

Abstract
ObjectiveTo characterize the circadian clock in murine cartilage tissue and identify tissue-specific clock target genes, and to investigate whether the circadian clock changes during aging or during cartilage degeneration using an experimental mouse model of osteoarthritis (OA).
MethodsCartilage explants were obtained from aged and young adult mice after transduction with the circadian clock fusion protein reporter PER2::luc, and real-time bioluminescence recordings were used to characterize the properties of the clock. Time-series microarrays were performed on mouse cartilage tissue to identify genes expressed in a circadian manner. Rhythmic genes were confirmed by quantitative reverse transcription–polymerase chain reaction using mouse tissue, primary chondrocytes, and a human chondrocyte cell line. Experimental OA was induced in mice by destabilization of the medial meniscus (DMM), and articular cartilage samples were microdissected and subjected to microarray analysis.
ResultsMouse cartilage tissue and a human chondrocyte cell line were found to contain intrinsic molecular circadian clocks. The cartilage clock could be reset by temperature signals, while the circadian period was temperature compensated. PER2::luc bioluminescence demonstrated that circadian oscillations were significantly lower in amplitude in cartilage from aged mice. Time-series microarray analyses of the mouse tissue identified the first circadian transcriptome in cartilage, revealing that 615 genes (∼3.9% of the expressed genes) displayed a circadian pattern of expression. This included genes involved in cartilage homeostasis and survival, as well as genes with potential importance in the pathogenesis of OA. Several clock genes were disrupted in the early stages of cartilage degeneration in the DMM mouse model of OA.
ConclusionThese results reveal an autonomous circadian clock in chondrocytes that can be implicated in key aspects of cartilage biology and pathology. Consequently, circadian disruption (e.g., during aging) may compromise tissue homeostasis and increase susceptibility to joint damage or disease.
The circadian clock governs ∼24-hour cycles in physiology through rhythmic control of tissue-specific sets of clock-controlled genes (CCGs), which allows precise orchestration of organ function (1). In mammals (including humans), the circadian system is organized in a hierarchical manner. The suprachiasmatic nuclei of the hypothalamus receive information from external time cues (predominantly, the light/dark cycle) and synchronize peripheral clocks in most major body organs through neuronal or systemic factors (2–3). The molecular basis of the circadian clock relies on the rhythmic activity of evolutionarily conserved clock genes and proteins, including those for transcriptional activators (Clock/Npas2 and Bmal1), transcriptional repressors (Per1/2 and Cry1/2), and nuclear hormone receptors (Nr1d1/2 [Rev-Erbα/β] and Rora/Rorg) (1). This transcriptional–translational feedback loop controls expression of downstream CCGs to regulate tissue-specific physiology. Disruption of the circadian rhythm (attributable to, for example, effects of aging or shift work) has been correlated with an increased risk of various human diseases, including obesity, diabetes, cardiovascular disease, and cancer (4).
Articular cartilage is a specialized load-bearing tissue comprising an abundant extracellular matrix, which is produced and maintained by a sparse population of chondrocytes. As the only cell type residing in this tissue, chondrocytes have a poor capacity for endogenous repair, and there is little evidence of cell division throughout adult life (5). Deterioration in chondrocyte function and survival accompanies the damage and loss of cartilage, a common outcome in degenerative and inflammatory joint diseases such as osteoarthritis (OA) and rheumatoid arthritis (RA).
Several physiologic processes in cartilage exhibit diurnal variation, including the processes of matrix synthesis, the growth rate in the growth plate, and mineralization (6,7). Mice with mutations in clock genes show altered regulation of bone volume (9), retarded long bone growth (10), and increased susceptibility to inflammatory arthritis (11), providing in vivo evidence for the importance of the molecular clock in the skeletal system. Moreover, in patients with OA and those with RA, there is a clear circadian rhythm in the severity of pain and stiffness and the extent of manual dexterity (12–13). Expression of clock genes has been shown in bone and joint tissues (11,14) and in isolated chondrocytes (10–16), but no studies have directly examined the detailed mechanisms, inputs, or targets of the cartilage circadian clock, and none have examined how the clock changes during aging and in the pathogenesis of joint disease.
In the present study, we demonstrate the presence of self-sustained, functional circadian clocks in mouse cartilage tissue, primary chondrocytes, and a human chondrosarcoma cell line. Circadian transcriptome profiling revealed rhythmic expression of ∼3.9% of the genes expressed in mouse cartilage, including key genes associated with tissue homeostasis. In addition, circadian rhythms in the cartilage from aged mice were significantly dampened. Finally, the expression of key circadian clock genes was altered in articular cartilage from mice with experimentally induced OA. Taken together, our results implicate chondrocyte circadian clocks as a key regulator of cartilage homeostasis, and suggest that changes in circadian regulation may play a role in the pathogenesis of joint disease.
MATERIALS AND METHODS
Animals
All animal studies were performed in accordance with the 1986 UK Home Office Animal Procedures Act. Approval was provided by local ethics review or by the Murdoch Children's Research Institute Animal Ethics Committee.
PER2::luc mice carry the firefly luciferase gene fused in-frame with the 3′ end of the Per2 gene, creating a fusion protein reporter (17). ClockΔ19 mice harbor a deletion in exon 19 of the Clock gene (18), producing a dominant-negative mutant protein. ClockΔ19 mice were crossed onto a PER2::luc background. All mice were bred in-house at the University of Manchester. In addition, 6-week-old male C57BL/6J wild-type (WT) mice were purchased from Harlan Laboratories for use in the time-course microarrays.
Cartilage cultures and bioluminescence recording
Cartilage cultures from PER2::luc mice were prepared by dissection of the cartilaginous portion of the xiphoid process, by dissection of the femoral head cartilage from 6–10-day-old mice, or by scalpel microdissection of the articular cartilage from the femoral condyle and tibial plateau surfaces of 4.4-month-old mice. Cartilage was cultured on 0.4-μm cell culture inserts (Millipore), and bioluminescence was recorded in real time using photomultiplier tube (PMT) devices (19) or a LumiCycle apparatus (Actimetrics). Baseline subtraction was carried out using a 24-hour moving average. The circadian period was calculated using LumiCycle analysis software (Actimetrics) or using the RAP algorithm (19). Cultures were also visualized using a self-contained Olympus Luminoview LV200 microscope and recorded using a cooled Hamamatsu ImageEM C9100-13 EM-CCD camera. Images were obtained every hour for 6 days, and results were combined in ImageJ.
For temperature entrainment studies, xiphoid tissues were cut into halves and cultured under PMT recorders in separate incubators at 37°C. After 4 days, alternating 12-hour square-wave temperature cycles of 38.5°C/36°C were applied; the temperature protocols of the 2 incubators were in antiphase. After 3 full temperature cycles, cultures were returned to 37°C. For temperature compensation studies, xiphoid cultures were incubated under PMT devices at a constant temperature of 29°C, 32°C, 37°C, and 39°C (n = 3, n = 4, n = 7, and n = 3, respectively). The mean period (p) was determined at each temperature (T). The frequency (reaction rate [R]) was calculated as 1/p, and the temperature coefficient (Q10°) was worked out using the following equation: Q10° = (R2/R1)†10/(T2 − T1).
Aged animals
Male PER2::luc mice were aged to 20–24 months (n = 10) under a 12-hour light/dark cycle. Young control mice were aged to ∼2–4 months (n = 7). Animals were transferred into a state of constant darkness for 20 days. All animals were killed in the dark at their individual 12-hour cull time (CT12) (i.e., predicted onset of activity for each animal). Amplitude was calculated as the peak-trough difference in bioluminescence of the second peak, with subtraction of baseline data. A minimum of 3 peaks were used to determine the circadian period. Phase was determined by plotting the time at which the first peak occurred in culture, relative to CT12. For dexamethasone treatment, a final concentration of 100 nM was applied directly to the recording medium. Amplitude was calculated from the second peak posttreatment.
Histology
Xiphoid cartilage was fixed for 24 hours in 4% paraformaldehyde, followed by decalcification in 0.8M EDTA (pH 7.4). Tissue was embedded in wax blocks prior to sectioning and hematoxylin and eosin staining using a Shandon Varistain 24-4 automated staining system.
Time-series microarrays
C57BL/6J mice were entrained to a 12-hour light/dark cycle before release into constant darkness for 39 hours prior to collection of xiphoid cartilage. Tissues were collected every 4 hours, commencing at CT3, for 40 hours. Tissue was pooled (5 animals per pooled sample, 3 samples per time point) and snap frozen in liquid nitrogen. One sample per time point was used for microarray; the remainder of the samples were used for quantitative reverse transcription–polymerase chain reaction (qRT-PCR) validation.
Frozen tissue was disrupted using Mikro-Dismembrator S (Satorius Stedim Biotech). Extraction of RNA was carried out using RNeasy Fibrous Tissue Mini/Micro kits (Qiagen). Affymetrix Mouse430_2 GeneChips were run according to the manufacturer's instructions. Technical quality control and outlier analysis were performed using Affymetrix dChip software (version 2005). Background correction, quantile normalization, and gene expression analysis were performed using robust multiarray average. Raw data were deposited in Array Express (accession no. E-MEXP-3780). Genes with an average expression >2 SD higher than the mean background of all GeneChips were classified as expressed. CircWave Batch version 5 (provided by Dr. Roelof Hut, University of Groningen) was used to fit a sine-wave with 24-hour periodicity to each gene expression data set. The local false discovery rate was calculated using the fdrtool in the R program (results available from the corresponding author upon request) (20).
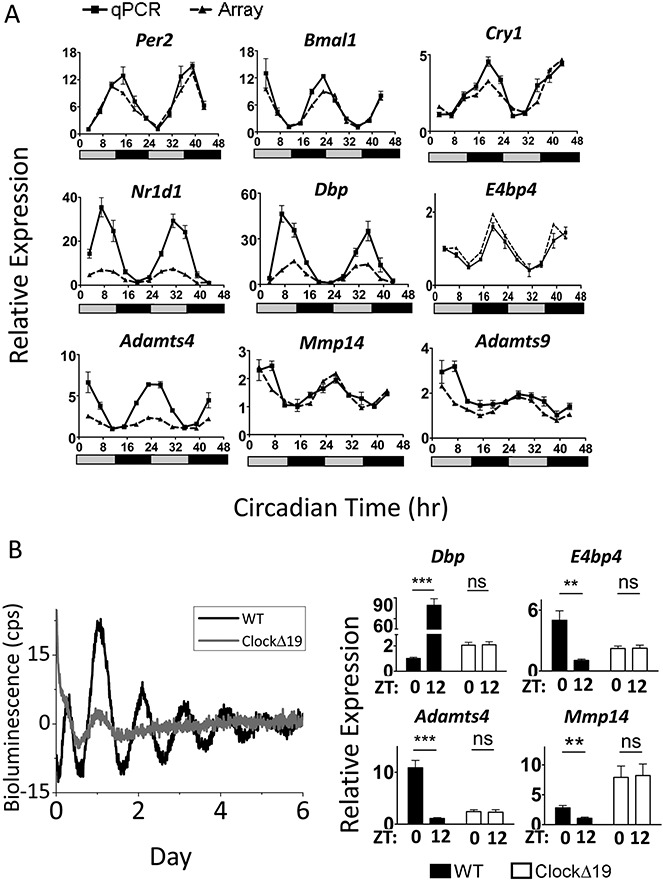
Using known clock genes as a guide, a cutoff point of q < 0.1 was arbitrarily assigned to determine circadian gene expression. JTKCycle (21) was also used to identify circadian transcripts. A Bonferroni-adjusted P value of 0.05 was arbitrarily set as the cutoff for significance. Functional annotation analysis was carried out on overlapping genes, using DAVID algorithms. To model the data for clustering, we used a Gaussian process model (22). To perform clustering, we specified a statistical model based on a Dirichlet process, a mixture of Gaussian processes (23), solved using approximate Bayesian inference (based on the approach described in ref.24). This allowed joint inference of clustering (i.e., which genes belong to which periodic function), alongside inference of the functions themselves. Validation of time-series arrays was carried out using TaqMan-based qRT-PCR (a list of the primers/probes used is shown in Supplementary Table 2, available on the Arthritis & Rheumatism web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38035/abstract), with results normalized to the values for β-actin expression, using the 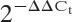 method.
method.
Primary chondrocyte culture
Cells were prepared from 5-day-old PER2::luc mice as described previously (25). Briefly, cartilage was dissected from the femoral heads, femoral condyles, and tibial plateaus of one litter of mice and sequentially digested in collagenase D solution (Roche) at 3 mg/ml and then 0.5 mg/ml. Cells were seeded directly into 35-mm recording dishes and grown until 90–100% confluent, prior to synchronization and PMT recording or RNA extraction (as described below). The chondrocyte phenotype was confirmed using immunofluorescence against type II collagen (Abcam catalog no. ab54236, with an Alexa 488–conjugated donkey anti-mouse antibody) and by staining of glycosaminoglycans in the extracellular matrix using 1% weight/volume Alcian blue 8GX.
SW-1353 cell culture and lentiviral transduction
The SW-1353 cells used herein constitutively overexpress Sox9 to drive Col2a1 expression and maintain a chondrocyte-like phenotype. For time-course experiments, confluent cells were synchronized with 100 nM dexamethasone for 1 hour. Samples were collected 24 hours postsynchronization. RNA samples were prepared using the RNeasy Mini kit (Qiagen). Gene expression was analyzed by TaqMan qRT-PCR (a list of the primers/probes used is shown in Supplementary Table 2, available on the Arthritis & Rheumatism web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38035/abstract), with results normalized to the values for GAPDH using the method. For lentiviral transduction, fragments of the mouse Per2 (−418 bp) and Bmal1 (−649 bp) promoters driving luciferase reporters in lentiviral transfer vectors (19) were cotransfected with lentiviral packaging plasmids into HEK 293FT packaging cells. Virus particles were concentrated in Vivaspin 20 columns (Satorius Stedim Biotech) and applied to cells prior to synchronization and subsequent PMT recording.
Induction of experimental arthritis
OA was induced in 10-week-old male C57BL/6 WT mice by destabilization of the medial meniscus (DMM) of the right knee (26), with sham operation of the left knee as a control. Animals were killed at 1, 2, and 6 weeks after surgery (n = 4 animals with sham/DMM-operated knees per time point). Tibial epiphyses were isolated, placed in RNALater (Ambion) containing 20% ETDA, and decalcified at 4°C for 72 hours. Total RNA was extracted from pooled laser-microdissected sections using TRIzol reagent (Invitrogen) and linearly amplified in 2 rounds using the MessageAmp aRNA kit (Ambion).
Labeled RNA was hybridized to duplicate whole-genome microarrays (G4122A; Agilent). Raw data were processed in R statistical language, using the limma package. Data were adjusted for multiple testing using Benjamini and Hochberg's method to control the false discovery rate. Raw data have been deposited in the Gene Expression Omnibus database (26). Microarrays were validated by SYBR Green qRT-PCR (a list of the primers used is shown in Supplementary Table 3, available on the Arthritis & Rheumatism web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38035/abstract), using the comparative Ct method. Data were normalized to the geometric mean expression values for 2 housekeeping genes, Atp5b and Rpl10.
RESULTS
An intrinsic molecular circadian clock in cartilage
To investigate circadian rhythms in cartilage tissue, we monitored the dynamics of PER2::luc protein expression ex vivo in xiphoid (adult mouse) cartilage and femoral head (6-day-old mouse) cartilage from PER2::luc reporter mice, using bioluminescence photon counting. This revealed robust circadian oscillations in both types of cartilage (period ∼24.5 hours), which persisted for 1–3 weeks (Figures 1A and B). (Further details are available from the corresponding author upon request.) Treatment of cartilage samples (n = 3) with 100 nM dexamethasone (a known synchronizer of peripheral clocks) immediately reinitiated a robust rhythm (P = 0.018 versus untreated control) (Figure 1A).
Figure 1.
Evidence of an autonomous circadian clock in mouse cartilage tissue. A, Representative bioluminescence traces (in counts per second) in cultures of xiphoid cartilage (mean ± SD period 24.67 ± 0.13 hours; n = 7) and articular cartilage from the femoral head (mean ± SD period 23.7 ± 0.6 hours; n = 4) of PER2::luc mice. Arrow indicates the time of treatment with 100 nM dexamethasone. B, Hematoxylin and eosin (H&E) staining (left) and high-sensitivity EM-CCD camera images (right) of xiphoid tissue. Left, A representative H&E–stained tissue section demonstrates typical morphologic features of the xiphoid cartilage (first panel; bar = 500 μm). The second panel is a higher-magnification view of the boxed area in the first panel (bar = 100 μm). Right, EM-CCD images of the xiphoid tissue were obtained at 32, 44, and 56 hours post–dexamethasone synchronization (bars = 300 μm). C, Temperature entrainment. Two xiphoid cartilage cultures (represented by red and blue lines) from the same animal were held under antiphase temperature cycles (alternating 12-hour cycles of 38.5°C/36°C; baseline temperature of 37°C). Left, Detrended trace (in counts per minute); shaded area represents enforcement of the temperature protocol. Right, Time of peak circadian expression on day 1 and day 7; CT0 = circadian time 0 (start of recording). On day 7, the 2 cultures had significantly different peak times (P = 0.0006 by paired t-test). D, Temperature compensation. Left, Representative detrended traces (in counts per second) in cartilage (n = 4) over a 10°C temperature range. Right, Frequency (reaction rate), calculated as 1/period. Temperature coefficient (Q10) = 0.9, line of best fit y = −0.00039x + 0.05422, R2 = 0.68. Results are the mean ± SEM.
Bioluminescence of the xiphoid tissue was also visualized using a high-sensitivity EM-CCD camera. This revealed rhythmic PER2::luc activity from the cartilaginous tissue (Figure 1B) (see also Supplementary Video, available on the Arthritis & Rheumatism web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38035/abstract). There were no differences in the clock properties between the femoral head cartilage and the xiphoid cartilage (mean ± SD period 24.18 ± 0.3 hours and 23.4 ± 0.5 hours, respectively; P = 0.2 by t-test). Microdissected articular cartilage from the knee joints of adult PER2::luc mice also demonstrated circadian oscillations (results available from the corresponding author upon request). However, the small tissue size meant that cultures did not produce robust enough rhythms for further study. Therefore, xiphoid tissue was used as a convenient and more amenable model in subsequent studies.
Because cartilage is avascular, with no direct innervation, we hypothesized that body temperature oscillations controlled by the central brain clock could be important in entraining local rhythms in this tissue. Indeed, when PER2::luc mouse cartilage was incubated under alternating 12-hour temperature cycles of 38.5°C/36°C, oscillation amplitude was induced (Figure 1C). Incubating cultures of cartilage tissue from the same animal under antiphase temperature cycles drove the tissue into antiphase (Figure 1C). Altered phase was maintained under subsequent conditions of constant temperature, thus demonstrating that the observed effect on the circadian rhythm was a clock-based change rather than a passive response to changes in temperature.
Recent evidence suggests that temperature fluctuations will target circadian clock genes at the transcriptional level (3–27). To this end, we lentivirally transduced a human chondrosarcoma cell line with clock promoter luciferase reporters, and then administered single 6-hour pulses of heat (+2.5°C) or cool (−2.5°C). Consistently, we found that the transcriptional levels of Per2:luc and Bmal1::luc were altered (results available from the corresponding author upon request).
A defining feature of a circadian oscillator is temperature compensation, the maintenance of robust periodicity over a broad temperature range (28). Indeed, our data demonstrated that the circadian rhythm in cartilage was effectively temperature compensated. The circadian period ranged from 23.5 hours to 26.5 hours over a 10°C temperature range (temperature coefficient Q10 = 0.9) (Figure 1D). Taken together, these findings establish the presence of a temperature-compensated, autonomous circadian clock in cartilage tissue and suggest that this clock senses suprachiasmatic nuclei–driven daily changes in body temperature as one means of maintaining synchrony with the rest of the body.
Impact of aging on the cartilage circadian rhythm
With aging, systemic circadian rhythms in body temperature and hormone release are altered (29). This may consequently alter the cartilage clock. To test this, we compared PER2::luc rhythms in cartilage from aged mice (ages 20–24 months) and young adult mice (ages 2–4 months). Although there were no significant changes in the circadian period or phase, oscillation amplitude was significantly reduced (reduction of ∼40%) in aged mouse cartilage (Figures 2A–C). Interestingly, upon dexamethasone treatment, robust rhythmicity was reinitiated to a similar extent (increase of ∼5–10–fold) in both aged and young adult mouse cartilage tissue (Figure 2D), indicating that intrinsic pacemaking mechanisms within aged cartilage may still be intact. These studies provide evidence that the circadian rhythm in cartilage loses its robustness with advanced age.
Figure 2.

A less robust circadian clock in mouse cartilage during aging. A, Representative traces of PER2::luc bioluminescence from xiphoid cartilage of young adult mice (ages 3–4 months; n = 7) and aged mice (ages 20–24 months; n = 9). Arrow indicates time of treatment with 100 nM dexamethasone (Dex) (n = 4 young mice, n = 3 aged mice). B, Oscillation amplitude (left) and circadian period (right) in cartilage tissue from aged mice compared to young mice. ∗ = P = 0.015 by t-test. NS = not significant. C, Circadian phase at the time between 12-hour cull and the first peak of bioluminescence in culture. Vertical lines represent the mean for each group (P = 0.46). D, Similar extent of induction of oscillation amplitude by dexamethasone treatment in young and aged mouse cartilage tissue; amplitude is expressed as a percentage of the amplitude of the young tissue before treatment. Results are the mean ± SEM. Data were analyzed by two-way analysis of variance. There were no significant effects of age group (P = 0.77), and interactions were not significantly different (P = 0.9992). ∗∗ = P = 0.003.
Identification of the circadian transcriptome in cartilage
To gain insight into the function of the circadian clock in cartilage, we aimed to identify its tissue-specific targets. To this end, we performed a time-series microarray using xiphoid tissue from mice kept in constant darkness. This eliminated the effect of the light/dark cycle, and allowed us to reveal endogenously circadian genes. Time points were every 4 hours for 40 hours. To identify circadian transcripts, we used 2 well-recognized algorithms based on different statistical models. With use of the CircWave Batch algorithm, we identified 896 genes oscillating with a period of ∼24 hours (q < 0.1). Parallel analysis was performed using the JTKCycle method (21), which identified 1,574 rhythmic genes with a 20–28-hour period (adjusted P < 0.05). To be stringent, we considered only those genes identified by both methods as circadian. Thus, we identified 615 genes with rhythmic expression and a circadian period of ∼24 hours (3.9% of expressed genes, represented by 704 probes) (Figure 3A and Supplementary Table 1, available on the Arthritis & Rheumatism web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38035/abstract).
Figure 3.

Identification of the circadian transcriptome in mouse cartilage. A, Heat map depicting the expression level of the 615 circadian genes (represented by 705 probe sets), showing that 3.9% of the cartilage transcriptome is circadian, as identified using CircWave Batch (local false discovery rate < 0.1, equivalent to approximate R2 > 0.7) and JTKCycle (Bonferroni-adjusted P < 0.05). Genes are organized according to timing of peak expression. B, Clustering and identification of periodic functions. Each panel represents a single cluster, with normalized log-expression levels marked as connected points for each gene over time. Colored lines represent the inferred cluster-wise periodic function, with 2 standard deviations marked by the shaded area. Clusters fall into 4 broad categories, based on the time of peak expression. In A and B, gray-shaded bars represent subjective day, and solid black bars represent subjective night. C, Tissue specificity of cartilage clock-controlled genes. Venn diagram shows a comparison of cartilage circadian genes with previously published gene lists from studies of the liver and skeletal muscle of mice. The total number of genes identified as circadian in each tissue is shown in parentheses; areas of overlap indicate common genes.
Rhythmic cartilage genes were clustered according to their inferred periodic functions, determined using a Gaussian process model. Genes fell into 11 distinct clusters, which were grouped into 4 categories based on the phase of their peak expression (Figure 3B). Most genes peaked during subjective daytime (Figure 3A and results available from the corresponding author upon request). Functional annotation and gene ontology (GO)–term analysis using DAVID revealed several overrepresented functional groups (a full list is available from the corresponding author upon request), with the top overrepresented groups being “purine/ATP binding,” “circadian rhythm,” “extracellular matrix location/microfibril,” “negative regulator of (macromolecule) biosynthetic processes,” “proteolysis,” and “apoptosis.” Interestingly, 81.3% of genes related to “negative regulation of biosynthesis” and 78.6% of genes related to “proteolysis” peaked during the animals' resting phase (results available from the corresponding author upon request).
We compared our gene list to published sequential chromatin immunoprecipitation assay data on the expression of circadian clock transcription factor Bmal1 in the liver (30), and we found that 132 of our rhythmic genes had previously determined Bmal1 binding sites (see Supplementary Table 1, available on the Arthritis & Rheumatism web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38035/abstract). Only 19 circadian genes were common (Figure 3C) between our gene expression data set from mouse cartilage and previously published data from the liver (31) and skeletal muscle (32) of mice, most of which were core clock genes, supporting the concept of tissue-specific clock function.
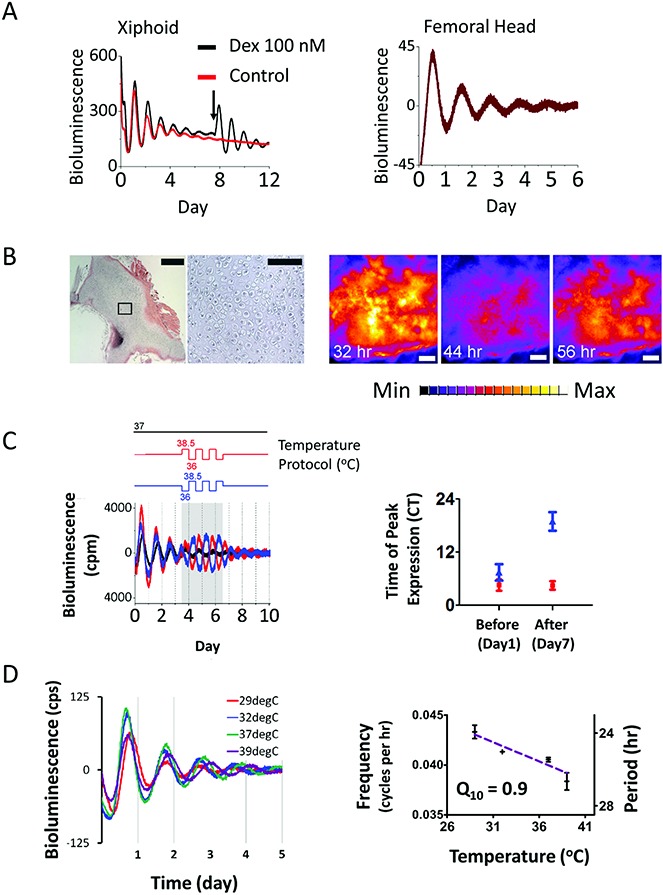
The expression profiles of canonical clock genes (Bmal1, Per2, Cry1, Nr1d1, Dbp, and E4bp4) and selected cartilage genes (Adamts4, Adamts9, and Mmp14) were validated by temporal qRT-PCR in mouse xiphoid tissue (Figure 4A). To confirm the importance of the molecular clock in controlling cartilage gene rhythmicity, we used ClockΔ19 mutant mice on a PER2::luc background. The expression and behavior of genes in ClockΔ19 mice are known to be arrhythmic (18). We established that PER2::luc rhythmicity was lost in the mutant mouse cartilage, and that time-dependent expression of the circadian output genes Dbp and E4bp4 was abolished (Figure 4B). Importantly, time-dependent expression of the cartilage genes Adamts4 and Mmp14 was similarly abolished in the ClockΔ19 mouse cartilage (Figure 4B), supporting the conclusion that these genes are clock-controlled.
Figure 4.
Validation of circadian genes in mouse cartilage. A, Relative expression of a panel of clock genes and putative clock-controlled genes in mouse xiphoid tissue, as determined by microarray and validated by quantitative reverse transcription–polymerase chain reaction (qRT-PCR). Data were normalized to the nadir of expression. All genes tested followed the expression pattern identified by the array; when analyzed using one-way analysis of variance, all genes showed significant change with time (P < 0.0001; for Mmp14, P = 0.0039). Gray-shaded bars denote subjective day, and solid black bars denote subjective night. Results are the mean ± SEM (n = 3 replicates, pooled from 5 animals per replicate). B, Abolition of circadian rhythmicity in ClockΔ19 mutant animals. Left, Xiphoid tissue cultures from ClockΔ19 mice on a PER2::luc background showed no molecular rhythmicity, as compared to wild-type (WT) mice, in cultures (results representative of 2 independent experiments; bioluminescence expressed in counts per second). Right, Time-dependent expression of clock output genes was abolished in ClockΔ19 animals. The qRT-PCR data were normalized to the nadir of expression in WT mice. Results are the mean ± SEM (n = 5). ∗∗ = P < 0.01; ∗∗∗ = P < 0.001, by t-test. NS = not significant; ZT = zeitgeber time; ZT0 = lights-on.
Cell-autonomous circadian rhythms in chondrocytes
To confirm that chondrocytes harbor cell-autonomous circadian clocks, we isolated immature articular chondrocytes from 5-day-old PER2::luc mice. PMT recording revealed self-sustained circadian oscillations resembling those from the cartilage tissue (Figure 5A). The mouse cells demonstrated typical morphologic features of chondrocytes, characterized by Alcian blue staining of glycosaminoglycans in the extracellular matrix and high Col2 expression (Figure 5B). Furthermore, clock-synchronized primary chondrocytes time-dependently expressed the core clock genes Nr1d1, Per2, Bmal1, and Cry1 (Figure 5C).
Figure 5.

Cell-autonomous circadian clocks in chondrocytes. A, Representative circadian oscillations of bioluminescence from immature articular chondrocytes isolated from 5-day-old PER2::luc mice (n = 5). B, Confirmation of the chondrocyte phenotype. Left, Cell morphology under phase contrast. Bar = 100 μm. Middle, Alcian blue staining of glycosaminoglycans in the extracellular matrix. Bar = 50 μm. Right, Immunofluorescence for type II collagen (green) with DAPI nuclear counterstain (blue). Bar = 50 μm. C, Time-dependent expression of clock genes in primary chondrocytes. Cells were synchronized and gene expression was assayed by quantitative reverse transcription–polymerase chain reaction (qRT-PCR) at 6-hour intervals. Data were normalized to the nadir of expression. Results are the mean ± SEM (n = 3 technical replicates). P < 0.001 for all genes, by one-way analysis of variance (ANOVA). D, Expression of Per2:luc and Bmal1:luc reporters in separate cultures of SW-1353 cells after lentiviral delivery. Cells were synchronized by treatment with dexamethasone for 1 hour before bioluminescence recording. Representative detrended traces are shown (mean ± SEM period 24.1 ± 0.3 hours; n = 4). E, Time-dependent expression of cartilage clock targets in human chondrosarcoma cells. Cells were synchronized, and gene expression was assayed by qRT-PCR at 4-hour intervals. Data were normalized to the nadir of expression. Results are the mean ± SEM (n = 4). P < 0.05 for all genes, by one-way ANOVA.
To test whether human chondrocytes also exhibit circadian rhythmicity, we used lentiviral delivery to stably transduce a human chondrosarcoma cell line (SW-1353) with luciferase reporters driven by promoter fragments of the clock genes Per2 or Bmal1. Both reporters oscillated robustly over several days (Figure 5D), in antiphase to one another. Similar to that observed in mouse cells, the clock genes Nr1d1, Per2, and Bmal1 were time-dependently expressed in SW-1353 cells (Figure 5E). Interestingly, putative clock target genes (Adamts4/9, Mmp14, and Gnl3) were also expressed in a time-dependent manner (Figure 5E), although Adamts4 expression was less robust.
Taken together, these data support the concept that chondrocytes contain a cell-autonomous molecular clock capable of driving downstream targets, extending our findings from a mouse model into human cells. This implies that there is a possible conservation of clock mechanisms in chondrocytes from different species.
Clock gene changes in articular cartilage in an experimental mouse model of OA
We next sought to address whether joint articular cartilage expresses clock genes, and whether the expression of clock genes is altered in a surgical DMM model of OA. Laser microdissection allowed us to focus on lesion sites, to study factors initiating damage. One week post-DMM, cartilage tissue damage was minimal; 2 weeks post-DMM, tissue exhibited early focal degeneration; 6 weeks post-DMM, tissue was more extensively damaged, with fibrillation.
Our microarray data set of the gene networks involved in mouse OA is presented elsewhere (26). We interrogated these data and found that the canonical clock genes Bmal1, E4bp4, Nr1d1, Per2, Cry1, Dbp, and Rora were indeed expressed in the DMM articular cartilage. Bmal1 was expressed at a significantly higher level in DMM cartilage than in sham control cartilage at 1, 2, and 6 weeks post-DMM (P < 0.05) (Figure 6). The clock output genes Nr1d1, Dbp, Rora, and E4bp4 were also significantly altered 2 weeks post-DMM (P < 0.05) (Figure 6), consistent with the time point at which the greatest change in Bmal1 expression was observed. Changes in clock gene expression were validated using qRT-PCR (results available from the corresponding author upon request). Taken together, these data indicate that clock gene expression changes early in the progression of experimental OA.
Figure 6.
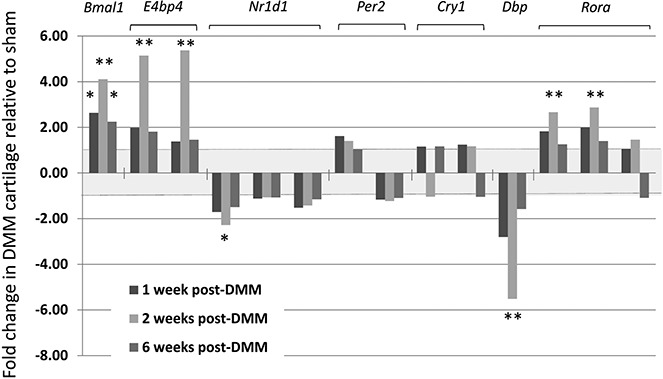
Deregulation of clock genes in an experimental mouse model of osteoarthritis. Articular cartilage mRNA expression of core clock and clock output genes was determined by microarray analysis. Results of all probes for each gene are shown, with findings expressed as the fold up-regulation or down-regulation of gene expression in joints subjected to destabilization of the medial meniscus (DMM) compared to sham-operated joints in mice at 1, 2, or 6 weeks post–DMM surgery. Values are the mean fold change in 4 mice. The shaded region between +1-fold and −1-fold represents no differential expression. ∗ = adjusted P < 0.05; ∗∗ = adjusted P < 0.01.
DISCUSSION
Mice with mutations in clock genes show ectopic ossification of tendon and cartilage, altered regulation of bone volume, an increased susceptibility to inflammatory arthritis, and, as recently reported, defects in long bone growth through clock control of chondrocyte differentiation (9–33). These studies highlight the potential importance of circadian clocks in the skeletal system. However, to the best of our knowledge, no studies have demonstrated functional and cell-autonomous circadian clocks in fully differentiated cartilage tissue. Therefore, we set out to address the following questions. 1) Do cartilage and chondrocytes contain self-sustained circadian clocks? 2) What factors entrain the cartilage clock? 3) What cartilage-specific genes/pathways are controlled by the circadian clock? 4) How does the cartilage circadian clock change with aging and during the pathogenesis of experimental arthritis?
Herein, we have demonstrated cell-autonomous circadian clocks in cartilage tissue and chondrocytes. Similar to previously described peripheral circadian clocks (3–28), the cartilage clock is entrained by temperature cycles approximating body temperature fluctuations and its periodicity is effectively maintained across a wide temperature range. Thus, sensing changes in body temperature may be one way for the cartilage clock to maintain synchronization to the rest of the body. Although chondrocytes are dispersed in the avascular and aneural cartilage, systemic factors freely diffuse and it is probable that multiple entraining factors, such as hormone signaling, act alongside temperature to entrain the clock.
Age is the major risk factor for developing OA. Its precise role in disease pathogenesis is unknown. Disruption to whole-body circadian rhythms through aging could alter systemic time cues and compromise cartilage clock function. Indeed, the results of the present study show that PER2::luc circadian rhythms were dampened in aged mouse cartilage tissue. We speculate that age-associated dampening in circadian rhythms could disrupt downstream OA-associated genes and increase disease susceptibility. Circadian amplitude in aged tissue could be restored to that of young cartilage by treatment with dexamethasone, suggesting that inherent changes in the molecular clock are unlikely. Instead, we hypothesize that age-related dampening of the cartilage clock may be a consequence of altered systemic time cues. This is consistent with a previous report showing no difference in the molecular clock of cultured human fibroblasts between old and young individuals, although their serum had different clock-synchronizing properties (34). Our data suggest that the aged cartilage clock might be re-tuned by factors strengthening systemic circadian rhythms, such as scheduled exercise (35), restricted meal times (36), or the scheduled warming and cooling of joints; this may have significant impact on the future management of joint diseases such as OA.
Peripheral circadian clocks temporally regulate tissue-specific functions through control of downstream CCGs. Indeed, 3–15% of total transcripts are expressed rhythmically, depending on the tissue, sampling frequency, and analysis stringency (31–37). Herein, we have identified the first circadian transcriptome profile in cartilage tissue. Our results revealed rhythmic expression of ∼3.9% of all expressed genes, with the majority of the genes being tissue specific. By selecting genes identified by both CircWave and JTKCycle, we generated a relatively conservative circadian gene list. However, it is likely that some of the genes identified are directly responsive to time cues, such as body temperature, rather than being local clock driven. Studies in other tissues (38–39) have demonstrated only a small number of such system-driven genes (10–16%). Indeed, we demonstrated cell-autonomous oscillations of clock genes and putative targets in cells lacking systemic time cues.
The rhythmic transcripts identified in cartilage exhibited 11 distinct patterns of expression. We predict that genes within each cluster are coregulated by similar clock proteins and tissue-specific transcription factors. Interestingly, in our mouse cartilage data set, early-day transcripts included several extracellular matrix components (fibrillins, laminin, collagens, and netrin), and almost one-half of the rhythmic proteolysis genes peaked at this early-day time point. Restricting genes controlling macromolecule turnover to the resting phase may confer a selective advantage, allowing more effective tissue remodeling and repair following (nocturnal) activity bouts. It is possible that disruption to this time-coordinated homeostasis during aging increases OA susceptibility.
Disruptions in several of the rhythmic genes/pathways identified herein have previously been linked to joint diseases (40–44). These include genes involved in catabolic signaling (Adamts4, Adamts9, and Mmp14), anabolic signaling (Tgfβr3 and Timp4), and the oxidative defense pathway (Gclc and Gstm1), as well as apoptosis-related genes (Xiap). Some genes have even been suggested as drug targets, for example, the Adamts family of aggrecanases (45). Interestingly, a newly identified OA susceptibility gene, Gnl3 (42), was also rhythmic. The circadian nature of these genes may help inform appropriate targeting strategies, including, for example, timed drug administration (chronotherapy), with the aim of improving efficacy and reducing toxicity (46). Our study also raises the possibility of targeting the gene activity of chondrocytes through the use of circadian clock–acting compounds (47–48), which may prove of use in the treatment of diseases such as OA.
It should be noted that we used xiphoid tissue as a model for mature cartilage in the time series, due to limitations involved in microdissecting articular cartilage, but that both tissues exhibit circadian rhythms (results available from the corresponding author upon request). Furthermore, we demonstrated that both mouse primary chondrocytes and human chondrosarcoma-derived SW-1353 cells rhythmically express clock genes in vitro (Figure 5), although we recognize that the latter cell line is a limited model for primary cells or tissue (49).
Results of previous studies have suggested that clock genes change during inflammatory joint disease (11–15), but no studies have addressed whether clock genes in articular cartilage change during OA initiation/progression. Herein, we have shown that clock genes are indeed expressed in articular cartilage. Moreover, during the development of OA in a mouse DMM model, the expression of these genes was disrupted. Interestingly, most of the changes were seen during the early stage of the disease (2 weeks post-DMM), not when the disease phenotype was more established (6 weeks post-DMM). This could mean that clock gene changes contribute to the disease phenotype through the disruption of CCGs, rather than being a consequence of tissue damage. Due to technical limitations, we compared expression changes of clock genes at a single daytime time point (10:00 AM–noon). Future studies with more time points will be required to determine changes in the period or amplitude of these genes.
Taken together, our data provide substantial insight into the circadian regulation of cartilage biologic processes. These results provide a firm base upon which to further explore the link between the circadian clock, aging, cartilage homeostasis, and OA.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Drs. Boot-Handford and Meng had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design. Gossan, Boot-Handford, Meng.
Acquisition of data. Gossan, Hughes, Bateman, Rowley, Little, Meng.
Analysis and interpretation of data. Gossan, Zeef, Hensman, Hughes, Bateman, Piggins, Rattray, Boot-Handford, Meng.
Acknowledgments
The authors thank the Genomic Technologies, the Histology, the Photographics, and the Bioimaging Core Facilities in the Faculty of Life Sciences (University of Manchester) for providing technical support/advice. We also thank Dr. Michael Hughes (Yale University School of Medicine) for providing the JTKCycle script, Dr. Roelof Hut (University of Groningen) for the CircWave software, Prof. Joseph Takahashi (University of Texas Southwestern Medical Center) for the PER2::luc and ClockΔ19 mice, Ding Jin (University of Manchester) for technical support, Dr. Louise Kung (University of Manchester) for technical advice, Dr. Daniele Belluoccio for help with cartilage microdissection, Dr. Katrina Bell (Murdoch Children's Research Institute, Bioinformatics) for assistance with the mouse OA array analysis, and Dr. Patrick Nolan (Murdoch Children's Research Institute, Mammalian Genetics Unit), Prof. Robert Lucas, and Prof. Tim Hardingham (University of Manchester) for comments on the manuscript.
Additional Supporting Information may be found in the online version of this article.
REFERENCES
- Reppert SM, Weaver DR. Coordination of circadian timing in mammals. Nature. 2002;418:935–41. doi: 10.1038/nature00965. [DOI] [PubMed] [Google Scholar]
- Hastings MH, Reddy AB, Maywood ES. A clockwork web: circadian timing in brain and periphery, in health and disease. Nat Rev Neurosci. 2003;4:649–61. doi: 10.1038/nrn1177. [DOI] [PubMed] [Google Scholar]
- Buhr ED, Yoo SH, Takahashi JS. Temperature as a universal resetting cue for mammalian circadian oscillators. Science. 2010;330:379–85. doi: 10.1126/science.1195262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi JS, Hong HK, Ko CH, McDearmon EL. The genetics of mammalian circadian order and disorder: implications for physiology and disease. Nat Rev Genet. 2008;9:764–75. doi: 10.1038/nrg2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhosale AM, Richardson JB. Articular cartilage: structure, injuries and review of management. Br Med Bull. 2008;87:77–95. doi: 10.1093/bmb/ldn025. [DOI] [PubMed] [Google Scholar]
- Igarashi K, Saeki S, Shinoda H. Diurnal rhythms in the incorporation and secretion of 3H-proline and 3H-galactose by cartilage cells and osteoblasts in various bone-forming sites in growing rats. Orthodontic Waves. 2013;72:11–15. [Google Scholar]
- Stevenson S, Hunziker EB, Herrmann W, Schenk RK. Is longitudinal bone growth influenced by diurnal variation in mitotic activity of chondrocytes of the growth plate? J Orthop Res. 1990;8:132–5. doi: 10.1002/jor.1100080117. [DOI] [PubMed] [Google Scholar]
- Russell JE, Grazman B, Simmons DJ. Mineralization in rat metaphyseal bone exhibits a circadian stage dependency. Proc Soc Exp Biol Med. 1984;176:342–5. doi: 10.3181/00379727-176-41880. [DOI] [PubMed] [Google Scholar]
- Maronde E, Schilling AF, Seitz S, Schinke T, Schmutz I, Horst van der G, et al. The clock genes Period 2 and Cryptochrome 2 differentially balance bone formation. PLoS One. 2010;5:e11527. doi: 10.1371/journal.pone.0011527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takarada T, Kodama A, Hotta S, Mieda M, Shimba S, Hinoi E, et al. Clock genes influence gene expression in growth plate and endochondral ossification in mice. J Biol Chem. 2012;287:36081–95. doi: 10.1074/jbc.M112.408963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashiramoto A, Yamane T, Tsumiyama K, Yoshida K, Komai K, Yamada H, et al. Mammalian clock gene Cryptochrome regulates arthritis via proinflammatory cytokine TNF-α. J Immunol. 2010;184:1560–5. doi: 10.4049/jimmunol.0903284. [DOI] [PubMed] [Google Scholar]
- Bellamy N, Sothern RB, Campbell J, Buchanan WW. Circadian rhythm in pain, stiffness, and manual dexterity in rheumatoid arthritis: relation between discomfort and disability. Ann Rheum Dis. 1991;50:243–8. doi: 10.1136/ard.50.4.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellamy N, Sothern R, Campbell J, Buchanan W. Rhythmic variations in pain, stiffness, and manual dexterity in hand osteoarthritis. Ann Rheum Dis. 2002;61:1075–80. doi: 10.1136/ard.61.12.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengatto CM, Mussano F, Honda Y, Colwell CS, Nishimura I. Circadian rhythm and cartilage extracellular matrix genes in osseointegration: a genome-wide screening of implant failure by vitamin D deficiency. PLoS One. 2011;6:e15848. doi: 10.1371/journal.pone.0015848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas S, Straub RH. Disruption of rhythms of molecular clocks in primary synovial fibroblasts of patients with osteoarthritis and rheumatoid arthritis, role of IL-1β/TNF. Arthritis Res Ther. 2012;14:R122. doi: 10.1186/ar3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanbe K, Inoue K, Xiang C, Chen Q. Identification of clock as a mechanosensitive gene by large-scale DNA microarray analysis: downregulation in osteoarthritic cartilage. Mod Rheumatol. 2006;16:131–6. doi: 10.1007/s10165-006-0469-3. [DOI] [PubMed] [Google Scholar]
- Yoo SH, Yamazaki S, Lowrey PL, Shimomura K, Ko CH, Buhr ED, et al. PERIOD2:LUCIFERASE real-time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proc Natl Acad Sci U S A. 2004;101:5539–46. doi: 10.1073/pnas.0308709101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitaterna MH, King DP, Chang AM, Kornhauser JM, Lowrey PL, McDonald JD, et al. Mutagenesis and mapping of a mouse gene, Clock, essential for circadian behavior. Science. 1994;264:719–25. doi: 10.1126/science.8171325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Meng QJ, Tyler NJ, Stokkan KA, Loudon AS. A circadian clock is not required in an arctic mammal. Curr Biol. 2010;20:533–7. doi: 10.1016/j.cub.2010.01.042. [DOI] [PubMed] [Google Scholar]
- Strimmer K. fdrtool: a versatile R package for estimating local and tail area-based false discovery rates. Bioinformatics. 2008;24:1461–2. doi: 10.1093/bioinformatics/btn209. [DOI] [PubMed] [Google Scholar]
- Hughes ME, Hogenesch JB, Kornacker K. JTK_CYCLE: an efficient nonparametric algorithm for detecting rhythmic components in genome-scale data sets. J Biol Rhythms. 2010;25:372–80. doi: 10.1177/0748730410379711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen CE, Williams CK. Gaussian processes for machine learning. Cambridge (MA): MIT Press; 2006. [Google Scholar]
- Dunson DB. Nonparametric Bayes applications to biostatistics. In: Hjort NL, Holmes C, Muller P, Walker SG, editors. Bayesian nonparametrics. Cambridge (UK): Cambridge University Press; 2010. pp. 223–73. [Google Scholar]
- Hensman J, Rattray M, Lawrence ND. 2012. Fast variational inference in the conjugate exponential family. arXiv: 1206.5162 [cs.LG]. URL: http://arxiv.org/abs/1206.5162.
- Gosset M, Berenbaum F, Thirion S, Jaques C. Primary culture and phenotyping of murine chondrocytes. Nat Protoc. 2008;3:1253–60. doi: 10.1038/nprot.2008.95. [DOI] [PubMed] [Google Scholar]
- Bateman JF, Rowley L, Belluoccio D, Chan B, Bell K, Fosang AJ, et al. Transcriptomics of wild-type mice and mice lacking ADAMTS-5 activity identifies genes involved in osteoarthritis initiation and cartilage destruction. Arthritis Rheum. 2013;65:1547–61. doi: 10.1002/art.37900. [DOI] [PubMed] [Google Scholar]
- Morf J, Rey G, Schneider K, Stratmann M, Fujita J, Naef F, et al. Cold-inducible RNA-binding protein modulates circadian gene expression posttranscriptionally. Science. 2012;338:379–83. doi: 10.1126/science.1217726. [DOI] [PubMed] [Google Scholar]
- Francois P, Despierre N, Siggia ED. Adaptive temperature compensation in circadian oscillations. PLoS Comput Biol. 2012;8:e1002585. doi: 10.1371/journal.pcbi.1002585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SA, Pagani L, Cajochen C, Eckert A. Systemic and cellular reflections on aging and the circadian oscillator: a mini-review. Gerontology. 2011;57:427–34. doi: 10.1159/000320673. [DOI] [PubMed] [Google Scholar]
- Rey G, Cesbron F, Rougemont J, Reinke H, Brunner M, Naef F. Genome-wide and phase-specific DNA-binding rhythms of BMAL1 control circadian output functions in mouse liver. PLoS Biol. 2011;9:e1000595. doi: 10.1371/journal.pbio.1000595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes ME, DiTacchio L, Hayes KR, Vollmers C, Pulivarthy S, Baggs JE, et al. Harmonics of circadian gene transcription in mammals. PLoS Genetics. 2009;5:e1000442. doi: 10.1371/journal.pgen.1000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy JJ, Andrews JL, McDearmon EL, Campbell KS, Barber BK, Miller BH, et al. Identification of the circadian transcriptome in adult mouse skeletal muscle. Physiol Genomics. 2007;31:86–95. doi: 10.1152/physiolgenomics.00066.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunger MK, Walisser JA, Sullivan R, Manley PA, Moran SM, Kalscheur VL, et al. Progressive atrophy in mice with a targeted disruption of the Mop3/Bmal-1 locus. Genesis. 2005;41:122–32. doi: 10.1002/gene.20102. [DOI] [PubMed] [Google Scholar]
- Pagani L, Schmitt K, Meier F, Izakovic J, Roemer K, Viola A, et al. Serum factors in older individuals change cellular clock properties. Proc Natl Acad Sci U S A. 2011;108:7218–23. doi: 10.1073/pnas.1008882108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AT, Piggins HD. Feedback actions of locomotor activity to the circadian clock. Prog Brain Res. 2012;199:305–36. doi: 10.1016/B978-0-444-59427-3.00018-6. [DOI] [PubMed] [Google Scholar]
- Feillet CA, Albrecht U, Challet E. “Feeding time” for the brain: a matter of clocks. J Physiol Paris. 2006;100:252–60. doi: 10.1016/j.jphysparis.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Panda S, Antoch MP, Miller BH, Su AI, Schook AB, Straume M, et al. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell. 2002;109:307–20. doi: 10.1016/s0092-8674(02)00722-5. [DOI] [PubMed] [Google Scholar]
- Kornmann B, Schaad O, Bujard H, Takahashi JS, Schibler U. System-driven and oscillator-dependent circadian transcription in mice with a conditionally active liver clock. PLoS Biol. 2007;5:e34. doi: 10.1371/journal.pbio.0050034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes ME, Hong HK, Chong JL, Indacochea AA, Lee SS, Han M, et al. Brain-specific rescue of Clock reveals system-driven transcriptional rhythms in peripheral tissue. PLoS Genet. 2012;8:e1002835. doi: 10.1371/journal.pgen.1002835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson RK, Waters JG, Kevorkian L, Darrah C, Cooper A, Donell ST, et al. Expression profiling of metalloproteinases and their inhibitors in synovium and cartilage. Arthritis Res Ther. 2006;8:R124. doi: 10.1186/ar2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swingler TE, Waters JG, Davidson RK, Pennington CJ, Puente XS, Darrah C, et al. Degradome expression profiling in human articular cartilage. Arthritis Res Ther. 2009;11:R96. doi: 10.1186/ar2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- arcOGEN Consortium, arcOGEN Collaborators. Identification of new susceptibility loci for osteoarthritis (arcOGEN): a genome-wide association study. Lancet. 2012;380:815–23. doi: 10.1016/S0140-6736(12)60681-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeston J, Wainwright S, Hughes C, Caterson B. The regulation of the ADAMTS4 and ADAMTS5 aggrecanases in osteoarthritis: a review. Clin Exp Rheumatol. 2008;26:139–45. [PubMed] [Google Scholar]
- Cawston TE, Wilson AJ. Understanding the role of tissue degrading enzymes and their inhibitors in development and disease. Best Pract Res Clin Rheumatol. 2006;20:983–1002. doi: 10.1016/j.berh.2006.06.007. [DOI] [PubMed] [Google Scholar]
- Flannery CR. Novel therapies in OA. Curr Drug Targets. 2010;11:614–9. doi: 10.2174/138945010791011884. [DOI] [PubMed] [Google Scholar]
- Levi F, Schibler U. Circadian rhythms: mechanisms and therapeutic implications. Annu Rev Pharmacol Toxicol. 2007;47:593–628. doi: 10.1146/annurev.pharmtox.47.120505.105208. [DOI] [PubMed] [Google Scholar]
- Solt LA, Wang Y, Banerjee S, Hughes T, Kojetin DJ, Lundasen T, et al. Regulation of circadian behaviour and metabolism by synthetic REV-ERB agonists. Nature. 2012;485:62–8. doi: 10.1038/nature11030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota T, Lee JW, St John PC, Sawa M, Iwaisako K, Noguchi T, et al. Identification of small molecule activators of cryptochrome. Science. 2012;337:1094–7. doi: 10.1126/science.1223710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebauer M, Saas J, Sohler F, Haag J, Soder S, Pieper M, et al. Comparison of the chondrosarcoma cell line SW1353 with primary human adult articular chondrocytes with regard to their gene expression profile and reactivity to IL-1β. Osteoarthritis Cartilage. 2005;13:697–708. doi: 10.1016/j.joca.2005.04.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.