

Abstract
A new type of polymer nanoparticle (PNP) containing a high density of covalently linked doxorubicin, attached via a non-cleavable amine linkage (amine-linked Dox-PNP) was prepared. Together with a previously reported cleavable carbamate-linked Dox-PNP, this new amine-linked Dox-PNP was subsequently evaluated against free doxorubicin for its cytotoxicity and inhibitory effects on SKNSH wild-type and SKrDOX6 doxorubicin-resistant human neuroblastoma cell lines. Analogous cholesterol-containing PNPs (Chol-PNPs) and indomethacin-containing PNPs (IND-PNPs) were also synthesized and used as the non-cytotoxic controls. While neither cell line was affected by Chol-PNPs or IND-PNPs, SKrDOX6 doxorubicin-resistant cells exhibited similar cytotoxic responses to free doxorubicin and both amine- and carbamate-linked Dox-PNPs, suggesting that doxorubicin or the doxorubicin-containing polymer must be the active agent in the latter case. SKNSH wild-type cells also responded to both Dox-PNPs, albeit at a higher apparent concentration than free doxorubicin alone. The growth of SKNSH wild-type cells was significantly inhibited upon incubation with carbamate-linked Dox-PNPs, as with free doxorubicin, over a 7-day period. In comparison to free doxorubicin, carbamate-linked Dox-PNPs produced a longer (72-h) period of initial inhibition in SKrDOX6 doxorubicin-resistant cells.
Introduction
Small-molecule chemotherapeutics are known to infiltrate both cancerous and healthy cells, resulting in many undesirable side effects.1 Thus, the ability to selectively deliver drugs to diseased cells while bypassing healthy cells, greatly increasing the therapeutic efficiency of the drug, has been a long sought-after goal. One of the most promising strategies for achieving this type of selective delivery has been through the use of nanoscale particles that can encapsulate a high dose of cytotoxic agents and release them at the disease sites.2-4 If such drug delivery vehicles also have increased retention time, half-life, and bioavailability as well as higher specificity for cancer cells, the toxic side effects of their drug cargo might be lowered significantly.5-11
Among the many delivery vehicles that have been proposed, the encapsulation of chemotherapeutic drugs into polymeric nanoparticles has shown enhanced selectivity toward solid tumor masses in vivo via the enhanced permeation and retention (EPR) effect.12-16 These polymer scaffolds hold great promise as a means to safely and effectively administer chemotherapeutic drugs. However, before they can be evaluated in vivo, some indication as to the in vitro uptake, release, dose-response, and efficacy must be obtained against the appropriate controls.
We previously reported two different drug-containing polymer nanoparticles (PNPs), one (carbamate-linked Dox-PNP) with covalently conjugated doxorubicin in its core and one (IND-PNP) with covalently conjugated indomethacin, both surrounded by a hydrophilic shell of poly(ethylene oxide).17, 18 The doxorubicin in the carbamate-linked Dox-PNPs is attached to the polymer backbone through an acid-labile carbamate linkage while the indomethacin is linked to the polymer via an amide linkage (See Schemes S1 and S2 in Electronic Supplementary Information (ESI†)). Both can be effectively released under bench-top acidic conditions that mimics those found in solid tumors.19 In this manuscript, we expanded this work to evaluate the in vitro efficacy of both Dox- and IND-PNPs vs. free doxorubicin as well as two other PNP systems: one possess inert cholesterol moieties (Scheme 1) to serve as an in vitro control and the other contains doxorubicin moieties that are linked to the polymer backbone through non-cleavable amine linkages (amine-linked Dox-PNPs, Scheme 2). While carbamate-linked Dox-PNPs exhibited good in vitro efficacy in SKNSH human neuroblastoma cell lines, as expected, we were surprised that the amine-linked Dox-PNPs also produced a cytotoxic effect in vitro. However, the cholesterol-containing PNPs showed no toxicity, suggesting that doxorubicin, either on or off the polymer, is the cytotoxic agent.
Scheme 1.

Synthesis of cholesterol-conjugated polymer nanoparticles from PEO6-b-CHOL30. Cholesterol was deprotonated with NaH in refluxing THF to generate the cholesterol alkoxide which was then added to bromoxylene norbornene monomer (7) to generate the cholesterol-conjugated norbornene monomer 1. Monomer 3 was first polymerized using Grubbs first-generation catalyst 2. After the polymerization was complete, monomer 1 was added to generate the second block, and the polymerization was quenched with ethyl vinyl ether. The amphiphilic block copolymer was dissolved in THF and nanoparticle formation was induced by addition of water dropwise to the vigorously stirring solution. Excess THF was removed via dialysis against nanopure water, yielding narrowly dispersed polymer nanoparticles.
Scheme 2.

Synthesis of amine-linked doxorubicin-conjugated polymer nanoparticles. Monomer 3 was polymerized using Grubbs first-generation catalyst (2). After the polymerization was complete, amine-linked doxorubicin-conjugated norbornene (4) was added to generate the second block, and the polymerization was terminated with ethyl vinyl ether. The amphiphilic block copolymer was dissolved in DMSO, and nanoparticle formation was induced by addition of water dropwise to the vigorously stirring solution. Excess DMSO was removed via dialysis against ultrapure deionized water, yielding narrowly dispersed polymer nanoparticles.
Results and Discussion
Synthesis of Chol-PNPs and amine-linked Dox-PNPs
To form good nanoparticles using the procedure described in the Materials and Methods section (vide infra), the block copolymer must be a solid. Given our previous experience,18 the PEO15-b-CHOL15 and PEO12-b-CHOL25 (Scheme 1, as well as the corresponding CHOL25-b-PEO12) polymers, which are viscous liquids at room temperature, should not afford well-defined PNPs. For the PEOm-b-CHOLn and CHOLn-b-PEOm polymers reported in Table 1, this observation dictates that the cholesterol-containing block must be several times longer than the PEO block for the block copolymer to be a solid. This is indeed the case.
Table 1.
Characterization data for cholesterol-containing amphiphilic block copolymers.
| Polymer | Mn (Daltons) |
Mw (Daltons) |
PDI | Theoretical Mn (Daltons) |
|---|---|---|---|---|
| PEO15-b-CHOL15 | 13807 | 16900 | 1.22 | 16690 |
| PEO6-b-CHOL30 | 21551 | 22234 | 1.03 | 21096 |
| PEO9-b-CHOL28 | 18980 | 19509 | 1.02 | 21424 |
| PEO12-b-CHOL25 | 21798 | 22862 | 1.04 | 21153 |
| CHOL30-b-PEO6 | 20152 | 21792 | 1.08 | 21096 |
| CHOL28-b-PEO9 | 20375 | 23592 | 1.08 | 21424 |
| CHOL25-b-PEO12 | 19111 | 21102 | 1.10 | 21153 |
Both PEO6-b-CHOL30 and PEO9-b-CHOL28 (as well as the analogous CHOLn-b-PEOm copolymers) are white solids with critical water concentrations (∼15 wt% for a 0.01wt% polymer solution in THF) in a similar range as that observed for the PEO15-b-DOX15 used in making the carbamate-linked Dox-PNPs.17 As such, PNP fabrication from PEO6-b-CHOL30 proceeded smoothly to give well-defined cholesterol-containing PNPs that are similar in size to the carbamate-linked Dox-PNPs (Fig. 1).
Fig. 1.
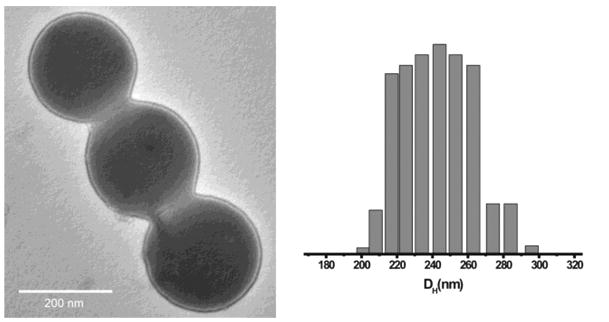
Transmission electron microscopy (TEM) image (left) and the size distribution plot (right, obtained through dynamic light scattering (DLS) measurements) of Chol-PNPs.
In the design of the new amine-linked doxorubicin conjugated copolymers (Scheme 2), the size of the blocks was initially kept the same as the previously synthesized carbamate-linked Dox-PNPs. However, the resulting amine-linked PEO15-b-DOX15 polymer is a viscous liquid polymer at room temperature.
We suspect that replacing the carbamate linkage of the doxorubicin to the polymer in our original design17 with a xylylic amine group would change the ability of the adjacent drug groups to hydrogen bond to one another of (Fig. S1 in ESI†) and thus may decrease the “packing ability” the polymer. To compensate for this effect, the ratio of the blocks was reproportioned to increase the hydrophobicity and weight of the doxorubicin-containing block; the resulting PEO10-b-DOX20 copolymer is indeed a dark red solid that can be used successfully in the PNP synthesis (Scheme 2).
Preparation of PNPs for Cell Studies
For the control experiments, Chol-PNPs and IND-PNPs were prepared in nanopure water, transferred into cell media, and used immediately. Initial attempts were made to formulate Dox-PNPs as suspensions in DMEM media for cell testing. Although the PNPs remain intact after 2 weeks in this media,they do not appear to have long-term stability—after 2 months, dynamic light scattering (DLS) measurements and TEM showed a significant increase in size and dispersity of the nanoparticles (Fig. S2 in ESI†). Hence, for the purpose of this study, we fractioned water suspensions of the Dox-PNPs into Eppendorf tubes and lyophilized them to yield bright red powder samples that can be re-suspended into nanopure water immediately before each cell testing.
Cytotoxic potency of doxorubicin-conjugated PNPs in comparison to free doxorubicin
Anthracycline derivatives such as doxorubicin are frequently used to treat numerous human malignancies including neuroblastoma, the most prevalent solid tumor of the peripheral sympathetic nervous system in young children.20, 21 However, the therapeutic efficacy of this relatively insoluble small-molecule drug is limited by its low bioavailability and short-plasma half-life, necessitating frequent dosing.22 In addition, the often life-threatening side effects (cardiotoxicity,23 bone marrow suppression,24, 25 infertility,26 etc.) that accompany the use of doxorubicin, including latent development of leukemia after prolonged treatment,27 deem this an undesirable chemotherapeutic agent. Furthermore, resistance to doxorubicin can develop even after a single treatment, thus retarding not only its effectiveness, but the effectiveness of similar drugs.28
We hope to remedy the aforementioned shortcomings by packaging a high density of doxorubicin into nanoscale vessels that can be preferentially delivered to tumor sites via the EPR effect (vide supra), without affecting healthy tissues. In addition, the poly(ethylene oxide) shells of our PNPs are well-known to mask the particles against the reticuloendothelial (RES) system29 and should also increase the plasma half-life of doxorubicin. Finally, the slow release of doxorubicin, through acid-labile linkages,17 inside acidic tumor tissues and endosomes should give our PNPs an additional selective edge over the free drug.
To investigate the ability of the covalent linkage to the polymer to affect the in vitro efficacy of the doxorubicin moieties, we designed two different Dox-PNPs. As previously reported, carbamate-linked Dox-PNPs release doxorubicin slowly at pH 5 and below. However, amine-linked doxorubicin PNPs do not release under clinically relevant acidic conditions (see ESI†). Because it has been reported for a PK2 analog that the therapeutic efficacy of doxorubicin is lost when the drug is not released from the polymer,30 we did not expect amine-linked Dox-PNPs to show any cytotoxic activity. We also designed and synthesized an “inert” PNP with cholesterol in the core, linked to the polymer backbone via a non-degradable ether linkage. These chol-PNPs serve as control particles and should theoretically mimic, in vitro, the behavior of the amine-linked Dox-PNPs. Finally, we employed IND-PNPs,8 conjugated via a cleavable amide linkage, as a control for the carbamate-linked Dox-PNPs in vitro.
The uptake of the both Dox-PNPs by SKNSH cells, presumably through endocytosis,31 occurred relatively slowly compared to the free doxorubicin which occurs in about 2 h— nanoparticles can be clearly observed inside the cell after 24 h using both optical and fluorescent microscopy (Fig. 2). Interestingly, the uptake appears to be non-uniform with some cells accumulating more Dox-PNPs than others. This may be due in part to the self aggregation of PNPs as they slowly settle out of solution over several hours,32 causing enhanced accumulation into one cell and leaving neighboring cells less affected.
Fig. 2.
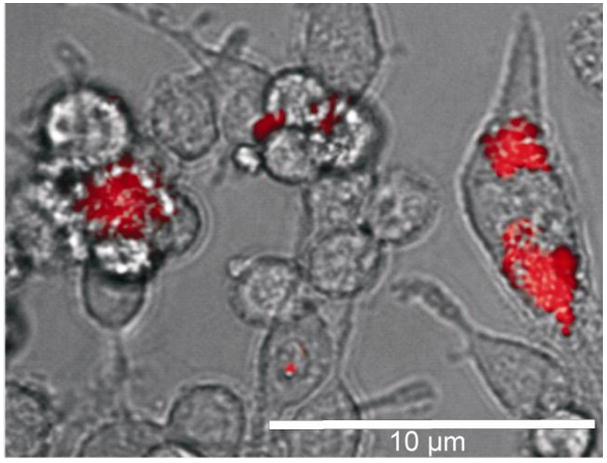
Differential contrast image with fluorescence overlay of Dox-PNPs in SKNSH wild-type neuroblastoma cells. Scale bar is 10 μm.
The effect of both Dox-PNPs on neuroblastoma cell viability was evaluated by incubating SKNSH and SKrDOX6 cells with increasing concentrations (10-9 to 10-5 M) of Dox-PNPs and free doxorubicin for 96 h, at which time the viability of cells was determined by MTT assay. Notably, carbamate-linked Dox-PNPs have an IC50 of 10-5 M for the SKrDOX6 doxorubicin-resistant cells (Fig. 3a), that is very similar to that of the free drug.33 As expected,33, 34 the cytotoxicity of free doxorubicin was concentration-dependent, with an IC50 of 5×10-7 M in SKNSH cells (Fig. 3b). In contrast, Dox-PNPs only show significant effect toward the more doxorubicin-sensitive SKNSH cells at the relatively higher IC50 of 5×10-6 M. This higher IC50 can be explained by comparing the cytotoxicity profiles of Dox-PNPs for both SKNSH and SKrDOX6 cells: neither is concentration-dependent up to 10-5 M, suggesting an imperfect uptake of the nanoparticles into the cells below this threshold, as shown in Figure 2 (vide supra).
Fig. 3.
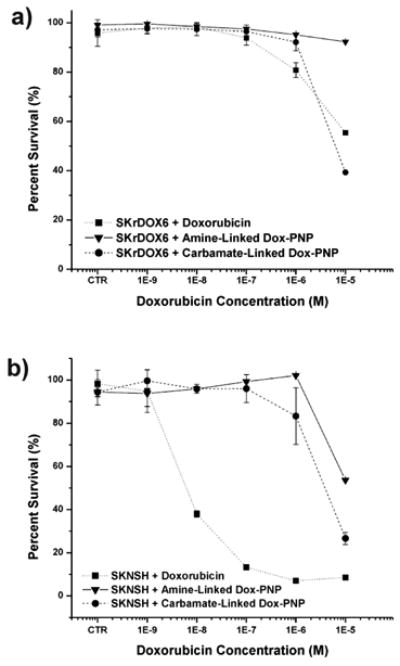
Cytotoxicity profile (10-9 to 10-5 M) of amine-linked and carbamate-linked doxorubicin-conjugated polymer nanoparticles (Dox-PNP) compared to free doxorubicin in a) neuroblastoma drug-sensitive (SKNSH) and b) drug-resistant (SKrDOX6) cell lines. Data were taken 96 h after incubation. CTR = 0 M. For bench-top release profile of carbamate-linked Dox-PNP, please see reference 17. The amine-linked Dox-PNP does not release Dox even after 9 days of incubation in acidic media.
The apparent drug concentration reported for all of the in vitro studies performed herein is based on a theoretical 100% release of the drug from the PNPs, even though we have noted that only a very small fraction of doxorubicin is released from the carbamate-linked Dox-PNPs after 24 h and no doxorubicin from the amine-linked PNPs after extensive (9 days) incubation at pH 4 (see ESI†).17 Although the pH of the cell media remains neutral (7.4 - 7.6, see ESI†) over the time of the experiment, the known acidic environment inside the endosome,35 formed during an endosomal uptake process, would induce doxorubicin cleavage from the polymer backbone. If PNPs were indeed being taken up into the cells via endocytosis, release of the cleavable drug, or the partially digested particle, from the endosome may then occur through rupture of the endosomic lamellar membrane, releasing the cytotoxic agents into the cytoplasm.36
Surprisingly, amine-linked Dox-PNPs, possessing non-cleavable doxorubicin moieties, also exhibit a cytotoxic effect on SKNSH cells (IC50 = 1 × 10-5 M), although at a lower degree than either the carbamate-linked Dox-PNPs or free doxorubicin alone. Recently Říhová et al.37 reported that doxorubicin-linked HPMA (N-(2-hydroxypropyl)methacrylamide, a PK1 analog) through a non-cleavable bond does exhibit cytotoxicity, albeit at a lower efficacy than traditional PK1 formulations. This observed cytotoxicity was attributed to damage to cytoplasmic, endosomal, and lysosomal membranes from unconjugated peptide linkages on the polymer backbone that can cause cell death. However, this cannot be the case in our formulations, which has no free peptide linkages. Additionally, because the cholesterol and indomethacin PNPs of the same size as our amine-linked Dox-PNPs do not show any significant effect on cell populations (Fig. S4 in ESI†), there should not be any cytotoxicity that involve the polymer backbones or the nanoparticles. We speculate that our amine-linked Dox-PNPs can undergo slow disassembly in the cellular environment to release doxorubicin-containing polymer chains that have DNA-accessible drug moieties. While this is an interesting observation, the higher IC50 value of the carbamate-linked Dox-PNPs prompted us to further examine their efficacy via cell-growth inhibition studies.
Effects of carbamate-linked doxorubicin-containing PNPs on cell-growth inhibition
The effect of carbamate-linked Dox-PNPs on cell growth was determined by incubating them with SKNSH and SKrDOX6 cells at 10-6 M for 168 h, and counting viable cells at 4, 8, 16, 24, 48, 72, 96, 120, and 168 h time intervals (Table S2 in ESI†). Significant cell growth inhibition was observed for SKNSH cells when incubated with carbamate-linked Dox-PNPs (Fig. 4a). Beyond the initial uptake period (∼ 16 h), the inhibition profile of carbamate-linked Dox-PNPs is similar in trend to free doxorubicin, clearly demonstrating a cytotoxic effect on the SKNSH cells (Fig. 4a). While free doxorubicin begins to kill off cells at 24 h, carbamate-linked Dox-PNPs begin to exert significant cytotoxicity ∼ 96 h.
Fig. 4.
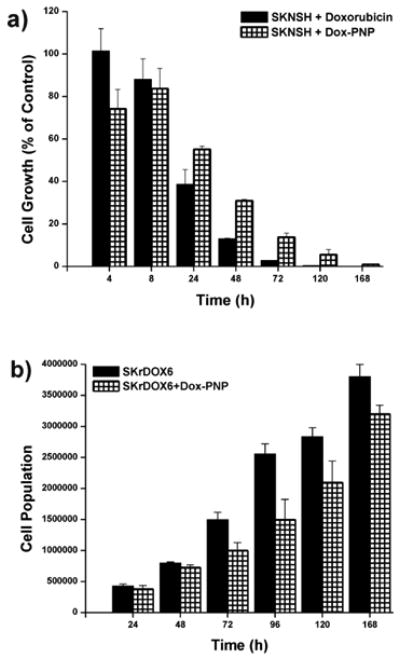
Growth inhibition of doxorubicin-conjugated polymer nanoparticles (carbamate-linked Dox-PNP at 10-6 M concentration of drug) in: a) drug-sensitive neuroblastoma (SKNSH) cell lines compared to free doxorubicin as a percentage of non-interrupted cell growth and b) drug-resistant neuroblastoma (SKrDOX6) cell lines compared to uninhibited growth.
Also consistent with the above observations, carbamate-linked Dox-PNPs inhibit the growth of SKrDOX6 cells after the initial uptake period (Fig. 4b). This inhibition reaches a maximum at 72 h (see ESI†, this is slower than free doxorubicin which reaches its maximum at 48 h) and gradually decreases. That our carbamate-linked Dox-PNPs show analogous cytotoxicity behavior compared to free doxorubicin suggests that the drug can be released from PNPs and works in an unhindered fashion. The uneven uptake of the PNPs, which may lead to non-uniform concentrations of the drug inside different cells, is not surprising in a relative static environment such as a small-volume cell culture where colloidal macromolecular constructs can potentially settle and localize anisotropically compared to soluble small-molecule drugs (vide supra). This in turn would result in an efficacy bias that favors the free drug during the short time of our experiment. In addition, because cell cultures are poor imitations of solid tumors, physiological effects such as increased acidity and the EPR effect are not present, potentially stifling the designed advantages of our PNP platform. Indeed, previous research into dendrimer-based drug delivery vehicles has shown that a modest in vitro efficacy does not necessarily imply limited in vivo therapeutic activity.38
Conclusions
In conclusion, we have demonstrated through in vitro cell studies that doxorubicin-containing PNPs can be viable chemotherapeutic agents. Not only is the polymer platform non-cytotoxic, the PNPs can be readily taken up by the cells and exert a signigicant cytotoxic response due to the doxorubicin payload. While the “formal” release of doxorubicin from the polymer backbone, as in the case of amine-linked Dox-PNPs, may not be necessary to induce a cytotoxic response, the release of the drug from carbamate-linked Dox-PNP definitely increase the efficacy of the platform.
As an alternative to traditional small-molecule chemotherapy, encapsulated nanoscale platforms such as our Dox-PNPs have excellent potential to reduce life-threatening side effects of toxic chemotherapeutic agents while increasing both the therapeutic efficacy of the drug payload and a patient's quality of life. In vivo studies will be needed to fully evaluate the scope of these nanoscale drug delivery agents. These are currently underway.
Materials and Methods
Reagents
Dulbecco's modified Eagle's medium (DMEM), penicillin, streptomycin and fetal bovine serum (FBS) were purchased from HyClone (Logan, UT). Media 1 contains DMEM supplemented with 10% FBS, penicillin (100 units/mL medium), and streptomycin (100 μg/mL medium). Media 2 contains DMEM supplemented with 10% FBS, penicillin (100 units/mL medium), streptomycin (100 μg/mL medium), and doxorubicin (10-6 M). Doxorubicin for the control cell-exposure experiment and 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl tetrazolium bromide (MTT) were purchased from Sigma (St. Louis, MO).
5-(4-{2-Exo-[2-(2-{-2[2-(2-methoxy-ethoxy)-ethoxy]-ethoxy}-ethoxy)-ethoxy]-ethoxymethyl}-benzyloxy)-bicyclo[2.2.1]hept-2-ene (PEO monomer 3) and α-bromo-α′-(exo-5-norbornene-2-ol)-p-xlyene (bromoxylene monomer 7) were prepared according to literature procedure.39 Carbamate-linked doxorubicin- and indomethacin-containing polymer nanoparticles were prepared as previously described (Schemes S1 and S2 in ESI†).17, 18 The apparent doxorubicin concentration of the Dox-PNPs solution was calculated based on the amount of doxorubicin present in the PNP and assuming 100% theoretical release. Please see ESI† for other details.
Synthesis of norborne-p-benzylether-xylenyl 3′-amine-linked doxorubicin (4)
Into a foil-wrapped 50-mL Schlenk flask were added 7 (153 mg, 5.21×10-4 mol) and doxorubicin•HCl (200 mg, 3.44 × 10-4 mol). The flask was placed under nitrogen and dry DMF (15 mL) was added via cannula, followed by the dropwise addition of triethylamine (96 μL, 6.89 × 10-4 mol) via syringe. The solution was stirred for 18 h at room temperature and concentrated on a rotary evaporator. The residue was partially redissolved in methylene chloride (40 mL) and the resulting solution was filtered over a Buchner funnel to remove any insoluble doxorubicin starting material. The collected filtrate was concentrated on the rotary evaporator to afford a dark red-orange residue, which was purified via column chromatography (5 vol% MeOH: CH2Cl2) to yield a red-orange solid (200 mg, 2.69 × 10-4 mol, 78% yield). For 1H NMR, 13C NMR, and MS data, please see ESI†.
Synthesis of cholesterol-containing control monomer (1)
A 50-ml Schlenk flask equipped with a magnetic stir bar was charged with cholesterol (770 mg, 1.9 mmol) and NaH (7.17 mg, 2.2 mmol). The flask was placed under nitrogen and anhydrous THF (25 mL) was added to the reaction via cannula. The reaction was refluxed under nitrogen for 24 h. In a separate 50-mL Schlenk flask, bromoxylene monomer 7 (489 mg, 1.6 mmol) was placed under nitrogen and dissolved in THF (10 mL). This solution was then transferred via cannula to the stirring reaction mixture and refluxed under nitrogen for 3 days. The reaction was allowed to cool to room temperature and quenched with wet methanol. Solvents were removed from the reaction under reduced pressure to yield a white solid. This crude product was taken up in a minimal amount of CH2Cl2 and purified via column chromatography (70:30 v/v hexanes:CH2Cl2). The product was isolated as a pure white solid (670 mg, 1.1 mmol, 67% yield). For 1H NMR, 13C NMR, and MS data, please see ESI†.
Synthesis of amine-linked PEO10-b-DOX20
In an inert-atmosphere glovebox, monomer 3 (13.7 mg, 2.69 × 10-5 mol) was weighed into a 20-mL scintillation vial equipped with a magnetic stirring bar. A 9:1 v/v mixture of anhydrous CDCl3:CD3OD (2 mL) was added, followed by a solution of catalyst 2 (2.2 mg, 2.69 × 10-6 mol) in anhydrous CDCl3:CD3OD (9:1 v/v, 1 mL). (The CD3OD was added to maintain the solubility of the copolymer. While the monomers are soluble in CDCl3, the Dox-containing block of the copolymer is not. We minimized the amount of CD3OD because Dinger et al.40, 41 have noted that its use will eventually lead to catalyst decomposition). The mixture was stirred for 45 min at room temperature. An aliquot (100 μL) was removed, quenched with ethyl vinyl ether, and analyzed by GPC (Mn = 5200, PDI = 1.19). Subsequently, a solution of monomer 4 (40 mg, 5.39 × 10-5mol) in 9:1 v/v anhydrous CDCl3:CD3OD (1.5 mL) was added and stirred for an additional 60 min before the polymerization was terminated with the addition of ethyl vinyl ether (1 mL). The copolymer 310-b-420 was isolated as a dark red solid by precipitation into cold pentane (200 mL), filtering, and washing with fresh pentane (3×50 mL). GPC: Mn = 18818, PDI = 1.15.
Synthesis of PEOm-b-CHOLn and CHOLn-b-PEOm block copolymers
In an inert-atmosphere glovebox, PEO monomer 3 (5.27 mg, 0.01 mmol) was weighed into a 20-mL scintillation vial equipped with a magnetic stir bar. Dry THF (4 mL) was added, followed by a solution of the Cl2(PCy3)2Ru=CHPh catalyst 2 (1.42 mg, 1.7 μmol) in THF (1 mL). The solution was stirred for 1 h at room temperature. Subsequently, a solution of cholesterol monomer 1 (30 mg, 0.05 mmol) in THF (1 mL) was added and the solution was allowed to stir for an additional 1 h. The polymerization was terminated with the addition of ethyl vinyl ether (1 mL). The copolymer PEO6-b-CHOL30 (28 mg, 80%) was isolated as a white solid by precipitation into cold pentane (200 mL), filtering, and washing with fresh pentane (3 × 50 mL).
The CHOLn-b-PEOm copolymers were synthesized in the manner as described above but with the reverse addition of monomers: 1 then 3.
General procedure for the preparation of Dox PNPs
Carbamate-linked Dox-PNPs were prepared as previously reported.17 For the amine-linked materials, a similar procedure was followed. A stock solution of the copolymer in DMSO (0.01 wt%) was stirred for 4 h at room temperature to ensure complete polymer solubilization. An aliquot (2.5 mL) was transferred to a 4-mL scintillation vial and set to stir vigorously. Nanopure water was added to the stirring copolymer solution at a rate of 1 drop (10 μL, 0.35 wt%) per every 10 s using a micro-pipette until the solution contained 15 wt% water. The resulting aggregate solution was placed in a dialysis tube (Spectra/Por RC, 3-mL Float-a-lyzer, MWCO = 3500, Spectrum Laboratories) and dialyzed against ultrapure water in a 500-mL Erlenmeyer flask with the dialysis solution changed every 30 min. Complete removal of the organic solvent (DMSO) from the filtrate after 48 h was verified by UV-vis spectroscopy as indicated by the disappearance of the UV cut-off (DMSO at 260 nm).
General procedure for the preparation of Chol-PNPs
A stock solution of the copolymer in THF (0.01 wt%) was stirred for 4 h at room temperature to ensure complete polymer solubilization. An aliquot (2.5 mL) was transferred to a 4-mL scintillation vial and set to stir vigorously. Nanopure water was added to the stirring copolymer solution at a rate of 1 drop (10 μL, 0.35 wt%) per every 10 s using a micro-pipette until the solution contained 18 wt% water (> the critical water concentration of the least hydrophobic polymer PEO15-b-CHOL15, as determined by static light scattering on a Wyatt Dawn Instrument). The resulting aggregate solution was placed in a dialysis tube (Spectra/Por RC, 3-mL Float-a-lyzer, MWCO = 3500, Spectrum Laboratories) and dialyzed against ultrapure water in a 500-mL Erlenmeyer flask with the dialysis solution changed every 30 min. Complete removal of the organic solvent (THF) from the filtrate after 48 h was verified by UV-vis spectroscopy as indicated by the disappearance of the UV cut-off (THF at 212 nm).
Preparation of PNPs in DMEM media for cell studies
Colloidal solutions of the PNPs, as prepared in nanopure water, were transferred to 2-mL Eppendorf tubes and centrifuged at 10K rpm for 30 minutes. The supernatant was removed and replaced with a similar volume of DMEM and the resulting samples were stored for 4 °C and used for cell testing within a few days. Particle integrity was confirmed by TEM after 2 months of storage (see ESI†).
Preparation of lyophilized PNPs for cell studies
Colloidal solutions of the Dox-PNPs, as prepared in nanopure water, were transferred in 1-mL aliquots into 1.5-mL Eppendorf tubes. Water was removed via lyophilization, leaving the PNPs as a bright red powder. Lyophilized PNPs prepared in this way can be stored in the dark at 25 °C for several months. Immediately before use, nanopure water (100 μL) was added to the Eppendorf tube and vortexed vigorously to resuspend the particles as a 1-mM colloidal solution of doxorubicin. Particle integrity was confirmed by TEM after 2 months of storage (see Fig. S3 in ESI†).
Cell Culture and Cytotoxicity Assay
Cell cultures were maintained in a 40% humidity atmosphere containing 5% CO2. Human neuroblastoma (SKNSH) cells were maintained at 37 °C in Media 1. SKrDOX6 doxorubicin-resistant cells were maintained at 37 °C in Media 2.
The cytotoxic activities of both types of Dox-PNPs, Chol-PNPs, IND-PNPs, and free doxorubicin toward both cell lines were quantitatively determined using MTT colorimetric assay. Cells were seeded at 104 cells/well in 96-well plates and maintained in culture for 24 h at 37 °C in Media 1. The doxorubicin-containing media (Media 2) used to maintain SKrDOX6 doxorubicin-resistant cells was replaced with fresh Media 1 twenty-four hours before testing was initiated. Free doxorubicin, Dox-PNPs, and Chol-PNPs, at 10-9 to 10-5 M concentrations, were then added to designated wells and the whole plate was incubated for an additional 96 h. MTT (10 μL of a 5 mg/ml solution) was added to each well, followed by incubation for 4 h at 37 °C. To each well was then added 0.5-N aqueous HCl containing 10 wt% sodium dodecyl sulfate (100 μL) and the whole plate was incubated for 15 h at 37°C to completely solubilize the cells. The absorbance of the resulting solution in the 570-650 nm range was measured, and the cell survival was determined by comparison of optical densities with untreated control cell cultures.
Cell-Growth-Inhibition Assay
5 Cells were seeded at 50,000 cells/well in 6-well plates and the inhibition activities of carbamate-linked Dox-PNPs and free doxorubicin were quantitatively determined by visual cell counting using a Neubauer-type hemocytometer chamber. The doxorubicin-containing media (Media 2) used for the growth of doxorubicin-resistant cells (SKrDOX6) was replaced with fresh Media 1 twenty-four hours before testing was initiated. Free doxorubicin and carbamate-linked Dox-PNPs (10-6 M) were then added to designated wells and the whole plate was incubated for an additional 168 h. Cell counting was performed at 4, 8, 16, 24, 48, 72, 96, 120, and 168 h. The extent of cell inhibition was determined by cell population counts and compared with untreated control cell cultures.
Supplementary Material
Acknowledgments
We thank all coworkers in the Nguyen and Mirkin groups for support and fruitful discussions during the preparation of this manuscript. We are indebted to Prof. Mary Beth Madonna, M.D. for reviewing the final manuscript before publication. Financial support for this work was provided by the National Institute of Health (NCI 1U54 CA119341-01 through the CCNE program), the National Science Foundation (DMR CAREER grant 0094347, EEC-0647560 through the NSEC program, and DMR-0520523 through the MRSEC program). Additional financial support was provided by the John W. Anderson Foundation, North Suburban Medical Research Junior Board, Medical Research Junior Board Foundation, Medical Research Institute Council, R. Wile Foundation, and the Children's Memorial Research Center Program in Cancer Biology and Epigenomics. DS is a Malkin Family Foundation Fellow and an ARCS Scholar. Characterization of the PNPs was performed in the NUANCE, Keck, and IMSERC facilities at Northwestern University (supported by NSF-NSEC, NSF-MRSEC, Keck Foundation, the State of Illinois, and Northwestern University).
Footnotes
In memoriam of Bernard L. Mirkin (1938-2007)
Electronic Supplementary Information (ESI) available: General descriptions of materials and instrumentation, characterization data for the monomers and polymers, data from control experiments, and additional data as referenced in text.
Contributor Information
Sandra H. Clark, Email: s-clark@northwestern.edu.
SonBinh T. Nguyen, Email: stn@northwestern.edu.
Notes and references
- 1.Duncan R. Nat Rev Drug Discovery. 2003;2:347–360. doi: 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]
- 2.Davis ME, Chen Z, Shin DM. Nat Rev Drug Discovery. 2008;7:771–782. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 3.Heath JR, Davis ME. Annu Rev Med. 2008;59:251–265. doi: 10.1146/annurev.med.59.061506.185523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang L, Chan JM, Gu FX, Rhee JW, Wang AZ, Radovic-Moreno AF, Alexis F, Langer R, Farokhzad OC. ACS Nano. 2008;2:1696–1702. doi: 10.1021/nn800275r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bartlett DW, Davis ME. Biotechnol Bioeng. 2008;99:975–985. doi: 10.1002/bit.21668. [DOI] [PubMed] [Google Scholar]
- 6.Uchino H, Matsumura Y, Negishi T, Koizumi F, Hayashi T, Honda T, Nishiyama N, Kataoka K, Naito S, Kakizoe T. British J Cancer. 2005;93:678–687. doi: 10.1038/sj.bjc.6602772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chavanpatil MD, Khdair A, Gerard B, Bachmeier C, Miller DW, Shekhar MPV, Panyam J. Mol Pharm. 2007;4:730–738. doi: 10.1021/mp070024d. [DOI] [PubMed] [Google Scholar]
- 8.Feng SS. Biomaterials. 2008;29:4146–4147. doi: 10.1016/s0142-9612(08)00562-0. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Z, Lee SH, Gan CW, Feng SS. Pharm Res. 2008;25:1925–1935. doi: 10.1007/s11095-008-9611-6. [DOI] [PubMed] [Google Scholar]
- 10.Sun B, Ranganathan B, Feng SS. Biomaterials. 2007;29:475–486. doi: 10.1016/j.biomaterials.2007.09.038. [DOI] [PubMed] [Google Scholar]
- 11.Smith D, Pentzer EB, Nguyen ST. Polym Rev. 2007;47:419–459. [Google Scholar]
- 12.Yu JJ, Lee HA, Kim JH, Kong WH, Kim Y, Cui ZY, Park KG, Kim WS, Lee HG, Seo SW. J Drug Targeting. 2007;15:279–284. doi: 10.1080/10611860701357235. [DOI] [PubMed] [Google Scholar]
- 13.Kommareddy S, Amiji M. J Pharm Sci. 2006;96:397–407. doi: 10.1002/jps.20813. [DOI] [PubMed] [Google Scholar]
- 14.Shenoy DB, Chawla JS, Amiji MM. In: Nanoscale Materials Science in Biology and Medicine. Laurencin CT, Botchwey EA, editors. Materials Research Society; Boston, Massachusetts: 2005. pp. 369–373. [Google Scholar]
- 15.Mitra S, Gaur U, Ghosh PC, Maitra AN. J Controlled Release. 2001;74:317–323. doi: 10.1016/s0168-3659(01)00342-x. [DOI] [PubMed] [Google Scholar]
- 16.Gu F, Zhang L, Teply BA, Mann N, Wang A, Radovic-Moren AF, Langer R, Farkokhzad O. Proc Natl Acad Sci U S A. 2008;105:2586–2591. doi: 10.1073/pnas.0711714105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bertin PA, Smith D, Nguyen ST. Chem Commun. 2005:3793–3795. doi: 10.1039/b504643b. [DOI] [PubMed] [Google Scholar]
- 18.Bertin PA, Watson KJ, Nguyen ST. Macromolecules. 2004;37:8364–8372. [Google Scholar]
- 19.Tannock IF, Rotin D. Cancer Res. 1989;49:4373–4384. [PubMed] [Google Scholar]
- 20.Bernstein ML, Leclerc JM, Bunin G, Brisson L, Robison L, Shuster J, Byrne T, Gregory D, Hill G, Dougherty G. J Clin Oncol. 1992;10:323–329. doi: 10.1200/JCO.1992.10.2.323. [DOI] [PubMed] [Google Scholar]
- 21.Society AC. What are the key statistics about neuroblastoma? [Accessed June 15, 2008]; http://www.cancer.org/docroot/CRI/content/CRI_2_4_1X_What_are_the_key_statistics_about_neuroblastoma_31.asp?rnav=cri.
- 22.Twelves CJ, Dobbs NA, Aldhous M, Harper PG, Rubens RD, Richards MA. Cancer Chemother Pharmacol. 1991;28:302–307. doi: 10.1007/BF00685539. [DOI] [PubMed] [Google Scholar]
- 23.Sperber AD, Cantor AA, Biran H, Keynan A. Israel J Med Sci. 1987;23:896–899. [PubMed] [Google Scholar]
- 24.Harris PA, Garai AS, Valenzuela MA. J Pharm Sci. 1975;64:1574–1576. doi: 10.1002/jps.2600640939. [DOI] [PubMed] [Google Scholar]
- 25.Alberts DS, Salmon SE. Cancer Chemother Rep. 1975;59:345–350. [PubMed] [Google Scholar]
- 26.Longhi A, Vitali G, Macchiagodena M, Bacci G. In: Trends in Bone Cancer Research (Horizons in Cancer Research, Volume 24) Birch EV, editor. Nova Science Publishers, Inc.; New York: 2006. pp. 247–264. [Google Scholar]
- 27.Andre M, Mounier N, Leleu X, Sonet A, Brice P, Henry-Amar M, Tilly H, Coiffier B, Bosly A, Morel P, Haioun C, Gaulard P, Reyes F. Blood. 2004;103:1222–1228. doi: 10.1182/blood-2003-04-1124. [DOI] [PubMed] [Google Scholar]
- 28.Chang B, Clark SH, Mirkin BL. unpublished results. 2007 [Google Scholar]
- 29.Bhadra D, Bhadra S, Jain P, Jain NK. Pharmazie. 2002;57:5–29. [PubMed] [Google Scholar]
- 30.Rihova B, Kopecek J. J Controlled Release. 1985;2:289–310. [Google Scholar]
- 31.Allen C, Yu Y, Eisenberg A, Maysinger D. Biochim Biophys Acta, Biomembr. 1999;1421:32–38. doi: 10.1016/s0005-2736(99)00108-x. [DOI] [PubMed] [Google Scholar]
- 32.Smith D, Nguyen ST. unpublished results. 2007 [Google Scholar]
- 33.Emran MA, Rebbaa A, Mirkin BL. Cancer Lett. 2002;182:53–59. doi: 10.1016/s0304-3835(02)00062-9. [DOI] [PubMed] [Google Scholar]
- 34.Rebbaa A, Chou PM, Emran MA, Mirkin BL. Cancer Chemo Pharmacol. 2001;48:423–428. doi: 10.1007/s002800100375. [DOI] [PubMed] [Google Scholar]
- 35.Overly CC, Lee KD, Berthiaume E, Hollenbeck PJ. Proc Natl Acad Sci U S A. 1995;92:3156–3160. doi: 10.1073/pnas.92.8.3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Labat-Moleur F, Steffan AM, Brisson C, Perron H, Feugeas O, Furstenberger P, Oberling F, Brambilla E, Behr JP. Gene Ther. 1996;3:1010–1017. [PubMed] [Google Scholar]
- 37.Rihova B, Strohalm J, Hovorka O, Subr V, Etrych T, Chytil P, Pola R, Plocova D, Boucek J, Ulbrich K. J Controlled Release. 2008;127:110–120. doi: 10.1016/j.jconrel.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 38.Lee CC, Gillies ER, Fox ME, Guillaudeu SJ, Frechet JMJ, Dy EE, Szoka FC. Proc Natl Acad Sci U S A. 2006;103:16649–16654. doi: 10.1073/pnas.0607705103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Watson KJ, Anderson DR, Nguyen ST. Macromolecules. 2001;34:3507–3509. [Google Scholar]
- 40.Dinger MB, Mol JC. Eur J Inorg Chem. 2003:2827–2833. [Google Scholar]
- 41.Dinger MB, Mol JC. Organometallics. 2003;22:1089–1095. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.