

Abstract
Protein kinase Cι (PKCι) is an oncogene in lung and ovarian cancers. PKCι is an attractive therapeutic target for treatment of lung cancer, particularly those whose tumors express elevated PKCι. However, it is unknown whether PKCι is a viable therapeutic target in the ovary, and virtually nothing is known about the mechanism by which PKCι drives ovarian tumorigenesis. Here, we demonstrate that PKCι maintains a tumor-initiating cell (TIC) phenotype that drives ovarian tumorigenesis. A highly tumorigenic population of cells from human ovarian cancer cell lines exhibit properties of cancer stem-like TICs including self-renewal, clonal expansion, expression of stem-related genes, enhanced transformed growth in vitro, and aggressive tumor-initiating activity in vivo. Genetic disruption of PKCι inhibits the proliferation, clonal expansion, anchorage-independent growth and enhanced tumorigenic properties of ovarian TICs. Biochemical analysis demonstrates that PKCι acts through its oncogenic partner Ect2 to activate a Mek-Erk signaling axis that drives the ovarian TIC phenotype. Genomic analysis reveals that PRKCI and ECT2 are coordinately amplified and overexpressed in the majority of primary ovarian serous tumors, and these tumors exhibit evidence of an active PKCι-Ect2 signaling axis in vivo. Finally, we demonstrate that auranofin is a potent and selective inhibitor of oncogenic PKCι signaling that inhibits the tumorigenic properties of ovarian TIC cells in vitro and in vivo. Our data demonstrate that PKCι is required for a TIC phenotype in ovarian cancer, and that auranofin is an attractive therapeutic strategy to target deadly ovarian TICs in ovarian cancer patients.
Introduction
Ovarian cancer is the fifth leading cause of cancer-related death in women in the United States owing largely to late diagnosis and a high relapse rate after initial response to conventional therapy. As a result of this clinical course, the overall 5-year survival rate of ovarian cancer is only 15%–30% (1). Clinical relapse is thought to be due to the survival and regrowth of highly tumorigenic ovarian cancer stem-like or tumor-initiating cells (TICs) after chemotherapy.
TICs are a relatively rare subpopulation of cells within the bulk tumor mass that has the ability to self-renew and give rise to the heterogeneous cancer cell lineages that comprise the tumor. The characteristic properties of TICs include increased lifespan, self-renewal, expression of stem-related genes, clonal expansion, enhanced transformed growth, resistance to chemotherapy and the ability to efficiently initiate tumors in immune-compromised mice. These properties allow TICs to survive conventional platinum- and/or taxane-based therapy and cause relapse. However, the cellular mechanisms underlying the oncogenic behavior associated with ovarian TICs are still poorly understood.
Protein kinase Cι (PKCι), a member of the protein kinase C family, was found to be a target for frequent tumor-specific gene amplification in multiple human cancer types, including cancers of the lung, stomach, head and neck, colon, breast, and ovary (2–9). In these tumor types, PKCι gene amplification drives PKCι expression which in turn is associated with poor prognosis. PKCι confers resistance to chemotherapy-induced apoptosis of human leukemia cells, and inhibition of PKCι expression or activity sensitizes chronic myelogenous leukemia cells to chemotherapeutic agent-induced apoptosis (10). Furthermore, over-expression of kinase-deficient PKCι or knockdown of PKCι expression blocked anchorage-independent growth and invasion of human non-small cell lung cancer (NSCLC) cells and human pancreatic ductal adenocarcinoma (PDAC) cells (2, 11, 12), showing that PKCι is also required for maintenance of the transformed phenotype of cancer cells. In addition, expression of constitutively-active PKCι in the intestinal epithelium led to an increase in carcinogen-induced colon tumors, whereas expression of kinase-deficient PKCι had the opposite effect (4), demonstrating that PKCι promotes intestinal tumorigenesis in vivo.
In the lung, PKCι forms an oncogenic complex with its binding partners Par6 and the Rho family guanine nucleotide exchange factor, Ect2 (13, 14). The formation of this complex activates a downstream signaling axis consisting of Rac1, Pak, Mek, and Erk, which drives oncogenic growth and tumor formation in NSCLC and PDAC cells (2, 13). Based on a genomic analysis of NSCLC, matrix metalloproteinase 10 (MMP10; stromelysin 2) was identified as a transcriptional target and downstream effector of the oncogenic PKCι signaling axis (11). Interestingly, MMP10 was found to promote the expansion of Kras-mediated bronchioalveolar stem cells (BASCs) and to be required for BASC maintenance, tumor initiation, and metastatic potential in a murine model of Kras-mediated lung adenocarcinoma (15, 16). Taken together, these results indicate a strong functional link between PKCι and oncogenic TIC behaviors. Based on these results, we hypothesized that PKCι may drive cancer formation through the maintenance of a TIC phenotype in human tumors.
We recently identified the anti-rheumatic gold compounds aurothiomalate and aurothioglucose as small molecule inhibitors of oncogenic PKCι that can inhibit oncogenic PKCι signaling in lung cancer cells (14, 17). These compounds function by interacting with a critical cysteine residue within the PKCι PB1 domain thereby disrupting binding of Par6 to PKCι, leading to inhibition of oncogenic PKCι signaling (14). Our preclinical studies in the lung indicate that tumor PKCι expression is a critical determinant of response to treatment with these gold-containing compounds (17). In the present study, we demonstrate for the first time that PKCι plays a pivotal role in promoting the oncogenic behavior of ovarian TICs including clonal expansion, enhanced transformed growth and tumor initiation in vivo. We also demonstrate for the first time that auranofin (ANF), a gold compound in the same chemical class as aurothiomalate and aurothioglucose that is still available for clinical use, is an effective inhibitor of of PKCι signaling and the ovarian TIC phenotype. Our data provide a compelling rationale for an ongoing clinical trial to assess dosing and initial efficacy of ANF for treatment of serous ovarian cancer patients in a maintenance setting.
Materials and Methods
Cell lines and antibodies
Human SKOV3 and mouse ID8 cells were provided by the tissue core resource of the Mayo Clinic Ovarian SPORE (directed by Dr. K. Knutson, Mayo Clinic, Rochester, MN). SKOV3 and ID8 cells were cultured in DMEM + 4.5 g/L D-glucose + L-glutamine (Life technologies, Grand Island, NY) supplemented with sodium pyruvate, non-essential amino acids (NEAA, Life technologies), and 10% fetal bovine serum (FBS, Life technologies). ES2 cells were obtained from the American Type Culture Collection, Manassas, VA and cultured in McCoy’s 5A + L-glutamine (Life technologies) supplemented with NEAA (Life technologies), 15 mM HEPES, and 10% FBS. PKCι antibody was purchased from BD Biosciences (San Jose, CA). phospho-Mek, Mek, phospho-Erk, Erk, and GAPDH antibodies were purchased from Cell Signaling Technology (Danvers, MA). FLAG antibody was purchased from Sigma-Aldrich (St. Louis, MO). Phospho-T328-Ect2 (pECT2) antibody was produced and characterized as described previously (16). HRP-conjugated secondary antibodies were purchased from KPL (Gaithersburg, MD).
Enrichment and culture of ovarian cancer oncospheres
Oncospheres were enriched from SKOV3, ES2 and ID8 ovarian cancer cell lines by culturing adherent cells (50,000 cells/ml) in ultra-low attachment culture flasks (Sigma-Aldrich, St. Louis, MO) with DMEM-F12 medium containing 50 μg insulin (Sigma-Aldrich), 0.4% albumin bovine fraction V (Sigma-Aldrich), N-2 plus media supplement (Life technologies), B-27 supplement (Life technologies), 20 μg/ml EGF (PeproTech, Rocky Hill, NJ), and 20 μg/ml FGF (PeproTech) for 4 weeks. Oncospheres were passaged by trypsinization and resuspension of single cells in non-adherent stem cell culture medium. For re-differentiation, oncospheres were collected and re-plated in adherent cell culture conditions (10,000 cells/ml) for 2 weeks using growth media containing 10% FBS as described above for each cell line.
Lentiviral RNAi constructs, cell transduction and immunoblot analysis
A validated and characterized lentiviral construct containing RNAi sequence against the 3′ untranslated region of human PKCι was obtained from the Sigma-Aldrich Mission shRNA library (St Louis, MO) and packaged into lentiviruses as described previously (11). A lentiviral vector containing a short hairpin RNAi which recognizes no human genes was used as a non-target control (NT-RNAi). RNAi infections were performed as described previously (11). At 48 hours post infection, the infected cells were treated with 5 μg/ml of puromycin for selection of a stably transduced cell population.
Cellular proliferation, clonal expansion and anchorage-independent growth assays
Cellular proliferation was assessed using the MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) reduction assay. In short, ovarian cancer cells were trypsinized and plated in 96-well cell culture plates (5,000 cells/well). Cells were collected and assayed for proliferation every 24 hours according to the standard protocol provided by the manufacturer (Promega, Madison, WI). Clonal expansion of oncosphere cells was assessed by isolating single cells in 96-well ultra-low attachment plates by serial dilution (one cell/well). The expansion of single cells was monitored by phase contrast microscopy over a 15 day period. Oncospheres containing more than 10 cells at day 15 were considered to have undergone expansion. Anchorage-independent growth of ovarian cancer cells grown in adherent culture (referred to as parental cells), and of oncosphere cells, was assayed as described previously (12). Cell colonies were fixed, stained with Giemsa (EMD Chemicals, Gibbstown, NJ) and quantified using ImagePro Plus 7.0 (Media Cybernetics, Inc.). All experiments were independently repeated at least three times.
Tumorigenicity in mice
The ability of ovarian cancer cells to grow as orthotopic tumors was assessed in immune-deficient nude mice (Harlan-Sprague-Dawley, Indianapolis, IN). Briefly, 5–7-week-old female mice were anesthetized with Ketamine/Xylazine, a small midline abdominal incision was made, the left ovary was externalized, and 20 μl containing 1,000 viable ovarian cancer cells previously transfected with an expression plasmid containing firefly luciferase suspended in sterile PBS were orthotopically injected into the left ovary under a dissecting microscope. SKOV3 or ES2 cells were injected into immune-deficient nude mice, whereas ID8 cells, an ovarian cancer cell line derived from a C57 mouse, were injected into syngeneic C57 BL/6J mice. Tumor growth was monitored using the IVIS imaging system (Caliper Life Sciences, Hopkinton, MA). Final tumor dimensions were determined with calipers as described previously (12). Tumor volume was calculated using the formula, (Width)2 × Length/2. Tissues were formalin-fixed, sectioned and stained with hematoxylin and eosin for histological analysis. Samples were analyzed by immunohistochemistry using pEct2 antibody as described previously (16). For the experiments involving ANF treatment, ES2 oncosphere cells were injected into the left ovaries of mice as described above. Once tumors were detectable by IVIS (day 11), mice were randomly assigned to receive either auranofin (ANF; 12 mg/kg body weight, six days/week) dissolved in 100% ethanol and diluted in saline (0.9%) for injection, or diluent. Tumor growth was monitored by IVIS imaging every 3–4 days as described above. Mice were harvested at day 27 at which time diluent-treated mice began exhibiting signs of morbidity. Tumor tissues were harvested and processed for analysis as described above.
Results
Ovarian cancer oncospheres exhibit a tumor-initiating cell (TIC) phenotype
Given the potential importance of tumor initiating cells in ovarian tumorigenesis, we enriched for cells exhibiting a highly tumorigenic TIC phenotype by culturing ovarian cancer cells in defined serum-free stem cell medium under low-attachment conditions. Two human ovarian cancer cell lines (SKOV3 and ES2) were used for these experiments. Both cell lines readily formed oncospheres when placed in stem cell culture conditions (Fig. 1A). To investigate whether these cell cultures are enriched in stem-like cells, we assessed expression of a panel of stem cell markers in parental, oncosphere, and oncosphere cultures that were placed back into adherent culture to allow them to re-differentiate (Fig. 1B). ES2 oncospheres expressed elevated levels of the stem-related genes ALDH1, NANOG, CD44, ABCG2, CD133 (Prom1) and STELLAR (18), whereas SKOV3 oncospheres expressed elevated ALDH1 and CD44 but not the other stem-related genes. In both cell lines, the elevated expression of these stem-related genes was lost when oncosphere cells were returned to adherent culture (Fig. 1B). Interestingly, both ES2 and SKOV3 oncospheres exhibited elevated expression of MMP10, a gene that we previously demonstrated was a transcriptional target of PKCι signaling in lung cancer cells (11), and which we demonstrated is important for Kras-mediated lung tumor initiation in mice (19).
Figure 1. Ovarian cancer oncospheres exhibit a tumor-initiating cell (TIC) phenotype.

A) Phase contrast photographs of ovarian cancer cells (ES2 and SKOV3) grown in adherent culture (upper panel) and in non-adherent culture as oncospheres (lower panel). B) Oncospheres from SKOV3 and ES2 cells express elevated levels of subsets of stem cell markers including ALDH1, NANOG, ABCG2, CD44, MMP10, PROM1 and STELLAR. Data are expressed as mean fold-change from parental cells +/− SEM. n=3; *p<0.05. C) Ovarian oncospheres exhibit enhanced anchorage-independent growth. Re-diff.: re-differentiated oncospheres. Data are expressed as mean fold-change from parental cells +/− SEM. n=5; *p<0.001. D) Oncospheres from ES2 cells exhibit enhanced tumor-initiating potential. Photographs of representative tumors were taken 27 days after orthotopic injection of 1,000 parental or oncosphere cells.
To investigate whether oncosphere cultures acquire aggressive oncogenic behavior associated with a stem-like phenotype, we performed soft agar assays to assess anchorage-independent growth of parental, oncosphere and re-differentiated oncosphere cells (Fig. 1C). We found that oncospheres from both cell lines exhibited increased anchorage-independent growth when compared to control parental cultures from which they were derived (Fig. 1C). Interestingly, when oncospheres were placed back into adherent culture in the presence of serum-containing media, the cells lose their enhanced soft agar colony formation and resemble parental control cells (Fig. 1C). Finally, we assessed the ability of parental and oncosphere cells to initiate tumors in vivo. For this purpose, we developed an orthotopic ovarian tumor model in which ovarian tumor cells are injected orthotopically into the capsule of the ovary of immune-deficient nude mice. Initial experiments determined that a minimum of 100,000 parental SKOV3 or ES2 cells were required to establish tumors with 100% tumor take (data not shown). In contrast, injection of as few as 1,000 ES2 or SKOV3 oncosphere cells led to 100% tumor take and development of large tumors within the injected ovary (Fig. 1D) and injection of 100 ES2 or SKOV3 oncosphere cells yield a tumor take of 40% (2/5) and 20% (1/5) respectively; therefore, 1,000 cells were used as the inoculum size for subsequent studies. In contrast to oncosphere cells, 1,000 parental ES2 cells failed to form tumors, demonstrating the enhanced tumorigenic, tumor-initiating properties of ES2 oncosphere cells (Fig. 1D). Given the tumor-initiating properties of oncospheres, we will henceforth refer to these cells as tumor-initiating cells (TICs).
Ovarian TICs require PKCι for clonal expansion and enhanced anchorage-independent growth
Our finding that expression of MMP10, a gene that we previously identified as a transcriptional target and downstream effector of oncogenic PKCι signaling in the lung (11), was elevated suggested that PKCι signaling is activated in, and important for the maintenance of, TIC cultures. Consistent with this hypothesis, immunoblot analysis demonstrated that PKCι expression is higher in ES2 oncosphere cells when compared to parental cells, whereas that of the related PKCζ was not (Suppl. Fig. 1A). To directly assess the role of PKCι in ovarian TIC proliferation and transformed growth, we performed PKCι knockdown (PKCι KD) utilizing a previously characterized and validated lentiviral-based shRNA construct targeting PKCι (11). The efficiency of PKCι knockdown was confirmed by Q-PCR and immunoblot analysis (Fig. 2A). We next examined whether PKCι is important for clonal expansion of the ovarian TICs. Single TICs (NT or PKCι KD) isolated from oncosphere cultures were plated as single cells in non-adherent culture in individual wells of 96 well plates and their ability to clonally expand into oncospheres was followed over a 15-day period (Fig. 2B). While NT TICs efficiently expanded into large oncospheres, the majority of PKCι KD TICs from both ES2 and SKOV3 failed to expand with the majority of clones remaining as single cells (Fig. 2B). Quantitative analysis of individual clones demonstrated that whereas NT TICs exhibits a very high clonal expansion efficiency (~93% for SKOV3 and ~67% for ES2), PKCι KD TICs from both cell lines exhibited a significant reduction in clonal expansion efficiency (~13% for SKOV3 and ~7% for ES2) (Fig. 2C). Thus, PKCι plays a critical role in clonal expansion of ovarian TICs in vitro.
Figure 2. PKCι is required for clonal expansion and anchorage-independent growth of ovarian TICs in vitro.

A) Immunoblot and Q-PCR analysis demonstrating efficient knockdown of PKCι in ES2 and SKOV3 ovarian cancer cell lines. Cells were transduced with recombinant lentivirus containing shRNA targeting PKCι (PKCι KD) or non-target shRNA (NT) followed by selection in puromycin for 2 weeks. B) Knockdown of PKCι inhibits clonal expansion of ovarian TICs. Individual ES2 and SKOV3 TICs were photographed at day 0 and day 15 in non-adherent culture in 96 well plates. C) Statistical analysis of clonal expansion in NT and PKCι KD ES2 and SKOV3 TICs. PKCι KD significantly inhibits clonal expansion of ES2 and SKOV3 TICs. *p<0.001. D) PKCι KD inhibits anchorage-independent growth of ovarian TICs in soft agar. Both TICs (black bars) and parental cells (white bars) exhibit PKCι-dependent soft agar colony formation. Data are expressed as mean fold-change from NT parental cells +/− SEM. n=5; *p<0.05 compared to NT parental cells; **p<0.05 compared to NT TICs.
To determine the role of PKCι in the enhanced anchorage-independent growth exhibited by TIC cultures, we performed soft agar assays on NT and PKCι KD TICs (black bars) and parental cells (white bars) (Fig. 2D). PKCι KD significantly inhibits soft agar colony formation in both SKOV3 (left panel) and ES2 (right panel) TICs and parental cells when compared to NT KD cells. The specificity of the effect of PKCι KD was confirmed by expressing exogenous PKCι in PKCι KD cells, which signficiantly restored soft agar colony formation (Suppl. Fig. 1B).
Ovarian TICs exhibit PKCι-dependent tumor-initiating activity in vivo
To evaluate the effect of PKCι on the tumor initiating potential of TICs in vivo, we used the ovarian cancer orthotopic mouse model described in Materials and Methods. SKOV3 NT parental cells, NT TICs and PKCι KD TICs (1,000 viable cells/injection) expressing firefly luciferase were injected into the ovary of immune-deficient nude mice and tumor growth was monitored using IVIS imaging (Fig. 3A). At 60 days post-injection, imaging revealed large tumors in mice injected with NT oncosphere cells (left panel) but either very small or no detectable tumor formation in mice injected with either PKCι KD TICs (middle panel) or NT parental cells (right panel). The results of IVIS imaging over the 60 day period revealed that only mice injected with NT TICs formed large tumors whereas both PKCι KD TICs and NT parental cells failed to initiate and maintain tumors (Fig. 3B). Similar results were obtained when parental ES2 or ES2 TICs were injected (Fig. 3C). Thus, PKCι is required for the tumor-initiating potential of human ovarian cancer TICs in vivo.
Figure 3. PKCι is required for the tumor initiating activity of ovarian TICs in vivo.
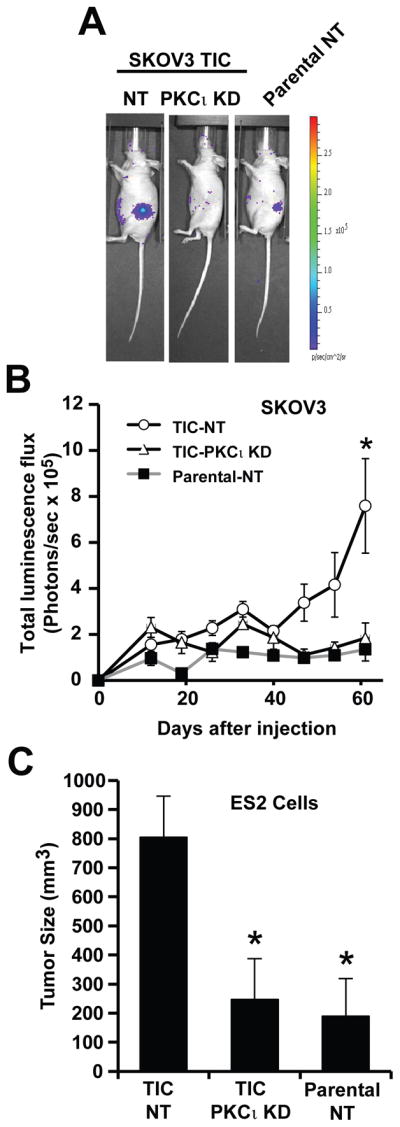
A. In vivo luminescence (IVIS) imaging of representative mice 60 days after injection orthotopically with 1,000 viable cells derived from NT parental SKOV3 cells, NT SKOV3 TICs or PKCι KD SKOV3 TICs. TICs exhibit enhanced tumor-initiating ability compared to parental cells. PKCι KD inhibits the enhanced tumor initiating properties of SKOV3 TICs. B) SKOV3 NT parental cell, NT TIC and PKCι KD TIC tumor growth was monitored by IVIS at the indicated time points after injection of 1,000 viable cells into recipient mice. Data are expressed as mean of total luminescence flux +/− SEM. n=4; * p<0.05 compared to parental NT (P-NT) or PKCι KD TICs. C) Orthotopic ES2 tumors were assessed for tumor size after injection of 1,000 ES2 NT parental, NT TICs and PKCι KD TICs as described in Materials and Methods. Data are expressed as mean tumor size (mm3) +/− SEM. n=6; *p<0.05 compared to NT TICs.
PKCι plays a critical role in the murine ovarian tumor-initiating cell phenotype
To determine whether PKCι also plays a role in the TIC phenotype of murine ovarian cancer, we assessed the role of PKCι in the behavior of TICs isolated from the murine ovarian tumor cell line ID8 (Fig. 4). ID8 was isolated from an ovarian carcinoma tumor in a C57BL/6J mouse. TIC cultures from ID8 cells were established by growing the cells in non-adherent conditions in defined serum-free stem cell medium as described in Materials and Methods. Similar to our results in human ovarian tumor cell lines, ID8 cells grew efficiently as oncospheres in non-adherent culture (Fig. 4A). ID8 oncospheres exhibited enhanced anchorage-independent growth that is lost upon re-differentiation of the oncosphere by returning them to adherent culture in serum-containing medium (Fig. 4B). Furthermore, efficient knock down of PKCι (Fig. 4C) led to a profound inhibition of clonal expansion (Fig. 4D and E), anchorage-independent growth (Fig. 4F) and tumor initiating activity of ID8 oncospheres in vivo (Fig. 4G and H). We conclude that PKCι is required for maintenance of the TIC phenotype in mouse ID8 cells, indicating that PKCι plays a general role in ovarian tumorigenesis.
Figure 4. PKCι is required for the tumor-initiating phenotype of murine ID8 ovarian cancer cells.

A) Phase contrast photographs of parental ID8 cells (upper panel) and oncospheres (lower panel). Scale bar = 100 μm. B) ID8 oncospheres exhibit enhanced anchorage-independent growth in soft agar. Re-diff: re-differentiated oncosphere cells. Data expressed as mean fold change +/− SEM compared to parental ID8 cells. n=5; *p<0.05. C) Immunoblot (upper panel) and Q-PCR analysis (lower panel) confirming efficient knockdown of PKCι in ID8 cells. Data expressed as mean % NT control +/− SEM. n=3; *p<0.05. D) Representive images of individual NT and PKCι KD ID8 TICs under clonal expansion conditions. E) Statistical analysis of clonal expansion of NT and PKCι KD ID8 TICs. PKCι KD significantly inhibits clonal expansion of ID8 TICs. *p<0.0001. F) PKCι KD significantly inhibits anchorage-independent growth of ID8 parental cells and TICs. Data expressed as mean fold change in colony numbers +/− SEM compared to parental NT cells. n=5; *p<0.05 compared to NT parental cells; **p<0.05 compared to their respective NT control cells. G) Representative IVIS images showing tumor growth of NT TIC, PKCι KD TIC and parental NT cells after orthotopic injection into syngeneic C57B6 mice. H) Growth of orthotopic ovarian ID8 tumors from TIC NT, TIC PKCι KD and parental NT cells. Data are expressed as mean total luminescence flux +/− SEM. n=6; *p<0.05 compared with parental NT and PKCι KD TICs.
The PKCι-Ect2 signaling axis is activated in ovarian TICs and primary ovarian tumors
We previously demonstrated that oncogenic PKCι signaling in the lung requires interaction of PKCι with its binding partner Par6, and that PKCι-Par6 binding recruits the Rho family GTPase GEF Ect2 to the complex (11, 13). PKCι directly phosphorylates Ect2 at T328 (16). PKCι-mediated Ect2 phosphorylation regulates the ability of Ect2 to activate Rac1 (16), which in turn activates a Mek-Erk signaling cascade that regulates the expression of MMP10 in a PKCι-dependent fashion (Fig. 5A) (11, 13). To assess whether this oncogenic PKCι signaling mechanism is operative in ovarian TICs, we assessed the effect of PKCι KD on the activity of key components of this signaling pathway (Fig. 5B). Immunoblot analysis of cellular extracts from NT and PKCι KD ES2 TICs demonstrated that PKCι KD had little or no effect on total Ect2 expression, but led to a significant loss of pEct2 in PKCι KD TICs when compared with NT TICs (Fig. 5B). PKCι KD also led to a commensurate decrease in both Mek and Erk phosphorylation levels (Fig. 5B) and to a decrease in MMP10 mRNA expression (Fig. 5C). To assess the functional role of the PKCι-Par6-Ect2-Mek-Erk-MMP10 signaling axis in TIC behavior, was assessed the effect of RNAi-mediated KD of Ect2 and MMP10, key effectors of this pathway downstream of PKCι, on TIC behavior (Suppl. Fig. 2). Ect2 KD in ES2 TICs led to a decrease in MMP10 expression, and both Ect2 and MMP10 KD led to a decrease in clonal expansion of ES2 TICs. Taken together, these data indicate that the oncogenic PKCι-Par6-Ect2-Mek-Erk-MMP10 signaling axis is active in ovarian TICs and is important for TIC behavior. Since the atypical PKC subfamily consists of two related isoforms, PKCι and PKCζ, we assessed whether PKCζ has a similar effect on ovarian TIC behavior and signaling. PKCζ KD in ES2 oncosphere cells, using our previously characterized shRNA lentiviral constructs (11), had little or no effect on clonal expansion or MMP10 expression, indicating that PKCζ does not play a major role in ovarian oncosphere behavior or PKCι signaling (Suppl. Fig. 3).
Figure 5. PKCι activates a PKCι-Par6-Ect2-Mek-Erk signaling cascade in ovarian TICs.

A) Schematic of the oncogenic PKCι signaling cascade identified in non-small cell lung cancer cells. B) PKCι KD in ES2 TICs causes a decrease in phosphorylation of T328 on Ect2, a previously characterized PKCι phosphorylation site on Ect2, and commensurate decreases in pMek and pErk levels. C) PKCι KD inhibits expression of MMP10, a transcriptional target of PKCι. Data are expressed as mean MMP10 RNA abundance normalized to NT control. n=3,*p<0.05 compared to NT control. D) PRKCI and ECT2 are co-amplified in primary ovarian serous carcinomas. Oncoprint readout of PRKCI and ECT2 gene copy number gains in primary ovarian serous carcinomas from The Cancer Gene Atlas (TCGA) dataset. Data reveal that PRKCI and ECT2 are virtually always co-amplified in ~80% ovarian serous carcinoma tumors. E) Analysis of PRKCI, ECT2 and MMP10 mRNA expression in primary ovarian serous tumors from the TCGA dataset reveal a strong positive correlation between expression of these three genes.
To assess whether the PKCι signaling pathway characterized above is relevant to primary ovarian tumors, we interrogated gene expression in a dataset consisting of 489 ovarian serous carcinoma cases within The Cancer Genome Atlas (TCGA). Analysis revealed that PRKCI and ECT2 exhibit coordinate gene copy number gains in ~80% of ovarian serous tumors as part of the chromosome 3q26 amplicon (Fig. 5D). Furthermore, gene expression analysis demonstrated a statistically significant and positive correlation between PRKCI, ECT2 and MMP10 mRNA levels in ovarian serous tumors (Fig. 5E). Taken together, these data demonstrate that PRKCI and ECT2 are genetically and biochemically linked in primary ovarian tumors, and suggest that in tumors harboring PRKCI and ECT2 copy number gains, the PKCι-Par6-Ect2–Mek-Erk-MMP10 signaling axis is activated.
The PKCι inhibitor auranofin potently inhibits PKCι signaling and ovarian TIC behavior
We recently identified the anti-rheumatoid gold compounds aurothiomalate and aurothioglucose as potent and selective inhibitors of oncogenic PKCι signaling that act by inhibiting the interacting between PKCι and Par6, thereby disrupting the PKCι-Par6-Ect2 signaling complex (14, 20). Unfortunately, therse compounds are no longer available for clincialuse. Therefore, we assessed the efficacy of auranofin (ANF), a gold compound in the same chemical class, to inhibit PKCι signaling. Given the critical role of PKCι signaling in ovarian TIC behavior, we assessed the effects of ANF on the oncogenic properties of ovarian TICs. Consistent with a role for the PKCι-Par6 complex in oncogenic PKCι signaling, we observed a dose-dependent inhibition of TIC proliferation in the presence of ANF with an apparent IC50 of ~200 nM (Fig. 6A). To assess whether the inhibitory effects of ANF on TIC growth is associated with inhibition of PKCι signaling, we assessed the effect of ANF on PKCι pathway intermediates (Fig. 6B and C). Expression of FLAG-Par6 in ovarian TICs allowed us to directly assess binding of endogenous PKCι to Par6 in the presence and absence of ANF. ANF treatment led to a loss of Par6-bound PKCι as determined by FLAG immunoprecipitation and PKCι immunoblot analysis (Fig. 6B, upper panels). Immunoblot analysis further revealed that ANF treatment inhibited pEct2, pMek and pErk levels (Fig. 6B, lower panels), and MMP10 expression (Fig. 6C) when compared to DMSO-treated control cells. Furthrmore, transient expression of an aurothiomalate-resistant PKCι mutant, PKCι C69I (14), in ES2 TICs can inhibit ANF-induced loss of MMP10 (Suppl. Fig. 4). Taken together, these data demonstrate that ANF inhibits the oncogenic PKCι-Par6-Ect2-Mek-Erk-MMP10 signaling axis in ovarian TICs.
Figure 6. Auranofin (ANF) inhibits PKCι signaling and the ovarian TIC phenotype in vitro.

A) The PKCι inhibitor auranofin (ANF) exhibits dose-dependent inhibition of ovarian TIC proliferation. ES2 TICs were grown in non-adherent oncosphere culture in the presence of the indicated concentration of ANF. MTT reduction was assessed after 5 days of ANF treatment. Data are expressed as MTT reduction relative to DMSO-treated control cells +/− SEM, n=3. Data are representative of three independent experiments. B) ANF inhibits PKCι signaling in ovarian TICs. ES2 TICs transfected with FLAG-Par6 plasmid were treated for 24 hours with DMSO or ANF (1 μM) and cellular lysates were subjected to FLAG immunoprcipitation followed by immunoblot analysis for Par6-bound PKCι and FLAG Par6 (upper panels). ANF-treated TICs exhibit a decrease in PKCι-Par6 binding. Total lysates from ES2 TICs revealed a decrease in pEct2, pMek and pErk in ANF-treated TICs when compared to DMSO-treated control TICs. C) Treatment of ES2 TICs with 1 μM ANF led to a decrease in MMP10 mRNA abundance as determined by Q-PCR analysis. Data are expressed as mean MMP10 mRNA abundance normalized to DMSO control cells. n=3. *p<0.01 compared to DMSO control cells. D) ANF inhibits clonal expansion of ES2 TICs. E) Representative photomicrographs of TIC oncospheres from single DMSO and ANF treated ES2 TICs after 15 days in oncosphere culture. F) ANF inhibits oncosphere growth. Data are expressed as mean oncosphere size (μm3) +/− SEM; n=19, 19, 7 for DMSO, 1 μM ANF, 3 μM ANF group, respectively; *p<0.01 compared with DMSO. G) ANF treatment for 24 hours inhibits expression of stem cell makers in ES2 TICs. Q-PCR results are expressed as mean fold change in mRNA abundance +/− SEM compared with DMSO treated cells. n=3; *p<0.05. H) ANF inhibits anchorage-independent growth of ES2 TICs in soft agar.
Given the ability of ANF to inhibit PKCι signaling in ovarian TICs, we next assessed the effect of ANF on the tumorigenic behavior of TICs. Treatment of ES2 TICs with ANF led to significant inhibition of clonal expansion of TICs (Fig. 6D). This inhibitory effect manifested itself in both a dose-dependent decrease in the number of individual TICs that clonally expand (Fig. 6E) and in the size of oncospheres in the presence of ANF (Fig. 6F). ANF treatment also led to a significant decrease in expression of stem cell markers (Fig. 6G) and anchorage-independent growth of TICs in soft agar (Fig. 6H). Taken together, these data demonstrate that pharmacologic inhibition of PKCι with ANF blocks the TIC phenotype in vitro.
To assess the ability of ANF to inhibit ovarian TIC-mediated tumor formation in vivo, we established orthotopic ES2 TIC tumors in immune-deficient nude mice and treated tumor-bearing mice with ANF as described in Materials and Methods. As expected, injection of 1,000 ES2 TICs led to efficient establishment of tumors that were detectable by bioluminescence imaging by day 11, at which time tumor-bearing mice were randomly placed into one of two treatment groups that received either ANF or diluent. Mice in the ANF treatment group exhibited significant inhibition of tumor growth when compared to diluent-treated animals (Fig. 7A). By day 27 after tumor injection, diluent-treated mice exhibited signs of morbidity that necessitated termination of the experiment, whereas ANF-treated mice showed no signs of morbidity. Analysis of tumor tissue from diluent and ANF-treated mice revealed a decrease in mitotic index of tumor cells from ANF-treated mice when compared to diluent control mice (Fig. 7B). Biochemical analysis revealed that ANF treatment led to a significant decrease in tumor pERK levels (Fig. 7C) and pEct2 staining (Fig. 7D and E) consistent with inhibition of the PKCι-Ect2-Mek-Erk signaling axis identified in TICs in vitro. Thus, ANF is a potent PKCι inhibitor that blocks ovarian TIC behavior in vitro and tumor formation in vivo.
Figure 7. Auranofin (ANF) inhibits PKCι signaling and ovarian tumor growth in vivo.

Orthotopic ES2 TIC tumors were established in immune-deficient nude mice by orthotopic injection of 1,000 ES2 TICs into the capsule of the ovary as described in Materials and Methods. At day 11, tumor-bearing mice were randomly assigned to receive either ANF (12 mg/kg/day/six days a week) or the same volume and frequency of vehicle solution (NaCl, 0.9%) for the duration of the experiment. A) Quantitative analysis of tumor growth by IVIS bioluminescence. The results are expressed as mean fold change of luminescence in each treated mouse compared to day 11 +/−SEM. n=5; *p<0.05. B) Sections from tumors were analyzed for mitotic index as described in Materials and Methods. Tumors from ANF-treated mice exhibited a decrease in mitotic index compared to diluent-treated control mice. n=5; *p<0.05. C) Immunoblot analysis (upper panel) of tumor lysates from diluent and ANF treated mice revealed a decrease in pERK levels in ANF-treated tumors. Quantitative analysis of pErk blots demonstrates a significant decrease in pErk levels in ANF-treated mouse tumors when compared to diluent-treated control mice. n=3. *p<0.05. D) Immunohistochemical staining of representative diluent- and ANF-treated tumors for pEct2. ANF treatment led to a decrease in pEct2 staining when compared to diluent control tumors. Staining was abolished by pre-incubation with Ect2 phospho-peptide antigen as described previously (16) indicating the specificity of the staining for pECt2 antigen. E) Quantitative analysis of pEct2 immunohistochemical staining reveals a significant decrease in pEct2 staining in ANF-treated tumors compared to diluent control tumors. n=5; *p<0.05.
Discussion
Ovarian cancer is the fifth leading cause of cancer death in women. A major contributing factor to the poor prognosis of ovarian cancer patients is the high relapse rate of tumors after initial response to platinum-based chemotherapy with resistant disease. Recent studies have indicated that relapse and acquisition of chemoresistance is likely due to the survival and subsequent outgrowth of a relatively rare population of highly malignant cells within the primary tumor termed cancer stem cells (CSCs) or tumor-initiating cells (TICs). Ovarian TICs are characterized by elevated expression of stem cell marker genes and the ability to grow as oncospheres in non-adherent culture in stem cell media. TICs also exhibit an increased ability to clonally expand, grow as colonies in soft agar, and initiate tumors in vivo (21). In addition, ovarian TICs possess resistance to chemotherapeutic agents such as cis-platin providing further support for their involvement in relapse (22, 23). Based on these considerations, TICs have been hypothesized to be a critical tumor cell population that must be therapeutically targeted in order to elicit lasting tumor responses and improved patient survival.
In the present study, we cultured adherent SKOV3 and ES2 human ovarian cancer cells, and ID8 murine ovarian cancer cells, in defined serum-free stem cell media under low attachment conditions to select for stem-like cell populations. Each of the cell lines were able to readily form oncospheres enriched in cells exhibiting characteristic features of TICs, including clonal expansion, expression of stem marker genes, enhanced anchorage-independent growth and efficient tumor-initiating activity when injected orthotopically into the ovary of recipient mice. An interesting aspect of our findings is that SKOV3 and ES2 TICs, while both expressing elevated stem-associated markers, expressed a distinct pattern of such markers, suggesting that these cell lines are maintained by distinct stem-like cell populations. These differences in stem cell marker gene expression patterns among oncosphere populations suggest the heterogeneous nature of ovarian cancer cells, and underscore the difficulty of identifying TICs based solely on expression of specific stem cell marker genes. Despite these differences, we did identify several genes whose expression was elevated in TICs from both cell lines including ALDH1, CD44 and MMP10. The finding that MMP10, which encodes the matrix metalloproteinase 10, is elevated in ovarian TICs, including in murine ID8 TICs (data not shown), is significant since we had previously identified MMP10 as a critical transcriptional target of PKCι in the lung (11). This observation led us to assess the role of PKCι in maintenance of the ovarian TIC phenotype.
We previously demonstrated that PKCι is required for tumor formation in the LSL-Kras model of lung adenocarcinoma (24). In this mouse model, the initiating step in tumor formation appears to be the activation of oncogenic Kras in, and the subsequent transformation and expansion of, BASCs. Thus, BASCs are thought to serve as TICs in this mouse model; our findings demonstrated that PKCι is essential for Kras-mediated transformation of BASCs, and suggested that PKCι may play a similar role in the maintenance of TICs in fully transformed human tumor cells. Our present results demonstrate that genetic disruption of PKCι leads to a significant inhibition of the TIC phenotype in both human and murine ovarian TICs. Our results indicate that oncogenic PKCι signaling represents an intrinsic mechanism by which ovarian TICs maintain their tumorigenic, stem-like properties.
In the lung, we identified an oncogenic PKCι-Par6-Ect2-Mek-Erk-MMP10 signaling axis that is required for the ability of PKCι to support transformed growth of lung cancer cells in vitro and in vivo (11–13). Our current studies are consistent with a key role of this signaling pathway in PKCι-dependent maintenance of an ovarian TIC phenotype; PKCι KD leads to loss of phosphorylated Ect2 (a direct PKCι substrate), pMek and pErk, as well as expression of MMP10. Furthermore, treatment of ovarian TICs with the selective PKCι inhibitor ANF, which inhibits PKCι signaling through disruption of the PKCι-Par6 oncogenic complex, leads to similar inhibition of the PKCι-Par6-Ect2-Mek-Erk-MMP10 signaling cascade and potent inhibition of oncogenic TIC behavior. Our data further demonstrate that oncogenic PKCι signaling is active in ovarian TICs in vivo and that either genetic or pharmacologic inhibition of PKCι leads to a significant blockade of tumor initiation in vivo.
We previously demonstrated that PKCι is overexpressed in primary non-small cell lung cancer (NSCLC) cell lines and primary tumors, and PKCι expression is associated with poor clinical outcome in NSCLC patients (3). In lung squamous cell carcinomas (LSCC), PKCι expression is driven by frequent chromosome 3q26 amplification (3). Similar findings have been reported in ovarian serous tumors (7, 8), and PKCι has been shown to be important for the transformed phenotype of ovarian cancer cell lines in vitro (7). However, little was known about the molecular mechanisms by which PKCι drives the oncogenic phenotype in ovarian cancer cells. We have validated and extended these published findings, showing that ~80% of ovarian serous tumors in the TCGA dataset exhibit gene copy number gains in PRKCI which is associated with elevated PRKCI mRNA expression. In the lung, PKCι is genetically, biochemically and functionally linked to the Rho family GTPase GEF Ect2 (13). Our current findings demonstrate that PRKCI and ECT2 are co-amplified and overexpressed in ovarian serous tumors as part of a 3q26 amplicon, and that PKCι and Ect2 are functionally linked to drive a PKCι-Par6-Ect2-Mek-Erk signaling pathway that is active in ovarian TICs. Analysis of the TCGA primary ovarian tumor data further demonstrate a strong positive correlation between the tumor expression of PKCι, Ect2 and MMP10 suggesting that the PKCι-Par6-Ect2-Mek-Erk-MMP10 signaling axis identified in ovarian TICs in vitro is activated in primary ovarian serous tumors. Taken together, our data indicate that PKCι promotes a PKCι-Par6-Ect2-Mek-Erk-MMP10 signaling axis that drives an ovarian TIC phenotype in vitro and in vivo. Furthermore, our data provide compelling evidence that PRKCI and ECT2 are genetically, biochemically and functionally linked in primary ovarian serous tumors. These data suggest a novel paradigm in which a single genetic event, chromosome 3q26 amplification, leads to coordinate amplification and overexpression of two co-operating oncogenes that together drive an ovarian TIC phenotype. Our findings further demonstrate that the selective PKCι inhibitor ANF can disrupt the TIC phenotype in vitro and in vivo. These data provide a compelling rationale of the use of ANF for treatment of ovarian cancer patients, particularly ovarian serous tumors harboring PRKCI and ECT2 gene copy number gains and activation of oncogenic PKCι signaling. We have initiated a phase I clinical trial to assess the feasibility of using ANF in ovarian serous cancer patients at high risk for relapse after initial therapy, a process likely driven by ovarian TICs that survive initial therapy.
Supplementary Material
Implications.
PKCι drives a tumor-initiating cell phenotype in ovarian cancer cells that can be therapeutically targeted with auranofin, a small molecule inhibitor of PKCι signaling.
Acknowledgments
The authors wish to thank and the Mayo Clinic Ovarian SPORE Tissue shared resource (directed by Dr. Keith Knudson) for providing ovarian cancer cell lines, Ms. Brandy Edenfield for fixation and histological staining of tumor tissues, Mr. Justin Weems and Ms. Capella Weems for technical assistance, and Dr. Nicole R. Murray and the members of the Fields laboratory for critical review of the manuscript. This work was supported in part by a Pilot Project grant from the Mayo Clinic Ovarian SPORE to A.P.F. and a post-doctoral fellowship award from the Ovarian Cancer Research Fund to Y.W. A.P.F. is the Monica Flynn Jacoby Professor of Cancer Research, an endowment fund that provides partial support for his research program.
Footnotes
The authors declare that they have no conflicts of interest
References
- 1.Cannistra SA. Cancer of the ovary. N Engl J Med. 2004;351:2519–29. doi: 10.1056/NEJMra041842. [DOI] [PubMed] [Google Scholar]
- 2.Scotti ML, Bamlet WR, Smyrk TC, Fields AP, Murray NR. Protein kinase Ciota is required for pancreatic cancer cell transformed growth and tumorigenesis. Cancer Res. 2010;70:2064–74. doi: 10.1158/0008-5472.CAN-09-2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Regala RP, Weems C, Jamieson L, Khoor A, Edell ES, Lohse CM, et al. Atypical protein kinase C iota is an oncogene in human non-small cell lung cancer. Cancer Res. 2005;65:8905–11. doi: 10.1158/0008-5472.CAN-05-2372. [DOI] [PubMed] [Google Scholar]
- 4.Murray NR, Jamieson L, Yu W, Zhang J, Gokmen-Polar Y, Sier D, et al. Protein kinase Ciota is required for Ras transformation and colon carcinogenesis in vivo. J Cell Biol. 2004;164:797–802. doi: 10.1083/jcb.200311011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takagawa R, Akimoto K, Ichikawa Y, Akiyama H, Kojima Y, Ishiguro H, et al. High expression of atypical protein kinase C lambda/iota in gastric cancer as a prognostic factor for recurrence. Ann Surg Oncol. 2010;17:81–8. doi: 10.1245/s10434-009-0708-x. [DOI] [PubMed] [Google Scholar]
- 6.Kojima Y, Akimoto K, Nagashima Y, Ishiguro H, Shirai S, Chishima T, et al. The overexpression and altered localization of the atypical protein kinase C lambda/iota in breast cancer correlates with the pathologic type of these tumors. Hum Pathol. 2008;39:824–31. doi: 10.1016/j.humpath.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 7.Zhang L, Huang J, Yang N, Liang S, Barchetti A, Giannakakis A, et al. Integrative genomic analysis of protein kinase C (PKC) family identifies PKCiota as a biomarker and potential oncogene in ovarian carcinoma. Cancer Res. 2006;66:4627–35. doi: 10.1158/0008-5472.CAN-05-4527. [DOI] [PubMed] [Google Scholar]
- 8.Eder AM, Sui X, Rosen DG, Nolden LK, Cheng KW, Lahad JP, et al. Atypical PKCiota contributes to poor prognosis through loss of apical-basal polarity and cyclin E overexpression in ovarian cancer. Proc Natl Acad Sci U S A. 2005;102:12519–24. doi: 10.1073/pnas.0505641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weichert W, Gekeler V, Denkert C, Dietel M, Hauptmann S. Protein kinase C isoform expression in ovarian carcinoma correlates with indicators of poor prognosis. Int J Oncol. 2003;23:633–9. [PubMed] [Google Scholar]
- 10.Murray NR, Fields AP. Atypical protein kinase C iota protects human leukemia cells against drug-induced apoptosis. J Biol Chem. 1997;272:27521–4. doi: 10.1074/jbc.272.44.27521. [DOI] [PubMed] [Google Scholar]
- 11.Frederick LA, Matthews JA, Jamieson L, Justilien V, Thompson EA, Radisky DC, et al. Matrix metalloproteinase-10 is a critical effector of protein kinase Ciota-Par6alpha-mediated lung cancer. Oncogene. 2008;27:4841–53. doi: 10.1038/onc.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Regala RP, Weems C, Jamieson L, Copland JA, Thompson EA, Fields AP. Atypical protein kinase Ciota plays a critical role in human lung cancer cell growth and tumorigenicity. J Biol Chem. 2005;280:31109–15. doi: 10.1074/jbc.M505402200. [DOI] [PubMed] [Google Scholar]
- 13.Justilien V, Fields AP. Ect2 links the PKCiota-Par6alpha complex to Rac1 activation and cellular transformation. Oncogene. 2009;28:3597–607. doi: 10.1038/onc.2009.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Erdogan E, Lamark T, Stallings-Mann M, Lee J, Pellecchia M, Thompson EA, et al. Aurothiomalate inhibits transformed growth by targeting the PB1 domain of protein kinase Ciota. J Biol Chem. 2006;281:28450–9. doi: 10.1074/jbc.M606054200. [DOI] [PubMed] [Google Scholar]
- 15.Regala RP, Justilien V, Walsh MP, Weems C, Khoor A, Murray NR, et al. Matrix metalloproteinase-10 promotes Kras-mediated bronchio-alveolar stem cell expansion and lung cancer formation. PLoS One. 2011;6:e26439. doi: 10.1371/journal.pone.0026439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Justilien V, Jameison L, Der CJ, Rossman KL, Fields AP. Oncogenic activity of Ect2 is regulated through protein kinase C iota-mediated phosphorylation. J Biol Chem. 2011;286:8149–57. doi: 10.1074/jbc.M110.196113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Regala RP, Thompson EA, Fields AP. Atypical protein kinase C iota expression and aurothiomalate sensitivity in human lung cancer cells. Cancer Res. 2008;68:5888–95. doi: 10.1158/0008-5472.CAN-08-0438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008;40:499–507. doi: 10.1038/ng.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Justilien V, Regala RP, Tseng IC, Walsh MP, Batra J, Radisky ES, et al. Matrix metalloproteinase-10 is required for lung cancer stem cell maintenance, tumor initiation and metastatic potential. PLoS One. 2012;7:e35040. doi: 10.1371/journal.pone.0035040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stallings-Mann M, Jamieson L, Regala RP, Weems C, Murray NR, Fields AP. A novel small-molecule inhibitor of protein kinase Ciota blocks transformed growth of non-small-cell lung cancer cells. Cancer Res. 2006;66:1767–74. doi: 10.1158/0008-5472.CAN-05-3405. [DOI] [PubMed] [Google Scholar]
- 21.Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, et al. Cancer stem cells--perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66:9339–44. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- 22.Hu L, McArthur C, Jaffe RB. Ovarian cancer stem-like side-population cells are tumourigenic and chemoresistant. Br J Cancer. 2010;102:1276–83. doi: 10.1038/sj.bjc.6605626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baba T, Convery PA, Matsumura N, Whitaker RS, Kondoh E, Perry T, et al. Epigenetic regulation of CD133 and tumorigenicity of CD133+ ovarian cancer cells. Oncogene. 2009;28:209–18. doi: 10.1038/onc.2008.374. [DOI] [PubMed] [Google Scholar]
- 24.Regala RP, Davis RK, Kunz A, Khoor A, Leitges M, Fields AP. Atypical protein kinase C{iota} is required for bronchioalveolar stem cell expansion and lung tumorigenesis. Cancer Res. 2009;69:7603–11. doi: 10.1158/0008-5472.CAN-09-2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.