

Abstract
Carbonic anhydrases (CAs) comprise a family of zinc-containing enzymes that catalyze the reversible hydration of carbon dioxide. CAs contribute to a myriad of physiological processes, including pH regulation, anion transport and water balance. To date, 16 known members of the mammalian alpha-CA family have been identified. Given that the catalytic family members share identical reaction chemistry, their physiologic roles are influenced greatly by their tissue and sub-cellular locations. CAVI is the lone secreted CA and exists in both saliva and the gastrointestinal mucosa. An alternative, stress-inducible isoform of CAVI (CAVI-b) has been shown to be expressed from a cryptic promoter that is activated by the CCAAT/Enhancer-Binding Protein Homologous Protein (CHOP). The CAVI-b isoform is not secreted and is currently of unknown physiological function. Here we use neuronal models, including a model derived using Car6 and CHOP gene ablations, to delineate a role for CAVI-b in ischemic protection. Our results demonstrate that CAVI-b expression, which is increased through CHOP-signaling in response to unfolded protein stress, is also increased by oxygen-glucose deprivation (OGD). While enforced expression of CAVI-b is not sufficient to protect against ischemia, CHOP regulation of CAVI-b is necessary for adaptive changes mediated by BDNF that reduce subsequent ischemic damage. These results suggest that CAVI-b comprises a necessary component of a larger adaptive signaling pathway downstream of CHOP.
Keywords: carbonic anhydrase, ischemia, neurosphere, unfolded protein response (UPR), oxygen glucose deprivation (OGD), brain-derived neurotrophic factor (BDNF)
1. INTRODUCTION
The mammalian alpha carbonic anhydrase (α-CA) family is comprised of thirteen active metalloenzymes (Sly and Hu, 1995) and three inactive isoforms which lack one or more of a triad of histidine residues that coordinate metal binding (Aspatwar et al., 2010). Active CA enzymes catalyze the reversible hydration of carbon dioxide, resulting in the production of bicarbonate and protons. The ability of CAs to facilitate this reaction has linked their activity to many physiological processes such as maintaining pH and CO2 homeostasis, electrolyte secretion, and a myriad of biosynthetic reactions (i.e.; gluconeogenesis, lipogenesis, and ureagenesis) and CA inhibitors have the therapeutic potential to treat disorders ranging from cancer to glaucoma to epilepsy (Supuran, 2008; Supuran, 2011).
CAs are ubiquitously expressed throughout eukaryotes and are found in the cytosol, on the plasma membrane, and in the mitochondrial matrix, with a single isoform, CAVI, secreted from salivary acinar cells (Parkkila, 2000; Sly and Hu, 1995). In the brain, CAs couple neuron-derived carbon dioxide with bicarbonate and proton efflux, which help to buffer the extracellular pH and can regulate post synaptic receptor function (Shah et al., 2005).
CAs can also respond to cell stress. Both CAIX and CAXII expression are regulated by hypoxia inducible factor (HIF) (Wykoff et al., 2000) and the CAIX protein is commonly found in cancers where the tumor microenvironment is hypoxic and HIF signaling is constitutively active (Ivanov et al., 2001; Wykoff et al., 2000). The finding that mice where the homologous Car9 gene has been genetically-ablated have a brain phenotype suggests a basal function, as well (Pan et al., 2012).
A third stress-inducible CA with as-of-yet unknown function is encoded by the murine Car6 gene. Activation of the endoplasmic reticulum unfolded protein stress response pathway (ERUPR) has been shown to lead to the expression of CAVI-b from a cryptic promoter whose use is regulated by the CCAAT/Enhancer-Binding Protein Homologous Protein (CHOP) (Sok et al., 1999). In general, activation of the ERUPR is adaptive in the short term, but aggravated or persistent stress can trigger apoptosis (Hung et al., 2003). CHOP acts in the ERUPR downstream of protein kinase RNA-like ER kinase (PERK) and activating transcription factor 4 (ATF4), and its recruitment is thought to lead to apoptosis (Oyadomari and Mori, 2004; Woo et al., 2009). In support of this idea, Chop−/− mice exhibit marked decreases in programmed cell death following induction of ER stress (Marciniak et al., 2004) as well as decreased neuronal injury following stroke (Tajiri et al., 2004). Hence, by extension the CHOP regulated induction of CAVI-b was originally hypothesized to be pro-apoptotic (Sok et al., 1999).
However, a recent report challenged this paradigm by demonstrating that the loss of CHOP attenuates the pro-survival effects of brain derived neurotrophic factor (BDNF) on hypoxic damage (Halterman et al., 2010). Further, a separate group reported that the loss of CHOP aggravates hippocampal neuronal injury and memory performance following induced ER stress (Chen et al., 2012). These findings together do not support the anticipated pathological role for CHOP and suggests a more complex function in cell death decisions.
In the current study, we investigated whether insults commonly associated with neurodegenerative disorders (ischemia and ER stress) caused up-regulation of stress-inducible CAVI-b. Further, we examined whether increased expression was necessary or sufficient to elicit changes in cell health following toxic insult. Our results demonstrate that CAVI-b expression on its own has little effect on cell viability or susceptibility to insult, but instead functions as a part of the CHOP signaling cascade to mediate the benefits of BDNF treatment on neurons' ability to survive ischemia. We conclude that CAVI-b can function as part of the pro-survival branch of the CHOP signaling cascade and is necessary for conveying the beneficial effects of BDNF on hypoxic cell survival.
2. RESULTS
2.1. CAVI-b is retained within the cell and partitions between the nucleus and cytoplasm
Subcellular targeting is an important determining factor for CA function. However, preliminary testing indicated that commercially available anti-mouse CAVI antibodies were insufficient for immunodetection techniques (data not shown), and most clinically utilized anti-human CAVI antibodies do not cross react with mouse CAVI. Hence, a custom rabbit anti-mouse CAVI antibody was derived by our laboratory.
Protein lysates from HEK293 cells that had been transfected with vectors expressing V5 epitope fusion proteins for CAVI-b, CAV1-a, or CAIX were probed with anti-V5 antibody, resulting in the detection of bands of the predicted molecular weight (Fig.1A). The custom anti-CAVI antibody recognized proteins on the blot corresponding in size to those expected for recombinant CAVI-a and CAVI-b, but did not react with recombinant CAIX, suggesting some specificity (Fig. 1A). Despite this specificity, unfortunately the antibody was unable in our hands to detect CAVI at endogenous expression levels in native mouse tissues (data not shown).
Figure 1. CAVI-b protein expression and localization.

(A) Schematics of mouse CAVI-b, CAVI-a and CAIX fusion proteins expressed in frame with the V5 epitope denote their expected positions on a Western blot of lysates from transiently transfected HEK293 cells probed with anti-V5 antibody. Labeled arrows (to the right of the image) denote CAVI-a and CAVI-b as detected by a custom generated anti-CAVI antibody raised against a peptide epitope found in both isoforms. (B) Fluorescence images taken of mCherry in live cells that had been transfected with control or CAVI fusion constructs as shown schematically, with the lower images showing DAPI staining of nuclei in the same cells. (C) An anti-mCherry antibody detects the same protein as an anti-CAVI antibody in transiently transfected cells expressing CAVI-b. The negative control shows a field which contains CAVI-b∷mCherry positive cells, processed without primary anti-CAVI antibody.
As an alternative approach, recombinant protein targeting was assessed in live HEK293 cells expressing CAVI∷mCherry fusions (Fig. 1B), and these results were compared to protein distributions in fixed CN1.4 cells (which we use as a model in this work) using both anti-mCherry and anti-CAVI antibodies (Fig. 1C). The CAVI-b∷mCherry fusion was distributed between the nucleus and cytoplasm, while the CAVI-a∷mCherry fusion appeared to reside predominantly in the secretory apparatus, consistent with overexpression of a normally secreted protein. Although the mCherry tag could potentially cause some mis-localiztion, immunostaining with anti-V5 antibody of V5 epitope tagged CAVI-a, CAVI-b and CAIX transfected HEK293 cells mimicked the localization patterns observed with mCherry fusion proteins (data not shown). Together, these results suggest a cytoplasmic/nuclear distribution of CAVI-b.
2.2. ER stress up-regulates CAVI in a neuronal model
Previously, CAVI-b was shown to be expressed in fibroblasts in response to tunicamycin (Tu) treatment through CHOP-C/EBP heterodimer signaling at a cryptic Car6 promoter element contained within the first intron of the canonical gene product (Sok et al., 1999). Here, we tested whether the same was true in the neuronal CN1.4 cell line. Consistent with its depending upon ER stress for induction, Car6-b message was undetectable by RT-PCR under basal conditions (Fig. 2A), even after extensive amplification. However, both Tu and thapsigargin (Tg) treatment (12hrs), which are expected to induce a ERUPR, resulted in increased BiP message (data not shown) as well as increased Car6-b mRNA abundance (Fig. 2A). Unlike Car6-b, Chop mRNA levels were detectable under resting conditions, and quantitative RT-PCR (qPCR) results indicated that they were also elevated following treatment (Fig. 2A and 2B). Consistent with ER stress leading to cell toxicity, measurements of lactose dehydrogenase (LDH) release showed that treatment with Tu and Tg induced significant cell death compared to untreated cells (Fig. 2C).
Figure 2. ER stress and OGD up-regulate Car6-b mRNA expression in a neuronal model.

(A and D) Representative RT-PCR analysis of Car6-b, Chop and Actin mRNA expression in CN1.4 cortical neuronal cells as a function of 12 hours of treatment with 250 nM Tu or Tg or after 12 hours of OGD, followed by 6 hours of reperfusion. Car6-b was not detectable under control conditions. (B and E) Quantitation of Chop mRNA induction by qPCR following treatment as indicated. A similar fold-induction of Car6-b mRNA caused by Tu, Tg, or OGD was impossible to calculate accurately, given our inability to detect message prior to induction of the ERUPR. (C and F) Quantitation of LDH release following Tu, Tg or OGD, as described above. The results shown in panels B–F represent the mean ±S.D. of three independent experiments. ANOVA was performed for statistical analysis. *P<0.05, **P<0.01, ***P<0.001 compared with untreated or normoxic cells.
Oxygen-glucose deprivation (OGD) was then used to model ischemia. As with Tg and Tu, OGD has been shown to up-regulate ER stress related proteins downstream of the CHOP signaling cascade (Badiola et al., 2011). We found that Chop mRNA was reliably increased by OGD in CN1.4 cells (Fig. 2D and 2E), and that OGD also resulted in increased expression of Car6-b mRNA (Fig. 2D). LDH assays confirmed that OGD resulted in significant cell death (Fig. 2F). These data suggest that both CHOP and CAVI-b are regulated in response to ER stress and ischemia in our neural cell line (Sok et al., 1999).
2.3. Exogenously expressed CAVI does not exacerbate cell death
Because CHOP expression is typically associated with activation of the pro-apoptotic arm of the UPRER and is also necessary for CAVI-b expression, we tested whether CAVI-b expression alone is sufficient to lead to cell death. A V5-tagged CAVI-b fusion protein was transiently expressed in CN1.4 cells and cell survival was measured using LDH assays. Enforced expression in CN1.4 cells had little effect on either basal cell survival or stress-induced cell death in response to chemical toxins or simulated ischemia (Fig. 3A). These results suggest that exogenous CAVI-b alone is not sufficient to either induce or exacerbate cell death.
Figure 3. Car6-b expression is not sufficient to cause cell death.

(A) Quantitation of LDH release in cortical CN1.4 cells transiently expressing a CAVI-b∷V5 transgene 24 hours following transfection. Transfection efficiencies ranged from 60% to 70%. (B) Representative RT-PCR analysis of Car6-b mRNA expression as a function of 12 hours of exposure to Tg, at the concentrations indicated (in nM). (C) Quantitation of LDH release as a function of Tg concentration. The results represent the mean ±S.D. of three independent experiments. ANOVA was performed for statistical analysis. **P<0.01, shown representatively; not all statistically significant comparisons are shown.
A similar conclusion was reached when comparing a Tg dose-response with respect to Car6-b mRNA expression and cell death. CN1.4 cells were subjected to differing concentrations of Tg, ranging from a dose that caused maximal cell death to concentrations that were without effect on cell viability. Substantial increases in Car6-b mRNA levels were induced with only 0.49 nM Tg (Fig. 3B), but LDH release did not become significantly elevated until concentrations of 7.80 nM and above were applied (Fig. 3C).
The increased expression of Car6-b at concentrations of Tg that did not affect cell viability suggested an intriguing possibility. Mild, non-toxic stress can often up-regulate signaling cascades that subsequently protect cells from damage induced by more toxic stimuli (Hung et al., 2003). While our data clearly show that CAVI-b is not sufficient to alter life-death balance, within this context it seemed reasonable to test the possibility that CAVI-b may function in adaptive processes.
2.4. CHOP induction of CAVI-b is necessary for the adaptive effects of BDNF
Neurospheres consist of free-floating clusters of neural precursor cells that replicate in culture and can be differentiated to form neurons, astrocytes, and oligodendrocytes (Reynolds and Weiss, 1992). They are relatively easy to cultivate and do not require lengthy differentiation processes prior to their use. Because they can be derived from genetically modified mouse strains, they provide the means to cleanly address the relative contributions CHOP and/or CAVI to physiologic stress. Car6−/− mice are viable (Pan et al., 2011) and thus far, the only overt phenotype that has been reported relates to salivary function (Culp et al., 2011). Here, neurosphere cultures were derived from embryonic brains harvested from WT, Car6−/− and Chop−/− mice as a model to study adaptive stress responses.
As observed for CN1.4 cells, both Car6-b and Chop expression was induced in WT neurospheres by exposure to Tu, Tg, or OGD (Fig. 4). Unlike the WT cells, Car6−/− neurospheres did not exhibit Car6-b expression in response to stress, despite an apparent increase in Chop message (Fig. 4). However, a missense non-coding transcript that has been shown to be expressed from the Car6 locus in the knockout mouse (Culp et al., 2011) could be detected (data not shown). This observation confirmed that the Car6−/− neurospheres have an intact signaling pathway through which stress mediated regulation can occur, but ultimately lack CAVI-b itself. The hypothesis that CHOP is one of the principal components of this signaling pathway was tested by measuring Car6-b induction by Tu, Tg or OGD in Chop−/− neurospheres. As predicted, Chop−/− neurospheres exhibited neither increased Chop nor Car6-b message levels in response to this repertoire of stressors (Fig. 4). Finally, the induction of BiP, a canonical downstream marker of the ERUPR, suggested that Tu, Tg, and OGD were eliciting a typical ER stress response (Fig. 4).
Figure 4. Characterization of WT, Car6−/− and CHOP−/− neurosphere models.
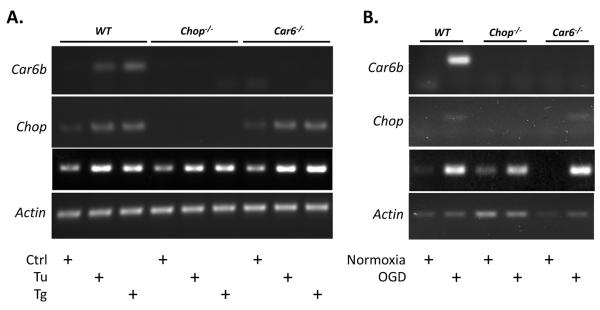
Representative RT-PCR of CHOP, Car6, BiP and Actin mRNA following 12 hours of treatment with (A) 250 nM Tu or Tg or (B) after 12 hours of OGD, followed by 6 hours of reperfusion.
CHOP is known to be induced by BDNF and required for adaptation to hypoxia elicited by its exposure (Halterman et al., 2010). Here, we asked whether BDNF's protective effect also requires Car6 (Fig. 5A). Nuclear pyknosis, which is a reliable indicator of cell damage (Fig. 5B), was assessed as a function of OGD and/or pre-exposure to BDNF in each of the three lines of neurospheres. Our results demonstrate a slight increase in pyknotic cells in Chop−/− and Car6−/− cultures compared to WT following index hypoxia. In agreement with previous observations, loss of Chop resulted in a near complete suppression of BDNF-mediated protection. Interestingly, loss of Car6 also suppressed the benefits of BDNF. These data strongly support a role for CAVI-b as a downstream effecter of cell viability in adaptive BDNF-CHOP coupling.
Figure 5. Both CHOP and Car6 are required for BDNF protection from hypoxic cell damage.

(A) The data represents counts of pyknotic nuclei in fixed, DAPI-labeled WT, Chop−/−, and Car6−/− neurospheres under normoxic conditions or following 1 hour of OGD, as a function of BDNF pretreatment. Results are presented as the average +/− S.E.M. for four independent experiments. **P<0.01. Significance was determined by ANOVA. (B) Images of DAPI-stained nuclei following normoxia or OGD, as indicated. Under basal conditions (to the left), cells exhibit very few pyknotic nuclei (blue arrows pointing up) with occasional cells that appear to be entering pyknosis (blue arrows pointing down). However, many of the cells that were exposed to OGD (to the right) exhibit pyknosis.
3. DISCUSSION
The observation that ER stressors can increase the expression of neuronal CAVI-b in a CHOP dependent fashion are particularly interesting considering a core theme associated with neurodegenerative disorders involves protein aggregation (Forman et al., 2003). ER stress markers including PERK levels and caspase-4 activation have been reported as increased in the brain of patients with Alzheimer's disease (AD) (Hoozemans et al., 2005; Onuki et al., 2004), while the brains of patients diagnosed with the extensively studied neurodegenerative disorder Parkinson's disease (PD) also exhibit markers of ER stress and up-regulated levels of ER chaperones (Conn et al., 2004). Moreover, Parkinsonism-inducing neurotoxins 6-hydroxydopamine and 1-methyl-4-phenylpyridinium are able to trigger the ERUPR and consequent death of dopaminergic neurons (Conn et al., 2004; Holtz and O'Malley, 2003). There is also evidence of ER stress in the pathogenesis of poly-glutamine diseases, prion diseases and amyotrophic lateral sclerosis (ALS) (Yoshida, 2007). Further, cerebral ischemia, one the most common disorders associated with stroke in adults, has also been shown to induce ER stress in neurons activating the ER unfolded protein response (ERUPR) (Tajiri et al., 2004). Hence, the ERUPR has been well studied in the context of neurodegenerative disease where CHOP is recognized to play a role in cell death decisions following toxic ERUPR activation (for a review, see (Oyadomari and Mori, 2004)).
CAs are widely expressed, with a variety of isoforms found localized to specific subcellular locations in neural tissue (for review, see (Kallio et al., 2006; Parkkila, 2000)). Two of the most well recognized of these are the membrane-bound CAs IV and XIV, which are GPI anchored proteins that contribute to extracellular pH buffering within the central nervous system (Shah et al., 2005). The functions of other CA isoforms expressed in the brain are not as clear. Even CA isoforms with well-defined roles in other tissues may act differently when expressed in neurons. For example, CA III, whose expression is generally considered to be restricted to muscle tissue, is unlikely to have exactly the same role in microglia cells as muscle (Nogradi et al., 1993). Further, immunohistochemical analyses show that CAV is expressed in astrocytes and neurons (Dodgson, 1991). Yet this CA isoform has an undetermined role in the brain despite being functionally-defined in other tissues. Hence, there exists a vast gap in our knowledge of how neuronal CAs influence brain physiology.
CAs may be relevant to either physiologic or pathophysiologic stress responses in the brain. For instance, CAIX and CAXII expression is HIF-regulated with both proteins being commonly found in cancers that occur throughout the body, including brain tumors (Chiche et al., 2009; Ivanov et al., 2001; Wykoff et al., 2000). Elevated CAIX and CAXII levels in tumor cells have been attributed to their ability to withstand hypoxia and resist subsequent acidosis, suggesting a functional role in tumor development. This idea is generally supported by experiments showing that knockdown of these genes can greatly reduce tumor volume (Chiche et al., 2009). In addition to hypoxia-induced up-regulation of CAIX, acidosis has also been shown to elevate human CAIX expression levels in a neuronal cell model (Ihnatko et al., 2006). Moreover, although very low levels of murine Car9 and Car12 message are detectable in wild-type mouse brains (Pan et al., 2006), nonetheless, Car9 KO mice present with a brain phenotype including increased vacuolar degeneration and deficits in both locomotor activity and memory (Pan et al., 2012). These results suggest that CAIX may directly or indirectly have a basal role in brain tissue apart from its stress-induced expression. Our work presented here expands upon both the range of CA genes shown to be expressed in the brain, as well as the functional role of CAs in neuronal physiology.
Over a decade ago, a unique stress-inducible CA isoform was shown to be coded for by the Car6 gene in mice (Sok et al., 1999). Transcription initiation from a cryptic promoter element contained in the first intron of the canonical gene structure was shown to be driven by the stress-responsive C/EBP transcription factor CHOP (Sok et al., 1999). CHOP is positioned along the PERK/ATF4 branch of the ERUPR signaling pathway and inhibits the expression of anti-apoptotic Bcl2 family members while promoting the expression of pro-apoptotic family members. Under non-stress conditions, expression of the type A gene product, CAVI-a, which represents the only secreted isozyme in the CA family, is restricted to salivary acinar cells (Parkkila, 2000). Under stress conditions where CHOP is activated, a truncated CAVI-b isozyme is generated that lacks a secretion signal (Sok et al., 1999).
Our work confirms that the CAVI-b isoform is retained in the cell, where it is distributed between the nucleus and the cytoplasm (Fig. 1). Most CAs are small enough to passively enter the nucleus, and since CAVI-b lacks a canonical nuclear localization sequence, the distribution is likely to reflect size rather than targeting. We found that CAVI-b was induced by ER stress (Fig. 2), and that the stress induction of CAVI-b in neurons was absolutely dependent upon CHOP (Fig. 4). However, in contrast to an expected role in the apoptotic branch of the UPRER, recombinant over-expression of CAVI-b did not affect survival of neurons exposed to OGD (Fig. 3), and instead functioned as an essential part of an adaptive BDNF-CHOP axis (Fig. 5) to promote cell viability. These results are consistent with previously reported protective effects of BDNF-CHOP coupling in primary cortical neurons (Halterman et al., 2010), as well as a more recent report that CHOP protects against retinal degeneration due to protein misfolding (Nashine et al., 2013). Since CAVI-b is insufficient to protect cells from hypoxia on its own (Fig. 3), other components of the BDNF-CHOP signaling axis, including perhaps PI3K/Akt and MAPK signal transduction pathways (Almeida et al., 2009; Li et al., 2007) are likely to function in tandem to elicit protection.
In general, CAs are considered to be cytoprotective and help maintain acid-base homeostasis. Since cellular acidification contributes to ischemia-reperfusion injury, and CAIX and CAXII helps tumor cells to resist extracellular acidosis, it would be reasonable to predict that CAVI-b suppresses injury by regulating pH balance. However, the catalytic rate of recombinant CAVI-b appears to be quite low compared to CAVI-a (data not shown). This is likely due to the lack of a small but well-conserved region in the N-terminus of CAVI-b. Hence, the role of CAVI-b is probably not related to acid-base balance.
As an alternative, a non-canonical role may be physiologically significant. Of the sixteen mammalian CA isoforms, three have been shown to be acatalytic. However, their mechanisms of action and precise roles have yet to be defined. The loss of one of the acatalytic enzymes, CAVIII, has been associated with ataxia, mild mental retardation and quadrupedal gait in humans and with a lifelong gait disorder in mice (Jiao et al., 2005). Since CAVIII appears to bind to the inositol tri-phosphate receptor and may regulate its substrate affinity, these phenotypes have been suggested to be related to calcium signaling dysfunction (Hirota et al., 2003). While the same is unlikely to be true for CAVI-b, nevertheless, a better understanding of the proteins that it interacts with may provide further insight.
In conclusion, we have shown that the canonical pro-apoptotic role of CHOP activation following unfolded protein stress is balanced by an adaptive role that involves the expression of CAVI-b. Interestingly, CHOP has been reported to be required for both the UPRER and the mitochondrial UPR (UPRmt) (Horibe and Hoogenraad, 2007; Zhao et al., 2002). Because CHOP can heterodimerize with other bZip transcription factors, its output is likely to be context dependent. Our cell culture models hold promise to serve as an important investigative tool in decrypting the `molecular switch' and signaling mechanisms responsible for adaptation through BDNF-CHOP-CAVI-b coupling.
4. EXPERIMENTAL PROCEDURES
4.1. Reagents
DMSO, thapsigargin, tunicamycin, human brain-derived neurotropic factor, poly-D-lysine and DAPI were purchased from Sigma-Aldrich (St. Louis, MO). Cell culture grade 0.05% and 0.025% trypsin-EDTA, Dulbecco's modified Eagle's medium (DMEM), 100x penicillin/streptomycin, L-glutamine, gentamicin, natural mouse laminin, dPBS [+Mg2+/Ca2+], dPBS [−Mg2+/Ca2+] and HBSS were obtained from Invitrogen (Grand Island, NY). Mouse NeuroCult NSC basal media, NeuroCult proliferation supplements, NeuroCult differentiation supplements and recombinant human (rh) EGF were obtained from StemCell Technologies (Vancouver, B.C., Canada). Fetal bovine serum was purchased from Atlanta Biologicals (Lawrenceville, GA) and used at a final concentration of 10% in medium, where indicated. The anti-V5 epitope antibody was from Invitrogen (Grand Island, NY), a polyclonal antibody generated in rabbit and targeted at the mouse Car6 peptide sequences CHNNNTIQNGYRSTQPNN was contracted from GenTel Laboratories (Madison, WI), goat anti-mouse HRP and goat anti-rabbit HPR secondary antibodies were obtained from Abcam (Cambridge, MA). Alexa Fluor 594 conjugated goat anti-rabbit was from Invitrogen and goat anti-mouse DyLight 488 was from Jackson ImmunoResearch (West Grove, PA).
4.2. Cell Culture
All cell culture models were maintained in a water-jacketed incubator at 37°C and 5% O2, unless otherwise stated. NSPHs were cultured in T25 flasks with Mouse NeuroCult media containing proliferation supplements. The neural CN1.4 cell line was grown in DMEM at 33°C and moved to 39°C 24 hours prior to experimentation to slow proliferation and induce differentiation (Bongarzone et al., 1998). CN1.4 cells were cultured in DMEM High Glucose and supplemented with FBS, 0.1% gentamicin, 4 mM glutamine and penicillin/streptomycin. For transient expression, cells were transfected with FuGENE 6 (Promega, Madison, WI), as per the manufacturer's instructions.
For oxygen-glucose deprivation (OGD), glucose-free DMEM containing 0.2% FBS, acclimated at N2/CO2/O2 (94%/5%/1%) for 12 hours prior to experiment, was used to wash cells twice before incubation for 12 hours in this same oxygen-free N2/CO2/O2 (94%/5%/1%) atmosphere via a triple gas controlled environment glove box (Coy Lab Products, Grass Lake, MI). Thereafter, the medium was replaced by standard DMEM containing both 5% glucose and 0.2% FBS. Cells were collected at 6 hours after reoxygenation for mRNA analysis and LDH release assays.
4.3. Western Analysis
Protein was collected by the addition of 150 uL per well of ice-cold: 30 mM Tris-HCl pH 7.5, 150 mM NaCl, 20 mM Mg-Acetate with 0.1% Triton-X 100, 1X protease inhibitor cocktail, 1X PSMF added immediately prior to use. Protein concentrations were measured using a BCA protocol. Samples were centrifuged at 14,000 rpm for 5 minutes at 4°C in a bench top centrifuge, and supernatants were stored at −80°C. Aliquots of equal volume were resolved by SDS-PAGE (12.5%), transferred to nitrocellulose membranes (Millipore, Bedford, MA) and blocked for 60 minutes at room temperature in wash buffer (50 mM Tris, 0.9% NaCl, 0.05% Tween 20) containing 5% non-fat dry milk. Primary antibodies were added with constant agitation overnight at 4°C, blots were rinsed three times in wash buffer, incubated with secondary antibodies (diluted 1:2500) for 2 hours, washed, and detected by ECL (Renaissance, Amersham Biosciences, Piscataway, NJ).
4.4. Lactate Dehydrogenase Assay
The release of the intracellular enzyme lactate dehydrogenase (LDH) into the medium was used as a quantitative measurement of cell viability. The measurement of LDH was carried out as described previously (Decker and Lohmann-Matthes, 1988) by using the Cytotoxicity Detection LDH Kit (Roche Applied Science, Indianapolis, IN).
4.5. Reverse-Transcription PCR
Total RNA was obtained using the RNA extraction kit (Qiagen, Valencia, CA) following the manufacturer's protocol. cDNA was synthesized from 1 μg total RNA using SuperScript First-Strand Synthesis System for RT-PCR Kit (Invitrogen, Grand Island, NY) following the manufacturer's instructions. Standard PCR reactions were carried out in a BioRad thermal cycler using RT-PCR primers: CHOP (C/EBP homologous protein) (F, 5'-TATCTCATCCCCAGGAAACG-3' R, 5'-GGGCACTGA CCACTCTGTTT-3'), Car6 [CN1.4/N2A] (F, 5'-TGGAAGCCAATTTCCTGAACGTC-3' R, 5'-AGCAAGGAGACCCACATTCCAG-3'); Car6 [NSPH] (F, 5'-GAGCTTGGTGAACTATGAGAAGG-3' R, 5'-GGCCTTTGAGATGAACTCAGTG-3'); BiP (F, 5'-GTTTGCTGAGGAAGACAAAAAGCTC-3' R, 5'-CACTTCCATAGAGTTTGCTGATAATTG-3'), and actin (F, 5'-GGCCGCCCTAGGCACCAG-3' R 5'-GGGTCATCTTTTCACGGTTGG-3'). Real time analyses of gene expression levels were performed using first-strand cDNA samples generated with iQ™ SYBR® Green Supermix (Bio-Rad) and amplification in a Bio-Rad real time PCR detection system. CT calculations were applied to determine abundances relative to a standard curve.
4.6. Neurosphere Generation
Neural stem cells (NSCs) were generated from the developing midbrain of embryonic C57/BL6J mice at 13.5 days postcoitus in agreement with NIH guidelines, the approval of the University of Rochester's Committee on Animal Resources (UCAR) and according to the protocol previously reported by Ahlenius et al. (Ahlenius and Kokaia, 2010). The Car6−/− (Car6tm1Pwp) mice used in these studies were obtained from Dr. S. Parkkila (University of Tampere, Finland) and are derived from those originally reported by Pan et al. (Pan et al., 2011). The CHOP−/− (B6.129S-Ddit3tm1Dron/J) mice are available from Jackson Labs. All knockout strains have been outcrossed against C57/BL6J at least three times. Following isolation of the embryos and dissection of the midbrain, the meninges were removed and the medial ganglionic eminences (MGEs) were separated and transferred to an Eppendorf tube containing L-15 media. MGEs from six embryos were combined and L-15 media was replaced with NeuroCult media. Tissue samples were gently triturated with a sterile fire-polished glass pipette until a single cell suspension was made. Cells were subsequently plated in NeuroCult media supplemented with NeuroCult Mouse Proliferation Supplement and 20ng/ml rhEGF at a density of 10–15 cells/μl on uncoated culture flasks. Cells were subsequently cultured in a 37°C humidified incubator with 5% CO2 until the formation of Neurospheres (NSPHs).
4.7. Coverslip Preparation for Neurosphere Cultures
Sterile 12 mm circular glass coverslips were acid-washed with 1M HCl overnight, subsequently rinsed with sterile H20, 95% EtOH and allowed to dry. The coverslips were then flame sterilized using ethanol and transferred to 6-well plates: 2 coverslips per well. Plates were further sterilized by 30 minutes exposure to UV radiation.
4.8. Coating Plates for Neurosphere Differentiation
Culture dishes used for NSPH experiments underwent a coating process according to the following protocol. Poly-D-lysine (0.1mg/mL) was added to plate and incubated at 37°C for 4 hours. Following incubation, the lysine was aspirated and wells thoroughly rinsed with sterile ddH2O (3X). Wells were allowed to dry completely following the last wash. Subsequently, mouse laminin/PBS [+Mg2+/Ca2+] solution (1ug/mL) was added to the plate and incubated for 12 hours at 37°C. Laminin solution was removed; wells were rinsed with HBSS (3X), never allowing laminin-coated surfaces to dry. A final volume of HBSS was added to each plate and stored at 37°C until ready for use.
4.9. Analysis of Nuclear Pyknosis
Neurospheres were plated in NeuroCult proliferation media overnight at 37°C then changed to NeuroCult differentiation media for 24 hours also at 37°C. These cells – maintained under ambient oxygen conditions – were exposed to 50nM BDNF or control (DMSO) for 12 hours in DMEM (+ glucose). Cells were then subject to OGD as described above for one hour. Samples were not reperfused prior to analysis. To assess nuclear morphology, cells were fixed in 4% paraformaldehyde in PBS (pH 7.4) for 15 minutes at room temperature, counterstained with DAPI, and mounted in Fluoromount-G. Nuclear pyknosis was assessed by an observer blinded to cell genotype and to experimental manipulations who counted 5–6 non-overlapping fields per coverslip, with a total cell count of 100–200 cells.
4.10. Statistical analysis
All data is expressed as the mean of at least three independent experiments (± standard error of the mean) unless otherwise stated. Statistical comparisons between two treatment groups were performed using Student's t-test while one-way ANOVA was used to compare variations across multiple measures. All statistical data was calculated with GraphPad Prism (GraphPad Software, Inc., La Jolla, California).
Ischemia induces the expression of an alternative, intracellular CAVI variant.
Expression of this variant relies upon CHOP signaling.
Loss of either CHOP or CAVI prevents BDNF mediated protection from ischemia.
Acknowledgments
This work was supported in part by USPHS R01 NS064945 (K.N.) and an UNCF/Merck Postdoctoral Fellowship Award to T.A.M. We also acknowledge Rita Giuliani for technical expertise in developing the neurosphere model and Jonathan Malecki for valuable technical contributions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Ahlenius H, Kokaia Z. Isolation and generation of neurosphere cultures from embryonic and adult mouse brain. Methods Mol Biol. 2010;633:241–52. doi: 10.1007/978-1-59745-019-5_18. [DOI] [PubMed] [Google Scholar]
- Almeida S, et al. BDNF regulates BIM expression levels in 3-nitropropionic acid-treated cortical neurons. Neurobiol Dis. 2009;35:448–56. doi: 10.1016/j.nbd.2009.06.006. [DOI] [PubMed] [Google Scholar]
- Aspatwar A, Tolvanen ME, Parkkila S. Phylogeny and expression of carbonic anhydrase-related proteins. BMC Mol Biol. 2010;11:25. doi: 10.1186/1471-2199-11-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badiola N, et al. Induction of ER stress in response to oxygen-glucose deprivation of cortical cultures involves the activation of the PERK and IRE-1 pathways and of caspase-12. Cell Death Dis. 2011;2:e149. doi: 10.1038/cddis.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bongarzone ER, et al. Two neuronal cell lines expressing the myelin basic protein gene display differences in their in vitro survival and in their response to glia. J Neurosci Res. 1998;54:309–19. doi: 10.1002/(SICI)1097-4547(19981101)54:3<309::AID-JNR2>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Chen CM, et al. C/EBP homologous protein (CHOP) deficiency aggravates hippocampal cell apoptosis and impairs memory performance. PLoS One. 2012;7:e40801. doi: 10.1371/journal.pone.0040801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiche J, et al. Hypoxia-inducible carbonic anhydrase IX and XII promote tumor cell growth by counteracting acidosis through the regulation of the intracellular pH. Cancer Res. 2009;69:358–68. doi: 10.1158/0008-5472.CAN-08-2470. [DOI] [PubMed] [Google Scholar]
- Conn KJ, et al. Identification of the protein disulfide isomerase family member PDIp in experimental Parkinson's disease and Lewy body pathology. Brain Res. 2004;1022:164–72. doi: 10.1016/j.brainres.2004.07.026. [DOI] [PubMed] [Google Scholar]
- Culp DJ, et al. Oral colonization by Streptococcus mutans and caries development is reduced upon deletion of carbonic anhydrase VI expression in saliva. Biochim Biophys Acta. 2011;1812:1567–76. doi: 10.1016/j.bbadis.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker T, Lohmann-Matthes ML. A quick and simple method for the quantitation of lactate dehydrogenase release in measurements of cellular cytotoxicity and tumor necrosis factor (TNF) activity. J Immunol Methods. 1988;115:61–9. doi: 10.1016/0022-1759(88)90310-9. [DOI] [PubMed] [Google Scholar]
- Dodgson SJ. Why are there carbonic anhydrases in the liver? Biochem Cell Biol. 1991;69:761–3. doi: 10.1139/o91-116. [DOI] [PubMed] [Google Scholar]
- Forman MS, Lee VM, Trojanowski JQ. `Unfolding' pathways in neurodegenerative disease. Trends Neurosci. 2003;26:407–10. doi: 10.1016/S0166-2236(03)00197-8. [DOI] [PubMed] [Google Scholar]
- Halterman MW, et al. The endoplasmic reticulum stress response factor CHOP-10 protects against hypoxia-induced neuronal death. J Biol Chem. 2010;285:21329–40. doi: 10.1074/jbc.M109.095299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota J, et al. Carbonic anhydrase-related protein is a novel binding protein for inositol 1,4,5-trisphosphate receptor type 1. Biochem J. 2003;372:435–41. doi: 10.1042/BJ20030110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtz WA, O'Malley KL. Parkinsonian mimetics induce aspects of unfolded protein response in death of dopaminergic neurons. J Biol Chem. 2003;278:19367–77. doi: 10.1074/jbc.M211821200. [DOI] [PubMed] [Google Scholar]
- Hoozemans JJ, et al. The unfolded protein response is activated in Alzheimer's disease. Acta Neuropathol. 2005;110:165–72. doi: 10.1007/s00401-005-1038-0. [DOI] [PubMed] [Google Scholar]
- Horibe T, Hoogenraad NJ. The chop gene contains an element for the positive regulation of the mitochondrial unfolded protein response. PLoS One. 2007;2:e835. doi: 10.1371/journal.pone.0000835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung CC, et al. Protection of renal epithelial cells against oxidative injury by endoplasmic reticulum stress preconditioning is mediated by ERK1/2 activation. J Biol Chem. 2003;278:29317–26. doi: 10.1074/jbc.M302368200. [DOI] [PubMed] [Google Scholar]
- Ihnatko R, et al. Extracellular acidosis elevates carbonic anhydrase IX in human glioblastoma cells via transcriptional modulation that does not depend on hypoxia. Int J Oncol. 2006;29:1025–33. [PubMed] [Google Scholar]
- Ivanov S, et al. Expression of hypoxia-inducible cell-surface transmembrane carbonic anhydrases in human cancer. Am J Pathol. 2001;158:905–19. doi: 10.1016/S0002-9440(10)64038-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y, et al. Carbonic anhydrase-related protein VIII deficiency is associated with a distinctive lifelong gait disorder in waddles mice. Genetics. 2005;171:1239–46. doi: 10.1534/genetics.105.044487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallio H, et al. Expression of carbonic anhydrases IX and XII during mouse embryonic development. BMC Dev Biol. 2006;6:22. doi: 10.1186/1471-213X-6-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, et al. Downregulation of Bim by brain-derived neurotrophic factor activation of TrkB protects neuroblastoma cells from paclitaxel but not etoposide or cisplatin-induced cell death. Cell Death Differ. 2007;14:318–26. doi: 10.1038/sj.cdd.4401983. [DOI] [PubMed] [Google Scholar]
- Marciniak SJ, et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18:3066–77. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nashine S, et al. Ablation of C/EBP Homologous Protein Does Not Protect T17M RHO Mice from Retinal Degeneration. PLoS One. 2013;8:e63205. doi: 10.1371/journal.pone.0063205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogradi A, Kelly C, Carter ND. Localization of acetazolamide-resistant carbonic anhydrase III in human and rat choroid plexus by immunocytochemistry and in situ hybridisation. Neurosci Lett. 1993;151:162–5. doi: 10.1016/0304-3940(93)90011-9. [DOI] [PubMed] [Google Scholar]
- Onuki R, et al. An RNA-dependent protein kinase is involved in tunicamycin-induced apoptosis and Alzheimer's disease. EMBO J. 2004;23:959–68. doi: 10.1038/sj.emboj.7600049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381–9. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- Pan P, et al. Carbonic anhydrase gene expression in CA II-deficient (Car2−/−) and CA IX-deficient (Car9−/−) mice. J Physiol. 2006;571:319–27. doi: 10.1113/jphysiol.2005.102590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan PW, et al. Gene expression profiling in the submandibular gland, stomach, and duodenum of CAVI-deficient mice. Transgenic Res. 2011;20:675–98. doi: 10.1007/s11248-010-9441-2. [DOI] [PubMed] [Google Scholar]
- Pan PW, et al. Brain phenotype of carbonic anhydrase IX-deficient mice. Transgenic Res. 2012;21:163–76. doi: 10.1007/s11248-011-9520-z. [DOI] [PubMed] [Google Scholar]
- Parkkila S. An overview of the distribution and function of carbonic anhydrase in mammals. EXS. 2000:79–93. doi: 10.1007/978-3-0348-8446-4_4. [DOI] [PubMed] [Google Scholar]
- Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 1992;255:1707–10. doi: 10.1126/science.1553558. [DOI] [PubMed] [Google Scholar]
- Shah GN, et al. Carbonic anhydrase IV and XIV knockout mice: roles of the respective carbonic anhydrases in buffering the extracellular space in brain. Proc Natl Acad Sci U S A. 2005;102:16771–6. doi: 10.1073/pnas.0508449102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sly WS, Hu PY. Human carbonic anhydrases and carbonic anhydrase deficiencies. Annu Rev Biochem. 1995;64:375–401. doi: 10.1146/annurev.bi.64.070195.002111. [DOI] [PubMed] [Google Scholar]
- Sok J, et al. CHOP-Dependent stress-inducible expression of a novel form of carbonic anhydrase VI. Mol Cell Biol. 1999;19:495–504. doi: 10.1128/mcb.19.1.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov. 2008;7:168–81. doi: 10.1038/nrd2467. [DOI] [PubMed] [Google Scholar]
- Supuran CT. Carbonic anhydrase inhibitors and activators for novel therapeutic applications. Future Med Chem. 2011;3:1165–80. doi: 10.4155/fmc.11.69. [DOI] [PubMed] [Google Scholar]
- Tajiri S, et al. Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death Differ. 2004;11:403–15. doi: 10.1038/sj.cdd.4401365. [DOI] [PubMed] [Google Scholar]
- Woo CW, et al. Adaptive suppression of the ATF4-CHOP branch of the unfolded protein response by toll-like receptor signalling. Nat Cell Biol. 2009;11:1473–80. doi: 10.1038/ncb1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wykoff CC, et al. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res. 2000;60:7075–83. [PubMed] [Google Scholar]
- Yoshida H. ER stress and diseases. FEBS J. 2007;274:630–58. doi: 10.1111/j.1742-4658.2007.05639.x. [DOI] [PubMed] [Google Scholar]
- Zhao Q, et al. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002;21:4411–9. doi: 10.1093/emboj/cdf445. [DOI] [PMC free article] [PubMed] [Google Scholar]