

Abstract
The inability of mineralocorticoid receptor (MR) blockade to reduce hypertension associated with high Angiotensin (Ang) II suggests direct actions of Ang II to regulate tubular sodium reabsorption via the epithelial Na+ channel (ENaC) in the aldosterone-sensitive distal nephron (ASDN). We used freshly isolated ASDN from mice to delineate the synergism and primacy between aldosterone and Ang II in controlling functional ENaC activity. Inhibition of MR specifically prevented the increased number of functionally active ENaC but not ENaC open probability elicited by a low sodium diet. In contrast, we found no functional role of glucocorticoid receptors (GR) in the regulation of ENaC activity by dietary salt intake. Simultaneous inhibition of MR and Ang II type 1 receptors (AT1R) ameliorated the enhanced ENaC activity caused by low dietary salt intake and produced significantly greater natriuresis than either inhibitor alone. Chronic systemic Ang II infusion induced more than two times greater increase in ENaC activity than observed during dietary sodium restriction. Importantly, ENaC activity remained greatly above control levels during maximal MR inhibition. We conclude that during variations in dietary salt intake both aldosterone and Ang II contribute complementarily to the regulation of ENaC activity in the ASDN. In contrast, in the setting of Ang II-dependent hypertension, ENaC activity is up-regulated well above the physiological range and is not effectively suppressed by inhibition of the aldosterone-MR axis. This provides a mechanistic explanation for the resistance to MR inhibition that occurs in hypertensive subjects having elevated intrarenal Ang II levels.
Keywords: collecting duct, connecting tubule, distal nephron, renal sodium handling, hypertension
INTRODUCTION
Sodium reabsorption in the aldosterone-sensitive distal nephron (ASDN) is of great importance for proper renal sodium handling and maintenance of whole body sodium homeostasis1. Dysfunction of sodium transport at this site has been tightly linked to disturbances in circulating volume and blood pressure abnormalities1,2. Activity of the Epithelial Na+ channel (ENaC) underlies electrogenic Na+ reabsorption at ASDN, which includes the connecting tubule (CNT) and the cortical collecting duct (CCD)3–6. The relevance of ENaC function for blood pressure control in humans is unequivocally supported by the fact that genetic disorders affecting blood pressure, such as Liddle's syndrome and Type I pseudohypoaldosteronism (PHA), arise from gain-of-function and loss-of-function mutations in ENaC, respectively7–11.
ENaC activity in the ASDN is inversely related to dietary sodium intake12–14. Activation of the Renin-Angiotensin-Aldosterone system (RAAS) in response to sodium restriction increases ENaC activity and expression via aldosterone-mineralocorticoid receptor (MR) pathway3–5,9,15–17. However, the existence of the “aldosterone paradox”, where elevations in circulating aldosterone in response to hypovolemia and hyperkalemia result in drastically different patterns of urinary sodium and potassium excretion18,19, points to a role for other signaling components of RAAS, most likely Angiotensin (Ang) II, in the regulation of ENaC in response to volume contraction.
Ang II is the principal effector of RAAS20,21. Increases in circulating Ang II levels induce vasoconstriction, secretion of aldosterone and elevations in systemic blood pressure22. In the kidney, Ang II promotes antinatriuresis, in part, by stimulating luminal Na+ entry and tubular Na+ reabsorption in the distal nephron23–25. Virtually all Ang II actions to enhance renal sodium transport are mediated by Ang II type 1 receptors (AT1R). AT1R are abundantly expressed on both apical and basolateral sides of epithelial cells along the whole length of the renal nephron from the proximal tubule to the collecting duct26,27. We28 and others25, 29 have documented that Ang II acutely increases ENaC activity and open probability (Po) in freshly isolated split-opened mouse and rat ASDN. The signaling pathway involves AT1R-dependent activation of NADPH oxidase and generation of reactive oxygen species, likely superoxide and peroxide, to stimulate ENaC28 and to diminish inhibitory actions of the arachidonic acid metabolites29. Of interest, acute stimulation of ENaC Po by Ang II persists during saturation28 and inhibition29 of MR cascade indicating aldosterone-independent nature of this regulation19. It is also interesting that prolonged treatment of isolated murine ASDN with Ang II causes translocation of αENaC to the apical plasma membrane and increases the number of functionally active channels28. It remains unclear, whether physiologically relevant changes in Ang II levels have their own non-redundant contribution to regulation of ENaC activity in ASDN in response to changes in dietary sodium intake.
In experimental animal models of Ang II-induced hypertension30,31, intrarenal Ang II levels become much higher than those in plasma due to activation of the intrarenal and intratubular renin angiotensin system (RAS)21. Inappropriately stimulated intrarenal RAS leads to excessive sodium retention, in part, due to possible activation of ENaC in ASDN32–34. Cumulative evidence suggests that effects of elevated Ang II levels on ENaC expression and sodium reabsorption in the ASDN cannot be solely explained by Ang II-induced aldosterone secretion. Thus, mice with global knockout of the major subtype of AT1 receptors, (AT1a) exhibit a marked reduction in αENaC abundance in the kidney despite slightly elevated aldosterone levels35. Furthermore, the effect of AT1R blockade on αENaC expression was not prevented by spironolactone, suggesting a direct role of the AT1R in regulation of αENaC gene expression36. Systemic infusion of Ang II increases αENaC protein abundance in rat kidney cortex36. It is unclear, though, whether elevated circulating and intrarenal Ang II levels translate into direct changes in functional ENaC activity. Intriguingly, inhibition of MR with spironolactone in rat and mouse models of Ang II-dependent hypertension produced very mild and transient hypotensive effects32,33,37. In contrast, direct ENaC blockade with amiloride does attenuate blood pressure in Ang II infused rats34. This may indicate a dominant role of Ang II-driven aldosterone-independent ENaC activation in Ang II-infused models of hypertension38.
In the current study, we employ systemic pharmacological inhibition of MR and AT1R with direct assessment of ENaC activity using patch clamp electrophysiology in freshly isolated split-opened ASDN of mice, to test synergism and primacy in aldosterone and Ang II signals to ENaC during the physiological response to changes in dietary sodium intake and in the pathophysiology of the Ang II-induced hypertension. We found that, by controlling functional ENaC expression (number of active channels on the apical plasma membrane), the aldosterone cascade has relatively greater contribution in stimulating ENaC during conditions of dietary sodium restriction than Ang II signaling, which is responsible for regulation of ENaC Po. In contrast, chronic Ang II infusion over-stimulates ENaC beyond the physiological range of ENaC regulation. We propose that this, in part, contributes to the excessive renal sodium conservation and hypertension in response to chronic Ang II infusion. Importantly, we report here that blockade of aldosterone-MR cascade is insufficient to effectively suppress excessively activated ENaC when Ang II levels are elevated.
METHODS
Reagents and animals
All chemicals and materials were from Sigma (St. Louis, MO), VWR (Radnor, PA), and Tocris (Ellisville, MO) unless noted otherwise and were at least of reagent grade. Animal use and welfare adhered to the NIH Guide for the Care and Use of Laboratory Animals following protocols reviewed and approved by the Animal Care and Use Committee of the University of Texas Health Science Center at Houston and the Tulane University School of Medicine. For experiments, male C57BL/6J mice (Charles River Laboratories, Wilmington, MA) 6–8 weeks old, were used. Animals had free access to tap water.
Research diets and Treatments
To examine effects of salt intake, animals were provided diets containing nominally free (<0.01% Na+, TD.90228), regular (0.32% Na+, TD.7912), and high (2% Na+, TD.92034) sodium for one week. All diets were purchased from Harlan Teklad (Madison, WI, USA). Spironolactone (30 mg/kgBW), mifepristone (30 mg/kgBW), losartan (10 mg/kgBW) were added to drinking water for 3, 7, or 14 days depending on experimental design. As necessary for some protocols, mice were injected with Deoxycorticosterone acetate (DOCA) for 3 consecutive days (2.4 mg/injection/animal) prior to the experimentation similarly to what we have done previously39.
Male 6 weeks C57BL/6 mice (approximately 25 g) were infused with Ang II (Sigma) dissolved in 5% acetic acid (VWR) via subcutaneous osmotic minipumps (Alzet model 1002, Alza, Palo Alto, CA) at concentration 400ng/kgBW/min for 2 weeks closely following well-established protocols30,31.
Mice were subjected to inhaled anesthesia with isofluorane, and a catheter connected to a radiotelemetry device was inserted into the left carotid artery to monitor systolic blood pressures using telemetry (model PA-C10; Data Sciences International, St. Paul, MN) in conscious and unrestrained conditions. After a 5-days recovery phase, baseline systolic blood pressure levels were measured, and the mice were then after subjected to a similar anesthesia for osmotic mini-pump subcutaneous implantation. Mice were infused with Ang II (400 ng/kgBW/min) for 14 days. Systolic blood pressure data were collected in sham and Ang II-infused mice and analyzed using Dataquest A.R.T, Software 4.0 (Data Sciences International).
Electrophysiology
The procedure for isolation of the aldosterone-sensitive distal nephrons (ASDNs) suitable for electrophysiology has been described previously28,40,41. ENaC activity in principal cells was determined in cell-attached patches on the apical membrane made under voltage-clamp conditions (−Vp=−60mV) using standard procedures (see online supplement for more details).
Immunofluorescent microscopy
Kidneys were fixed in 10% formalin at 4°C overnight, processed for paraffin embedding, sectioning (3 μm), and sequentially immunostained using rabbit anti-αENaC and goat anti-AQP2 antibodies as described previously42. The primary antibodies used were anti-αENaC (SPC-403D; StressMarq Biosciences Inc.) at 1:400 dilution and anti-AQP2 (sc-9882, Santa Cruz Biotechnology) at 1:200 dilution for overnight and 90 minutes incubations, respectively. For immunofluorescent detection, we used donkey anti-rabbit or donkey anti-goat secondary IgG antibodies with Alexa Fluor 488 (green) or 594 (red) conjugates (Invitrogen) and counterstaining of the tissue sections for nuclei visualization with 4,6-diamidino-2-phenylindole (DAPI; D1306, Invitrogen). The immunofluorescence images were captured with a Nikon DS-Digital camera attached to a 50i Eclipse Fluorescence microscope from Nikon using objectives of 40× and 100× (oil immersion) magnification. Negative controls were obtained by omission of the specific primary antibody.
Western blotting
Immediately after dissection, kidneys were frozen in liquid nitrogen and stored there for further use. Prior to experimentation the kidneys were homogenized in 3 volumes of ice-cold hypotonic lysis buffer containing 50mmol/L Tris, 1% Triton X-100, 5mmol/L EDTA (pH=7.4) supplemented with 1 mM PMSF and 2 mg/ml protease inhibitor cocktail (Complete mini, Roche Diagnostics, Germany). Homogenates were centrifuged at 1000 g for 15 min at +4 C. The supernatants were split in two parts to assess total cellular proteins and plasma membrane proteins using plasma membrane protein extraction kit (Biovision Inc, USA) following the manufacturer's protocol. Protein concentration in the homogenates was determined with a Bradford assay. The samples (25 μg/lane) were separated on 9% polyacrylamide gels at 150 V for 1 h 15 min and transferred to nitrocellulose membranes at 100 V for 1 h 45 min. The membranes were blocked for 1 h at RT in 5% nonfat milk in TBS-T (150 mmol/L NaCl, 50 mmol/L Tris-HCl pH=7.4, 0.1% Tween 20). Subsequently the membranes were probed with anti-αENaC primary rabbit antibodies (1:1000, Stressmarq Biosciences, Canada) followed by peroxidase-conjugated goat anti-rabbit secondary antibodies (1:20000, Bio-Rad, USA) for 1 hour at RT. The membranes were re-probed with anti-α-tubulin (1:200, Abcam, UK) primary rabbit antibodies and peroxidase-conjugated goat anti-rabbit secondary antibodies (1:20000, Bio-Rad, USA). When total cellular αENaC expression was assessed, αENaC band intensities were normalized to the intensities of the corresponding α-tubulin bands. For membrane enriched fractions, band intensities were normalized to the intensity of the first αENaC band from the first control sample. All experiments were repeated at least 3 times.
Urinary sodium excretion
Urine was collected from bladders of sacrificed animals and frozen at −20°C until analyzed. The time of urine sampling was the same (10AM–11AM) for all animals. Urinary sodium excretion was calculated as the ratio of urinary sodium to creatinine concentrations. Urinary sodium concentration was measured using PFP7 Flame photometer (Techne, Burlington, NJ). Urinary creatinine concentration was assessed with Hitachi 7000 HPLC System (Pleasanton, CA, USA).
Data analysis
All summarized data were reported as mean ± SEM. Data from different experimental sets were compared using a Student's (two-tailed) t-test or an One-Way ANOVA as appropriate. P ≤ 0.05 was considered significant.
RESULTS
ENaC activity is indistinguishable in CNT and CCD
We first performed a detailed characterization of ENaC activity in the structurally different CNT and CCD in mice kept on a regular salt intake (0.32% Na+). As illustrated in Figure 1A, we mechanically isolated bifurcations of the distal nephrons representing merging of two connecting tubules (CNT) into a cortical collecting duct (CCD). ENaC activity was surveyed using patch clamp electrophysiology in cell attached configuration within split-opened areas located up to 100 μm above (CNT region) and 300 μm below (CCD region) the bifurcations. To reliably assess ENaC activity (fNPo), the mean NPo was corrected to a frequency of observing patches with active channels (f = number of patches with channels/total number of patches). As summarized in Figure 1B, ENaC activity was nearly identical within the designated area. We also did not detect any differences in ENaC gating properties (open probability, Po) and the number of functional ENaC within a patch (N) in both CNT and CCD (data not shown). These results suggest that ENaC activity is uniform in the cortical region of the classical ASDN representing transition of CNTs into CCDs. Thus, we did not further discriminate ENaC activity in CNT and CCD.
Figure 1. ENaC activity is similar in CND and CCD.

(A) A representative micrograph of freshly isolated murine ASDN split-opened at CCD (bottom) and at the right branch of CNT (top) used for patch clamp evaluation of ENaC activity. Scale bar is 40 μm. (B) Summary of ENaC activity (fNPo) in CNT and CCD of ASDN from mice kept on a regular salt regimen (0.32% Na+). Here and below the number of individual patch recordings are shown.
Mineralocorticoid but not glucocorticoid receptors are involved in regulation of ENaC activity by aldosterone in murine ASDNs
To address the relative contribution of aldosterone-independent mechanisms, such as Ang II, in regulation of functional ENaC expression in the ASDN, we first inhibited MR with spironolactone (30 mg/kgBW for 7 days) in mice maintained on a regular salt intake. This treatment significantly diminished but did not abolish ENaC activity (fNPo) from 0.40±0.03 (n=223) to 0.21±0.05 (n=52). To ensure that the concentration of spironolactone is sufficient for complete inhibition of MR, we injected mice with deoxycorticosterone acetate (DOCA) for 3 consecutive days prior to the assessment. Saturation of mineralocorticoid status significantly increased ENaC activity to 0.61±0.08 (n=54) in ASDNs from control mice but had no effect on ENaC (0.23±0.04, n=58) in ASDNs from mice treated with spironolactone (supplementary Figure S1).
Elevated circulating aldosterone levels can potentially affect ENaC function via glucocorticoid receptors (GR) particularly during sodium restricted conditions. To test this possibility, we performed detailed analysis of ENaC activity in mice kept on different salt diets for one week (low, <0.01%; regular, 0.32%; and high, 2% Na+) in the absence and presence of systemic inhibition of GR with mifepristone (30 mg/kgBW, for 7 days). This concentration is similar to that used previously for GR inhibition43. ENaC activity was augmented during low Na+ diet and diminished under high Na+ diet (Figure 2A, see also Figure 3, top row for representative traces of ENaC activity). This involved changes in both functional ENaC expression (Figure 2B) and ENaC Po (Figure 2C). As shown, mifepristone treatment fails to alter ENaC activity (Figure 2A), functional ENaC expression (Figure 2B), and ENaC Po (Figure 2C) under all tested experimental conditions (see supplementary Table S1 for the absolute values).
Figure 2. Glucocorticoid receptors do not contribute to regulation of ENaC by systemic salt intake.
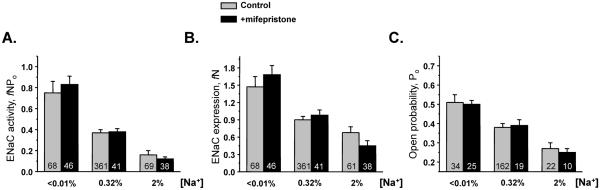
Summary graph of ENaC activity, fNPo (A); functional ENaC expression, fN (B); and ENaC open probability, Po (C) in mice kept on low (<0.01%), regular (0.32%), and high (2%) Na+ diets for one week, respectively, in the control (light grey bars) and after treatment with glucocorticoid receptor (GR) antagonist, mifepristone (black bars).
Figure 3. Coordinated regulation of ENaC activity by aldosterone and Ang II signaling cascades.

Representative continuous current traces from cell-attached patches monitoring ENaC activity in mice on low and high Na+ diets in the control (top row), after treatment with MR antagonist spironolactone (middle row) and after combined treatment with spironolactone and AT1R blocker, losartan for one week (bottom row). Patches were held at a test potential of Vh= − Vp= − 60 mV. Inward Li+ currents are downward. Dashed lines indicate the respective current states marked as oi and a c denotes the closed state.
Overall, we concluded that aldosterone utilizes activation of MR but not GR to modulate ENaC activity in ASDN.
Inhibition of MR does not abolish regulation of ENaC by systemic salt status
We next probed how inhibition of aldosterone-MR axis affects regulation of ENaC by variations in dietary salt intake. Using a similar strategy as discussed above, mice were fed with various Na+ diets for one week and were supplemented with MR inhibitor spironolactone (30 mg/kgBW). As summarized in Figure 4A, spironolactone greatly attenuated ENaC activity under low salt intake while had no significant effect of ENaC in mice kept on a high (2%) salt diet (see Figure 3, middle row for representative traces). Nevertheless, we observed modest but significant augmentation of ENaC activity when comparing high and low sodium diets in mice receiving spironolactone treatment (Figures 3, 4A). Detailed patch clamp analysis revealed that inhibition of MRs prevented changes in functional ENaC expression in response to dietary salt variations (Figure 4B, see also supplementary Table S1). Furthermore, we did not observe changes in the frequency of patches with active channels on low and high sodium diets (25% and 24%, respectively) in spironolactone treated animals. For control animals frequencies were 50% and 31%, respectively. These results indicate a dominant role of aldosterone-MR axis in regulation of functional ENaC expression in response to systemic physiological cues, such as dietary sodium variations. In contrast, we found that regulation of ENaC Po by dietary salt intake remained intact after spironolactone treatment (Figure 4C, see also supplementary Table S1). Thus, we concluded that aldosterone-independent stimulation of ENaC activity by dietary salt restriction involves augmentation of ENaC open probability.
Figure 4. Inhibition of MR and AT1R abolishes stimulation of ENaC activity by dietary sodium restriction.

Summary graphs of ENaC activity, fNPo (A); functional ENaC expression, fN (B); and ENaC open probability, Po (C) in mice kept on low (<0.01%), regular (0.32%), and high (2%) Na+ diets for one week, respectively, in the control (light grey bars) and after treatment with mineralocorticoid receptor (MR) antagonist, spironolactone (dark grey bars). * - significant decrease versus respective control values; # - significant increase versus spironolactone 2% Na+. Summary graphs of ENaC activity, fNPo (D); functional ENaC expression, fN (E); and ENaC open probability, Po (F) in mice kept on low (<0.01%), regular (0.32%), and high (2%) Na+ diets for one week, respectively, in the control (light grey bars) and after combined treatment with spironolactone and AT1R blocker, losartan (shaded dark grey bars). * -significant decrease versus respective control values.
Ang II signaling is critical for increased ENaC Po by low sodium diet
The observation that ENaC Po responds to variations in systemic salt status in the absence of aldosterone-MR cascade (Figure 4C) argues for the involvement of additional mechanisms responsible for this regulation. We have recently documented that Ang II via activation of AT1R is capable of increasing ENaC Po and this regulation persists even in the presence of saturated mineralocorticoid status28. Thus, we next tested whether Ang II signaling contributes to regulation of ENaC Po by dietary salt intake. Mice were placed on different salt diets for 1 week and treated simultaneously with spironolactone and AT1R antagonist, losartan (10 mg/kgBW). We found that, in contrast to the treatment with spironolactone alone, ENaC activity was no longer significantly different in animals kept on low and high sodium diets (Figure 4D, see also Figure 3 bottom row). Absolute values are reported in supplementary Table S1. Consistently, we observed that functional ENaC expression was not different between diets with losartan having no further inhibitory effect (Figure 4E). Importantly, we found that with both MR and AT1R blockade, ENaC Po was only slightly different in mice placed on a low and high sodium diets and this difference did not reach significance (Figure 4F). Therefore, we conclude that Ang II and aldosterone cascades contribute complementarily to stimulation of ENaC activity during dietary salt restriction. Aldosterone signaling, by specifically controlling the number of functional ENaCs, has a relatively greater contribution than Ang II, which is mainly responsible for stimulation of ENaC open probability.
Inhibition of aldosterone-MR and Ang II-AT1R cascades additively produces natriuresis
We next probed if coordinated activation of ENaC activity by aldosterone and Ang II signaling cascades parallels the changes in renal sodium handling. For this, we assessed snapshot urinary sodium excretion at day 3 in mice on low sodium regimen. Systemic treatment with losartan (10 mg/kgBW, for 3 days) and spironolactone (30 mg/kgBW, for 3 days) significantly increased urinary sodium excretion (Figure 5A) but had no effect on urinary creatinine levels (Figure 5B). Importantly, combined treatment with both inhibitors caused a modestly greater natriuresis compared to that produced by either substance alone. In contrast, there were no measurable changes in urinary sodium excretion in mice kept on a high sodium diet for 3 days with any of the treatments (Figure 5C). Consistently, urinary creatinine values were also unaffected (Figure 5D). The results in Figure 5, while indicative, support the concept that aldosterone and Ang II signaling cascades stimulate ENaC activity in a complementary manner to maximally increase distal nephron sodium reabsorption specifically under conditions of dietary salt restriction. It is recognized, that changes in urinary sodium excretion during pharmacological inhibition of Ang II and aldosterone signaling pathways also reflect possible changes in activity of other Na+-transporting systems in the kidney, most notably the sodium chloride cotransporter (NCC)44,45.
Figure 5. Aldosterone and Ang II signaling cascades additively contribute to regulation of urinary Na+ excretion during low sodium intake.

Summary graphs of urinary Na+ excretion normalized to creatinine levels in mice kept on low (A) and high (C) sodium diets for 3 days in the control and after systemic treatment with losartan, spironolactone, losartan+spironolactone. Summary graphs of urinary creatinine levels in mice kept on low (B) and high (D) sodium diets for 3 days in the control and after systemic treatment with losartan, spironolactone and losartan+spironolactone. * - significant increase versus control; ** - significant increase versus losartan and spironolactone.
ENaC activity is greatly augmented in Ang II-infusion induced hypertension
Substantial experimental evidence suggests marked upregulation of intrarenal Ang II concentration during many pathological states, including hypertension30,31 and diabetes mellitus46,47. Studies in both rats and mice have demonstrated that chronic Ang II infusions lead to increased urinary levels of angiotensinogen and Ang II21,38. These data suggest that augmented intratubular Ang II signaling enhances renal sodium reabsorption, in part, by over-stimulating ENaC. Thus, we directly assessed changes in blood pressure (Figure 6A) and ENaC activity (Figure 6B) in mice kept on a normal salt regimen (0.32% Na+) in response to chronic infusion of Ang II (400ng/kgBW·min for 14 days). Mice infused with Ang II displayed significant elevated systolic blood pressures compared with the vehicle infused control group after Day 3 of Ang II infusions (129±5 vs.115±2 mm Hg; P=0.06). By Day 5, systolic blood pressures were augmented in mice receiving Ang II (144±4 vs. 117±1 mm Hg; P<0.05) and continued to increase by Day 9 (151±3 vs. 118±2 mm Hg; P<0.001) and Day 13 (157±9 vs. 120±1 mm Hg; P<0.05) compared to the control group. The time course of developing hypertension is consistent with previous reports using a similar experimental strategy30,33.
Figure 6. Chronic Ang II infusion elevates systemic blood pressure and drastically increases functional ENaC activity.
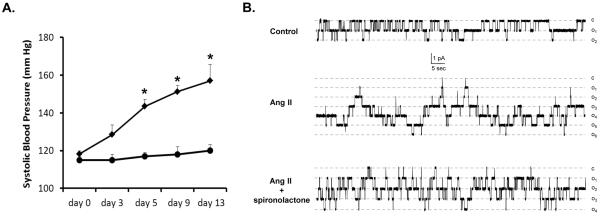
(A) The time courses of changes in systolic blood pressure in response to chronic infusion of vehicle (circles) and 400 ng/kgBW/min of Ang II (diamonds) as determined by telemetry. * - significant increase versus respective systolic blood pressure values in control vehicle infused mice. (B) Representative continuous current trace from cell-attached patches monitoring ENaC activity in mice after 2 weeks infusion of vehicle (top row), Ang II (middle row) and Ang II in the present of spironolactone (bottom row). All other conditions are identical to that described in Figure 4.
As visualized by the representative traces of ENaC activity (Figure 6B) and summarized in Figure 7A, chronic Ang II infusion leads to a robust (approximately 6 times) elevation of ENaC activity compared to that in vehicle infused animals. Of note, this elevation was twice the activity than that observed in response to strong physiological stimuli, such as dietary salt restriction (see Figures 3, 4 for comparison). Specifically, we found that Ang II infusion increases all components of functional ENaC activity: the frequency of observing patches with active channels (Figure 7B), functional ENaC expression (Figure 7C), and ENaC Po (Figure 7D). The absolute values of ENaC activity in vehicle infused and Ang II infused animals are reported in supplementary Table S2.
Figure 7. MR blockade fails to restore ENaC activity during chronic Ang II infusion.

(A) Summary graph of ENaC activity, fNPo, in mice kept on regular salt intake after 2 weeks infusion of vehicle (Control), Ang II and Ang II in the presence of MR inhibition with spironolactone. Dashed line indicates the level of ENaC activity after spironolactone treatment in mice kept on regular salt intake (see also Figures 2, 3). (B) Pie charts representing the frequency of observing patches with active channels (f) for the conditions described in (A). For comparison, the frequency of observing patches with active channels for condition of low sodium diet is also included. Summary graphs of functional ENaC expression (C) and ENaC Po (D) after 2 weeks infusion of vehicle (Control), Ang II and Ang II in the presence of MR inhibition with spironolactone. Dashed line indicates the respective levels observed during regular salt intake + spironolactone. * - significant increase versus Control.
To assess relative contribution of aldosterone-dependent and -independent mechanisms in activation of ENaC activity in Ang II-infused animals, mice were provided with spironolactone for the 14 day period of the treatment. Inhibition of MR did attenuate the stimulatory effect of Ang II on ENaC (Figure 6B). However, ENaC activity remained more than 4 times higher than in control animals treated with spironolactone (Figure 7A, shown with a dash line). Inhibition of MR failed to return the functional ENaC expression to the levels observed in control animals treated with spironolactone (Figure 7C, shown with a dash line). Furthermore, spironolactone did not diminish the frequency of observing patches with active channels (Figure 7B), and had no measurable effect on ENaC Po in Ang II-infused mice (Figure 7D). Supplementary Table S2 contains absolute values of ENaC activity, ENaC Po and functional ENaC expression in Ang II infused animals treated with spironolactone. We conclude that chronic elevation of Ang II levels increases ENaC activity greatly above the levels observed during routine changes in dietary salt amount. Inhibition of mineralocorticoid receptors fails to efficiently suppress ENaC activity thus suggesting a critical contribution for Ang II-mediated mechanisms independent of aldosterone.
Regulation of plasma membrane αENaC levels in Ang II-infused mice
We finally probed whether Ang II infusion results in augmentation of ENaC protein abundance at the plasma membrane. For this, we visualized the spatial subcellular αENaC distribution in kidney sections at the cortex (Figures 8A, B) and inner medulla (Figures 8C, D) regions of control and Ang II-infused mice with immunofluorescent microscopy. No fluorescent signal was detected in the absence of primary but in the presence of secondary antibodies (data not shown). Using a higher magnification, we observed that αENaC fluorescent signal displays a predominant apical distribution in the collecting ducts from kidney sections from Ang II-infused mice compared to control mice (Figures 8E, F). The αENaC immunoexpression was present in the principal cells co-stained with a specific marker AQP-2 (Figures 8G–J).
Figure 8. Chronic Ang II infusion promotes apical ENaC distribution in principal cells of the collecting duct.

Representative micrographs of αENaC immunofluorescent staining in 3 μm kidney sections from control (A) and chronic Ang II-infused mice (B) in the cortex region as well as in the inner medulla regions (C, D). High resolution images of the collecting duct of a kidney section from control and Ang II-infused mice demonstrating expression of αENaC (E, F), a marker of principal cells, AQP2 (G, H) and the respective merged micrographs (I, J). Note the lack of merging colors in the apical aspect in most of the principal cells of the collecting duct from the control mouse kidney section (G) compared to the Ang II-infused mouse (J). Fluorescent signals reporting αENaC and AQP2 expression are shown with preudocolor green and red, respectively. Nuclei are stained using 4,6-diamidino-2-phenylindole (DAPI, pseudocolor blue). Respective scale bars are also shown.
Since immunofluorescent microscopy provides only qualitative evidence of the subcellular ENaC redistribution in Ang II-infused mice, we further monitored the full-length 95 kDa αENaC in whole kidney homogenates (total cell fraction) and plasma membrane enriched fraction using a sucrose gradient-based membrane protein extraction kit (see concise methods). For the experiments, kidneys from the same animals as in Figures 6 and 7 were used. As demonstrated on the representative Western blot in Figure 9A, the membrane enriched fraction nearly lacked α-tubulin staining. Importantly, we observed a shift of αENaC molecular weight suggesting that plasma membrane αENaC is glycosylated. This is consistent with previously published results that cell surface αENaC in MDCK cells is almost exclusively glycosylated48. The glycosylated αENaC in the total cell fraction was barely detectable (data not shown). This suggests that only a minor fraction of the total cell full length αENaC protein pool resides on the plasma membrane. Chronic Ang II infusion had only a tendency to increase αENaC abundance in whole kidney homogenates (Figures 9B, D), whereas plasma membrane αENaC levels were significantly higher in Ang II-treated mice (Figure 9C, E). Administration of MR inhibitor spironolactone decreased αENaC abundance in total cell (Figures 9B, D) and plasma membrane enriched fraction (Figure 9C, E) of Ang II-infused animals below the values observed in control mice. We concluded that chronic Ang II-infusion increases plasma membrane αENaC protein levels in an aldosterone-dependent manner. Furthermore, the observed augmentation of functional ENaC activity in Ang II-infused animals under spironolactone treatment (Figures 6, 7) is likely due to activation of inactive “silent” ENaCs on the apical plasma membrane and is not associated with augmented levels of the channel.
Figure 9. Chronic Ang II infusion increases αENaC plasma membrane abundance in an aldosterone-dependent manner.

(A) Representative Western blot from whole kidney lysates (total cell fractions and membrane enriched fractions) probed with anti-αENaC and anti-α tubulin antibodies. (B) Representative Western blot of total cell fractions from control, Ang II-infused, and Ang II-infused + spironolactone (spir) mice probed with anti-αENaC and anti-α tubulin antibodies. Each band represents individual animal. The mice were the same as used for Figures 6 and 7. (C) Representative Western blot of plasma membrane enriched fractions from control, Ang II-infused, and Ang II-infused + spironolactone (spir) mice probed with anti-αENaC and anti-α tubulin antibodies. All other conditions are the same as in (B). (D) Summary graph comparing total cell αENaC expression in the control, after Ang II-infusion, and after Ang II infusion + spironolactone from Western blots similar to that shown in (B). * -significant decrease versus control. (E) Summary graph comparing αENaC expression in membrane enriched fraction in the control, after Ang II-infusion, and after Ang II infusion + spironolactone from Western blots similar to that shown in (C). * - significant decrease versus control.
DISCUSSION
This study was undertaken to examine how signals from two major hormones responsible for sodium homeostasis, Ang II and aldosterone, modulate ENaC activity in the distal nephron in response to systemic signals. We found that during physiological alterations in dietary sodium, aldosterone cascade plays a conclusive role by controlling the number of functionally active ENaC channels. Ang II signaling, by shaping gating properties of ENaC, has a lesser contribution. In contrast, aldosterone-independent mechanisms of ENaC activation become dominant during the pathology of Ang II-dependent hypertension. This involves augmentation of the number of functional ENaC and ENaC Po and cannot be effectively suppressed by inhibition of aldosterone-MR axis.
In the current study, we found that inhibition of GR with mifepristone had minor effect on ENaC activity during all tested dietary salt intakes (Figure 2) suggesting a minor role of glucocortioid signaling mechanisms in setting basal ENaC activity during different sodium diets. In agreement, a recent study reports that systemic administration of dexamethasone, while increasing αENaC abundance in rat kidneys in an aldosterone-independent manner, fails to activate Na+ currents measured in isolated split-open collecting ducts49. We also demonstrate that maximal stimulation of mineralocorticoid signaling in DOCA-treated mice does not increase ENaC activity when MR are inhibited with spironolactone (supplementary Figure S1). This argues against the possibility that elevated circulating aldosterone levels, particularly during volume depleted states, stimulate GR to affect ENaC. Therefore, the mounting evidence points to an almost exclusive role of MR as mediators of steroid-induced ENaC activation in the ASDN during physiological responses to dietary sodium supplementations. However, over-stimulation of GR might contribute to the development of abnormal sodium retention in the distal nephron. Thus, a cumulative role of both GR and MR in stimulation of ENaC activity has been proposed in patients with Cushing syndrome and animal models with infusion of adrenocorticotropic hormone (ACTH) as a possible mechanism of hypertension50.
Using a physiologically relevant object, split-opened murine ASDN, we demonstrate that inhibition of aldosterone-MR signaling fails to suppress regulation of functional ENaC activity by variations in dietary sodium regimen (Figures 4A–C). This unequivocally points to an important role of aldosterone-independent mechanisms for proper control of ENaC activity by systemic salt status. A previous report demonstrates that spironolactone administration blocked the increase in αENaC protein abundance in response to dietary NaCl restriction51. However, ENaC redistribution to the apical plasma membrane was not prevented by inhibition of MR51. We did not detect significant changes in the number of functionally active ENaC in spironolactone-treated mice kept on high and low sodium diet suggesting that alterations in subcellular ENaC localization do not transform to increased functional ENaC expression. We did observe, however, that regulation of ENaC gating properties (i.e. Po) by dietary sodium is not affected by MR inhibition. In contrast, combined pharmacological blockade of AT1R and MR nearly abolished changes in ENaC Po when dietary Na+ was manipulated (Figures 3, 4). This argues that Ang II cascade has non-redundant stimulatory actions on ENaC activity via control of channel gating. Consistently, we28 and others29 recently documented that acute stimulation of ENaC Po by Ang II in split-opened ASDNs is independent of MR status.
Augmented circulating Ang II levels in the models of Ang II-induced hypertension result in marked decreases in renal sodium excretion and likely increases in distal nephron sodium reabsorption33. Systemic Ang II infusion also increases αENaC protein abundance in rat renal cortex36. Paradoxically, inhibition of MR with spironolactone33 and eplerenone32 fails to correct renal sodium retention and hypertension in response to systemic Ang II infusion. It was proposed that increased function of NCC but not ENaC could be a key mechanism in Ang II-dependent hypertension37. Using direct measurements of ENaC activity in native ASDNs with patch clamping, we demonstrate a robust excessive ENaC activation in Ang II-infused mice to the values exceeding the physiological range of ENaC regulation by dietary salt (Figure 6B). Spironolactone in the concentration sufficient to completely abolish stimulatory actions of aldosterone on ENaC (Figure S1) was ineffective to suppress ENaC to the control values. The number of active ENaC on the apical plasma membrane and ENaC Po remained elevated upon spironolactone treatment (Figure 7). The stimulatory effect of Ang II infusion on ENaC Po in the presence of MR inhibition was anticipated28,29 and consistent with aldosterone-independent contribution of Ang II cascade to regulation of ENaC Po by dietary salt intake (Figure 4). Several studies suggested that Ang II might also play a direct role in regulation of ENaC expression. Thus, AT1R −/− mice have decreased αENaC abundance in the presence of elevated circulating aldosterone levels35. AT1R blockade also results in a fall in αENaC mRNA abundance, and downregulation of αENaC expression was not blocked by spironolactone36. In the current study, we probed whether the increased number of functional ENaC on the apical plasma membrane in Ang II-infused animals after spironolactone is associated with aldosterone-independent ENaC expression and/or trafficking. While there was only a tendency of increasing αENaC levels in whole kidney homogenates of Ang II infused mice, the stimulatory effect of Ang II on αENaC was significant for the plasma membrane fraction (Figure 9). Consistently, our immunofluorescent studies demonstrate accumulation of αENaC predominantly in the apical region in principal cells of Ang II-infused mice (Figure 8). Strikingly, spironolactone treatment decreased αENaC protein abundance on the plasma membrane below the control levels in Ang II infused mice (Figure 9C). Therefore, the observed elevation of functionally active ENaC with patch clamping in Ang II-infused animals after spironolactone is not due to increased protein levels but rather stimulation of “silent” channels residing on the apical plasma membrane.
The important observation of this study is that systemic infusion of Ang II increases ENaC activity in the ASDN to levels greatly exceeding those observed during physiological conditions associated with volume contraction (Figures 6B, 7). Accumulating experimental evidence suggests a critical role of intrarenal reninangiotensin system (RAS) in development of excessive sodium retention and Ang II-dependent hypertension21,52. Intrarenal synthesis of Ang II appears to be sufficient to affect sodium handling in the distal nephron, since luminal perfusion of cortical collecting ducts with Ang I stimulates apical sodium entry in an ACE-dependent manner53. In contrast to the systemic RAS where elevations in circulating Ang II inhibit renin secretion from the granular cells in the juxtaglomerular apparatus, augmented distal nephron Ang II concentration further leads to increased renin mRNA and protein levels in principal cells of CNT and CCD54,55. This feed-forward mechanism is thought to underlie inappropriate augmentation of sodium reabsorption in the distal nephron38,56. Indeed, it was recently reported that systemic infusion of Ang II results in increased urinary Angiotensinogen (AGT) levels indicating intratubular RAS activation57. In contrast, dietary sodium restriction does not lead to augmentation of urinary AGT and Ang II excretion rates in rats and thus to appreciable intratubular RAS activation58. Since principal cells express angiotensin receptors at both apical and basolateral sides, it is likely that circulating and tubular Ang II can separately regulate ENaC-mediated sodium reabsorption during different physiological and pathological conditions. Specifically, activation of basolateral receptors by renal interstitial Ang II levels will play a dominant role in regulation of ENaC open probability during dietary sodium restriction. During Ang II-dependent hypertension, the additional stimulation of intratubular RAS leads to further ENaC over-activation via mechanisms that are independent of mineralocorticoid status.
PERSPECTIVES
It is now becoming recognized that inputs from coordinated but discrete converging signals are critical for proper control of renal sodium transport. This is known to be essential for normal blood pressure control. ENaC-mediated Na+-reabsorption in the distal nephron finalizes adjustments of renal sodium excretion to match dietary sodium intake and maintain sodium balance. The current study documents that aldosterone and Ang II signals provide integrated control of ENaC-mediated sodium reabsorption in the distal nephron. The aldosterone cascade prevails during adaptations to dietary sodium intake. In contrast, augmentation of circulating Ang II levels leads to activation of intrarenal RAS and switches regulation of ENaC activity to a largely aldosterone-independent mode. From a clinical standpoint, increased urinary excretion rates of Ang II or its precursor AGT in the setting of essential hypertension might be useful as indicators of increased ENaC-mediated sodium absorption in the distal nephron via mechanisms independent of classic aldosterone actions. This, in turn, may predict resistance to antihypertensive strategy of MR blockade.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What is new
Coordinated actions of aldosterone and Ang II cascades underlie respective increases in the number of functionally active ENaC channels and channel open probability when dietary sodium intake is reduced.
During the pathology of Ang II-dependent hypertension, ENaC is over-active and is no longer effectively regulated by the aldosterone-MR axis.
What is relevant
ENaC activity in the distal part of the renal nephron serves as a final regulator of urinary sodium excretion. Therefore, revealing the cellular and molecular mechanisms modulating ENaC activity during systemic inputs is directly relevant to understanding normal blood pressure control and pathophysiology of hypertension. These data provide a mechanistic explanation for the resistance to MR inhibition that occurs in hypertensive subjects having elevated intrarenal Ang II levels.
Summary
Direct regulation of ENaC activity by Ang II signaling has its own non-redundant role in adaptation of ENaC-mediated sodium reabsorption in the ASDN to variations in dietary salt intake.
Chronic Ang II infusion over-stimulates ENaC beyond the physiological range of ENaC regulation contributing to the excessive renal sodium conservation and hypertension. Blockade of aldosterone-MR cascade is insufficient to suppress excessively activated ENaC when Ang II levels are elevated.
ACKNOWLEDGEMENTS
Research reported in this publication was supported by the NIH-NIDDK DK095029 (to O. P.), NIH-NIGMS P30GM103337 (to L.G.N.), S&R Foundation Ryuji Ueno award (to O.P.) and AHA-GIA-13GRNT16220002 (to O.P.)
Footnotes
DISCLOSURES None.
REFERENCES
- 1.Staruschenko A. Regulation of transport in the connecting tubule and cortical collecting duct. Compr Physiol. 2012;2:1541–1584. doi: 10.1002/cphy.c110052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khawaja Z, Wilcox CS. Role of the kidneys in resistant hypertension. Int J Hypertens. 2011;2011:143471. doi: 10.4061/2011/143471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eaton DC, Malik B, Saxena NC, Al-Khalili OK, Yue G. Mechanisms of aldosterone's action on epithelial Na+ transport. J Membr Biol. 2001;184:313–319. doi: 10.1007/s00232-001-0098-x. [DOI] [PubMed] [Google Scholar]
- 4.Garty H, Palmer LG. Epithelial sodium channels: Function, structure, and regulation. Physiol Rev. 1997;77:359–396. doi: 10.1152/physrev.1997.77.2.359. [DOI] [PubMed] [Google Scholar]
- 5.Hummler E. Epithelial sodium channel, salt intake, and hypertension. Curr Hypertens Rep. 2003;5:11–18. doi: 10.1007/s11906-003-0005-1. [DOI] [PubMed] [Google Scholar]
- 6.Schild L, Kellenberger S. Structure function relationships of enac and its role in sodium handling. Adv Exp Med.Biol. 2001;502:305–314. doi: 10.1007/978-1-4757-3401-0_20. [DOI] [PubMed] [Google Scholar]
- 7.Hansson JH, Nelson-Williams C, Suzuki H, Schild L, Shimkets R, Lu Y, Canessa C, Iwasaki T, Rossier B, Lifton RP. Hypertension caused by a truncated epithelial sodium channel gamma subunit: Genetic heterogeneity of liddle syndrome. Nat Genet. 1995;11:76–82. doi: 10.1038/ng0995-76. [DOI] [PubMed] [Google Scholar]
- 8.Hansson JH, Schild L, Lu Y, Wilson TA, Gautschi I, Shimkets R, Nelson-Williams C, Rossier BC, Lifton RP. A de novo missense mutation of the beta subunit of the epithelial sodium channel causes hypertension and liddle syndrome, identifying a proline-rich segment critical for regulation of channel activity. Proc Natl Acad Sci USA. 1995;92:11495–11499. doi: 10.1073/pnas.92.25.11495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schild L. The epithelial sodium channel: From molecule to disease. Rev.Physiol Biochem Pharmacol. 2004;151:93–107. doi: 10.1007/s10254-004-0023-7. [DOI] [PubMed] [Google Scholar]
- 10.Shimkets RA, Warnock DG, Bositis CM, Nelson-Williams C, Hansson JH, Schambelan M, Gill JR, Jr., Ulick S, Milora RV, Findling JW. Liddle's syndrome: Heritable human hypertension caused by mutations in the beta subunit of the epithelial sodium channel. Cell. 1994;79:407–414. doi: 10.1016/0092-8674(94)90250-x. [DOI] [PubMed] [Google Scholar]
- 11.Schafer JA. Abnormal regulation of enac: Syndromes of salt retention and salt wasting by the collecting duct. Am J Physiol Renal Physiol. 2002;283:F221–F235. doi: 10.1152/ajprenal.00068.2002. [DOI] [PubMed] [Google Scholar]
- 12.Frindt G, McNair T, Dahlmann A, Jacobs-Palmer E, Palmer LG. Epithelial na channels and short-term renal response to salt deprivation. Am J Physiol Renal Physiol. 2002;283:F717–F726. doi: 10.1152/ajprenal.00379.2001. [DOI] [PubMed] [Google Scholar]
- 13.Frindt G, Palmer LG. Surface expression of sodium channels and transporters in rat kidney: Effects of dietary sodium. Am J Physiol Renal Physiol. 2009;297:F1249–F1255. doi: 10.1152/ajprenal.00401.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pochynyuk O, Bugaj V, Rieg T, Insel PA, Mironova E, Vallon V, Stockand JD. Paracrine regulation of the epithelial Na+ channel in the mammalian collecting duct by purinergic P2Y2 receptor tone. J Biol Chem. 2008;283:36599–36607. doi: 10.1074/jbc.M807129200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hummler E. Implication of enac in salt-sensitive hypertension. J Steroid Biochem Mol Biol. 1999;69:385–390. doi: 10.1016/s0960-0760(99)00073-4. [DOI] [PubMed] [Google Scholar]
- 16.Stockand JD. New ideas about aldosterone signaling in epithelia. Am J Physiol Renal Physiol. 2002;282:F559–F576. doi: 10.1152/ajprenal.00320.2001. [DOI] [PubMed] [Google Scholar]
- 17.Verrey F. Transcriptional control of sodium transport in tight epithelial by adrenal steroids. J Membr Biol. 1995;144:93–110. doi: 10.1007/BF00232796. [DOI] [PubMed] [Google Scholar]
- 18.Arroyo JP, Ronzaud C, Lagnaz D, Staub O, Gamba G. Aldosterone paradox: Differential regulation of ion transport in distal nephron. Physiology.(Bethesda.) 2011;26:115–123. doi: 10.1152/physiol.00049.2010. [DOI] [PubMed] [Google Scholar]
- 19.Zaika O, Mamenko M, Staruschenko A, Pochynyuk O. Direct activation of ENaC by angiotensin II: Recent advances and new insights. Curr Hypertens Rep. 2013;15:17–24. doi: 10.1007/s11906-012-0316-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crowley SD, Coffman TM. Recent advances involving the renin-angiotensin system. Exp Cell Res. 2012;318:1049–1056. doi: 10.1016/j.yexcr.2012.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Navar LG, Kobori H, Prieto MC, Gonzalez-Villalobos RA. Intratubular renin-angiotensin system in hypertension. Hypertension. 2011;57:355–362. doi: 10.1161/HYPERTENSIONAHA.110.163519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zaman MA, Oparil S, Calhoun DA. Drugs targeting the renin-angiotensin-aldosterone system. Nat Rev Drug Discov. 2002;1:621–636. doi: 10.1038/nrd873. [DOI] [PubMed] [Google Scholar]
- 23.Seva PB, van der LN, Verdonk K, Roks AJ, Hoorn EJ, Danser AH. Key developments in renin-angiotensin-aldosterone system inhibition. Nat Rev Nephrol. 2013;9:26–36. doi: 10.1038/nrneph.2012.249. [DOI] [PubMed] [Google Scholar]
- 24.van der LN, Zietse R, Hoorn EJ. Effects of angiotensin II on kinase-mediated sodium and potassium transport in the distal nephron. Curr Opin Nephrol Hypertens. 2013;22:120–126. doi: 10.1097/MNH.0b013e32835b6551. [DOI] [PubMed] [Google Scholar]
- 25.Peti-Peterdi J, Warnock DG, Bell PD. Angiotensin II directly stimulates ENaC activity in the cortical collecting duct via AT1 receptors. J Am Soc Nephrol. 2002;13:1131–1135. doi: 10.1097/01.asn.0000013292.78621.fd. [DOI] [PubMed] [Google Scholar]
- 26.Harrison-Bernard LM, Navar LG, Ho MM, Vinson GP, El-Dahr SS. Immunohistochemical localization of ang ii at1 receptor in adult rat kidney using a monoclonal antibody. Am J Physiol. 1997;273:F170–F177. doi: 10.1152/ajprenal.1997.273.1.F170. [DOI] [PubMed] [Google Scholar]
- 27.Miyata N, Park F, Li XF, Cowley AW., Jr. Distribution of angiotensin AT1 and AT2 receptor subtypes in the rat kidney. Am J Physiol. 1999;277:F437–F446. doi: 10.1152/ajprenal.1999.277.3.F437. [DOI] [PubMed] [Google Scholar]
- 28.Mamenko M, Zaika O, Ilatovskaya DV, Staruschenko A, Pochynyuk O. Angiotensin ii increases activity of the epithelial Na+ channel (ENaC) in distal nephron additively to aldosterone. J Biol Chem. 2012;287:660–671. doi: 10.1074/jbc.M111.298919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun P, Yue P, Wang WH. Angiotensin ii stimulates epithelial sodium channels in the cortical collecting duct of the rat kidney. Am J Physiol Renal Physiol. 2012;302:F679–F687. doi: 10.1152/ajprenal.00368.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gonzalez-Villalobos RA, Seth DM, Satou R, Horton H, Ohashi N, Miyata K, Katsurada A, Tran DV, Kobori H, Navar LG. Intrarenal angiotensin II and angiotensinogen augmentation in chronic angiotensin II-infused mice. Am J Physiol Renal Physiol. 2008;295:F772–F779. doi: 10.1152/ajprenal.00019.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gonzalez-Villalobos RA, Satou R, Ohashi N, Semprun-Prieto LC, Katsurada A, Kim C, Upchurch GM, Prieto MC, Kobori H, Navar LG. Intrarenal mouse reni-nangiotensin system during Ang II-induced hypertension and ace inhibition. Am J Physiol Renal Physiol. 2010;298:F150–F157. doi: 10.1152/ajprenal.00477.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ortiz RM, Graciano ML, Seth D, Awayda MS, Navar LG. Aldosterone receptor antagonism exacerbates intrarenal angiotensin II augmentation in ang ii-dependent hypertension. Am J Physiol Renal Physiol. 2007;293:F139–F147. doi: 10.1152/ajprenal.00504.2006. [DOI] [PubMed] [Google Scholar]
- 33.Zhao D, Seth DM, Navar LG. Enhanced distal nephron sodium reabsorption in chronic angiotensin II-infused mice. Hypertension. 2009;54:120–126. doi: 10.1161/HYPERTENSIONAHA.109.133785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gonzalez AA, Liu L, Lara LS, Seth DM, Navar LG, Prieto MC. Angiotensin II stimulates renin in inner medullary collecting duct cells via protein kinase c and independent of epithelial sodium channel and mineralocorticoid receptor activity. Hypertension. 2011;57:594–599. doi: 10.1161/HYPERTENSIONAHA.110.165902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brooks HL, Allred AJ, Beutler KT, Coffman TM, Knepper MA. Targeted proteomic profiling of renal Na+ transporter and channel abundances in angiotensin II type 1a receptor knockout mice. Hypertension. 2002;39:470–473. doi: 10.1161/hy02t2.102959. [DOI] [PubMed] [Google Scholar]
- 36.Beutler KT, Masilamani S, Turban S, Nielsen J, Brooks HL, Ageloff S, Fenton RA, Packer RK, Knepper MA. Long-term regulation of enac expression in kidney by angiotensin II. Hypertension. 2003;41:1143–1150. doi: 10.1161/01.HYP.0000066129.12106.E2. [DOI] [PubMed] [Google Scholar]
- 37.Ashek A, Menzies RI, Mullins LJ, Bellamy CO, Harmar AJ, Kenyon CJ, Flatman PW, Mullins JJ, Bailey MA. Activation of thiazide-sensitive co-transport by angiotensin II in the cyp1a1-ren2 hypertensive rat. PLoS One. 2012;7:e36311. doi: 10.1371/journal.pone.0036311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prieto MC, Gonzalez AA, Navar LG. Evolving concepts on regulation and function of renin in distal nephron. Pflugers Arch. 2013;465:121–132. doi: 10.1007/s00424-012-1151-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pochynyuk O, Rieg T, Bugaj V, Schroth J, Fridman A, Boss GR, Insel PA, Stockand JD, Vallon V. Dietary Na+ inhibits the open probability of the epithelial sodium channel in the kidney by enhancing apical P2Y2-receptor tone. FASEB J. 2010 doi: 10.1096/fj.09-151506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mamenko M, Zaika O, Doris PA, Pochynyuk O. Salt-dependent inhibition of epithelial Na+ channel-mediated sodium reabsorption in the aldosterone-sensitive distal nephron by bradykinin. Hypertension. 2012;60:1234–1241. doi: 10.1161/HYPERTENSIONAHA.112.200469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zaika O, Mamenko M, O'Neil RG, Pochynyuk O. Bradykinin acutely inhibits activity of the epithelial Na+ channel in mammalian aldosterone-sensitive distal nephron. Am J Physiol Renal Physiol. 2011;300:F1105–F1115. doi: 10.1152/ajprenal.00606.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gonzalez AA, Luffman C, Bourgeois CR, Vio CP, Prieto MC. Angiotensin II-independent upregulation of cyclooxygenase-2 by activation of the (pro)renin receptor in rat renal inner medullary cells. Hypertension. 2013;61:443–449. doi: 10.1161/HYPERTENSIONAHA.112.196303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rearte B, Maglioco A, Balboa L, Bruzzo J, Landoni VI, Laborde EA, Chiarella P, Ruggiero RA, Fernandez GC, Isturiz MA. Mifepristone (ru486) restores humoral and t cell-mediated immune response in endotoxin immunosuppressed mice. Clin Exp Immunol. 2010;162:568–577. doi: 10.1111/j.1365-2249.2010.04262.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.San-Cristobal P, Pacheco-Alvarez D, Richardson C, Ring AM, Vazquez N, Rafiqi FH, Chari D, Kahle KT, Leng Q, Bobadilla NA, Hebert SC, Alessi DR, Lifton RP, Gamba G. Angiotensin II signaling increases activity of the renal Na-Cl cotransporter through a wnk4-spak-dependent pathway. Proc Natl Acad Sci USA. 2009;106:4384–4389. doi: 10.1073/pnas.0813238106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sandberg MB, Riquier AD, Pihakaski-Maunsbach K, McDonough AA, Maunsbach AB. Ang ii provokes acute trafficking of distal tubule Na+-Cl− cotransporter to apical membrane. Am J Physiol Renal Physiol. 2007;293:F662–F669. doi: 10.1152/ajprenal.00064.2007. [DOI] [PubMed] [Google Scholar]
- 46.Peti-Peterdi J. High glucose and renin release: The role of succinate and gpr91. Kidney Int. 2010;78:1214–1217. doi: 10.1038/ki.2010.333. [DOI] [PubMed] [Google Scholar]
- 47.Kobori H, Kamiyama M, Harrison-Bernard LM, Navar LG. Cardinal role of the intrarenal renin-angiotensin system in the pathogenesis of diabetic nephropathy. J Investig Med. 2013;61:256–264. doi: 10.231/JIM.0b013e31827c28bb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hanwell D, Ishikawa T, Saleki R, Rotin D. Trafficking and cell surface stability of the epithelial Na+ channel expressed in epithelial madin-darby canine kidney cells. J Biol Chem. 2002;277:9772–9779. doi: 10.1074/jbc.M110904200. [DOI] [PubMed] [Google Scholar]
- 49.Frindt G, Palmer LG. Regulation of epithelial Na+ channels by adrenal steroids: Mineralocorticoid and glucocorticoid effects. Am J Physiol Renal Physiol. 2012;302:F20–F26. doi: 10.1152/ajprenal.00480.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bailey MA, Mullins JJ, Kenyon CJ. Mineralocorticoid and glucocorticoid receptors stimulate epithelial sodium channel activity in a mouse model of cushing syndrome. Hypertension. 2009;54:890–896. doi: 10.1161/HYPERTENSIONAHA.109.134973. [DOI] [PubMed] [Google Scholar]
- 51.Nielsen J, Kwon TH, Masilamani S, Beutler K, Hager H, Nielsen S, Knepper MA. Sodium transporter abundance profiling in kidney: Effect of spironolactone. Am J Physiol Renal Physiol. 2002;283:F923–F933. doi: 10.1152/ajprenal.00015.2002. [DOI] [PubMed] [Google Scholar]
- 52.Navar LG, Prieto MC, Satou R, Kobori H. Intrarenal angiotensin II and its contribution to the genesis of chronic hypertension. Curr Opin Pharmacol. 2011;11:180–186. doi: 10.1016/j.coph.2011.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Komlosi P, Fuson AL, Fintha A, Peti-Peterdi J, Rosivall L, Warnock DG, Bell PD. Angiotensin I conversion to angiotensin II stimulates cortical collecting duct sodium transport. Hypertension. 2003;42:195–199. doi: 10.1161/01.HYP.0000081221.36703.01. [DOI] [PubMed] [Google Scholar]
- 54.Prieto-Carrasquero MC, Harrison-Bernard LM, Kobori H, Ozawa Y, Hering-Smith KS, Hamm LL, Navar LG. Enhancement of collecting duct renin in angiotensin ii-dependent hypertensive rats. Hypertension. 2004;44:223–229. doi: 10.1161/01.HYP.0000135678.20725.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Prieto-Carrasquero MC, Botros FT, Pagan J, Kobori H, Seth DM, Casarini DE, Navar LG. Collecting duct renin is upregulated in both kidneys of 2-kidney, 1-clip goldblatt hypertensive rats. Hypertension. 2008;51:1590–1596. doi: 10.1161/HYPERTENSIONAHA.108.110916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nesterov V, Dahlmann A, Krueger B, Bertog M, Loffing J, Korbmacher C. Aldosterone-dependent and -independent regulation of the epithelial sodium channel (ENaC) in mouse distal nephron. Am J Physiol Renal Physiol. 2012;303:F1289–F1299. doi: 10.1152/ajprenal.00247.2012. [DOI] [PubMed] [Google Scholar]
- 57.Gonzalez-Villalobos RA, Janjoulia T, Fletcher NK, Giani GF, Nguyen M, Riquier-Brison AD, Seth D, Fuchs S, Eladari D, Picard N, Bachmann S, Delpire E, Peti-Peterdi J, Navar LG, Bernstein KE, McDonough AA. The absence of intrarenal ace protects against hypertension. J Clin Invest. 2013;123:2011–2023. doi: 10.1172/JCI65460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shao W, Seth DM, Prieto MC, Kobori H, Navar LG. Activation of the renin-angiotensin system by a low salt diet does not augment intratubular angiotensinogen and angiotensin II in rats. Am J Physiol Renal Physiol. 2013;304:F505–F514. doi: 10.1152/ajprenal.00587.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.