

SUMMARY
Dissemination of Acinetobacter baumannii strains harboring class D β-lactamases producing resistance to carbapenem antibiotics severely limits our ability to treat deadly Acinetobacter infections. Susceptibility determination in the A. baumannii background and kinetic studies with a homogeneous preparation of OXA-23 β-lactamase, the major carbapenemase present in A. baumannii, document the ability of this enzyme in manifesting resistance to the last-resort carbapenem antibiotics. We also report three x-ray structures of OXA-23; apo OXA-23 at two different pH values, and wild-type OXA-23 in complex with meropenem, a carbapenem substrate. This represents the first structure of a wild-type class D carbapenemase with a carbapenem antibiotic in its active site. The structures and dynamics simulations reveal an important role for Leu166, whose motion regulates the access of a hydrolytic water molecule to the acyl-enzyme species in imparting carbapenemase activity.
Keywords: Antibiotic resistance, β-lactamase, carbapenemase, crystal structure
INTRODUCTION
Regarded as of little clinical importance merely a decade ago, Acinetobacter baumannii has evolved to become a major opportunistic pathogen. It has spread rapidly throughout the world, and 50–70% of all A. baumannii clinical isolates are multi-drug-resistant (Kallen et al., 2010, Rosenthal et al., 2010). A subclass of β-lactam antibiotics, the carbapenems, which include the archetypical imipenem, along with meropenem and doripenem (Figure S1A), have been successfully utilized as last resort antibiotics for treatment of multi-drug-resistant Acinetobacter infections. Clinical selection and dissemination of the carbapenem-resistant A. baumannii constitutes a serious threat, as the mortality rates from infections by such resistant strains are high, often exceeding 50% (Abbo et al., 2007, Livermore et al., 2010).
The major mechanism of resistance to β-lactam antibiotics in A. baumannii, and in other Gram-negative bacteria, is the production of β-lactamases, which hydrolyze the β-lactam moiety of the antibiotics and render them inactive. Based on their amino-acid sequences, β-lactamases are divided into four classes (Massova and Mobashery, 1998). Classes A, C, and D are active-site-serine β-lactamases, whereas those of class B are zinc-dependent. Although carbapenemase activity could emerge from any of the four classes of β-lactamases, in the case of A. baumannii it is the production of class D enzymes (also known as the OXA-type β-lactamases) (Queenan and Bush, 2007, Walther-Rasmussen and Hoiby, 2006).
Currently over 250 individual class D β-lactamases have been reported. They are characterized by a high degree of structural diversity and a wide-ranging antibiotic-resistance spectrum. Although the active site of the class D enzymes strongly resembles that of the class A enzymes, with an active-site serine experiencing acylation by the β-lactam antibiotic, and a structurally conserved lysine residue nearby that promotes this process, the mechanisms of β-lactam hydrolyses by the two classes of enzymes differ markedly. Both acylation and deacylation steps are facilitated by the conserved lysine residue, which is N-carboxylated in a posttranslational step (Golemi et al., 2001, Schneider et al., 2009). Class D enzymes that are capable of hydrolyzing carbapenem antibiotics are referred to as carbapenem-hydrolyzing class D β-lactamases or CHDLs. Several CHDLs, including OXA-23, OXA-40 (previously known as OXA-24), OXA-51 and OXA-58 enzymes, have been identified in A. baumannii (Afzal-Shah et al., 2001, Bou et al., 2000, Corvec et al., 2007, Poirel et al., 2005). Among them, the OXA-23 β-lactamase is recognized as the major source of carbapenem resistance in this deadly pathogen due to the worldwide dissemination of the OXA-23-producing Acinetobacter isolates (Mugnier et al., 2010, Poirel et al., 2010).
Despite their immense clinical importance, CHDLs from A. baumannii are poorly studied and currently structural information is available for only one enzyme OXA-24. Based on the structure of apo-OXA-24, it was proposed that the ability of the enzyme to hydrolyze carbapenems is facilitated by a tunnel-like entrance to the active site formed by the side chains of residues Tyr112 and Met223 (OXA-24 residue numbering) (Santillana et al., 2007). It was suggested that this tunnel structure forms a hydrophobic barrier, which gives access to the active site only to certain substrates. The structures of two deacylation-deficient mutants of OXA-24 in complex with carbapenem antibiotic doripenem have subsequently demonstrated that this tunnel-like structure remains largely unchanged upon binding of the antibiotic (Schneider et al., 2011). Comparison of these structures with that of a non-carbapenemase OXA-1 complexed with doripenem have shown that the pyrroline ring of carbapenem antibiotics in OXA-1 and OXA-24 are present in two alternative tautomeric forms. The two tautomers are the Δ2-tautomer with the pyrroline ring in an enamine form (Figure S1B), and a Δ1-tautomer where the pyrroline ring is in an imine form (Figure S1C and S1D). This information prompted the authors to conclude that the tunnel-like structure of OXA-24 facilitates the formation of the Δ2-tautomer of the pyrroline ring of doripenem, which could be the catalytically competent isomer. There could be two potential reasons for this. One is a more favorable spatial disposition of the hydroxyethyl moiety that allows the ingress of a water molecule into the active site. Or, alternatively, the existence of the enamine moiety, which can serve as a proton shuttle in promotion of a water molecule for deacylation (Schneider et al., 2011). However, this tunnel is absent in the OXA-48 apo enzyme from Klebsiella pneumoniae, the only other class D carbapenemase whose X-ray structure is currently available (Docquier et al., 2009). These findings raise a question as to whether class D enzymes utilize common structural elements such as the hydrophobic tunnel, to hydrolyze carbapenem antibiotics, or whether carbapenemase activity in various members of this β-lactamase family has evolved independently (Docquier et al., 2009).
To gain further insights into the structural basis of the carbapenemase activity in class D β-lactamases, we determined three X-ray crystal structures for the major carbapenemase of A. baumannii, OXA-23. Here, we report the structures of apo-OXA-23 crystallized at pH values of 4.1 and 7.0, and the OXA-23 complex with meropenem. This is the first structure of a wild-type class D carbapenemase with a clinically important carbapenem antibiotic, which allows us to observe the interaction between the substrate and a non-mutant enzyme. The kinetics of turnover of carbapenems by OXA-23, the respective x-ray structures and the associated molecular-dynamics simulations provide the first detailed characterization of this clinically important enzyme from a problematic pathogen.
RESULTS AND DISCUSSION
Resistance profile and enzyme kinetics of OXA-23
The OXA-23 β-lactamase has recently emerged as the major class D carbapenemase of A. baumannii, a notorious nosocomial pathogen. Rapid spread of the OXA-23-producing pathogens in the clinics worldwide constitutes a serious threat to human health. OXA-23 confers resistance to the last resort carbapenem antibiotics and thus significantly compromises the ability to treat deadly A. baumannii infections. Despite its significant clinical importance, the enzyme has not been studied in detail. To evaluate the ability of OXA-23 to produce resistance in A. baumannii, we cloned its gene into a shuttle vector and introduced it into the susceptible A. baumannii ATCC 17978 strain and subsequently evaluated the MICs of four clinically used carbapenem antibiotics. Production of OXA-23 in the background strain of A. baumannii ATCC 17978 increased MICs of imipenem and meropenem 128-fold and those of doripenem and ertapenem 64-fold (Table 1). The resulting MIC value for imipenem (16 μg/ml) corresponds to that reported for the clinical isolate in which the OXA-23 β-lactamase was identified for the first time (Paton et al., 1993). The MICs for three other antibiotics conferred by OXA-23 were even higher; 32 μg/ml for doripenem and 64 and 256 μg/ml for meropenem and ertapenem, respectively. All these MIC levels are clinically important and define the host strain producing OXA-23 as resistant to carbapenems. As these high MIC values were observed in the susceptible A. baumannii ATCC 17978 strain, existence of additional resistance mechanisms such as low outer-membrane permeability or efflux pumps in various clinical Acinetobacter isolates would even further elevate resistance, indicating that carbapenem antibiotics cannot be used for treatment of infections caused by the OXA-23-producing A. baumannii.
Table 1.
MICs of carbapenem antibiotics against A. baumannii
| Antibiotic | MIC (μg/ml)
|
||
|---|---|---|---|
|
A. baumannii ATCC 17978 |
A. baumannii ATCC 17978 + OXA-23 |
Fold increase | |
| Imipenem | 0.125 | 16 | 128 |
| Meropenem | 0.5 | 64 | 128 |
| Ertapenem | 4 | 256 | 64 |
| Doripenem | 0. 5 | 32 | 64 |
Homogeneously pure enzyme was used to evaluate the steady-state kinetic parameters for turnover of these carbapenems by OXA-23. The catalytic efficiency (kcat/Km) of the enzyme against meropenem was ≥6.8 × 104 M−1s−1, and against three other carbapenems was in the range of 4.2 × 104 to 7.4 × 104 M−1s−1 (Table 2). OXA-23 had 5- to 16-fold higher turnover numbers (kcat) and 7-fold or more higher Km values for imipenem than for the three other carbapenem antibiotics. A. baumannii exhibits low permeability of its envelope for various antibiotics (Clark, 1996, Vila et al., 2007). This low permeability combined with the levels of catalytic activity that we measured for turnover of imipenem, meropenem, doripenem and ertapenem translates into high-level clinical resistance of the bacterium to carbapenem antibiotics.
Table 2.
Kinetic parameters for hydrolysis of carbapenems by OXA-23
| Parameter | Imipenem | Meropenem | Ertapenem | Doripenem |
|---|---|---|---|---|
| kcat (s−1) | 0.35 ± 0.01 | 0.068 ± 0.001 | 0.021 ± 0.001 | 0.036 ± 0.001 |
| Km (μM) | 4.8 ± 0.3 | ≤1 | 0.50 ± 0.10 | 0.70 ± 0.09 |
| kcat/Km (M−1s−1) | (7.4 ± 0.4) × 104 | ≥6.8 × 104 | (4.2 ± 0.8) × 104 | (5.2 ± 0.7) × 104 |
Crystal structures of OXA-23 at low and neutral pH
Despite its recognized clinical importance, no structural information is available for OXA-23. We succeeded in determining three OXA-23 structures, two of apo-enzyme at pH values of 4.1 and 7.0, and a third of an acylated species with a carbapenem antibiotic (meropenem). The resolution of the structure at two different pH values was critical for the observation of the post-translational modification of the active-site Lys82. The side chain of this lysine in class D β-lactamases is N-carboxylated in vivo (Golemi et al., 2001), a modification that is critical for enzyme function. At pH 4.1, Lys82 should undergo complete N-decarboxylation (Golemi et al., 2001) and this is what is observed in our x-ray structure. At the higher pH of 7.0, the enzyme retains the N-carboxylation on the lysine, hence, the two structures provide information for both states of this catalytically important residue. Moreover, we were able to capture the substrate in the active site of OXA-23 by co-crystallization at low pH. At low pH the catalytic lysine is readily N-decarboxylated and thus the acyl-enzyme species is trapped and unable to efficiently undergo hydrolysis. This allowed us to obtain structural information on the acyl-enzyme species for the substrate meropenem bound covalently to the wild-type enzyme.
The OXA-23 protein folds as two non-contiguous domains, a mixed α/β domain with two α-helices and a six-stranded β-sheet, and an all-α domain, with the active site located at the junction of the two domains (Figure 1). The overall OXA-23 structure is similar to other members of the class D β-lactamases including the carbapenemases OXA-24 (Schneider et al., 2011) and OXA-48 (Docquier et al., 2009), and the non-carbapenemases OXA-1 (Schneider et al., 2009, Sun et al., 2003), OXA-2 (pdb code 1k38), OXA-10, the first of the class D enzymes whose structures were determined (Golemi et al., 2000, Paetzel et al., 2000), OXA-13 (Pernot et al., 2001) and OXA-46 (Docquier et al., 2010). Superimposition of the pH 7.0 form of OXA-23 with the class D β-lactamases whose structures have been determined, are summarized in Table S1. The root-mean-square deviations (rmsds) in the positions for all matching Cα atoms range from 0.6 Å for OXA-24 to 1.5 Å for OXA-1. Inspection of the superimposed structures shows that the differences in the rmsd values are primarily due to length and conformational differences in several external loops. When these loops are omitted, the fit is greatly improved in all cases (Table S1), indicating that the core regions of the OXA enzymes, comprising the domain 1 βsheet and the domain 2 α-helices, are structurally conserved. Moreover, the rmsd values for all of the OXA structures, with the exception of OXA-24, are all very similar at around 1 Å (Table S1). OXA-24 is significantly closer in structure to OXA-23 with an rmsd of 0.4 Å, consistent with the much higher sequence identity (65%) between these two A. baumannii enzymes when compared to the lower sequence identities (24–38%) with other class D enzymes from different bacterial species (Table S1). The regions of the superimposed structures that show the greatest conformational variation are in the C-terminal helix (α9), the loops between strands β1 and β2, strands β6 and β7, and between strand β9 and helix α9. Along with a long unstructured Ω-loop (which the class D enzymes have in common with the class A enzymes), these three loops are all adjacent to the active site. In OXA-24 the β6/β7 loop carries the methionine residue (Met223), which forms one side of the tunnel structure, and this residue is conserved in OXA-23 but in none of the other OXA enzymes. Moreover, this loop comprising six residues, has a conserved structure in the three carbapenemases, although OXA-48 lacks the conserved methionine and does not have the tunnel structure as has been noted previously (Docquier et al., 2009). In OXA-10 and OXA-13, the β6/β7 loop is three residues longer and folds away from the active site, while in OXA-2 and OXA-46 it contains only two residues. In a recent study with OXA-10 (De Luca et al., 2011), this loop was replaced with the corresponding loops from OXA-23, OXA-24 and OXA-48. All three loop mutants acquired significant activity towards imipenem, and the structures of the OXA-10 enzyme with loops from OXA-24 and OXA-48 showed that the β6/β7 loop adopted the conformation of the parental enzymes. The β9/α9 loops shows much greater variation in structure, and given its close proximity to the active site, may participate in anchoring the bound substrates.
Figure 1.
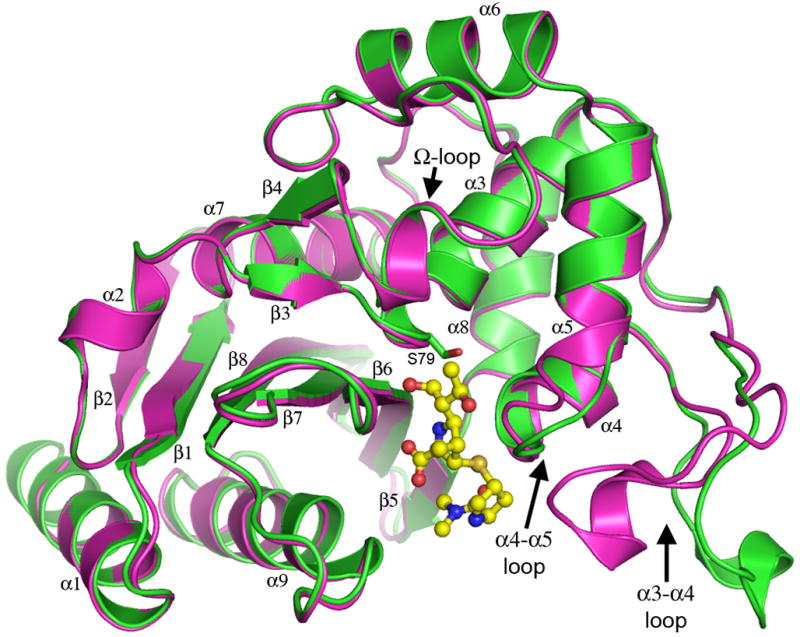
Ribbon representation of the superimposition of OXA-23 at pH 4.1 (green) and pH 7.0 (magenta). The loops that show large-scale structural rearrangements are shown at the lower right, in the “open” conformation at pH 4.1 and in the “closed” conformation at pH 7.0. The loop which corresponds topologically with the Ω-loop in the class A β-lactamases is also indicated. Meropenem (yellow ball-and-stick) is shown in the active site of OXA-23. See also Figure S1, Figure S4, Table S2 and Table S3)
The low pH form of OXA-23, refined at 1.15-Å resolution, has a fully occupied citrate anion at the position where a carbapenem substrate would be expected to bind. The two terminal carboxylate groups of the citrate occupy an oxyanion hole formed by the amide nitrogen atoms of the conserved active site serine Ser79 and Trp219 from strand β6, and the substrate carboxylate-binding site formed by the side chain of Arg259 (Figure S2A). As indicated above, this low pH form of the enzyme shows no evidence of post-translational N-carboxylation of the active-site Lys82 (Figure S3), typically observed in all OXA enzymes (Golemi et al., 2001). Attempts to displace the citrate from the low pH form by diffusing substrates into the pre-formed crystals proved unsuccessful, and this could be due to either tight binding of the citrate or the lack of the modified lysine residue, which is thought to be also responsible for catalyzing the acylation step. In contrast, analysis of the residual difference electron density in the active site of the neutral pH form showed that the active site was occupied only by water molecules, and there was clear evidence that the side chain of Lys82 was modified by N-carboxylation (Figure S2B).
Superimposition of the two apo OXA-23 structures shows that at low pH major structural rearrangements take place involving two loops adjacent to the active site. The loop between helices α4 and α5 (residues Leu125-Pro129) swings away from the active site by approximately 3.5 A. Subsequently, a large surface loop between helices α3 and α4 (residues Trp103-Met117) adopts an entirely different conformation, moving approximately 10–12 Å out into the solvent into an “open” conformation relative to its “closed” position in the neutral pH form (Figure S4). Although such major loop movements are unusual in the class D β-lactamases, they are not unprecedented since similar movements of the α3/α4 and α4/α5 loops were also observed in the OXA-46 structure (Docquier et al., 2010). Here the conformational changes appear to be in response to the presence in the active site of a tartrate anion, positioned in approximately the same location as the citrate in OXA-23.
The meropenem complex of wild-type OXA-23
The structure of the OXA-23-meropenem complex at pH 4.1 was solved by molecular replacement (MR) using both the low pH and neutral pH forms of apo-OXA-23 as starting models. The two MR solutions were comparable but upon inspection of the electron density it was noted that the mobile α3/α4 and α4/α5 loops were in the same “closed” conformation as seen in the neutral pH form, so this solution was chosen for subsequent refinement. Superimposition of the two independent molecules in the asymmetric unit gives an rmsd of 0.3 Å for 241 matching Cα atoms. There are no significant differences in the surface loops between the two molecules, although analysis of the crystal packing contacts of the two molecules shows that the specifics of the symmetry-related interactions differ somewhat. Although both molecules are in contact with a large solvent-filled cavity, molecule A is surrounded almost on all sides by eight symmetry-related molecules, whereas molecule B only makes contacts with four and appears much more solvent exposed. This variation in crystal contacts may account for the higher average atomic displacement parameters for molecule B compared to molecule A (Table S3). Inspection of the crystal packing also shows that the α3/α4 loop in both molecules is adjacent to the solvent-filled cavity, and modeling the “open” low-pH conformation of the loop taken from the apo-OXA-23 structure, onto the meropenem-OXA-23 structure indicates that there would be adequate space available for the complex to adopt the “open” form without steric clashes with neighboring molecules. Similarly, an analysis of the crystal packing in the low- and neutral-pH forms show that the conformation of the α3/α4 loop is not constrained in either structure by crystal packing. The mechanistic role of this loop motion is not clear at present and may simply be in response to the presence of the citrate in the active site as was suggested for OXA-46 where tartrate was bound in the active site (Docquier et al., 2010).
Meropenem was located in the OXA-23 active site in strong residual Fo-Fc difference electron density, which was continuous with the 2Fo-Fc density for the Ser79 side chain (Figure 2A). The meropenem is covalently bound to the Oγ atom of Ser79 and anchored by seven hydrogen-bonding interactions along with a number of hydrophobic packing contacts (Figure 2B). The C7 carbonyl oxygen, formerly the β-lactam carbonyl, is located in the oxyanion hole and is hydrogen bonded to the amide nitrogen atoms of Ser79 and Trp219, and the carboxylate forms three hydrogen bonds with the side chains of Thr217 and Arg259. There are two additional hydrogen bonds to well-ordered water molecules. Two conserved residues at the end of the long α3/α4 loop (Phe110 and Trp113) provide aromatic packing contacts with the pyrrolidine ring of meropenem. One of these residues, Phe110 participates in the formation of the tunnel-like structure, suggested to be important for the carbapenemase activity of the OXA-24 β-lactamase (Schneider et al., 2011).
Figure 2.

Meropenem binding to OXA-23. (A) Residual Fo-Fc electron density (pink) in the OXA-23 active site modeled with the final coordinates of meropenem (cyan ball-and-stick). Final 2Fo-Fc electron density for Ser79 is indicated. (B) Meropenem bound in the active site in the final model showing the hydrogen-bonding interactions (dashed black lines). See also Figure S2.
The quality of the electron density for the meropenem was such that all 26 non-hydrogen atoms of the substrate could be assigned unambiguously in the density, and even in the early stages of refinement it was evident that the C3 carbon of the pyrroline ring was sp3-hybridized with the sulfur on the Si face of the ring, such that the ring was in the Δ1-tautomeric state as the S stereoisomer (Figure S1D). This was in contrast to the configuration of the pyrroline ring in the OXA-24-doripenem complex (Schneider et al., 2011), where the Δ2-tautomer was observed (Figure S1B) with an sp2-hybridzed C3 carbon atom. It has been proposed that the tunnel-like structure over the active site of OXA-24 formed by residues Tyr112 and Met223 would interfere with formation of the Δ1-tautomer of carbapenem antibiotics (as observed in the OXA-1-doripenem complex) and thus is critical for the carbapenemase activity of OXA-24 (Schneider et al., 2011). In our OXA-23 structure a similar tunnel is formed by the side chains of Phe110 and Met221 (Figure 3), but this does not prevent the formation of the Δ1-tautomer, as is seen in the OXA-23-meropenem complex. Moreover, this OXA-23 Δ1-tautomer differs from the Δ1-tautomer described for the OXA-1-doripenem complex (Schneider et al., 2009), where the sulfur atom projected toward the opposite face of the pyrroline, giving the R stereoisomer at C3.
Figure 3.
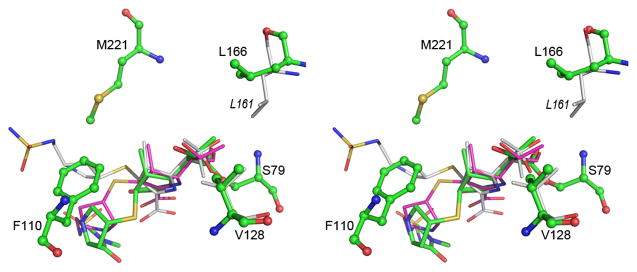
Stereoview of the superposition of the acyl-enzyme intermediate in OXA-23 (green), OXA-24 (magenta) and OXA-1 (white). The two hydrophobic residues which interact with the 6a-hydroxyethyl moiety of the intermediate are shown at upper right (Leu166 for OXA-23 and Leu161 for OXA-1) and lower right (Val128 for OXA-23). The difference in conformation of the leucine residue can be seen. The residues which constitute the tunnel-like structure in the OXA enzymes are indicated for OXA-23 (Phe110 and Met221). See also Figure S3 and Table S1.
On the opposite side of the pyrroline ring, the 6α-hydroxyethyl moiety at C6 is directed toward the side chains of two conserved hydrophobic residues, Val128 and Leu166 (Figure 3). Both residues are within the van der Waals contact distance of the terminal carbon of the ethyl group. The oxygen atom of the 6α-hydroxyethyl moiety is directed away from the active-site Ser79 residue and makes no hydrogen-bonding interactions with the protein. This is consistent with what was observed in the OXA-23-doripenem complexes, yet in contrast to the non-carbapenemase OXA-1-doripenem complex, where the 6α-hydroxyethyl is found in two different conformations, both having the hydroxyl oxygen directed toward the active-site serine such that it makes hydrogen-bonding interactions with the N-carboxylated Lys70. The lysine residue in OXA-1 forms a network of hydrogen bonds, both to the doripenem and to other amino acids (Ser120 and Trp160). Only one of these hydrogen-bonding interactions, that to the conserved Trp165 (equivalent to Trp160 on OXA-1), is observed in OXA-23. The equivalent residue to Ser120 in OXA-1 is a tyrosine (Tyr131) in OXA-23 and the side chain of this residue is directed away from the meropenem binding site such that no hydrogen-bonding interaction is possible. Thus in OXA-23, our structural data show that although a hydrophobic tunnel is formed by the side chains of Phe110 and Met221, it does not necessarily force the carbapenem substrate to adopt a Δ2-tautomer and a specific orientation of the 6-hydroxyethyl group, as has been suggested (Schneider et al., 2011). We now show that the same tunnel structure can lead to the formation of a Δ1-tautomer with the same 6-hydroxyethyl conformation as seen in OXA-24. There are clearly other factors which serve to present an acylated carbapenem in the correct orientation in the active site for efficient deacylation, and our data, along with the evidence from the OXA-48 structure that has no tunnel, would suggest that the tunnel might have less impact than previously suggested.
Comparison of the neutral pH apo OXA-23 structure with the OXA-23-meropenem complex shows that the aforementioned Leu166 undergoes a conformational change upon meropenem binding. In the apo structure, Leu166 is directed towards the Val128 side chain, and the two hydrophobic side chains are within 3.5 Å of each other. In the presence of meropenem the leucine swings away from the 6α-hydroxyethyl moiety by approximately 3 Å, rotating 90° about the Cα-Cβ bond and 60° about the Cβ-Cγ bond. This now places the leucine approximately 7 Å from Val128. In the OXA-1 complex with doripenem, the “6α-hydroxyethyl pocket” is also flanked by the conserved Val117 and Leu161. However, the OXA-1 Leu161 exists in the same conformation seen for Leu166 in apo OXA-23, such that there is a significant hydrophobic interaction with Val117. The difference in the positioning of the conserved leucine side chain in carbapenem complexes of OXA-1 and OXA-23 may play a critical role in allowing the access of the hydrolytic water to the N-carboxylated lysine to enable deacylation of the acyl-enzyme species. In OXA-1 the location of the Leu161 side chain would preclude the hydrolytic water from approaching N-carboxylated Lys70 or the ester carbon of the acylated doripenem, by effectively blocking access to the active site (Figure 4A), since the only other access to the carbamylated lysine from the external solvent is blocked by the hydroxyethyl group of the acyl-enzyme intermediate. In contrast, when the OXA-23 Leu166 side chain adopts the different conformation, this opens a channel to the corresponding N-carboxylated Lys82 and allows a water molecule to approach the acyl-enzyme species (Figure 4B).
Figure 4.
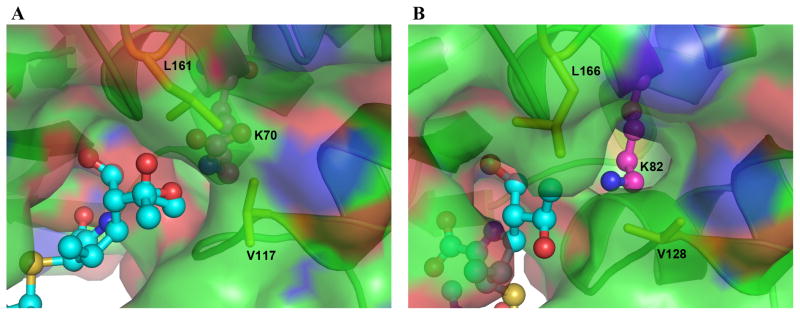
Molecular surface representation of (A) OXA-1 and (B) OXA-23. In both cases the bound substrate (doripenem and meropenem, respectively) are shown as cyan ball-and-stick. The motion of the Leu166 side chain in response to meropenem binding in OXA-23 opens a channel to the external solvent and allows access of a water molecule to the N-carboxylated Lys82.
This conserved leucine residue is in the Ω-loop, and further analysis of the OXA-1 and OXA-23 structures shows this loop has a slightly different structure in the two enzymes. There is an insertion of a glutamate (Glu162) in OXA-1 directly following Leu161, and this residue makes hydrogen-bonding interactions with the side chains of Asn226 and Asp68, which serves to pull the loop approximately 1 A closer towards the β-sheet than in OXA-23 and OXA-24. If the OXA-1 Leu161 residue were to swing upwards in the same way as we observe in OXA-23, there would be significant steric clash with the Asp68 side chain. Conversely, in OXA-23 the equivalent residue to this aspartate is a valine (Val67), Asn226 is replaced by another valine (Val227), and there is an adjacent hydrophobic residue (Ile223), the side chain of which occupies the site where the Glu162 residue was in OXA-1. These two hydrophobic residues (Ile223 and Val227) are from the β6/β7 loop, noted earlier as being important for the carbapenemase activity of the OXA enzymes (De Luca et al., 2011). In the two other known class D carbapenemases (OXA-24 and OXA-48) there are small hydrophobic residues which correspond spatially to Ile223 and Val227 in the OXA-23 β6/β7 loop, due to the similar fold of the loop in these three enzymes. The narrow-spectrum OXA enzymes with their widely varying β6/β7 loop structure do not retain these residues. The presence of these residues in the carbapenemases contribute to the formation of a hydrophobic pocket adjacent to Leu166 which would be attractive to the movement of the leucine side chain. None of the other OXA enzymes whose structures are known have as an extensive a hydrophobic pocket in this region and may not be capable of effecting the movement of the conserved leucine side chain in the same manner as we observe in OXA-23. In these narrow-spectrum enzymes, without such a side chain movement, it is unlikely that a channel to the external solvent can be opened, and the opportunity for a water molecule to enter the site adjacent to the carbamylated lysine and effect deacylation is minimized.
Molecular dynamics simulations of OXA-23 and OXA-1
These important experimental observations from the crystal structures were complemented by MD simulations. The OXA-23 and OXA-1 structures were acylated with meropenem and were N-carboxylated at their respective active-site lysine residues. Both the Δ1- and Δ2-tautomers of meropenem were generated independently for these MD simulations. Similar to what was observed in the crystal structure of OXA-23, we see a movement of the loop containing residues Trp103-Met117 during simulations. The movement is more pronounced with a Δ2-meropenem tautomer, and appears to be synchronized with the movement of the C2 substituent of the pyrroline ring. The maximal separation between the two loop conformations in the course of simulations was approximately 6 Å. The meropenem molecules do not experience any significant movement. Their average rmsds were approximately 0.8 Å and 1.3 Å for the OXA-1 and the OXA-23 simulations, respectively, with the bulk of the contribution coming from the more inherently flexible C2 substituent. This differs from what was observed in OXA-48, where in MD simulations the hydroxyethyl group of docked meropenem was seen to rotate about 30° such that the methyl group was accommodated in a hydrophobic pocket formed by Leu158 (equivalent to Leu166 in OXA-23) and Thr213 (equivalent to Met221), thus allowing a closer approach of a water molecule to the N-carboxylated lysine (Docquier et al., 2009).
The OXA-23 simulations show a large number of snapshots with a water molecule favorably positioned for deacylation (Figure 5). This would appear for two structural reasons. First, in OXA-23 the N-carboxylated lysine is held by only two hydrogen bonds, one with Trp165 and another with 6α-hydroxyethyl group of meropenem, whereas in OXA-1 one additional hydrogen bond to Ser120 (OXA-1 numbering) can be seen (Figure S5). The extra hydrogen bond in OXA-1 pulls the lysine approximately 1 Å in toward the active site serine compared to OXA-23, making it less accessible to water. In OXA-23 the displacement of the less-restrained lysine further from the Ser79 side chain serves to create space for an incoming water. Second, as seen in the crystal structures and discussed earlier, the positioning of the Leu166 side chain is different between OXA-23 and OXA-1 (Figure S5). In OXA-23 simulations, movement of the Leu166 side chain is observed such that it adopts an orientation that opens a channel through which a hydrolytic water molecule could pass and position itself for the deacylation step. In OXA-1 simulations this conserved leucine is not mobile and adopts a conformation that restricts access of water into the active site.
Figure 5.
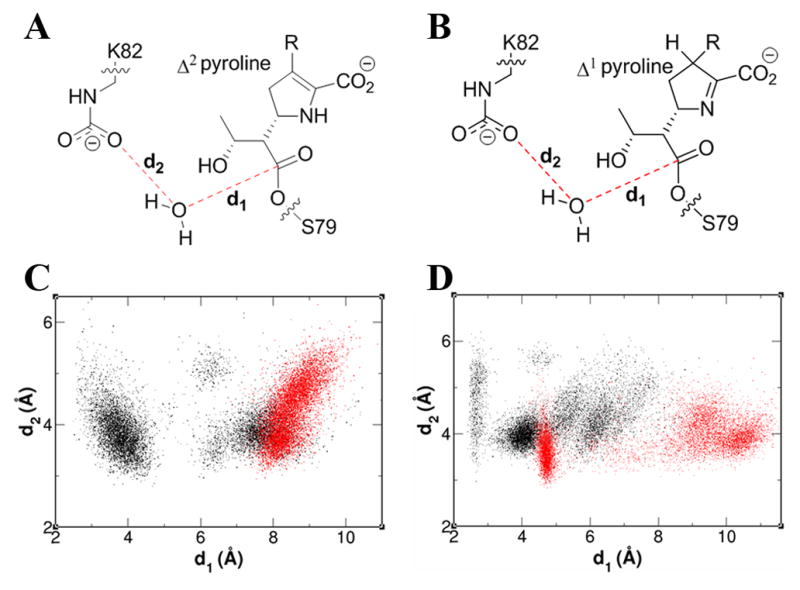
Schematic of the active site of the acyl-enzyme species of meropenem in (A) Δ2- and (B) Δ1-tautomeric forms. Distance distribution of the d1 and d2 during the 20 ns simulation of OXA-23 (black) and OXA-1 (red) acylated with meropenem in (C) Δ2- and (D) Δ1-tautomeric forms. See also Figure S5.
We have already noted that a comparison of the OXA-23 structure with that of non-carbapenemase OXA-1 also pinpoint these two significant structural differences in the active sites of these enzymes, and suggest that they could play a key role in the inability of OXA-1 to efficiently hydrolyze carbapenem antibiotics, in contrast to OXA-23. The additional hydrogen bond seen in the OXA-1 MD simulations between the N-carboxylated lysine and Ser120 is also observed in the OXA-1 structure, along with the shift in the lysine side chain relative to its position in OXA-23. This makes the OXA-1 N-carboxylated lysine less accessible to a hydrolytic water. The second important difference between the two enzymes is the spatial disposition of the conserved leucine (Leu166 in OXA-23 and Leu161 in OXA-1). As observed by both X-ray crystallography and MD simulation, binding of the carbapenem substrate in OXA-23 results in a significant movement of the conserved leucine, stabilized by the presence of an adjacent hydrophobic pocket created by the specific fold of the β6/β7 loop particular to the class D carbapanemases, which would then allow access of a water molecule into the active site of the enzyme. In both the apo OXA-1 structure and OXA-1 in complex with carbapenem antibiotic, this conserved leucine residue does not move and thus limits access of a hydrolytic water to the carbapenem-acylated active-site serine.
The ramification of this observation has direct bearing on whether an enzyme can function as a carbapenemase (as in OXA-23) or not (as in OXA-1). In the latter case, acylation takes place, but the complex experience deacylation only inefficiently. This case is very similar to that of the class A β-lactamases for which carbapenems are poor substrates, behaving transiently as inhibitors (Maveyraud et al., 1998). In evolution of the OXA-23 enzyme as a carbapenemase, it had to overcome energetic barriers to become catalytically more competent for turnover of these substrates. Our kinetic data and evaluation of antibiotic susceptibility patterns fully support this as the outcome in the case of OXA-23, but the enabling mechanism would not appear to be merely a single obvious mutation. It is likely to be the culmination of multiples of amino acid changes in the sequence of the protein. Nonetheless, the critical outcome, the motion of Leu166 to allow entry of the water molecule to the active site, may be an indispensable component of the function of this enzyme.
SIGNIFICANCE
Merely a decade ago carbapenems were the antibiotics of choice for treatment of infections caused by multi-drug-resistant Acinetobacter baumannii, a notorious nosocomial pathogen. However, resistance to carbapenem antibiotics now severely compromises therapeutic options against deadly Acinetobacter infections. Antibiotic susceptibility, kinetics and x-ray structures of OXA-23, the major carbapenemase in Acinetobacter, have been determined, giving us insight into the molecular mechanism of the carbapenemase activity for this significant antibiotic resistance enzyme in an important human pathogen.
EXPERIMENTAL PROCEDURES
Cloning
The gene encoding the class D β-lactamase OXA-23 from A. baumannii (GenBank accession number CAB69042.1) with its leader sequence and a promoter provided by the ISAbaI insertion sequence was custom synthesized and cloned into the shuttle vector, pNA1, which was constructed in our lab and is interchangeably useful for transformation of either Escherichia coli or A. baumannii. The construct was first introduced into the competent E. coli JM83 strain by transformation. The construct then was re-isolated from E. coli, sequenced and electroporated into the A. baumannii ATCC 17978 strain for subsequent antibiotic susceptibility testing.
For large-scale protein expression and purification, the gene for OXA-23 was optimized for expression in E. coli and custom synthesized. Nucleotides encoding the 17 amino acid leader sequence of OXA-23 were deleted and the unique NdeI and HindIII restriction sites were introduced at the 3′ and 5′ ends of the gene for the mature enzyme, respectively. The gene was subsequently cloned into the NdeI/HindIII restriction sites of the pET24a(+) vector (Invitrogen) and transformed into E. coli BL21 (DE3) competent cells.
Antibiotic susceptibility testing
Minimal inhibitory concentrations (MICs) of four carbapenem antibiotics, imipenem, meropenem, doripenem and ertapenem against the susceptible strain of A. baumannii ATCC 17978 and A. baumannii ATCC 17978 harboring shuttle vector expressing OXA-23 β-lactamase were determined according to the Clinical and Laboratory Standards Institute guidelines (Clinical and Laboratory Standards Institute, 2009). Bacteria were used at a final inoculum of 5 × 105 CFU/ml in Mueller-Hinton II broth (DIfco). The experiment was performed in triplicate and results interpreted after incubation at 37°C for 16–20 h.
Protein purification
Five ml of overnight culture of E. coli BL21 (DE3) harboring the pET24a(+) vector encoding the OXA-23 β-lactamase was inoculated into 500 ml of fresh LB broth supplemented with 60 μg/ml of kanamycin. The cells were grown at 37°C with shaking (180 rpm) until an OD600 of 0.8 was reached. The protein expression was induced by the addition of isopropyl β-D-thiogalactopyranoside to a final concentration of 1 mM. The cells were incubated at 22°C for 20 hours with shaking (180 rpm). pelleted by centrifugation at 18,000 × g for 30 minutes at room temperature, resuspended in 40 ml of 20 mM MES, pH 6.0 and disrupted by sonication. The cell extract was centrifuged at 18,000 × g for 1 hour at 4°C to remove insoluble debris. The supernatant was loaded onto a DEAE anion-exchange column (BioRad) equilibrated with the same buffer. The flow-through, containing the enzyme, was recovered and dialyzed against the 20 mM Tris buffer, pH 8.0. The flow-through was loaded onto a High Q anion-exchange column (BioRad) equilibrated with the same buffer. The column was washed with two volumes of 20 mM Tris pH 8.0 and the protein was eluted with a linear gradient of 0–500 mM NaCl. The fractions were analyzed for the presence of β-lactamase by the addition of the chromogenic substrate nitrocefin and subsequently by SDS-PAGE. The fractions containing the protein were pooled and dialyzed against HEPES buffer, pH 7.5. The protein purity was assessed by SDS-PAGE and was estimated to be more than 95%.
Enzyme kinetics
All kinetic data were obtained using either a Cary 50 or Cary 60 spectrophotometer (Varian or Agilent, respectively) at 22 °C. Nonlinear regression was performed to fit the data using Prisim (GraphPad Software, Inc.). Data were collected from at least three independent measurements. Reactions containing 100 mM NaPi (pH 7.0), 50 mM NaHCO3, and varying concentrations of carbapenem substrate (2–100 μM) were initiated by mixing with OXA-23 (0.1–2 μM in 20 mM HEPES (pH 7.5)). The absorbance was monitored to follow the course of each reaction at the following wavelengths: ertapenem (λ = 295 nm and Δε = −10,940 cm−1 M−1), doripenem (λ = 299 nm and Δε = −11,540 cm−1 M−1), imipenem (λ = 297 nm and Δε = −10,930 cm−1 M−1), and meropenem (λ = 298 nm and Δε = −7,200 cm−1 M−1). The linear portion of each reaction was used to determine the steady-state velocities (v). The steady-state parameters, kcat and Km, were calculated by plotting the observed rate constants (kobs = v/[E]) as a function of carbapenem concentration and fitting the data non-linearly to the Michaelis-Menten equation.
Crystallization and data collection
An initial screen was made using PEG/Ion Screens 1 and 2 (Hampton Research) using the sitting drop method and a protein concentration of 35 mg/ml. Crystals were obtained in a number of wells and subsequent refinement of the conditions gave diffraction quality crystals at low pH from 0.06 M citric acid/0.04 M bis-tris propane pH 4.1, 16 % PEG3350, and at neutral pH from 0.2 M succinic acid pH 7.0, 20% PEG3350. The OXA-23 meropenem complex was formed by incubating 950 μM of the enzyme with 10 mM meropenem at 4°C, and crystallizing the resultant complex from 0.06 M citric acid/0.04 M bis-tris propane pH 4.1, 16 % PEG3350. Complete diffraction datasets were collected from single crystals of OXA-23 at pH 4.1 and pH 7.0 using SSRL beam line BL12-2. The crystals diffracted to 1.15 Å and 2.4 Å resolution respectively. The pH 4.1 data set comprised 400 images with an oscillation angle of 0.5° with X-rays at 14000 eV (0.88557 Å) using a PILATUS 6M PAD detector running in shutterless mode. The pH 7.0 dataset also comprised 400 images with an oscillation angle of 0.5° with X-rays at 12568 eV (0.97950 Å). A single crystal of the OXA-23-meropenem complex diffracted to 2.15 Å in space group P21. A total of 500 images using an oscillation angle of 0.5° were collected at BL12-2. All images for the three structures were processed with XDS (Kabsch, 1993) and scaled with SCALA from the CCP4 suite of programs (Collaborative Computing Project, 1994). Final data collection statistics are given in Table S2.
Structure Solution and Refinement
The OXA-24 structure (Protein Data Bank code 3gp4) was used as the search model in the determination of the OXA-23 structure at pH 4.1, by molecular replacement (MR) using MOLREP from the CCP4 suite (Collaborative Computing Project, 1994). All solvent molecules were removed from the OXA-24 model prior to MR calculations, and the CCP4 program CHAINSAW was used to convert the OXA-24 model into OXA-23, whereby identical residues in the two sequences were retained and those which differed were truncated at the Cβ atom. A strong MR solution gave the position of the single OXA-23 molecule in the C2 asymmetric unit. The structure was refined for 15 cycles using REFMAC (Murshudov et al., 1999) and 2Fo-Fc and Fo-Fc electron density maps calculated. During the course of the refinement strong residual Fo-Fc density was observed in the active site, and inspection of the crystallization conditions suggested the presence of a citrate molecule which was subsequently added to the model. The fully refined OXA-23 pH 4.1 model was subsequently used to solve the structure of OXA-23 at pH 7.0, and both of these apo structures were used as MR models for the OXA-23-meropenem complex. In the latter, meropenem molecules were built into the available electron density in the active sites of both molecules in the asymmetric unit. Refinement of all three structures was completed with the PHENIX suite of programs (Adams et al., 2010), and manual building of the models using the molecular graphics program COOT (Emsley and Cowtan, 2004). Water molecules were added in structurally and chemically relevant positions in all three structures. The atomic displacement parameters (ADPs) for the protein atoms were refined isotropically for the pH 7.0 and meropenem structures, and anisotropically for the pH 4.1 structure. Final refinement statistics are given in Table S3. Figures were generated using PYMOL (DeLano, 2002).
Computational Methods
The OXA-23 crystal structure acylated with meropenem in Δ1-tautomeric form provided the initial atomic coordinates for the OXA-23 simulations. The Δ2-tautomer of meropenem was modeled in using Sybyl-X (Tripos Inc., St. Louis, MO). For OXA-1 simulations, a previous OXA-1 structure acylated with doripenem (PDB code: 3ISG) provided the initial atomic coordinates for the protein and Δ1- and Δ2-meropenem were modeled in based on the ligand conformations generated for OXA-23. Crystallographic waters of each enzyme were retained. The RESP methodology (Bayly et al., 1993) provided partial atomic charges for the acylated meropenem and serine, while Amber (University of California, San Francisco, CA) ff99 and gaff forcefields provided the rest of the partial atomic charges and all the forcefield parameters. Both simulation systems were solvated in a truncated octahedral box of explicit water molecules using the TIP3P water model (Jorgensen et al., 1983). The MD simulations were started with a 5000-step minimization of the water molecules. This was followed by a heating phase to 300 K and an equilibration stage for 200 ps. The data collection phase that followed was 20 ns long. The temperature was controlled using the Langevin method and isotropic position scaling provided pressure regulation. The simulations were run using the Amber 12 package and results were analyzed using its ptraj module and VMD (Humphrey et al., 1996).
Supplementary Material
HIGHLIGHTS.
OXA-23 is the major carbapenemase in Acinetobacter
Antibiotic susceptibility and enzyme kinetics of OXA-23 have been determined
The crystal structure of OXA-23 has been solved by X-ray crystallography
The first structure of a wild-type CHDL complexed with a carbapenem substrate is presented
Acknowledgments
Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource (SSRL), a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393) and the National Center for Research Resources (P41RR001209). We would like to thank SSRL Summer Interns Lyudmila Novikova and Chris Milianta for help with structure determination and refinement.
Footnotes
ACCESSION NUMBERS
The coordinates and structure factors for OXA-23-meropenem, OXA-23 pH 4.1, and OXA-23 pH 7.0 were deposited in the PDB with accession codes 4JF4, 4JF5 and 4JF6 respectively.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbo A, Carmeli Y, Navon-Venezia S, Siegman-Igra Y, Schwaber MJ. Impact of multi-drug-resistant Acinetobacter baumannii on clinical outcomes. Eur J Clin Microbiol Infect Dis. 2007;26:793–800. doi: 10.1007/s10096-007-0371-8. [DOI] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. 2010;D66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afzal-Shah M, Woodford N, Livemore DM. Characterization of OXA-25, OXA-26, and OXA-27, molecular class D β-Lactamases associated with carbapenem resistance in clinical isolates of Acinetobacter baumannii. Antimicrob Agents Chemother. 2001;45:583–588. doi: 10.1128/AAC.45.2.583-588.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayly CI, Cieplak P, Cornell WD, Kollman PA. A well-behaved electrostatic potential based method using charge restraints for determining atom-centered charges: The RESP model. J Phys Chem. 1993;97:10269–10280. [Google Scholar]
- Bou G, Oliver A, Martinez-Beltran J. OXA-24, a novel class D beta-lactamase with carbapenemase activity in an Acinetobacter baumannii clinical strain. Antimicrob Agents Chemother. 2000;44:1556–1561. doi: 10.1128/aac.44.6.1556-1561.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RB. Imipenem resistance among Acinetobacter baumannii: association with reduced expression of a 33–36 kDa outer membrane protein. J Antimicrob Chemother. 1996;45:245–251. doi: 10.1093/jac/38.2.245. [DOI] [PubMed] [Google Scholar]
- Clinical and Laboratory Standards Institute. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; Approved standard. 8. Wayne, PA: Clinical and Laboratory Standards Institute; 2009. [Google Scholar]
- Collaborative Computing Project No 4. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Corvec S, Poirel L, Naas T, Drugeon H, Nordmann P. Genetics and expression of the carbapenem-hydrolyzing oxacillinase gene blaOXA-23 in Acinetobacter baumannii. Antimicrob Agents Chemother. 2007;51:1530–1533. doi: 10.1128/AAC.01132-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL Molecular Graphics System. San Carlos, CA: 2002. [Google Scholar]
- De Luca F, Benvenuti M, Carboni F, Pozzi C, Rossolini GM, Mangani S, Docquier JD. Evolution of carbapenem-hydrolyzing activity in noncarbapenemase class D β-lactamase OXA-10 by rational protein design. Proc Natl Acad Sci. 2011;108:18424–18429. doi: 10.1073/pnas.1110530108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Docquier JD, Benvenuti M, Calderone V, Giuliani F, Kapetis D, De Luca F, Rossolini GM, Mangani S. Crystal structure of the narrow-spectrum OXA-46 class D beta-lactamase: relationship between active-site lysine carbamylation and inhibition by polycarboxylates. Antimicrob Agents Chemother. 2010;54:2167–2174. doi: 10.1128/AAC.01517-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Docquier JD, Calderone V, De Luca F, Benvenuti M, Giuliani F, Bellucci L, Tafi A, Nordmann P, Botta M, Rossolini GM, Mangani S. Crystal structure of the OXA-48 β-lactamase reveals mechanistic diversity among class D carbapenemases. Chem Biol. 2009;16:540–547. doi: 10.1016/j.chembiol.2009.04.010. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Golemi D, Maveyraud L, Vakulenko S, Samama JP, Mobashery S. Critical involvement of a carbamylated lysine in catalytic function of class D β-lactamases. Proc Natl Acad Sci. 2001;98:14280–14285. doi: 10.1073/pnas.241442898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golemi D, Maveyraud L, Vakulenko S, Tranier S, Ishiwata A, Kotra LP, Samama JP, Mobashery S. The first structural and mechanistic insights for class D β-lactamases: Evidence for a novel catalytic process for turnover of β-lactam antibiotics. J Am Chem Soc. 2000;122:6132–6133. [Google Scholar]
- Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph Model. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Crystallogr. 1993;26:795–800. [Google Scholar]
- Kallen AJ, Hidron AI, Patel J, Srinivasan A. Multidrug resistance among gram-negative pathogens that caused healthcare-associated infections reported to the National Healthcare Safety Network, 2006–2008. Infect Control Hosp Epidemiol. 2010;31:528–531. doi: 10.1086/652152. [DOI] [PubMed] [Google Scholar]
- Livermore DM, Hill RL, Thomson H, Charlett A, Turton JF, Pike R, Patel BC, Manuel R, Gillespie S, Balakrishnan I, Barrett SP, Cumberland N, Twagira M. Antimicrobial treatment and clinical outcome for infections with carbapenem- and multiply-resistant Acinetobacter baumannii around London. Int J Antimicrob Agents. 2010;35:19–24. doi: 10.1016/j.ijantimicag.2009.09.014. [DOI] [PubMed] [Google Scholar]
- Massova I, Mobashery S. Kinship and diversification of bacterial penicillin-binding proteins and β-lactamases. Antimicrob Agents Chemother. 1998;42:1–17. doi: 10.1128/aac.42.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maveyraud L, Mourey L, Kotra LP, Pedelacq JD, Guillet V, Mobashery S, Samama JP. Structural basis for clinical longevity of carbapenem antibiotics in the face of challenge by the common class A β-lactamases from antibiotic-resistant bacteria. J Am Chem Soc. 1998;120:9748–9752. [Google Scholar]
- Mugnier PD, Poirel L, Naas T, Nordmann P. Worldwide dissemination of the blaOXA-23 carbapenemase gene of Acinetobacter baumannii. Emerg Infect Dis. 2010;16:35–40. doi: 10.3201/eid1601.090852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Lebedev A, Wilson KS, Dodson EJ. Efficient anisotropic refinement of macromolecular structures using FFT. Acta Crystallogr. 1999;D55:247–255. doi: 10.1107/S090744499801405X. [DOI] [PubMed] [Google Scholar]
- Paetzel M, Danel F, de Castro L, Mosimann SC, Page MGP, Strynadka NCJ. Crystal structure of the class D β-lactamase OXA-10. Nature Struct Biol. 2000;7:918–924. doi: 10.1038/79688. [DOI] [PubMed] [Google Scholar]
- Paton R, Miles RS, Hood J, Amyes SGB. ARI-I: β-lactamase-mediated imipenem resistance in Acinetobacter baumannii. Int J Antimicrob Agents. 1993;2:81–88. doi: 10.1016/0924-8579(93)90045-7. [DOI] [PubMed] [Google Scholar]
- Pernot L, Frénois F, Rybkine T, L’Hermite G, Petrella S, Delettré J, Jarlier V, Collatz E, Sougakoff W. Crystal structures of the class D beta-lactamase OXA-13 in the native form and in complex with meropenem. J Mol Biol. 2001;310:859–874. doi: 10.1006/jmbi.2001.4805. [DOI] [PubMed] [Google Scholar]
- Poirel L, Marque S, Heritier C, Segonds C, Chabanon G, Nordmann P. OXA-58, a novel class D β-lactamase involved in resistance to carbapenems in Acinetobacter baumannii. Antimicrob Agents Chemother. 2005;49:202–208. doi: 10.1128/AAC.49.1.202-208.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirel L, Naas T, Nordmann P. Diversity, epidemiology, and genetics of class D β-lactamases. Antimicrob Agents Chemother. 2010;54:24–38. doi: 10.1128/AAC.01512-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queenan AM, Bush K. Carbapenemases: The versatile β-lactamases. Clin Microbiol Rev. 2007;20:440–458. doi: 10.1128/CMR.00001-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal VD, Maki DG, Jamulitrat S, Medeiros EA, Todi SK, Gomez DY, Leblebicioglu H, Abu Khader I, Miranda Novales MG, Berba R, Ramirez Wong FM, Barkat A, Pino OR, Duenas L, Mitrev Z, Bijie H, Gurskis V, Kanj SS, Mapp T, Hidalgo RF, Ben Jaballah N, Raka L, Gikas A, Ahmed A, Thu le TA, Guzman Siritt ME. International Nosocomial Infection Control Consortium (INICC) report, data summary for 2003–2008, issued June 2009. Am J Infect Control. 2010;38:95–104. doi: 10.1016/j.ajic.2009.12.004. [DOI] [PubMed] [Google Scholar]
- Santillana E, Beceiro A, Bou G, Romero A. Crystal structure of the carbapenemase OXA-24 reveals insights into the mechanism of carbapenem hydrolysis. Proc Natl Acad Sci. 2007;104:5354–5359. doi: 10.1073/pnas.0607557104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider KD, Bethel CR, Distler AM, Hujer AM, Bonomo RA, Leonard DA. Mutation of the active site carboxy-lysine (K70) of OXA-1 beta-lactamase results in a deacylation-deficient enzyme. Biochemistry. 2009;48:6136–6145. doi: 10.1021/bi900448u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider KD, Karpen ME, Bonomo RA, Leonard DA, Powers RA. The 1.4 Å crystal structure of the class D β-lactamase OXA-1 complexed with doripenem. Biochemistry. 2009;48:11840–11847. doi: 10.1021/bi901690r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider KD, Ortega CJ, Renck NA, Bonomo RA, Powers RA, Leonard DA. Structures of the class D carbapenase OXA-24 from Acinetobacter baumannii in complex with doripenem. J Mol Biol. 2011;406:583–594. doi: 10.1016/j.jmb.2010.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun T, Nukaga M, Mayama K, Braswell EH, Knox JR. Comparison of β-lactamases of classes A and D: 1.5-Å crystallographic structure of the class D OXA-1 oxacillinase. Protein Sci. 2003;12:82–91. doi: 10.1110/ps.0224303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila J, Martí S, Sánchez-Céspedes J. Porins, efflux pumps and multidrug resistance in Acinetobacter baumannii. J Antimicrob Chemother. 2007;59:1210–1215. doi: 10.1093/jac/dkl509. [DOI] [PubMed] [Google Scholar]
- Walther-Rasmussen J, Hoiby N. OXA-type carbapenemases. J Antimicrob Chemother. 2006;57:373–383. doi: 10.1093/jac/dki482. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.