

Abstract
OBJECTIVE
To characterize the phenotypic spectrum of males with bilaterally descended testes and a 45,X/46,X,(r)Y karyotype
DESIGN
Retrospective review of patient records; cytogenetic and molecular analysis
SETTING
Tertiary medical center setting
PARTICIPANTS
Five males, two prepubertal and three postpubertal, with a 45,X/46,X(r)Y karyotype and bilaterally descended testes
INTERVENTIONS
Linear growth evaluation, testicular endocrine and exocrine studies, cytogenetic and molecular analysis on each patient.
MAIN OUTCOME MEASURES
Clinical phenotype vs. genotype
RESULTS
Both prepubertal males had short stature and low testosterone. All three adults had normal puberty and normal testosterone levels. Two of the adults (one with short stature and one with normal stature) had elevated gonadotropins and azoospermia. The third adult had normal stature, severe oligospermia, normal gonadotropins, and normal serum testosterone.
CONCLUSIONS
The phenotypic spectrum of males with a 45,X/46,X(r)Y karyotype and bilaterally descended testes varies greatly from males with short stature and spermatogenic failure to males without short stature and less severely affected spermatogenesis. This broad spectrum of phenotypic findings needs to be taken into account when the clinical geneticist encounters a prenatal diagnosis of a 45,X/46,X(r)Y karyotype. This information will also be helpful for pediatric and reproductive endocrinologists in counseling males with bilaterally descended testes and a 45,X/46,X(r)Y karyotype.
Keywords: Mixed gonadal dysgenesis; 45,X/46,XY; Ring Y chromosome; azoospermia; severe oligospermia
Introduction
Most patients with a 45,X/46,XY or 45,X/47,XYY karyotype manifest gonadal failure and short stature, as well as an increased prevalence of cardiorenal malformations and germ cell tumors (1, 2). The gonadal phenotype in mos 45,X/46,XY patients, regardless of Y chromosome morphology, is consistently unpredictable and may range from bilateral non-functional rudimentary streak gonads to bilateral small scrotal testes. Intermediary between these gonadal phenotypes are patients with an ipsilateral testis and a contralateral rudimentary streak gonad (Table 1).
Table 1.
The phenotype of individuals with a 45,X/46,XY karyotype depends upon gonadal location (1).
| Phenotype | Gonad | Gonad |
|---|---|---|
| Sexually infantile female | Streak | Streak |
| Female with clitoromegaly | Streak | Intra-abdominal testis |
| Frank sexual ambiguity | Intra-abdominal testis | Scrotal testis |
| Male with short stature, elevated gonadotropins, and azoospermia | Scrotal testis | Scrotal testis |
Adult sexual phenotypes of mos 45,X/46,XY individuals are consistent with their gonadal phenotypes. Those with bilateral streak gonads are normal females, but completely lack puberty whereas those with ipsilateral testis are females with varying degrees of masculinization at birth and at puberty. Probably most mos 45,X/46,XY children with bilateral scrotal testes go unrecognized at birth and throughout childhood unless they have somatic features of Turner syndrome or significant growth retardation. Adults may be ascertained during an infertility evaluation when they demonstrate azoospermia and elevated serum gonadotropins consistent with testicular failure. Their short stature is frequently overlooked and peripheral blood karyotyping is infrequently performed, and even if performed, the laboratory may fail to identify the 45,X cell line when routine numbers of metaphases are counted.
To date males with bilaterally descended testes and a 45,X/46,XY karyotype have been described largely at a case report level (2–11). The following series of mos 45,X/46,XY patients with bilateral scrotal testes serves to challenge conventional thinking about the frequency and phenotypic features of this interesting subgroup of mos 45,X/46/XY subjects. In addition, the findings in this series of patients raise important questions about differing mechanisms leading to the varied clinical spectrum in mosaic 45,X/46,XY males with bilateral descended testes.
Cases (n=5)
Two of five males were ascertained prepubertally and three were identified at the time of infertility evaluation. All were phenotypic males with an apical, penile urethra and bilateral scrotal testes. Somatic features of Turner syndrome, except for short stature were conspicuously absent (Table 2). Their phenotypes ranged from prepubertal males identified because of short stature (n=2) to normal-appearing adults (n=3) ascertained because of infertility (Table 2). All karyotypes were based upon analysis of 50 metaphases unless otherwise stated. None of the patients had cardiovascular-renal anomalies or other affected relatives with the same karyotype. This study was approved by the Human Assurance Committee at MCG.
Table 2.
Phenotypes of five males with 45,X/46,X(r)Y karyotype.
| Pt | Age Race | Height Inches (centile) | Testes volume (R;L) | FSH | LH | T | Karyotype | Molecular Analysis | Sperm &/or testicular histology |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 8 AA |
45 (<5) |
1 mL 1 mL |
0.7 | 0.5 | 17 | Blood: mos 45,X[10]/46,X,r(Y)[40] L Testis: mos 45,X[18]/46,X,r(Y)[32] R Testis: mos 45,X[15]/46,X,r(Y)[35] |
PCR positive for SRY Southern blot positive for SRY, ZFY, DYZ3, pDP105 (12) |
Few germ cells No Leydig cells Bilateral Calcinosis |
| 2 | 7 C |
46 (<5) | 2 mL 2 mL |
NA | NA | <20 | Blood: mos 45,X[3]/46,X,r(?Y)[47] | Southern blot: positive for SRY, ZFY, DYZ3, pDP105 (12) Negative for 4B2 probe (distal interval 6) (19) |
NA |
| 3 | 29 AA |
48.5 (<3) |
3mL 3mL |
16.5 | 17 | 570 | Blood: mos 45,X[9]/46,X, r(Y)[41] Blood:mos 45,X[12]/46,X,+mar[37]/47,X,+mar,+mar[1] |
Not performed | Azoospermia Fibrosis L testis Biopsy |
| 4 | 36 C |
67.5 (25) |
8mL 8mL |
30 | 22 | 360 | Blood: mos 45,X[14]/46,X,r(?Y)[36] Skin: mos 45,X[5]/46,X,r(?Y)[45] |
Southern blot: positive for DYZ3 (20) | Azoospermia; Germinal aplasia; maturation arrest, Leydig cell hyperplasia |
| 5 | 32 C |
66 (10) |
15 mL 10 mL |
1.5 | 1.9 | 464 | Blood: mos 45,X[13]/46,X,r(?Y)[37] | ishr(Y)(wcpX+,wcpY+, DYZ3+, SRY+, DXZ1−) 18 STS: positive Yq11 Molecular mapping: all Y positive except Yq12 |
2×106 Spermatocyte arrest and sloughing |
AA= African American; C=Caucasian; NA=not available; R=right; L=left. T=testosterone in ng/dL; LH and FSH in mIU/mL. DYZ3 is a Y centromeric sequence, while Y97 is a Y centromere DNA probe. The number of cells analyzed for each karyotype is indicated in brackets according to standard nomenclature.
PREPUBERTAL CHILDREN
MALE # 1
An 810/12-year-old African American male was referred initially for possible bilateral cryptorchidism. General physical examination revealed a normal appearing male who was 45 inches tall (114 cm) and <5th centile for height. Genital examination revealed a normal penile urethra with palpable bilateral inguino-scrotal testes. The testes were difficult to measure accurately because they freely moved in and out of inguinal canal, but were estimated to be 1mL bilaterally. Serum gonadotropins and testosterone were consistent with those of a prepubertal child (Table 2). A peripheral blood karyotype revealed mos 45,X[10]/46,X,r(Y)[40].
The testes were extirpated because of the risk of tumor formation in the dysgenetic gonads. Microscopic examination of both testes revealed few germ cells, no Leydig cells, and multiple foci of degenerative changes (calcinosis) consistent with early bilateral gonadoblastoma. Leukocyte DNA was subjected to PCR and Southern blot analysis, which demonstrated the presence of Y chromosome targets for the Y short arm (SRY, ZFY), centromeric region (DYZ3), and low copy repeats were identified on Yp and Yq using the pDP105 probe provided by David Page (Table 2). The indication for karyotype was based upon the short stature and the small, unstable testes.
MALE # 2
An 8-year-old Caucasian male was 116.8cm (46 inches, 5th centile), being seen at 39/12 years-of-age because of short stature (Table 2). Growth delay was noted at 6–10 months-of-age but not clinically evaluated. In spite of a good growth hormone response (1.5ng/mL to 14.4ng/mL) to insulin induced hypoglycemia he had been on growth hormone therapy since 5 years-of-age. At his initial examination testicular volume was 1.5mL bilaterally, and later recorded as 2mL each at 8 years-of-age, at which time his serum testosterone was less than 20ng/dL. His peripheral blood karyotype was mos 45,X[3]/46,X,r(?Y)[47]. Confirmation of the ring Y chromosome was accomplished using PCR and Southern blot analysis (Table 2). These studies identified the presence of Y chromosome targets for Yp (SRY, ZFY), the centromeric region (DYZ3), and low copy repeats(pDP105) were identified on Yp and Yq using the pDP105 probe. Probe 4B-2 localized to the interphase between Yq11-Yq12 was the only negative result.
Except for short stature initially ascertained at 39/12 years-of-age the phenotype of this 8 year-old-male was normal. His final height at 157/12 years-of-age was 161.8cm (63.7 inches) (5th centile). Short stature at 8 years-of-age appears to be the primary indication for the cytogenetic studies. The patient was lost to follow up before further discussions could be initiated concerning the risk of gonadal tumors, testicular and endocrine and exocrine function.
ADULT MALES
All three adult patients presented with normal puberty, infertility, and normal adult male levels of testosterone (Table 2).
MALE # 3
A 29-year-old African American male presented with three years of infertility. On physical examination he was 144.1cm (56.75 inches, <3rd centile) with 3mL, firm testes. Serum pituitary gonadotropins were elevated (FSH=16.5mIU/mL; LH=17.1mIU/mL) and testosterone was normal (570 ng/dL).
Despite a normal serum testosterone, his facial hair was scanty and he had never shaved. His axillary and pubic hair development began at ages 14–15 and was normal. His three brothers and father were ≥175.5cm tall. Semen analysis revealed a volume of 1.5mL, but microscopic analysis revealed no mature sperm. He underwent a left testicular biopsy, demonstrating azoospermia and testicular fibrosis. An initial peripheral blood karyotype based on 106 metaphases revealed mos 45,X[9]/46,X,r(Y)[41). A repeat peripheral blood karyotype in a different laboratory was mos 45,X[12]/46.,X,+mar[37]/47,X,+mar,+mar[1].
It is interesting that this patient was one of “unlike sex twins.” His twin sister was healthy and normal. The patient also had some equivocal “soft signs” of gonadal dysgenesis on physical examination (bilateral short 4th metacarpals, bilateral cubitus valgus). The indications for karyotype in this patient seemed to be based on short stature, elevated gonadotropins, and azoospermia. The Y chromosome was truly a fragment that assumed a ring configuration in some metaphases.
MALE # 4
A 36-year-old Caucasian male presented with a 10-year history of infertility with normal libido, erections, and ejaculation. Semen analysis revealed a 3mL volume, but no mature sperm were present (Table 2). Physical examination revealed a height of 170cm (67 inches, 25th centile), testes volume=8mL bilaterally, and no somatic features of Turner syndrome. Serum gonadotropins were elevated (FSH=30.2mIU/mL; LH=21.6mIU/mL) and his serum testosterone was normal (378 and 360ng/dL). He had a history of mumps at 5 years-of-age without testicular involvement.
Testicular biopsy revealed incomplete spermatogenesis with areas of focal tubular sclerosis, Leydig cell hyperplasia, and maturation arrest (Table 2). His peripheral blood karyotype was mos 45,X[14]/46,X,r(?Y)[36], and skin biopsy revealed 45,X[5]/46,X,r(Y?)[45]. The suspected Y chromosome was confirmed by Southern blot analysis for centromeric sequences (DYZ3) (Table 2). This patient was karyotyped primarily because of testicular failure.
MALE # 5
A 32 year-old white male presented with infertility. He had normal puberty, libido, erections, and ejaculation. A prior semen analysis demonstrated severe oligospermia and a karyotype revealed mos 45,X/46,X,r(?Y)(p11.3 q12). When seen at MCG, he was 167cm (66 inches, 10th centile) tall with normal virilization and no Turner stigmata. Genital examination revealed a normal sized right testis (15mL) and a slightly small left testis, measuring 10mL (Table 2).
Laboratory studies demonstrated normal testosterone (464ng/dL), gonadotropins (FSH=1.5 mIU/mL; LH=1.9mIU/mL), alpha-fetoprotein, and an undetectable hCG level (Table 2). Two semen analyses revealed severe oligoasthenospermia, one with a concentration=2 million/mL and 0% motility and the other with only 1–2 motile sperm.
Karyotype demonstrated mos 45,X[13]/46,X,r(?Y)[37], and when repeated was 45,X(26%)/46,X r(Y)(74%), with marked variation in the size of the r(Y). (Figure 1A). FISH utilizing whole chromosome painting for the X and Y chromosomes, as well as centromeric, alphoid repeats and SRY probes confirmed the ring r(Y) chromosome--ishr(Y)(wcpX+,wcpY+,DYZ3+,SRY+,DXZ1−). A testicular biopsy demonstrated spermatocyte arrest with primary spermatocyte sloughing (Figure 1B), and no mature sperm were visualized. PCR of 18 sequence tagged sites of the AZFa-c region using the Promega Kit documented the presence of all fragments indicating that this portion of Yq was present in the ring (data not shown). Molecular Y chromosome mapping revealed that all sequences were present except the heterochromatic region Yq12 (DYZ1-negative).
Figure 1.
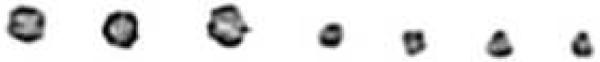

(Figure 1A) Ring chromosome for patient #5 varies in size.
(Figure 1B) Testicular biopsy shows seminiferous tubules (left) that when examined closer (right) demonstrated spermatocyte arrest with primary spermatocyte sloughing (indicated by arrows) without mature sperm.
Discussion
The most common presentation for individuals with a 45,X/46,XY karyotype is sexual ambiguity, accounting for ~60% (1, 2, 12). These patients have an intraabdominal streak and a testis (intraabdominal, inguinal, or labial in location), a uterus, and vagina. Most of the remainder are sexually infantile, phenotypic females with a uterus, vagina and bilateral streak gonads (1, 2, 12). The least common category of 45,X/46,XY patients consists of those with bilaterally descended testes, found in 11% (3/27) by Telvi et al (2) and 12% (3/25) by our group (1, 12). Unlike others with this karyotype, these patients do not have a uterus or vagina. Of interest, about half of 45,X/46,XY patients have an abnormal Y chromosome, including a ring Y, which may predispose to anaphase lag in a 46,XY male so that a 45,X cell line results. Ring chromosomes often form after breaks occur in each chromosome arm, resulting in the loss of chromosomal material with subsequent fusion (3). They may also form in other ways such as when telomeres join without chromosomal loss, by fusion of subtelomeric regions, or by the union of a broken chromosome with the opposite arm telomere. Ring Y chromosomes are relatively common although they are rarely inherited from father to son (3). A second monosomic cell line, such as 45,X, is often present because of meiotic instability of the ring (3).
The purpose of the present study was to determine the phenotype of five 45,X/46,XY males with bilaterally descended testes to evaluate the spectrum of abnormalities. All in our series had a r(Y) chromosome confirmed by molecular and/or FISH analysis when cytologically ambiguous. Interestingly, four of five had no Turner stigmata and none had cardiorenal defects, commonly seen in individuals with gonadal dysgenesis. Patient #3 may have had soft signs of Turner syndrome, which are subject to observer variation and anticipation bias. Two patients are prepubertal boys with characteristic short stature. Since gonadotropins are normally low in childhood, determination of testicular function is difficult at that time, although one had histologic evidence of impaired spermatogenesis. Testicular function may be normal in the first six months of life, which permits the diagnosis of hypergonadotropic and hypogonadotropic hypogonadism (13). Unfortunately, this short “window of opportunity” is usually overlooked and these males are ascertained once they manifest gonadal failure as adults, if at all. Short stature and/or the presence of Turner stigmata should trigger the need for a karyotype in these children.
The risk of testicular tumor formation in the dysgenetic testes, as in Male #1, who had calcinosis consistent with early gonadoblastoma, can be discussed before deciding upon extirpation or careful follow-up with regular physical exam, ultrasound Doppler, and tumor markers. Our findings indicate that testicular endocrine and exocrine function should be evaluated in early reproductive life in 45,X/46,XY males with bilaterally descended testes.
The findings of the three adults in our series challenge many of our assumptions about the basic mechanisms leading to the 45,X/46,XY genotype. Typically, these patients have short stature, elevated gonadotropins and azoospermia whether or not the Y chromosome is abnormal (12). However, contrary to the females with ovarian failure, most have normal puberty and serum testosterone levels probably due to operative negative feedback. Patient #3 fits most of these expectations since he has unequivocal short stature, spermatogenic failure (with elevated gonadotropins), but normal endocrine testicular function, although the length of time his gonadal function will remain normal is unknown. This patient is also interesting because he has three cell lines (mos 45,X(24%)/46,X,r(Y)(74%)/46 X,r(Y),r(Y)(2%), and shared his uterine environment with a "normal female" twin. The additional numbers of deleted Ys per cell reflect the basic mitotic instability of the structurally abnormal Y chromosome. Twinning, which may be a feature of the mosaic 45,X/46,XY or 45,X/46,XX genotypes, suggests that further investigation of his normal female twin might be valuable. Monozygotic isokaryotic 45,X/46,XY twins discordant for phenotypic sex have been reported (14).
Adult cases #4 and #5 provide a series of challenging contradictions. The most obvious and perplexing attribute that they share in common is normal stature. The 45,X cell line (25–28%), conspicuously present in both 45,X/46,XY males, did not limit linear growth. Similar to most 45,X/46,XY males, they all had small testes with spermatogenic failure and azoospermia. Complete testicular failure is not yet present in these two males as evidenced by their normal serum testosterone levels. Loss of testicular endocrine and exocrine function in 45,X/46,XY males is a consistent finding in true mosaic 45,X/46,XY males (2–11), which is probably mostly due to the presence of the 45,X cell line during early embryonic development and secondarily to loss of spermatogenesis genes on Yq.
As summarized in Table 3, phenotypic features of reported 45,X/46,XY males with bilaterally descended testes display unusual variability in height and in endocrine and exocrine testicular function. The males reported by Gasso-Matoses et al (4) and Tzancheva et al (10) are 31 and 19 years of age respectively with short stature, elevated gonadotropins, normal testosterone and azoospermia similar to our patient #3. Micic (6) reported one of his two patients with 45,X/46,Xdic(Yq) with normal stature (165 cm, 10th centile), elevated gonadotropins, normal testosterone and azoospermia similar to our Patient #4. Arnedo et al (3) reported a male with 45,X/46,Xr(Y) with normal stature (169 cm, 25th centile) and oligspermia (4.5 million/cc), like our Patient #5. However, this patient (45,X/46,XYdicYq) fathered a fetus with a 47,XX,r(Y)/46,XX karyotype at amniocentesis, although the karyotype after delivery was not reported. The extra X chromosome of the Klinefelter child was demonstrated to be of paternal origin by DNA analysis. FISH analysis of 187,126 sperm nuclei revealed an overall incidence of aneuploidy for all chromosomes of 12.4% as compared to 1.1% for control donors. Although less clear in 45,X/46,XY males with bilaterally descended testes, phenotypic 45,X females (regardless of mosaicism) have an increased risk of congenital anomalies, Down syndrome, fetal wastage, and 45,X offspring (15). Bettio et al (16) described three infertile males with 45,X/46,X,idic(Yq11), normal height (171,163, and 170 cm respectively), azoospermia, elevated gonadotropins in two patients, normal LH and testosterone in one patient. The normal stature in all three patients may be related to the presence of three copies of the statural determining gene SHOX in the isodicentric Y cells.
Table 3.
Previously reported 45,X/46,XY males with either a normal Y chromosome or a structurally abnormal Y chromosome (n=15).
| Study | Age | Y morphology | Height | FSH/LH | Testosterone | Sperm |
|---|---|---|---|---|---|---|
| Telvi (2) | NA | ? normal Y | −2 SD | NA | NA | Child |
| Telvi (2) | NA | ? normal Y | −7 SD | NA | NA | Child |
| Telvi (2) | NA | ? normal Y | −2 SD | NA | NA | Childa |
| Gasso-Matoses (4) | 31 | del(Y)(q11) | 157 cm | Elevated | Normal | Azoo |
| Tzancheva (10) | 19 | r(Y) | 140 cm | Exag to GnRH | Normal | Azoo |
| Tzancheva (10) | 13 | r(Y) | 131 cm | NA | Low | NA |
| Sher (9) | 7.5 | r(Y) | <5th centile | NA | NA | NA |
| Sher (9) | 11.7 | r(Y) | 5th centile | Low | NA | NA |
| Lin (5) | 38 | r(Y) | 145 cm | NA | NA | Azoo |
| Arnedo (3) | 33 | r(Y) | 169 cm | NA | NA | 4.5 million/cc |
| Micic (6) | 34 | dic(Yq11) | 165 cm | Elevated | Normal | Azoo |
| Micic (6) | 25 | r(Y);dic r(Y) | 148 cm | Elevated | Low | Azoo |
| Bettio (16) | 41 | idic(Yq11) | 171 cm | Elevated | N/A | Azoo |
| Bettio (16) | 35 | idic(Yq11) | 163 cm | Elevated | N/A | Azoo |
| Bettio (16) | 31 | idic(Yq11) | 170 cm | LH normal | Normal | Azoo |
No sperm on biopsy. dic=dicentric; idic=isodicentric; NA=not available; azoo=azoospermic.
Although some authors have suggested the phenotype may correlate with mosaicism in blood or in the testes (2–11), we did not observe any correlation of phenotype with the degree of mosaicism in peripheral blood leukocytes in these five patients nor did Telvi et al (2) when the patients were ascertained by amniocentesis. The percentage of 45,X cell lines in our report varied from 6–36%. Even patient #5, with relatively normal testicular function and the absence of short stature had 26% 45,X cells.
Striking differences in stature in 45,X/46,XY males are more difficult to reconcile, but may relate to absolute numbers of 45,X and 46,XY cells at different times in development, or different mechanisms may be operative in 45,X/46,XY males with scrotal testes. Marked delays in the mitotic error responsible for the origin of the 45,X cell line might lessen the impact of the 45,X cell line linear growth. Alternatively the mechanism(s) that generate the 45,X cell line may vary. For example it is conceivable that some tall individuals with a 45,X cell line are chimeras who received this cell line passively from an accompanying twin in-utero. Long-term follow up of two sexually discordant monozygotic 45,X/46,XY twins suggests that marked discrepancies in adult height in 45,X/46,XY individuals may occur by this mechanism (14).
Some investigators have suggested that when 45,X/46,XY is ascertained at amniocentesis about 90% of the children will be normal (17, 18). Perhaps the mosaic cell line becomes lost so that nearly all (or all) remaining cells are 46,XY. However, there is little long-term follow-up on these patients, so it is likely that prenatal ascertainment underestimates the proportion of patients with phenotypic abnormalities. Those studies reporting series of 45,X/46,XY patients after birth indicate phenotypic abnormalities (1, 2), which likely overestimates the severity since patients with a normal phenotype would go undetected. The clinician needs to exercise extreme caution in predicting the subsequent phenotype of patients with a 45,X/46,XY cell line.
The true incidence of a 45,X/46,XY karyotype in males with bilaterally descended testes is unknown. Most reproductive endocrinologists will consider a karyotype for an azoospermic male, particularly if gonadotropins are elevated. Two of our three adults had azoospermia and elevated gonadotropins and would have likely been detected. However, the male with normal stature, severe oligospermia, normal gonadotropins and testosterone might not have been detected except for the severe oligospermia. Short stature is an additional feature that is likely to trigger the performance of chromosomes in these patients. In our series, both prepubertal males and one adult male were very short, but two adults had normal stature.
In summary, 45,X/46,XY males (regardless of whether the Y chromosome is normal) with bilaterally descended testes usually present with short stature, infertility due to azoospermia or severe oligospermia, and elevated gonadotropins. Turner stigmata may or may not be present, but cardiac and renal anomalies must be excluded. Although testes may contain dysgenetic components (2), it is less clear in this subgroup of males whether the gonads should be removed, as is usually performed in other 45,X/46,XY individuals. It appears reasonable to follow these patients with regular physical exams, Doppler ultrasound, and serum tumor markers. Our findings from this enlightening series of 45,X/46,X(r)Y males highlights the tremendous phenotypic variation which may range from short stature and gonadal failure to more normal stature and some remaining gonadal function.
Acknowledgments
This work was supported by a grant from the U.S. Public Health Service-National Institute of Child Health and Human Development HD040287 (L.C.L.).
Footnotes
This work was presented in part at the Society for Gynecologic Investigation, March 2002.
References
- 1.Gantt PA, Byrd JR, Greenblatt RB, McDonough PG. A clinical and cytogenetic study of fifteen patients with 45,X/46XY gonadal dysgenesis. Fertil Steril. 1980;34:216–21. [PubMed] [Google Scholar]
- 2.Telvi L, Lebbar A, Del Pino O, Barbet JP, Chaussain JL. 45,X/46,XY mosaicism: report of 27 cases. Pediatrics. 1999;104:304–8. doi: 10.1542/peds.104.2.304. [DOI] [PubMed] [Google Scholar]
- 3.Arnedo N, Nogues C, Bosch M, Templado C. Mitotic and meiotic behaviour of a naturally transmitted ring Y chromosome: reproductive risk evaluation. Hum Reprod. 2005;20:462–8. doi: 10.1093/humrep/deh598. [DOI] [PubMed] [Google Scholar]
- 4.Gasso-Matoses M, Pico-Alfonso A, Fernandez-Garcia J, Lobato-Encinas J, Mira-Llinares A. 45,X/46,XY gonadal dysgenesis in an infertile adult male. Urol Int. 1992;48:239–41. doi: 10.1159/000282343. [DOI] [PubMed] [Google Scholar]
- 5.Lin YH, Lin YM, Chuang L, Wu SY, Kuo PL. Ring (Y) in two azoospermic men. Am J Med Genet A. 2004;128:209–13. doi: 10.1002/ajmg.a.30097. [DOI] [PubMed] [Google Scholar]
- 6.Micic M, Micic S, Babic M, Diklic V. Phenotype of two males with abnormal Y chromosomes. Clin Genet. 1990;37:321–6. doi: 10.1111/j.1399-0004.1990.tb03513.x. [DOI] [PubMed] [Google Scholar]
- 7.Newberg MT, Francisco RG, Pang MG, Brugo S, Doncel GF, Acosta AA, et al. Cytogenetics of somatic cells and sperm from a 46,XY/45,X mosaic male with moderate oligoasthenoteratozoospermia. Fertil Steril. 1998;69:146–8. doi: 10.1016/s0015-0282(97)00443-3. [DOI] [PubMed] [Google Scholar]
- 8.Pohlschmidt M, Rappold G, Krause M, Ahlert D, Hosenfeld D, Weissenbach J, et al. Ring Y chromosome: molecular characterization by DNA probes. Cytogenet Cell Genet. 1991;56:65–8. doi: 10.1159/000133051. [DOI] [PubMed] [Google Scholar]
- 9.Sher ES, Addelston MB, Plotnick L, Urban MD, Berkovitz GD. Molecular investigation of two male subjects with short stature and a 45,X/46,X,ring(Y) karyotype. Horm Res. 1998;49:46–50. doi: 10.1159/000023125. [DOI] [PubMed] [Google Scholar]
- 10.Tzancheva M, Kaneva R, Kumanov P, Williams G, Tyler-Smith C. Two male patients with ring Y: definition of an interval in Yq contributing to Turner syndrome. J Med Genet. 1999;36:549–53. [PMC free article] [PubMed] [Google Scholar]
- 11.Wegner RD, Scherer G, Pohlschmidt M, L'Allemand D, Gal A. Ring Y chromosome: cytogenetic and molecular characterization. Clin Genet. 1992;42:71–5. doi: 10.1111/j.1399-0004.1992.tb03142.x. [DOI] [PubMed] [Google Scholar]
- 12.Tho SP, Layman LC, Lanclos KD, Plouffe L, Jr., Byrd JR, McDonough PG. Absence of the testicular determining factor gene SRY in XX true hermaphrodites and presence of this locus in most subjects with gonadal dysgenesis caused by Y aneuploidy. Am J Obstet Gynecol. 1992;167:1794–802. doi: 10.1016/0002-9378(92)91777-8. [DOI] [PubMed] [Google Scholar]
- 13.Grumbach MM. A window of opportunity: the diagnosis of gonadotropin deficiency in the male infant. J Clin Endocrinol Metab. 2005;90:3122–7. doi: 10.1210/jc.2004-2465. [DOI] [PubMed] [Google Scholar]
- 14.Reindollar RH, Byrd JR, Hahn DH, Haseltine FP, McDonough PG. A cytogenetic and endocrinologic study of a set of monozygotic isokaryotic 45,X/46,XY twins discordant for phenotypic sex: mosaicism versus chimerism. Fertil Steril. 1987;47:626–33. doi: 10.1016/s0015-0282(16)59113-4. [DOI] [PubMed] [Google Scholar]
- 15.Kaneko N, Kawagoe S, Hiroi M. Turner's syndrome--review of the literature with reference to a successful pregnancy outcome. Gynecol Obstet Invest. 1990;29:81–7. doi: 10.1159/000293307. [DOI] [PubMed] [Google Scholar]
- 16.Bettio D, Venci A, Rizzi N, Negri L, Setti PL. Clinical and molecular cytogenetic studies in three infertile patients with mosaic rearranged Y chromosomes. Hum Reprod. 2006;21:972–5. doi: 10.1093/humrep/dei426. [DOI] [PubMed] [Google Scholar]
- 17.Hsu LY. Phenotype/karyotype correlations of Y chromosome aneuploidy with emphasis on structural aberrations in postnatally diagnosed cases. Am J Med Genet. 1994;53:108–40. doi: 10.1002/ajmg.1320530204. [DOI] [PubMed] [Google Scholar]
- 18.Hsu LY. Prenatal diagnosis of 45,X/46,XX. Am J Hum Genet. 1996;58:634–6. [PMC free article] [PubMed] [Google Scholar]
- 19.Tho SP, Behzadian A, Byrd JR, McDonough PG. Correlation of the testicular determinant factor sequence zinc finger Y with varying gonadal phenotypes in a series of 13 subjects with gonadal dysgenesis due to Y aneuploidy. Am J Obstet Gynecol. 1990;163:1968–75. doi: 10.1016/0002-9378(90)90782-3. [DOI] [PubMed] [Google Scholar]
- 20.Tho SP, Behzadian A, Byrd JR, McDonough PG. Use of human alpha-satellite deoxyribonucleic acid to detect Y-specific centromeric sequences. Am J Obstet Gynecol. 1988;159:1553–7. doi: 10.1016/0002-9378(88)90593-5. [DOI] [PubMed] [Google Scholar]