate is an important biological parameter necessary for proper implementation of conservation measures [12]. Thus it is important to get adequate information on population size and connectivity between fragments of populations for proper conservation and management of a species [13]. Lions just like other large terrestrial carnivores are usually very difficult to count due to their elusive behavior and ability to cover large home ranges. An effort to conduct complete counts of a lion population is thus likely to be both organizationally difficult and time consuming [14].The alternative is to interpolate population sizes using different sampling strategies.
The use of DNA from non-invasive samples such as faeces, saliva, hairs or feathers for individual genetic tags can provide useful information for population monitoring as well as contributing with important genetic parameters [15]. Non-invasive sampling is widely used in genetic studies of elusive animal populations [16]–[18].This method is of prime importance in conservation genetics and behavioral ecology because it allows for genetic studies of wild animals without having to catch or even directly observe the animals under study [19], thereby reducing the possible amount of stress and harm inflicted on the animal. Various studies have employed the use of non-invasive sampling to identify individuals in a population, estimate population size [20], [12], [21], [16], [22], to monitor population sizes over time [23]–[24] and to also estimate the home range of individuals [20]. By studying the appropriate nuclear markers (most often microsatellites) analysis of non-invasive genetic samples (e.g. faeces) collected opportunistically from the field can provide individual identification, adequate information on population size, sex identification as well as genetic polymorphism within and between populations [25]. Knowledge of past events in a population and genetic structure is very important to assess the risk of extinction and chances of regional persistence of lion populations in Nigeria and elsewhere. Conservation of the genetic diversity of a species is important for preserving endangered wildlife [26]. This is because genetic diversity is the raw material for evolutionary change; that will allow the population to evolve in response to catastrophic changes such as new disease outbreak, pests, competition and predators.
In a pilot study carried out on the lions in Yankari Game Reserve (YGR from here on), Central North-Eastern Nigeria, between 2007–2008, we employed a population survey using genetic analysis of faecal DNA and showed the feasibility and reliability of the method [27]. In the present study, we employed the same method of faecal sample DNA analyses increasing the number of loci from two to nine to improve the precision of individual assignment. We also extended the survey to Kainji-Lake National Park (KLNP from here on), western Nigeria, which is a protected area that contains the most closely located and presumably only other Nigerian lion population to Yankari. We aimed to identify individual lions in YGR and KLNP and to determine the degree of genetic variability within and between these populations. Because the lions in the two parks have been isolated for several years, we expect them to be genetically differentiated. Also, the small population sizes suggest that there is a substantial level of inbreeding, which should be reflected by a positive inbreeding coefficient.
Materials and Methods
Ethics
The study was carried out with permission from the National Park service in the case of KLNP. In YGR, the A.P. Leventis Ornithological Research Institute had a Memorandum of Understanding (MoU) with the Bauchi State government to conduct any type of ecological research. Faeces are not part of the animal and as such faecal samples are not banned by CITES from being transported between countries.
Study sites
Faecal samples were collected from two protected areas: Yankari Game Reserve (YGR) and Kainji-Lake National Park (KLNP) (Fig. 1).
Figure 1. Map of Nigeria with some major cities and position of the two survey sites.

Kainji-Lake National Park (KLNP) in black rectangle, Yankari Game Reserve (YGR) in black triangle.
The YGR is located in Central North-Eastern Nigeria with a landmass of 2,244 km2 (9° 50′N and 10° 30′E). Detail site description for YGR is contained in Tende et al. [27].
KLNP is located in the western part of Nigeria (10° 22′N 04° 33′E) and occupies a landmass of 5,340 km2. The vegetation is made up primarily of Guinea savanna woodland; common woodland species include Terminalia macroptera found along the Oli River which flows in the centre of the Park, Detarium microcarpum and Borkea africana woodland occupy about 70% of the Park area. Isoberlinia tomentosa woodland play vital role in providing shelter and cover for game. The mean annual rainfall is between 1000 and 1200 mm per year and occurs between April and October, with the highest peak of rain in September [28].
Sample collection and extraction
A total of 3724 hours were spent sampling in YGR during data collection between 2008 and 2012, and 294 hours were spent sampling in KLNP in 2009, 2010 and 2012. The Global Positioning System (GPS) was used to record the position of each sample collected. Field methods are described in detail in Tende et al. [27]. All samples were preserved in 95% ethanol at room temperature, thereafter taken to Lund University, Sweden and kept in a freezer at −40°C before DNA extraction. DNA was extracted from all samples collected from YGR (n = 836) and KLNP (n = 93). DNA extraction was carried out using the QIAamp® DNA Stool Mini Kit (Qiagen).
Contamination of DNA during extraction or the PCR process can be a major problem when using non-invasive DNA; this was carefully taken care of by conforming to guidelines to avoid this through the use of a blank to control for contamination during the extraction and PCR processes.
Mitochondrial DNA
A short (206 bp) portion of the mitochondrial cytochrome b gene was amplified and sequenced using PCR-based methods with locus specific primers to confirm samples that were from lions and to exclude a few cases of spotted hyena (Crocuta crocuta) or striped hyena (Hyaena hyaena). The primers LIHYF (5′- ATGACCAACATTCGAAAATCWC-3′) and LIHYR (5′-ATGTGGGTSACTGATGAG-3′) were designed to avoid amplification of human and ungulate DNA in general, in order to promote detection of the target species [27]. All amplifications were done using 2X Qiagen multiplex PCR kit in 10 µl reaction volume containing 5 µl Qiagen multiplex PCR buffer mix; 0.2 µM forward primer (Applied Biosystems), 0.2 µM reverse primer, 2.6 µl water and 2 µl of DNA extract with a hot start at 95°C for 15 minutes. PCR profiles consisted of 35 cycles as follows: 90°C for 30 seconds; annealing temperature of 52°C for 30 seconds with elongation period of 72°C for 30 seconds. A blank control (reagents only) from DNA extraction process was included in all PCRs to monitor for contamination. The results of the PCR were evaluated by electrophoresis using 2% agarose gels and GelRed™ (Biotium) staining. Positive samples were sequenced using LIHY forward primer (BigDye sequencing kit; Applied Biosystems) in an ABI Prism® 3100 capillary sequencer (Applied Biosystems).
The sequences were aligned against species reference sequences (Ascension numbers; EF437586.1, AJ809332.1 and EF107524.1) obtained from Genbank to determine species identity.
Microsatellite amplification and genotyping
All lion samples were scored for allelic variability at nine polymorphic microsatellite primers (FCA001, FCA008, FCA026, FCA031, FCA045, FCA077, FCA126, FCA506 and FCA567 [29]. PCR amplifications were performed in 6 µl reactions containing 0.12 µl (concentration: 10 µM) dye-labelled (6-Fam, Hex or Ned) F-primer, 0.12 µl unlabelled R-primer (concentration: 10 µM), 3 µl of 2X Qiagen Master mix, 0.76 µl double distilled water and 2 µl DNA extract. PCRs were done in a GeneAmp 9700 thermocycler (Applied Biosystems) with the following profiles: 95°C for 15 min; 40 cycles at 94°C 30 s, 52°C 90 s, and 72°C 90 s; followed by an elongation period at 72°C for 10 min. Primers were multiplexed together in batches based on differences in fragment length and dye. The primer combinations are as follows: FCA001-FCA026-FCA031, FCA008-FCA045-FCA126, and FCA077-FCA506-FCA567. After amplification, alleles of the PCR products of the multiplex 3 loci, labelled with different dyes and of different lengths were separated using capillary electrophoresis in an ABI PRISM 3730 Genetic Analyzer (Applied Biosystems). Alleles were sized relative to GS500 ROX size standard and proof read and scored in Geneious vs. 5.6.6 (Biomatters).
Molecular sexing
Sex of identified individuals were determined using X and Y specific primers (SMCX17 and DBY7); [30]. The primers have been designed to avoid non-target amplification [31]. PCR amplifications were performed in 6 µl reactions containing 0.12 µl F-primer (concentration: 10 µM), 0.12 µl R-primer (concentration: 10 µM), 3 µl of 2X Qiagen Master mixes, 0.76 µl double distilled water and 2 µl DNA extract). PCR profile conditions for the multiplex amplification of the SMCX17 and DBY7 fragments are as follows: An initial denaturation of 95°C for 15 min, 20 cycles of 94°C for 30 sec, 60°C for 40 sec (decreasing 0.5 per cycle) and 72°C for 90 sec, followed by 20 cycles of 94°C for 30 sec, 50°C for 40 sec and 72°C for 90 sec and a final elongation period of 72°C for 15 min. After amplification, 2.5 µl of each PCR product was evaluated using 2% agarose gel with GelRed™ (Biotium) staining and samples with one band were scored as females (XX) and samples with two bands as males (XY).
Allelic drop-out
Most non-invasive sampling studies are often confronted with low quantity and quality DNA (i.e. degraded DNA) making it ideal to use PCR primers that amplify short DNA fragments [32], [33]. Highly degraded DNA may cause allelic drop-out which results in heterozygotes being typed as homozygotes due to failure of amplification of one of the alleles. To minimize or avoid genotype errors due to allelic drop out and false alleles three independent PCRs were performed for each locus and samples as suggested by Taberlet et al. [34]. No alleles were retained in further analysis unless they had been detected at least twice.
Partial genotypes are assigned in some individuals at some loci where only one allele could be observed more than once. Although there is a possibility that the partial genotypes might belong to a new individual, this method of assigning them with matching samples ensure conservative population estimation [35] by minimizing individuals created through erroneous, multi-locus genotypes (non-existent individuals). Only samples that amplified at between 4 and 9 loci were included in further analysis. According to Murphy et al. [36] a minimum of four loci are sufficient for accurate individual identification.
We scored samples as being from the same individual if they had been scored as the same sex, identical genotypes at ≥ four loci, and if the mismatching locus could be explained by allelic dropout. Such analyses allowed us to discern both the number of unique individuals as well as the number of “re-captured individuals”. Faeces with the same multilocus genotypes are considered as recaptures.
Genetic Analysis
We used the identity analysis module in the program CERVUS [37] to identify individuals with unique genotypes within the data set. We also calculated number of alleles (K), allelic richness (A), observed (HOBS) and expected (HEXP) levels of heterozygosity, probability of observing identical genotypes by chance among unrelated samples (P(ID)) and probability of observing identical genotypes by chance among siblings (P(ID)sibs)from the microsatellite genotype data. The probability of identity, P(ID), describes the probability that two individuals which are drawn at random from a population will have the same genotypes at multiple loci [25]. The software program CREATE [38] was used to create input files for use in the software program FSTAT v2.93 [39]. Inbreeding coefficient (FIS), population fixation index (FST) and Jost's estimate of genetic differentiation (Dest) were calculated using FSTAT and GenAIEx 6.5 [40]. Test for deviations from Hardy-Weinberg equilibrium exact test within populations was calculated based on 1000 randomisations, bootstrapping over loci at 95% CI. The nominal statistical significance value of 5/1000 was adjusted for multiple comparisons using the Bonferroni correction to minimize possible type I error. FST is used instead of RST [41] because it is considered to be a more reliable estimate of genetic differentiation when using small data set with less than 20 loci.
Waypoints of sampled genotypes were laid out on site maps to provide an overview of areas they were sampled during the survey in the two study sites (Fig. 2a and b). The software package Wild1[42] in R was used to obtain Minimum Convex Polygon of all individuals and afterwards the extension Xtools (version 9.2) in ArcView 3.3 was used to calculate the area covered by each individual in order to have an overview of their movement patterns. For individuals that were encountered more than three times, their home ranges were estimated by using the minimum convex polygon method. Since at least a minimum of three points are needed to make a polygon, this was computed only for individuals that were encountered at least three times during the survey.
Figure 2.

a: All genotypes sampled along the core area of Yankari Game Reserve. b: All genotypes sampled along the core area of Kainji-Lake National Park.
Results
Out of 929 samples collected from the two protected areas (YGR n = 836 and KLNP n = 93), 713 were successfully amplified for the partial cytochrome b (YGR, n = 625 and KLNP n = 88). The 216 samples that did not amplify at all were excluded from further analyses. Hence, the overall amplification success rate was about 77%. This is higher than the success rate of 40% (108 samples) that was obtained in the 2008 data from YGR [27]. There was no observed difference in the number of samples that successfully amplified in YGR and KLNP.
We determined species identity by aligning our sequences to Genbank reference sequences of the mitochondrial cytochrome b gene of lions, spotted hyena and striped hyena. Based on five informative nucleotide positions, 300 (42%) sequences out of the 713 samples matched with spotted hyena, while the remaining samples (n = 413) were found to match with lion.
To get individual identification, the 413 samples found to contain lion DNA from the two areas (YGR n = 352 and KLNP n = 61) were genotyped at the nine polymorphic microsatellites loci.
For the nine loci we obtained complete genotypes (at all 9 loci) for 115 samples from YGR and for 39 samples from KLNP. Partial genotypes (<9 loci) were obtained from additional 185 and 22 samples from Yankari and Kainji, respectively. Fifty two samples that amplified at less than four markers (i.e. 0–3 loci) were discarded from further analyses. Hence, the total number of samples with informative genotypes (≥4 loci) was 361.
In YGR the nine loci had a mean of 3.33 alleles with an expected heterozygosity (HEXP) of 0.63 (assuming Hardy-Weinberg equilibrium) and an observed heterozygosity (HOBS) of 0.32, whereas KLNP had a mean of 6.44 alleles with an expected heterozygosity (HEXP) of 0.82 and observed heterozygosity (HOBS) of 0.51 (Table 1). The lion populations in Kainji and Yankari showed significant signs of inbreeding with FIS = 0.49 in YGR and FIS = 0.38 in KLNP (p<0.001, Table 1). The two populations were found to be genetically differentiated with FST = 0.17 and Dest = 0.65 (bootstrapping over loci the ±95% CI of FST was between 0.10–0.23, p = 0.004).
Table 1. Summary of genetic diversity; number of alleles (K), allelic richness (A), sample size (N), Observed and Expected heterozygosity (HOBS & HEXP) and inbreeding coefficient (FIS) in the two populations over the years.
| Locus | N | K | A | HOBS | HEXP | FIS | N | K | A | HOBS | HEXP | FIS |
| FCA001 | 8 | 4 | 3.61 | 0.50 | 0.60 | 0.29 | 5 | 4 | 4.00 | 0.25 | 0.82 | 0.72 |
| FCA008 | 5 | 3 | 2.77 | 0.20 | 0.51 | 0.63 | 10 | 6 | 4.06 | 0.40 | 0.77 | 0.50 |
| FCA026 | 8 | 3 | 2.88 | 0.37 | 0.62 | 0.41 | 10 | 9 | 5.66 | 0.70 | 0.90 | 0.23 |
| FCA031 | 4 | 2 | 2.00 | 0.50 | 0.65 | 0.14 | 10 | 6 | 4.71 | 0.20 | 0.83 | 0.76 |
| FCA045 | 8 | 2 | 2.00 | 0.00 | 0.53 | 1.00 | 8 | 6 | 4.49 | 0.57 | 0.79 | 0.29 |
| FCA077 | 8 | 6 | 4.37 | 0.70 | 0.80 | 0.07 | 8 | 6 | 4.61 | 0.83 | 0.87 | −0.05 |
| FCA126 | 8 | 2 | 2.00 | 0.00 | 0.53 | 1.00 | 8 | 5 | 3.80 | 0.37 | 0.71 | 0.49 |
| FCA506 | 7 | 3 | 2.44 | 0.14 | 0.69 | 0.86 | 8 | 7 | 5.05 | 0.50 | 0.85 | 0.43 |
| FCA567 | 8 | 5 | 3.93 | 0.50 | 0.72 | 0.32 | 10 | 9 | 5.43 | 0.80 | 0.87 | 0.08 |
| Mean | - | 3.33 | 2.88 | 0.32 | 0.63 | 0.49 | - | 6.44 | 4.64 | 0.51 | 0.82 | 0.38 |
Yankari Game Reserve (N = 8) Kainji-Lake National Park (N = 10)
A total of eight individuals (2 males, 3 females, 3 unknown) were identified in YGR if we assume that allelic drop-outs had affected the scored genotypes (Table 2), while ten individuals were identified in KLNP (7males, 3 females, Table 3). The 3 individuals in YGR whose sex could not be determined molecularly were due to shortage of DNA template. Based on the observed allele frequencies there was a low probability of observing identical genotypes P(ID) from two randomly sampled individuals from the same population in both YGR (P(ID) = 0.00000213, P(ID)sibs = 0.00259) and KLNP(P(ID) = 0.00000000150, P(ID)sibs = 0.000188). The mean polymorphic information content (PIC) values in both YGR (0.50) and KLNP (0.73) were high.
Table 2. Identified individuals in Yankari Game Reserve.
| SampleID | IndvID | Sex | #times sampled | FCA001 | FCA026 | FCA031 | FCA567 | FCA007 | FCA506 | FCA008 | FCA045 | FCA126 |
| YGR321 | Y#1 | - | 4 | 129/155 | 128/128 | ***/*** | 85/85 | 135/144 | 244/244 | ***/*** | 125/125 | 124/124 |
| YGR242 | Y#2 | F | 107 | 127/127 | 128/137 | 242/244 | 94/96 | 146/153 | 214/214 | 117/117 | 127/127 | 124/124 |
| YGR211 | Y#3 | - | 5 | 129/155 | 128/128 | ***/*** | 85/85 | 130/135 | 244/244 | ***/*** | 125/125 | 124/124 |
| YGRB1 | Y#4 | M | 6 | 127/129 | 128/130 | ***/*** | 85/85 | 135/141 | 244/244 | 124/124 | 125/125 | 124/124 |
| YGRN1 | Y#5 | M | 21 | 127/127 | 128/128 | 242/242 | 94/105 | 141/153 | 191/191 | 117/117 | 127/127 | 127/127 |
| YGR13 | Y#6 | F | 14 | 127/127 | 128/137 | 244/244 | 96/105 | 141/153 | 191/214 | 117/117 | 127/127 | 127/127 |
| YGR56 | Y#7 | F | 1 | 137/155 | 130/130 | ***/*** | 85/85 | 135/135 | ***/*** | ***/*** | 125/125 | 124/124 |
| YGR7 | Y#8 | - | 1 | 127/127 | 137/137 | 242/244 | 94/107 | 153/153 | 214/214 | 117/117 | 127/127 | ***/*** |
Shown are; Sample identity (SampleID), individual identity (IndvID), sex and allelic length at the nine scored loci, *** (indicates missing data).
Microsatellite Loci
Table 3. Identified individuals in Kainji-Lake National Park.
| Microsatellite loci | ||||||||||||
| SampleID | IndvID | Sex | #times sampled | FCA001 | FCA026 | FCA031 | FCA567 | FCA077 | FCA506 | FCA008 | FCA045 | FCA126 |
| KLNP13 | K#1 | M | 4 | 127/129 | 130/137 | 234/246 | 98/103 | 148/153 | 193/*** | 129/133 | 127/127 | ***/*** |
| KLNP18 | K#2 | M | 1 | ***/*** | 122/130 | 224/224 | 78/78 | ***/*** | 229/229 | 129/129 | ***/*** | 129/191 |
| KLNP19 | K#3 | M | 1 | ***/*** | 139/141 | 238/238 | 81/85 | 137/141 | 203/214 | 125/125 | 139/153 | 133/151 |
| KLNP4 | K#4 | M | 3 | ***/*** | 134/139 | 238/252 | 83/85 | 137/139 | 195/203 | 119/133 | 139/139 | 139/139 |
| KL26 | K#5 | M | 1 | 145/145 | 141/141 | 238/238 | 83/85 | 137/153 | 195/195 | 125/125 | 139/139 | 133/133 |
| KL18 | K#6 | F | 7 | ***/*** | 137/137 | 246/246 | 103/105 | 148/151 | 191/191 | 133/133 | 145/149 | 131/139 |
| KL11 | K#7 | F | 7 | 153/153 | 134/141 | 238/238 | 83/85 | 137/139 | 195/195 | 125/125 | 139/139 | 133/133 |
| KL9 | K#8 | M | 1 | 129/129 | 128/130 | 234/234 | 98/111 | 148/153 | 85/191 | 133/133 | ***/*** | 139/139 |
| KL33 | K#9 | F | 1 | 153/153 | 134/139 | ***/*** | 85/85 | 153/153 | 193/208 | 117/133 | ***/*** | 133/133 |
| K22 | K#10 | M | 1 | 145/153 | 144/148 | 244/244 | 83/94 | 137/*** | 197/203 | 127/129 | 149/153 | 133/*** |
Shown are: Sample identity (SampleID, individual identity (IndvID), sex and allelic length at the nine scored loci, *** (indicates missing data).
Within both survey areas, some of the identified individual genotypes were encountered in more than one year (YGR; n = 6, KLNP; n = 3), while some were encountered several times within a year and others were encountered only once or twice. In YGR individual Y#2 (female) and Y#5 (male) were observed to be present in the population all through the five year period. Individual Y#6 (female) was sampled from the beginning of the survey until 2011 (Fig. 3a). The combined genotypes of Y#2 and Y#5 are compatible with the hypothesis that the genotypes of Y#6 and Y#8 are their offspring (Table 2).
Figure 3.
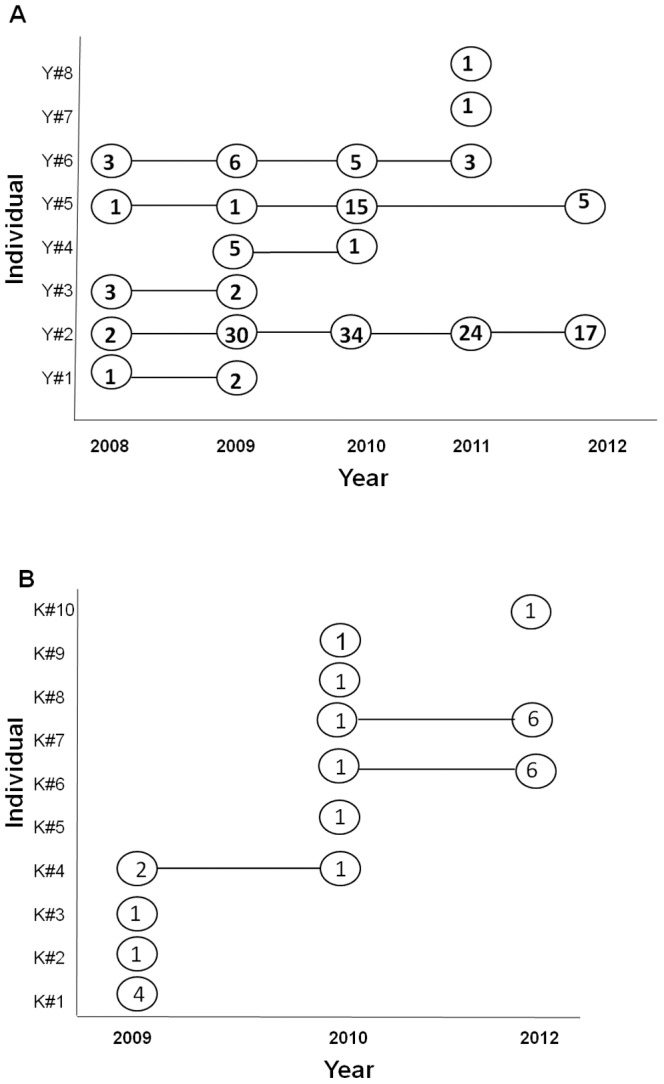
a: Sampling frequency per individual and year in Yankari Game Reserve. b: Sampling frequency per individual and year in Kainji-Lake National Park.
In both YGR and KLNP all individuals were sampled within the core area of the reserve (Fig. 2a and b). The home range analysis using the minimum convex polygon method gave an average home range of 11.91 km2, ±2.1 SD (1.71– 47.62 km2) in YGR (Appendix S1) and 26.75 km2 ± 1 SD (14.63– 39.37 km2) in KLNP (Appendix S2).
Discussion
Number of individuals
Based on the encounter of unique genotypes during the different survey years, a minimum of eight individuals was identified in YGR between 2008 and 2012, while ten individuals were identified in KLNP between 2009, 2010 and 2012. During the course of the survey there has been at least one observation of new cubs in the field in 2010 in YGR. Recently, there have also been sightings (between 8th and 12th March 2013) of 2 adult females with 3 cubs, 2 adult females without any cubs, 1 adult male with a mane together with 3 cubs in YGR. The sightings of these adults within YGR (1 male and 4 females, or 1 male and 2 females, (if we assume that it was the same females that were seen at different times) contradict the findings of Wildlife Conservation Society (WCS) who has reported, through call-up stations, that only two adult lions exist within the reserve [43]. These sightings are also confirmed by our molecular findings (Table 2), where females Y#2 and Y#6 and male Y#5 have been present most parts of the survey periods. Six of the eight identified individuals have been sampled multiple times (3-107 times) during the survey (Fig 3a). Our estimate gives the minimum number of lions that exist in both YGR and KLNP. Since several individuals have been encountered only once, especially in KLNP, the true number is potentially higher. The low estimates of probability of identity (P(ID) and P(ID)sibs) obtained in our study both in YGR and KLNP gives confidence to the identity of individuals identified in these two survey areas and support the uniqueness of the identified individuals. Waits et al. [44] suggested that these values should be between P(ID) = 0.01 – 0.0001 in studies estimating population size.
Despite the fact that there were fewer visits and also few samples collected from KLNP, we observed more individuals as well as a higher genetic diversity than in YGR. This suggests that there are and have been more individuals in the recent history in KLNP, which have contributed to a larger effective population size and a higher genetic diversity. Alternatively, the high diversity in KLNP may have resulted from connecting with gene flow with lions in the W-Arli-Pendjari complex, a vast protected area in Benin, Burkina Faso and Niger.
Comparison with previous study
A number of studies conducted on the natural populations of African lions have been carried out mostly in Eastern, Central and Southern Africa and in India. These studies have employed opportunistic field observation and/or genetic data to understand behavioral, social and breeding structure of the lions [45]–[53]. Researchers have incorporated molecular methods to understand the genetic status of the lions in their natural populations (c.f. [50], [54]), characterize their evolutionary history, as well as to study disease outbreaks [55]–[58]. Our study has employed non-invasive samples with aid of molecular techniques to make available information on the population size as well as the genetic status of the remaining relict wild lion populations in Nigeria. The method does not have any negative impact on the study species.
The number of individuals estimated in YGR in the present study is lower than the number recorded during the pilot survey conducted by Tende et al. [27] where eleven individuals were identified from two microsatellite loci. This difference could be due to either a disappearance from the population of some individuals due to natural deaths or to the activities of poachers within the reserve, or it could be an overestimation in our previous study due to the low number of loci used. The use of polyacrylamide gel to genotype individuals during our pilot study [27] could possibly also have biased our estimation due to bad gels where stutter bands might have been typed as true bands.
During the course of the laboratory analysis and efforts to optimize different primers for the study, we ran short of DNA template from most of the extracted samples from the first pilot study in YGR. Hence, not all samples analyzed in our 2008/2009 pilot survey could be rerun on the new microsatellite primers used. However, in some samples where we still had DNA template it was observed that certain individuals have persisted in the population throughout the survey years.
The higher amplification success rate attained in this study as compared to 2007/2008 could be attributed to the new PCR kit/technique (Qiagen multiplex PCR kit) that was employed and also we concentrated our sample efforts on fairly fresh faeces. The shorter time the faeces are left out in the environment, the less degradation of DNA and this is especially true in tropical environments where high temperature and UV radiation, can cause fast degradation of DNA [59].
Home range estimates
Many of the identified individual genotypes were encountered several times during the study. Lions are known to use large home ranges to satisfy energetic demands, but this can be limited where required resources have clumped distribution [50], [60]. This is the case during the dry season, for both study sites, when game concentrates close to rivers - Gaji River in YGR and Oli River in KLNP. Both rivers run through the core areas of the respective reserve/park and offer lush vegetation for prey species, as well as cover for the lions to rest and also stalk. It is also the main source of drinking water both for the predators and the prey. A similar survey by Spong et al. [50] to determine space use by lions in Selous Game Reserve, Tanzania, showed the most intensively used area to be small within the reserve and averaged only 11.7 km2, which is consistent with our finding in YGR where individuals were sampled mostly within the core areas which are intensively used and this averaged at 11.9 km2 (SD: ±2.1). They found out that prides often had close relatives in neighbouring prides but in areas with high prey abundance the home ranges between individuals, irrespective of relatedness tended to overlap more [50]. A survey by Lehmann et al. [60] on home range utilization of lions in Karongwe Game Reserve, South Africa, showed the home range used by a pride ranged between (10.3 – 64.4 km2) and for a single male ranged between (5.0 – 56.3 km2). Their findings about home range size are slightly larger than what we have obtained in our study (YGR: 1.71– 47.62 km2; KLNP: 14.63–39.37 km2). In our study a female (Y#2) was observed to have the largest home range (47.52 km2), this might probably be due to the need to hunt and feed the cubs and the male. This female has also been observed to persist in the population since the onset of this survey in 2007/2008. Both of the above studies [50], [60] employed the use of telemetry. Our study has shown that genotype data also can be used to determine the home range of lions, and possibly other mammals, enhancing sample size and reducing disturbance to the animal under study.
Inbreeding
The populations of lions in both KLNP and YGR exhibit significant signs of inbreeding. This is not surprising given their small population sizes. The inbreeding level found in YGR in the present study is in line with our pilot study conducted in 2008 [27] when the inbreeding coefficient was estimated to be 0.21, whereas in this present survey the value was found to be 0.49 (Table 1). The inbreeding levels in both YGR (0.49) and KLNP (0.38) are high and comparable to what has been recorded in some other carnivore species. For instance the estimated inbreeding coefficient in the Scandinavian wolf (Canis lupus) population was up to 0.41 [61], [62], [17] before the population was genetically rescued by one immigrant from Finland [63]. The arrival of this immigrant into the Scandinavian wolf population provided the possibility to avoid inbreeding, decrease the risk of inbreeding depression and cause population growth. High inbreeding coefficient reaching up to 0.37 has also been recorded in the brown bear (Ursus arctos) [64]. Inbreeding and subsequent negative effects of inbreeding have been reported in the lions in Ngorongoro Crater in Tanzania [65], [48]. The number of alleles at a set of microsatellite loci for the Etosha lion population (A = 4.6) reported by Lyke et al. [53], and that reported by Antunes et al. [58] (A = 4.4) is similar to that detected in KLNP (A = 4.6), but higher than in YGR (A = 2.8), where Lyke et al. [53] detected no sign of inbreeding in the Etosha population (FIS = 0.03). The inbreeding coefficient recorded in our study is higher than what has been recorded in the lion population in Etosha National Park in Namibia. Some studies (e.g., [4], [66]–[68]) have shown that if populations remain small and isolated for many generations they are bound to face increased inbreeding and gradual erosion of genetic variability. The lion populations in Nigeria are small, isolated and restricted to two protected areas (YGR and KLNP) where their populations may be declining. This is of course a threat to the long-term survival of these populations and to the lion population throughout West Africa [69].
Low genetic variability has been reported in lions in Ngorongoro Crater in Tanzania [48], [55], [70], [71]. Although there are recent observations of cubs within one of our study systems (YGR) the genetic status of this population as shown by our study is poor. This might affect individual survival and reproductive success in the long term if proper measures are not implemented. O'Brien [72] has observed that there is a strong correlation between genetic variation and reproductive parameters in lions. This means that with time the fitness of individuals within the lion populations in Kainji and Yankari will decline due to accumulation and expression of recessive and detrimental alleles with subsequent inbreeding depression, which might consequently drive the population to extinction.
Population structure
The YGR and KLNP populations were found to be genetically differentiated (FST = 0.17, Dest = 0.65). This is not surprising due to the fact that these populations are small and isolated from each other; about 1000 km apart and they are separated by dispersal barriers including highways, agricultural landscapes and cities. Without any corridor for dispersal, isolation is expected to build up the observed pattern of allelic differentiation between the two populations. Moreover, high human and livestock densities characterize most of the surroundings of these protected areas, which can increase mortality risk of cubs and “possible dispersers” because of overlap with human habitations and livestock. All these factors may act as barriers to gene flow between the two populations and possible populations in neighboring countries. Studies have shown that geographical and environmental features can influence gene flow and genetic variation within and between populations [73], [74]. Studies carried out in California on mountain lion (Puma concolor), and coyote (Canis latrans) and bobcat (Lynx rufus) to assess the level of gene flow and differentiation in these carnivores showed how anthropogenic obstacles such as roads, agriculture and urbanization were great barriers to dispersal and gene flow [75], [76]. It was shown how these barriers imposed artificial home range boundaries on territorial carnivores thereby decreasing genetically effective migration, which leads to population differentiation.
A study carried out by Antunes et al. [58] to assess genetic variation from 357 lions from most of its current range in Africa and Asia using microsatellite data revealed significant population structure (FST = 0.03–0.79) within East-African lion populations despite the low geographic distance within them. Their finding is in accordance with our finding about the two lion populations in Nigeria (Fst = 0.17).
The existence and maintenance of genetic diversity and good connectivity between subpopulations are essential factors for long-term viability of a population [77], which should serve as the primary target of any acceptable conservation management program. Genetic diversity is the raw material needed in order to evolve the ability to cope with environmental challenges such as disease outbreak and parasites [78].
Conservation genetics and management
The two lion populations are small and genetically different with signs of inbreeding within each population. This might elevate their risk of extinction in the face of sudden environmental catastrophes in the future. It has been shown that small populations are often faced with a higher risk of extinction from environmental catastrophes [3], [4], [5], [79]–[83] than large interconnected populations [84], [77], [85].
The main aim of a conservation genetic approach is to maintain diversity within and between populations so as to enhance the evolutionary potential of the population to cope with environmental changes. Laboratory and translocation experiments have indicated that small and inbred populations can be rescued by contribution of minimal number of immigrants [86]–[88], [62], [63]. This can help to decrease inbreeding and inbreeding depression [86], [89], and bring about profound changes in genetic structures [90], [91], [63]. Thus to mitigate the inbreeding condition and subsequent probable inbreeding depression in the lion population within Nigeria it might be important to transfer lions between parks or reserves or to reintroduce lions from the zoo back to the wild. This is necessary in order to enhance their genetic diversity for long term survival and reproduction. The bringing together of genetically dissimilar mates, hybrid vigor [89]–[91] can be advantageous because it will enhance reproductive success and also fitness [92]–[93], [68]. However, it would be recommended that lions that are genetically similar to the receiving population should be preferably used in any translocation program in order to avoid introduction of potentially non-locally adapted genotypes.
Supporting Information
Estimated home ranges for some individuals in Yankari Game Reserve.
(TIF)
Estimated home ranges for some individuals in Kainji-Lake National Park.
(TIF)
Acknowledgments
We will like to thank the following people for their enormous contributions to the success of this survey; the director and staff of A.P. Leventis Ornithological Research Institute (APLORI) Jos for all the logistic supports. A big thank you to all the staff of APLORI research centre Yankari Game Reserve and Taiwo Crossby Omotoriogun who was our faithful, hardworking and dedicated field assistant in 2009. Thanks to all friends and colleagues who helped to collect samples in Yankari: Onoja Joseph, Adetutu Ojomo, Himma Bakam, Shomboro Karau Dauda and Haladu Idi. We also thank Dr. Yahkat Barshep, Chima Nwogu, Martin Stervander and Onoja Joseph for helping with maps and analysis of the home range. Finally, we thank the management of Yankari Game Reserve and Kainji Lake National Park for allowing field work. This is contribution no. 68 from A.P. Leventis Ornithological Research Institute.
Funding Statement
Kolmården zoo conservation fund Sweden and the Leventis foundation provided the funds for the field and laboratory work. The study was supported by grants from the Swedish Research Council (621-2009-4945 to BH; 621-2010-5277 to SB). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Ceballos G, Ehrlich PR (2002) Mammalian population losses and the extinction crisis. Science 296 : ((5569)) 904–907. [DOI] [PubMed] [Google Scholar]
- 2. Frankham R (1995) Inbreeding and Extinction- a Threshold Effect. Conserv Biol 9 ((4)) 792–799. [Google Scholar]
- 3.Saccheri I, Kuussaari M, Kankare M, Vikman P, Fortelius W, et al. (1998) Inbreeding and extinction in a butterfly metapopulation. Nature 392: :491–494. [Google Scholar]
- 4. Bijlsma R, Bundgaard J, Boerema AC (2000) Does inbreeding affect the extinction risk of small populations? predictions from Drosophila. J Evol Biol 13: 502–514. [Google Scholar]
- 5. Higgins K, Lynch M (2001) Metapopulation extinction caused by mutation accumulation. Proc Natl Acad Sci USA 98: 2928–2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Crooks KR (2002) Relative sensitivities of mammalian carnivores to habitat fragmentation. Conserv Biol 16: 488–502. [Google Scholar]
- 7. Crooks KR, Soule ME (1999) Mesopredator release and avifaunal extinctions in a fragmented system. Nature 400: 563–566. [Google Scholar]
- 8. Morris DW, Kotler BP, Brown JS, Sundararaj V, Ale SB (2009) Behavioral indicators for conserving mammal diversity. Ann New York Acad Sci 1162: 334–356. [DOI] [PubMed] [Google Scholar]
- 9. Bauer H, Vander Merwe S (2004) Inventory of free ranging lions Panthera leo in Africa. Oryx 38: 26–31. [Google Scholar]
- 10.Nowell K, Jackson P, Compilers and editors (1996) Wild cats: Status survey and Conservation Action Plan. IUCN/SSC Cats Specialist Group. IUCN, Gland, Switzerland.
- 11.Wilson DE, Mittermeier RA, editors (2009) Handbook of the Mammals of the World.Vol.1. Carnivores. Lynx Ediciones. Barcelona.
- 12. Kohn MH, York EC, Kamradt DA, Haught G, Sauvajot RM, et al. (1999) Estimating population size by genotyping faeces. Proc R Soc Lond B 266: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Creel S, Spong G, Sands JL, Rotella J, Zeigle J, et al. (2003) Population size estimation in Yellowstone wolves with error-prone noninvasive microsatellite genotypes. Mol Ecol 12: 2003–2009. [DOI] [PubMed] [Google Scholar]
- 14. Balme GA, Hunter LTB, Slotow R (2007) Evaluating Methods for Counting Cryptic Carnivores. J Wildl Mgt 73 (3): 433–441. [Google Scholar]
- 15. Bellemain E, Taberlet P (2004) Improved non-invasive genotyping method: application to brown bear (Ursus arctos) faeces. Mol Ecol Notes 4: 519–522. [Google Scholar]
- 16. Ernest HB, Penedo MCT, May BP, Syvaren M, Boyce WM (2000) Molecular tracking of mountain lions in the Yosemite Valley region in California: genetic analysis using microsatellites and faecal DNA. Mol Ecol 9: 433–441. [DOI] [PubMed] [Google Scholar]
- 17. Bensch S, Andren H, Hansson B, Pedersen HC, Sand H, et al. (2006) Selection for heterozygosity gives hope to a wild population of inbred wolves. PLoS ONE 1(1): e72 doi:10.1371/journal.pone.0000072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang HM, Guo Y, Li DS, Wang PY, Fang SG (2009) Sixteen novel microsatellite loci developed for the giant panda (Ailuropoda melanoleuca). Conserv Genet 10: 589–592. [Google Scholar]
- 19. Taberlet P, Luikart G (1999) Non-invasive genetic sampling and Individual Identification. Biol J Lin Soc 68: 41–55. [Google Scholar]
- 20. Taberlet P, Camarra JJ, Griffin S, Uhres E, Hanotte O, et al. (1997) Nonivasive genetic tracking of the endangered Pyrenean brown bear population. Mol Ecol 6: 869–876. [PubMed] [Google Scholar]
- 21. Woods JG, Paetkau D, Lewis D, McLellan BN, Proctor M, et al. (1999) Genetic tagging free-ranging black and brown bears. Wildl Soc Bull 27: 616–627. [Google Scholar]
- 22. Waits LP, Luikart G, Taberlet P (2001) Estimating the probability of identity among genotypes in natural populations: cautions and guidelines. Mol Ecol 10: 249–256. [DOI] [PubMed] [Google Scholar]
- 23. Kendall KC, Metzgar LG, Patterson DA, Steele BM (1992) Power of sign surveys to monitor population trends. Ecol Appl 2: 422–430. [DOI] [PubMed] [Google Scholar]
- 24. Schwartz M, Tallmon D, Luikart G (1998) Review of DNA-based census and effective population size estimators. Anil Conserv 1: 293–299. [Google Scholar]
- 25. Kohn MH, York EC, Karmradt DA, Haught G, Sauvajot RM, et al. (1999) Estimating population size by genotyping faeces. Proc R Soc Lond B 266: 657–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Uphyrkina O, O'Brien SJ (2003) Applying molecular genetics tools to the conservation and action plan for the critically endangered Far Eastern leopard (Panthera pardus orientalis).Comp R Biol. 326: 93–97. [DOI] [PubMed] [Google Scholar]
- 27. Tende T, Ottosson U, Hansson B, Åkesson M, Bensch S (2010) Population size of lions in Yankari Game Reserve as revealed by faecal DNA sampling. J Afr Ecol 59: 7–12. [Google Scholar]
- 28. Afolayan TA (1978) The effect of fires on the vegetation in Kainji lake National Park, Nigeria. Oikos 33: 376–382. [Google Scholar]
- 29. Menotti RM, David VA, Lyons LA, Schaffer AA, Tomlin JF, et al. (1999) A genetic linkage map of microsatellites in the domestic cat (Felis catus). Genomics 57: 9–23. [DOI] [PubMed] [Google Scholar]
- 30. Hellborg L, Ellegren H (2004) Low levels of nucleotide diversity in mammalian Y chromosomes. Mol Biol Evol 21: 158–163. [DOI] [PubMed] [Google Scholar]
- 31. Seddon JM (2005) Canid-specific primers for molecular sexing using tissue or non-invasive samples. Conserv Genet 6: 147–149. [Google Scholar]
- 32. Taberlet P, Waits LP, Luikart G (1999) Noninvasive genetic sampling: look before you leap. Trends Ecol Evol 14(8): 323–327. [DOI] [PubMed] [Google Scholar]
- 33. Pompanon F, Bonin A, Taberlet P (2005) Genotyping errors: causes, consequences and solutions. Nature 6: 847–859. [DOI] [PubMed] [Google Scholar]
- 34. Taberlet P, Griffin S, Goossens B, Questiau S, Manceau V, et al. (1996) Reliable Genotyping of Samples With Very Low DNA Quantities Using PCR: Nucleic Acids Res. 24: 3189–3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Frantz AC, Pope LC, Carpenter PJ, Roper TJ, Wilson GJ, et al. (2003) Reliable microsatellite genotyping of the Eurasian badger (Meles meles) using faecal DNA. Mol Ecol 12: 1649–1661. [DOI] [PubMed] [Google Scholar]
- 36. Murphy MA, Waits LP, Kendall KC, Wasser SK, Higbee JA, et al. (2002) An evaluation of long-term preservation methods for brown bear (Ursus arctos) faecal DNA samples. Conserv Genet 3: 435–440. [Google Scholar]
- 37. Marshall TC, Slate J, Kruuk LEB, Pemberton JM (1998) Statistical confidence for likelihood-based paternity inference in natural populations. Mol Ecol 7: 639–655. [DOI] [PubMed] [Google Scholar]
- 38. Coombs JA, Letcher BH, Nislov KH (2008) CREATE: a software to create input files from diploid genotypic data for 52 genetic software programs. Mol Ecol Res 8: 578–580.Version.1.31. [DOI] [PubMed] [Google Scholar]
- 39.Goudet J (2002) FSTAT, a computer program to estimate and test gene diversities and fixation indices Version 2.9.3.2.
- 40. Peakall R, Smouse PE (2006) GenAlEx 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6: 288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Slatkin M (1995) A measure of population subdivision based on microsatellite allele frequencies. Gen Mol Biol 139: 457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sargeant GA (2011) wild1: R tools for wildlife research and management. R package version 1.09. Geological Survey Northern Prairie Wildlife Research Center, Jamestown, ND, USA.
- 43.Nyanganji G, Saidu Y, Henschel P, Dunn A (2012) 2011 survey of lion (Panthera leo) in Yankari Game Reserve and Kainji Lake National Park, Nigeria (A submitted report National Park service).
- 44. Waits LP, Luikart G, Taberlet P (2001) Estimating the probability of identity among genotypes in natural populations: cautions and guidelines. Mol Ecol 10: 249–256. [DOI] [PubMed] [Google Scholar]
- 45. Packer C, Pusey AE (1982) Cooperation and competition within coalitions of male lions: kin selection or game theory? Nature 296: 740–74. [Google Scholar]
- 46. Pusey AE, Packer C (1987) The evolution of sex-biased dispersal in lions. Behaviour 101: 275–310. [Google Scholar]
- 47. Gilbert DA, Packer C, Pusey AE, Stephens JC, O'Brien SJ (1991) Analytical DNA fingerprinting in lions: parentage, genetic diversity, and kinship. J Heredity 82: 378–386. [DOI] [PubMed] [Google Scholar]
- 48. Packer C, Pusey AE, Rowley H, Gilbert DA, Martenson J, et al. (1991) Case study of a population bottleneck: Lions of the Ngorongoro Crater. Conserv Biol 5 (2): 219–230. [Google Scholar]
- 49. Spong G, Creel S (2001) Deriving dispersal distances from genetic data. Proc R Soc Biol Sci Ser B 268: 2571–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Spong G, Stone J, Creel S, Björklund M (2002) Genetic structure of lions (Panthera leo L.) in the Selous Game Reserve: implications for the evolution of sociality. J Evol Biol 15: 945–953. [Google Scholar]
- 51. Kays RW, Patterson BD (2002) Mane variation in African lions and its social correlates. Can J Zool 80: 471–478. [Google Scholar]
- 52. Dubach J, Patterson BD, Briggs MB, Venzke K, Flamand J, et al. (2005) Molecular genetic variation across the southern and eastern geographic ranges of the African lion, Panthera leo. Conserv Genet 6: 15–24. [Google Scholar]
- 53.Lyke MM, Dubach J, Briggs MB (2013) A molecular analysis of the African lion (Panthera leo) mating structure and extra-group paternity in Etosha National Park. Mol Ecol DOI: 10. 1111/mec.12279. [DOI] [PubMed]
- 54. Shankaranarayanan P, Banerjee M, Kacker RK, Aggarwal RK, Singh L (1997) Genetic variation in Asiatic lions and Indian tigers. Electrophoresis 18: 1693–1700. [DOI] [PubMed] [Google Scholar]
- 55. Brown EW, Yuhki N, Packer C, O'Brien SJ (1994) A lion lentivirus related to feline immunodeficiency virus: epidemiologic and phylogenetic aspects. J Virol 68: 5953–5968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Roelke ME, Munson L, Packer C, Kock R, Cleaveland S, et al. (1996) A canine distemper virus epidemic in Serengeti lions (Panthera leo). Nature 379: 441–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Packer C, Altizer S, Appel M, Brown E, Martenson J, et al. (1999) Viruses of the Serengeti: patterns of infection and mortality in African lions. J Anil Ecol 68: 1161–1178. [Google Scholar]
- 58.Antunes A, Troyer JL, Roelke ME, Pecon-Slattery J, Packer C, et al. (2008) The Evolutionary Dynamics of the Lion Panthera leo Revealed by Host and Viral Population Genomics. Plos Gen 4 (11): e1000251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Piggott MP, Bellemain E, Taberlet P, Taylor AC (2004) A multiplex pre-amplification method that significantly improves microsatellite amplification and error rates for faecal DNA in limiting conditions. Conserv Genet 5: 417–420. [Google Scholar]
- 60. Lehmann MB, Funston PJ, Owen CR, Slotow R (2008) Home Range Utilization and Territorial Behaviour of Lions (Panthera leo) on Karongwe Game Reserve, South Africa. PLOS ONE3(12): e3998 doi:10.1371/journal.pone.0003998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Laikre L, Ryman N (1991) Inbreeding depression in a captive Wolf (Canis lupus) population. Conserv Biol 5: 33–40. [Google Scholar]
- 62. Liberg O, Andren H, Pedersen CH, Sand H, Sejberg D, et al. (2005) Severe inbreeding depression in a wolf Cani lupus population. Biol Lett 1: 17–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Vila C, Sundqvist AK, Flagstad Ø, Seddon J, Blörnerfeldt S, et al. (2003) Rescue of a severely bottlenecked wolf (Canis lupus) population by a single immigrant. Proc R Soc Lond B 270: 91–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Laikre L, Andren R, Lars H, Ryman N (1996) Inbreeding depression in Brown Bear (Ursus arctos). Biol Conserv 76: 69–72. [Google Scholar]
- 65. O'Brien SJ, Martenson JS, Packer C, Herbst L, de Vos V, et al. (1987) Biochemical genetic variation in a geographic isolates of African and Asiatic lions. Natl Geo Res 3: 114–124. [Google Scholar]
- 66. Lande R (1995) Mutation and conservation. Conserv Biol 9: 782–791. [Google Scholar]
- 67. Laikre L (1999) Conservation genetics of Nordic carnivores: lessons from zoos. Hereditas 130 (3): 203–216. [DOI] [PubMed] [Google Scholar]
- 68. Keller LF, Waller DM (2002) Inbreeding effects in wild populations. Trends Ecol Evol 17: 230–241. [Google Scholar]
- 69.Frankham R, Ballou JD, Briscoe DA (2002) Introduction to Conservation Genetics. Cambridge University Press, Cambridge, UK.
- 70. Munso L, Brown JL, Packer C, Janssen D, Reiziss SM, et al. (1996) Genetic diversity affects morphology in free-ranging lions (Panthera leo) of the Serengeti Plains and Ngorongoro Crater. J Rep Fer 108: 11–15. [DOI] [PubMed] [Google Scholar]
- 71. Wildt DE, Bush M, Goodrowe KL, Packer C, Pusey AE, et al. (1987) Reproductive and genetic consequences of founding isolated lion populations. Nature 329: 328–331. [Google Scholar]
- 72. O'Brien S (1994) A role for molecular genetics in biological conservation. Proc Natl Acad Sci USA 91: 5748–5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Manel S, Schwartz MK, Luikart G, Taberlet P (2003) Landscape genetics: combining landscape ecology and population genetics. Trends Ecol Evol 18: 189–197. [Google Scholar]
- 74. McRae BH, Beier P, Dewald LE, Huynh LY, Keim P (2005) Habitat barriers limit gene flow and illuminate historical events in a wide-ranging carnivore, the American puma. Mol Ecol 14: 1967–1977. [DOI] [PubMed] [Google Scholar]
- 75. Ernest HB, Boyce WM, Bleich VC, May B, Stiver SJ, et al. (2003) Genetic structure of mountain lion (Puma concolor) populations in California. Conserv Genet 4: 353–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Riley SPD, Pollinger JP, Sauvajot RM, York EC, Bromley C, et al. (2006) A southern California freeway is a physical and social barrier to gene flow in carnivores. Mol Ecol 15: 1733–1741. [DOI] [PubMed] [Google Scholar]
- 77. Frankham R (1996) Relationship of genetic variation to population size in wildlife. Conserv Biol 10: 1500–1508. [Google Scholar]
- 78.Frankham R, Kingslover JG (2004) Response to environmental change: Adaptation or extinction. In: Ferrier R., Dieckman U., Couvet D (Eds.) Evolutionary Conservation Biology. Cambridge University Press, Cambridge pp.85–100.
- 79. Frankel OH (1970) Variation, the essence of life. Proc Linn Soc New South Wales 95: 158–169. [Google Scholar]
- 80. Frankel OH (1974) Genetic conservation: our evolutionary responsibility. Genetics 78: 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lande R (1988) Genetics and demography in biological conservation. Science 214: 1455–1460. [DOI] [PubMed] [Google Scholar]
- 82. Woodroffe R, Ginsberg JR (1998) Edge effects and extinction of populations inside protected areas. Science 280: 2126–2128. [DOI] [PubMed] [Google Scholar]
- 83. Dunham J, Peacock M, Tracy CR, Nielsen J, Vinyard G (1999) Assessing extinction risk: integrating genetic information. Conserv Ecol [online] 3(1): 2 Available: http://www.consecol.org/vol3/iss1/art2/. [Google Scholar]
- 84. Lande R (1993) Risks of population extinction from demographic and environmental stochasticity and random catastrophes. Am Nat 142: 911–927. [DOI] [PubMed] [Google Scholar]
- 85. Lacy RC (2000) Considering threats to the viability of small populations using individual-based models. Ecol Bull 48: 39–52. [Google Scholar]
- 86. Spielman D, Frankham R (1992) Modelling problems in conservation genetics using captive Drosophila populations: improvement of reproductive fitness due to immigration of one individual into small partially inbred populations. Zoo Biol 11: 343–351. [Google Scholar]
- 87. Westemeier RL, Brawn JD, Simpson SA, Esker TL, Jansen RW, et al. (1998) Tracking the long-term decline and recovery of an isolated population. Science 282: 1695–1698. [DOI] [PubMed] [Google Scholar]
- 88. Madsen T, Shine R, Olsson M, Wittzell H (1999) Conservation biology: restoration of an inbred adder population. Nature 402: 34–35. [Google Scholar]
- 89. Ebert D, Haag C, Kirkpatrick M, Riek M, Hottinger JW, et al. (2002) A selective advantage to immigrant genes in a Daphnia meta population. Science 295: 485–488. [DOI] [PubMed] [Google Scholar]
- 90. Ball SJ, Adams M, Possingham HP, Keller MA (2000) The genetic contribution of single male immigrants to small, inbred populations: a laboratory study using Drosophila melanogaster. Heredity 84: 677–684. [DOI] [PubMed] [Google Scholar]
- 91. Saccheri IJ, Brakefield PM (2002) Rapid spread of immigrant genomes into inbred populations. Proc R Soc Lond B 269: 1073–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Amos W, WorthingtonWilmer J, Fullard K, Burg TM, Croxall JP, et al. (2001) The influence of parental relatedness on reproductive success. Proc R Soc Lond B 268: 2021–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Hedrick PW, Kalinowski ST (2000) Inbreeding depression in conservation biology. Ann Rev Ecol Syst 31: 139–162. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Estimated home ranges for some individuals in Yankari Game Reserve.
(TIF)
Estimated home ranges for some individuals in Kainji-Lake National Park.
(TIF)