

Abstract
Patients with chronic heart failure (CHF) suffer increased alveolar VD/VT (dead-space-to-tidal-volume ratio), yet they demonstrate augmented pulmonary ventilation such that arterial PCO2 (PaCO2) remains remarkably normal from rest to moderate exercise. This paradoxical effect suggests that the control law governing exercise hyperpnea is not merely determined by metabolic CO2 production (V̇CO2) per se but is responsive to an apparent (real-feel) metabolic CO2 load that also incorporates the adverse effect of physiological VD/VT on pulmonary CO2 elimination. By contrast, healthy individuals subjected to dead space loading also experience augmented ventilation at rest and during exercise as with increased alveolar VD/VT in CHF, but the resultant response is hypercapnic instead of eucapnic, as with CO2 breathing. The ventilatory effects of dead space loading are therefore similar to those of increased alveolar VD/VT and CO2 breathing combined. These observations are consistent with the hypothesis that the increased series VD/VT in dead space loading adds to as with increased alveolar VD/VT in CHF, but this is through rebreathing of CO2 in dead space gas thus creating a virtual (illusory) airway CO2 load within each inspiration, as opposed to a true airway CO2 load during CO2 breathing that clogs the mechanism for CO2 elimination through pulmonary ventilation. Thus, the chemosensing mechanism at the respiratory controller may be responsive to putative drive signals mediated by within-breath PaCO2 oscillations independent of breath-to-breath fluctuations of the mean PaCO2 level. Skeletal muscle afferents feedback, while important for early-phase exercise cardioventilatory dynamics, appears inconsequential for late-phase exercise hyperpnea.
Keywords: Chronic heart failure, Physiological dead space, Dead space loading, Alveolar dead space, Anatomical dead space, Series dead space, Parallel dead space, Whipp’s law, Comroe’s law, Fenn–Craig diagram, Exercise hyperpnea, Metabolic CO2 load, Airway CO2 load, CO2 breathing, Arterial PCO2 oscillations, Cognition, Perception
1. Laws and open questions on ventilatory control in health and in disease
1.1. Whipp’s law on ventilatory compensation for changes in physiological VD/VT
Despite more than a century of extensive and intensive research and continuing passionate debates, the mechanisms underlying the control of exercise hyperpnea in health and in disease remain far from clear. It is well established that in healthy subjects undergoing incremental exercise, the ventilatory response (in terms of total pulmonary ventilation, V̇E) increases with metabolic CO2 production (metabolic CO2 flow to the lungs, V̇CO2) according to a linear V̇E − V̇CO2 relationship over a wide range of mild-to-moderate work rates, such that arterial PCO2 (paCO2) and H+ concentration ([H+]a) are regulated homeostatically close to their resting levels throughout exercise (Wasserman, 1978; Wasserman et al., 1977, 2011). The regulation of PaCO2 by V̇E is given by the following metabolic hyperbola relationship (Table 1):
| (1) |
Table 1.
Glossary of key symbols.
| Symbol | Definition | |
|---|---|---|
| [H+]a | Arterial H+ concentration | |
| PaCO2 | Arterial PCO2 | |
| PI CO2 | Inspired PCO2 | |
| P̂I CO2 | Virtual (illusory) inspired PCO2 | |
| r | Fraction of metabolic CO2 load facing the controller that is properly attributed to the component | |
| (1 − r) | Fraction of metabolic CO2 load facing the controller that is misattributed to the component | |
| V̇CO2 | Metabolic CO2 production/metabolic CO2 flow to the lungs | |
|
|
Airway CO2 load | |
|
|
Virtual (illusory) airway CO2 load | |
|
|
Apparent (real-feel) metabolic CO2 load | |
|
|
Exogenous ‘metabolic CO2 flow to the lungs’ simulated by slug CO2 loading | |
| VD | Dead space | |
| V̇D | Wasted ventilation in the dead space | |
| VD/VT | Dead-space-to-tidal-volume ratio | |
| V̇E | Pulmonary ventilation | |
| V̇E/V̇CO2 | Ventilatory equivalent for CO2 | |
|
|
Apparent ventilatory equivalent for CO2 |
As enunciated by the late noted exercise physiologist B.J. Whipp (Whipp, 2008):
“PaCO2 regulation during exercise therefore depends on the relationship between two compound variables (the ventilatory equivalent for CO2 (V̇E/V̇CO2) and the physiological dead space fraction of the tidal volume (VD/VT), but only two! ……. In normal subjects (with little difference between anatomical (or series) and physiological dead space), VD normally increases as a linear function of VT with a positive intercept on the VT axis (Lamara et al., 1988). To regulate PaCO2 and pH, V̇E/V̇CO2, must decrease with an appropriately-proportional profile. This it does; note the positive intercept on the linear V̇E − VCO2 relationship in Fig. 1 (Whipp and Ward, 1991)! The linear V̇E − V̇CO2 relationship during exercise is therefore a result of the regulatory behavior and not a cause. In crude terms, the system seems to “know” that when VD/VT is reduced (making V̇E more efficient with respect to alveolar ventilation) V̇E “needs” to increase less per unit V̇CO2 to effect its regulatory function……. In 1991 Sue Ward and I (Whipp and Ward, 1991) thought that the appropriate core question to be resolved was that “……. although many mechanisms have been demonstrated which can increase ventilation during exercise, the essential challenge which remains is why, for moderate exercise, does ventilation only increase to levels commensurate with the level of pulmonary CO2 exchange?”……. It remains the unanswered question. Not providing the answer to the entire exercise hyperpnea but perhaps the crucial core or fundamental feature upon which factors such as volition, emotion, short-, and/or long-term potentiation, mechanical constraint and limitation, among others, provide modulating influences.”
Fig. 1.
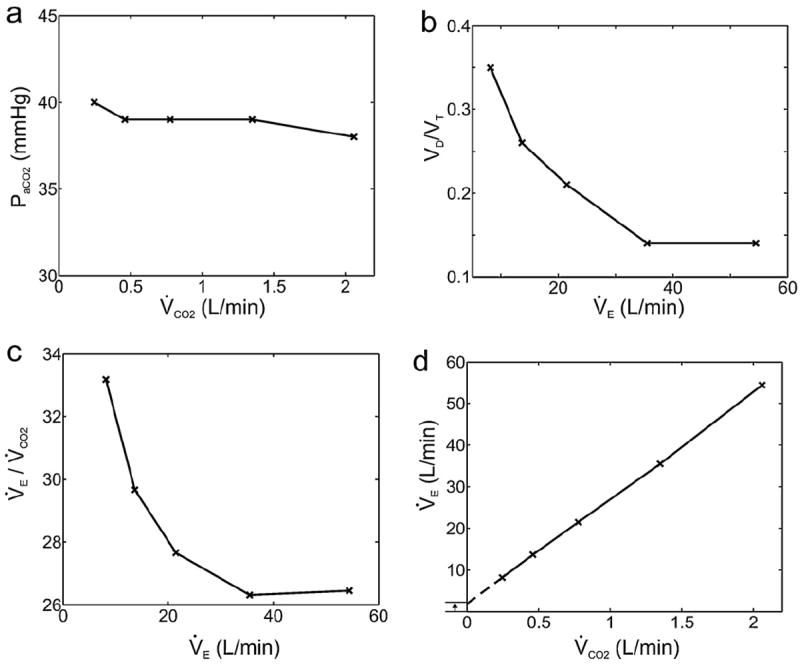
Whipp’s law for ventilatory compensation of changes in physiological VD/VT during exercise. Homeostatic regulation of PaCO2 during moderate exercise in healthy subjects (panel a) implies that decreases in physiological VD/VT with exercise (panel b) must be accompanied by corresponding decreases in V̇E/V̇CO2 (panel c). As a result, the V̇E − V̇CO2 relationship shows a positive intercept on the Y-axis (panel d). Data adapted from Table 1 in Whipp and Wasserman (1969).
Whipp’s remarks boil down to two key observations regarding PaCO2 regulation in moderate exercise: (i) V̇E seems to be controlled to compensate not only for the changes in V̇CO2 but also associated changes in physiological VD/VT; (ii) since physiological VD/VT typically decreases with increasing V̇E from rest to exercise (Asmussen and Nielsen, 1956; Jones, 1984; Lamara et al., 1988; Wasserman et al., 1967, 2005; Whipp and Wasserman, 1969), it follows that V̇E − V̇CO2 must also decrease accordingly, resulting in a positive Y-intercept in the V̇E − V̇CO2 relationship (Fig. 1). These observations are refreshing in that they represent a subtle departure from conventional wisdom. Although the interrelationships between V̇E, V̇CO2, VD/VT, PaCO2 and the slope and intercept of the V̇E − V̇CO2 relationship during exercise are well-known (Davis et al., 1980; Neder et al., 2001; Sun et al., 2002), the Y-intercept of the linear V̇E − V̇CO2 relationship has been traditionally thought of as an independent parameter that is integral to the control law for PaCO2 regulation in order to compensate for the “wasted ventilation” (V̇D = VD · f = V̇E · VD/VT, where f is respiratory frequency) (Davis et al., 1980). In contrast, Whipp (2008) cogently reckoned the positive Y-intercept as a dependent parameter that is secondary to a mechanistic coupling of V̇E to changes in physiological VD/VT during exercise and asked the critical “unanswered question” (herein referred to as Whipp’s law2; Table 2): why is V̇E always increased just enough to eliminate the CO2 produced during exercise in the face of the attendant changes in physiological VD/VT? This is an intriguing paradox as VD/VT is just a mathematical parameter that is dependent on controller output (in terms of VT) instead of input, and is not a physiologic signal per se. Unbeknownst to Whipp, however, the system’s seeming uncanny ability “to “know” that when VD/VT is reduced V̇E “needs” to increase less per unit V̇CO2” points squarely to the tantalizing possibility that emergent cognition and perception at the respiratory controller might indeed be part and parcel to ventilatory control. Core to Whipp’s question is hence: what exactly is the controller supposed to “know” and what it is not, and how so?
Table 2.
Ventilatory control laws for muscular exercise and CO2 breathing.
| Ventilatory control law | Statement of law |
|---|---|
| Whipp’s law (2008) | During moderate exercise, V̇E increases only to levels commensurate with the level of pulmonary CO2 exchange required for PaCO2 regulation. In particular, the system seems to “know” that when VD/VT is reduced, V̇E “needs” to increase less per unit V̇CO2 to effect its regulatory function |
| Comroe’s law (1965) | The lung is designed to eliminate CO2 in a CO2-free medium, air. When CO2 is added to the inspired air, it clogs the mechanism for CO2 elimination, and PaCO2 must rise |
1.2. Comroe’s law on clogging of CO2 elimination during CO2 breathing
In the 15 December 2011 issue of RPNB (179:2–3), Whipp’s unanswered question is revisited by several authors directly or indirectly, each bringing interesting new insights to the table yet all with divergent viewpoints. Contrasting the exquisite [H+]a/PaCO2 homeostasis in healthy subjects during exercise (Wasserman et al., 2011) with the apparent breakdown of such homeostatic regulation in the classic hypercapnic ventilatory response during CO2 breathing (Duffin, 2005), Poon (2011) suggests that the latter condition may reflect a shift of equilibrium in an underlying ‘homeostatic competition’ between the respiratory controller’s conflicting goals to minimize both the chemical and mechanical costs (or “discomforts”) of breathing (among other homeostatic and non-homeostatic goals that compete for use/disuse of the respiratory apparatus). A centerpiece of Poon’s proposition is a fundamental principle first put forward by the famed cardiorespiratory physiologist J.H. Comroe, Jr. (Comroe, 1965):
“The lung is designed to eliminate CO2 in a CO2-free medium, air. When CO2 is added to the inspired air, it clogs the mechanism for CO2 elimination, and arterial CO2 must rise.”
This principle (herein referred to as Comroe’s law, Table 2) delineates the all-too-obvious (yet oft-forgotten) adverse effects of inspired PCO2 (PI CO2) on CO2 elimination and PaCO2 homeostasis, as described by a more general form of Eq. (1) (for PI CO2 > 0):
| (2) |
In Eq. (2), PaCO2 is always augmented by the term PI CO2, which disrupts the normal regulation of PaCO2 through the coupling of V̇E to V̇CO2 (and VD/VT) when PI CO2 = 0. To understand how this disruption may clog the CO2 elimination mechanism, we propose a novel pulmonary CO2 exchange variable called airway CO2load , defined herein as airway CO2 flow to the lungs that clogs CO2 elimination through increased V̇E:
| (3) |
Equation (3) shows that for any PI CO2 > 0, increases directly with V̇E, making it harder and not as cost-effective to eliminate CO2 through pulmonary ventilation than when PI CO2 = 0. For low levels of inhaled CO2 (<1%) this CO2-clogging effect is negligible and PaCO2 can be effectively kept at close to the eucapnic level with only moderate increases in V̇E necessary (Anthonisen and Dhingra, 1978; Fordyce et al., 1984; Reischl and Stavert, 1982) especially with the expected simultaneous decrease in physiological VD/VT with increasing V̇E. However, for inhaled CO2 levels > ~3% the CO2-clogging effect becomes increasingly challenging and the controller must now balance the benefit of maintaining PaCO2 homeostasis against the mounting respiratory effort required to cope with the elevated level. For inhaled CO2 levels > ~5%, the prohibitive chemical constraints imposed by Eqs. (2) and (3) make it physically impossible for the controller to maintain PaCO2 homeostatically at the eucapnic level even with V̇E → ∞, hence PaCO2 must rise regardless of the level of respiratory effort (see Fig. 2 in (Poon, 2011)).
This observation led the venerable respiratory physiologist W.O. Fenn3 (Fenn and Craig, 1963) to view the conventional ‘CO2 response curve’ as ‘CO2 tolerance curve’ in that, rather than “responding” to a hypercapnic stimulus, the controller may simply choose to tolerate a rise of the PaCO2 and [H+]a levels in order to curb the excessive respiratory effort (or discomfort) necessary to restore eucapnia in the face of severe chemical constraints imposed by CO2 breathing (see also Section 4.3 below). From this perspective, the classic hypercapnic ventilatory response is at its core not a simple stimulus-response (dose-response) relationship in the Sherringtonian sense as traditionally thought (Cunningham et al., 1986; Grodins et al., 1954). Instead, it appears to reflect a prudent self-imposed “permissive hypercapnia” on the part of the controller with a measured breakdown of PaCO2 homeostasis in order to conserve the work of breathing in the face of severe CO2-clogging effects caused by CO2 breathing as per Comroe’s law, as described by the homeostatic competition model (Poon, 2009, 2010, 2011; Poon et al., 2007; Tin et al., 2010). Put in another way, not only does the controller seem to “know” that V̇E “needs” to track V̇CO2 and changes in VD/VT during exercise in order to regulate PaCO2 as per Whipp’s law, it also seems to “know” that whenever the CO2 elimination mechanism is clogged during CO2 breathing as per Comroe’s law, V̇E “needs” to increase less than required to maintain PaCO2 homeostasis in order to ease the work of breathing, as homeostatic regulation of PaCO2 would be difficult or no longer feasible in this case.
1.3. Ten open questions on ventilatory control in health and in disease
In the same issue of RPNB Paoletti et al. (2011) document the first attempt to correlate the exercise V̇E response and the severity of emphysema in patients with chronic obstructive pulmonary disease (COPD). They show that while these patients generally exhibited a steeper V̇E − V̇CO2 slope than normal, the V̇E − V̇CO2 slope decreased progressively as the emphysema became more severe. These authors postulate that heightened mechanical limitation in the more severe emphysema group (as indicated by a decreased forced expiratory volume in 1 s, FEV1, relative to the tidal volume attained at peak exercise) may have limited the ability to increase V̇E in response to the increasing metabolic demand during incremental exercise. In an accompanying commentary on the Paoletti et al. (2011) paper, Agostoni et al. (2011) point out that patients with chronic heart failure (CHF) also suffer decreased lung diffusion capacity and abnormal spirometry with impaired lung mechanics and expiratory flow limitation during exercise, but unlike COPD patients, the V̇E − V̇CO2 slope increases (instead of decreases) with increasing severity of the disease. Furthermore, they note that adding a large external dead space (dead space loading, or tube breathing) increases the Y-intercept of the V̇E − V̇CO2 relationship in CHF patients during exercise, whereas COPD patients with more severe emphysema also show a higher Y-intercept. They attribute the larger Y-intercepts in these cases to the corresponding larger wasted ventilation in the dead space. In contrast, Wood et al. (2011) report that the V̇E − V̇CO2 slope is increased under dead space loading in healthy women subjects, in accordance with previous findings in male subjects in whom PaCO2 was carefully measured to assess the effects of dead space loading and CO2 breathing on both the V̇E − V̇CO2 slope and Y-intercept (Poon, 1992b, 2008). More importantly, the study of Poon (1992b) showed that V̇E was higher and the V̇E − V̇CO2 slope steeper in dead space loading than in CO2 breathing at similar PaCO2 levels, while the Y-intercept was similarly increased in both.
On a separate front, Jensen et al. (2011) show that dead space loading in healthy subjects during exercise is associated with an earlier onset of intolerable dyspnea (increased exertional dyspnea with concomitant reductions in exercise tolerance) with consistent increases in PET CO2 (end-tidal PCO2), V̇E, VT and f, while the efficacy of neuromuscular and neuro-ventilatory coupling remaining relatively preserved. They attribute the dead space loading-induced increase in exertional dyspnea to increased corollary discharge reflecting the central motor command output to the respiratory muscles, or increased respiratory afferent feedback reflecting the ventilatory output and/or contractile respiratory muscle force/pressure generation, or both. In contrast, Izumizaki et al. (2011) argue that the threshold for dyspnea sensation during CO2 re-breathing is correlated to the response threshold of the breathing frequency instead of VT.
The embarrassment of riches in such extensive coverage on ventilatory control in health and in disease in a single issue of RPNB is remarkable but also leaves more questions than answers (Table 3). The ten open questions listed in Table 3 may be roughly divided into two classes: Q1-Q5 pertain to the controller’s response to disturbances primarily in pulmonary gas exchange (chemical plant) whereas Q6-Q10 involve significant disturbances in respiratory mechanics (mechanical plant) also. These critical questions defy satisfactory answers in terms of the classical chemoreflex model or homeostatic regulation model of ventilatory control in a consistent manner. In what follows, we present a general framework for chemical control of breathing that unifies and extends Whipp’s law and Comroe’s law regarding the effects of physiological VD/VT and inhaled CO2 on the homeostatic regulation of PaCO2. We show that Q1–Q5 can be satisfactorily and consistently explained within this new unifying framework. Our results provide strong evidence indicating that the chemosensing mechanism at the controller is endowed with cognition and perception capabilities that may be responsive to putative drive signals mediated by within-breath PaCO2 oscillations, a form of dynamic chemoreceptor signaling which has been suggested to play an important role in the control of V̇E independent of breath-to-breath fluctuations in the mean PaCO2 level (Band et al., 1980; Collier et al., 2008; Cross et al., 1982; Cunningham et al., 1973; Saunders, 1980; Yamamoto, 1960; Yamamoto, 1962). Preliminary findings of this work have been presented in abstract form (Poon, 2013a, b). Extension of the proposed framework to include mechanical plant abnormalities (Q6–Q9) will be addressed in sequel.
Table 3.
Open questions of ventilatory control in health and in disease.
| Chemical plant abnormalitiesa | |
| Q1 | Why is the V̇E − V̇CO2 slope higher in CHF than normal, more so with increasing severity of CHF? |
| Q2 | Why is the Y-intercept of the V̇E − V̇CO2 relationship unchanged with increasing severity of CHF? |
| Q3 | Why are both the V̇E − V̇CO2 slope and Y-intercept increased with dead space loading whereas only the V̇E − V̇CO2 slope is increased in CHF? |
| Q4 | Why are the resting and exercise ventilatory effects of CHF eucapnic whereas those of dead space loading hypercapnic with constant elevated PaCO2 from rest to exercise? |
| Q5 | Why is V̇E higher and the V̇E − V̇CO2 slope steeper in dead space loading than in CO2 breathing with PaCO2 held at similar hypercapnic levels? |
| Mechanical plant abnormalitiesb | |
| Q6 | How does the increased exertional dyspnea during dead space loading and CO2 breathing influence the control of exercise hyperpnea? |
| Q7 | Why is the V̇E − V̇CO2 slope increased in COPD, but it decreases with increasing severity of emphysema? |
| Q8 | Why does the Y-intercept of the V̇E − V̇CO2 relationship become higher with more severe emphysema? |
| Q9 | How do respiratory mechanical limitations influence the control of exercise hyperpnea in COPD? |
| Q10 | How do abnormal pulmonary gas exchange and abnormal respiratory mechanics conspire to modulate V̇E at rest and during exercise in COPD? |
Ventilatory control in patients with CHF is influenced primarily by increased physiological VD/VT (chemical plant abnormalities). Although CHF patients also suffer increased work of breathing and expiratory flow limitation (mechanical plant abnormalities), V̇E is well defended by switching to a rapid, shallow breathing pattern at rest and during exercise to maintain normal PaCO2 (Agostoni et al., 2002; Cross et al., 2012).
Ventilatory control in patients with COPD is influenced by both increased pulmonary ventilation/perfusion mismatch and increased work of breathing and expiratory flow limitation. In severe cases, the increases in work of breathing and expiratory flow limitation become so intense that V̇E can no longer be defended by switching to a rapid shallow breathing pattern at rest and during exercise, and hypercapnia ensues (Paoletti et al., 2011). In this event ventilatory control is influenced by abnormalities in both the chemical and mechanical plants.
2. Eucapnic augmented exercise hyperpnea in CHF: effect of apparent metabolic CO2 load
2.1. Exercise hyperpnea relationship in CHF
According to Whipp’s law, the regulation of PaCO2 (Fig. 1a) through the interplay between physiological VD/VT and V̇E/V̇CO2 (Figs. 1b, c) accounts for the small positive Y-intercept in the linear V̇E − V̇CO2 relationship in healthy subjects (Fig. 1d). By the same token, one would expect that under conditions where physiological VD/VT is increased (making V̇E less efficient with respect to alveolar ventilation) the controller would also “know” that V̇E “needs” to increase more per unit V̇CO2 to effect its regulatory function; hence the isocapnic V̇E − V̇CO2 slope should increase. This is precisely the case in CHF patients in whom physiological VD/VT is significantly increased as a result of increased pulmonary ventilation/perfusion mismatch and in some cases, also a result of the accompanying decreased VT with a tachypneic breathing pattern (Johnson, 2000, 2001b; Robertson, 2011; Sue, 2011; Wasserman et al., 1997; Woods et al., 2010) (Fig. 2 Fig. 2a1).4 In these patients, PaCO2 remains remarkably well regulated both at rest and during exercise through increases in V̇E/V̇CO2 and steepening of the V̇E − V̇CO2 slope that correlate directly with the severity of CHF (Buller and Poole-Wilson, 1990; Mezzani et al., 2009; Wasserman et al., 1997), with corresponding increases in alveolar (parallel) VD/VT accounting for much of the increases in V̇E − V̇CO2 slope while increases in anatomical (series) VD/VT (secondary to decreased VT) accounting for the remainder (Buller and Poole-Wilson, 1990; Wensel et al., 2004).5 It follows that question Q1 regarding the increased V̇E − V̇CO2 slope in CHF is a direct corollary to Whipp’s law under an increased (instead of decreased) physiological VD/VT. Such VD/VT -dependent augmentation of exercise hyperpnea is of considerable clinical significance, as a steep V̇E − V̇CO2 slope in moderate exercise or high V̇E/V̇CO2 ratio at maximal exercise has been suggested to be a strong predictor of poor prognosis in patients with CHF (Chua et al., 1997; Kleber et al., 2000; Ponikowski et al., 2001b).
Fig. 2.
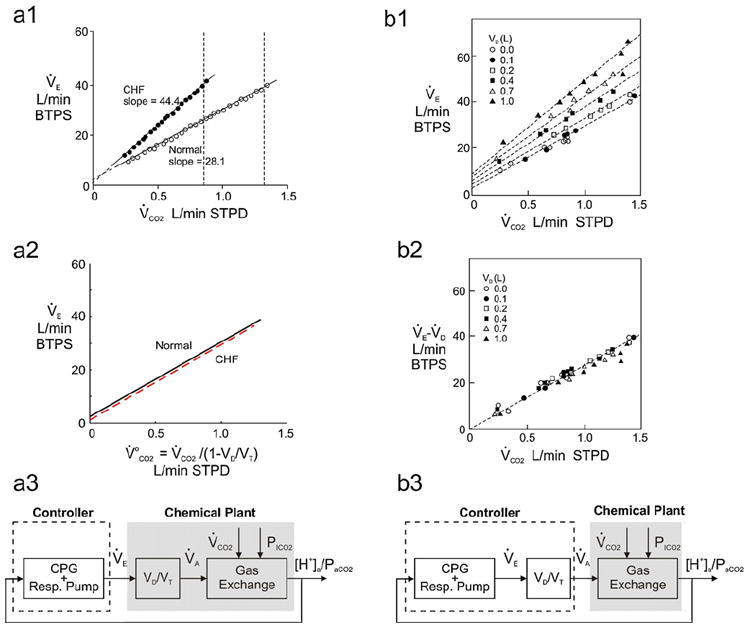
Apparent metabolic CO2 load in chronic heart failure (CHF) and dead space loading. a1: Linear V̇E − V̇CO2 relationship in a healthy subject and a CHF patient from rest to moderate exercise. The V̇E − V̇CO2 slope in CHF is significantly larger as a result of increased physiological (mainly alveolar) VD/VT, without any appreciable change in Y-intercept. Vertical broken lines indicate the corresponding respiratory compensation points when the linear V̇E − V̇CO2 relationship begins to curve upwards. Adapted from Mezzani et al. (2009) with permission. a2: In both healthy subject and CHF patient, exercise V̇E is tightly coupled to apparent metabolic CO2 load . a3: Control system block diagram corresponding to the control law shown in panel a2. b1: Dead space loading increases the slope as well as Y-intercept of the V̇E − V̇CO2 relationship in healthy subjects. b2: “Alveolar ventilation” (=V̇E − V̇D) is tightly coupled to V̇CO2 for varying sizes of external dead space. Panels b1 and b2 are adapted from Ward and Whipp (1980) with permission. b3: Control system block diagram corresponding to the control law shown in panel b2.
Furthermore, if the Y-intercept of the V̇E − V̇CO2 relationship in healthy subjects is indeed caused by a decrease in physiological VD/VT from rest to exercise (Whipp’s law) then one would expect that the Y-intercept should be small in cases where physiological VD/VT decreases to a lesser extent with increasing V̇E. This is indeed the case again for patients with CHF (Fig. 2a1) in whom the elevated physiological VD/VT decreases only slightly from rest to exercise (Sullivan et al., 1988; Wasserman et al., 2005). The relative stability of physiological VD/VT from rest to exercise in these patients is consistent with the fact that the elevated physiological VD/VT is dominated by the alveolar instead of anatomical component (Buller and Poole-Wilson, 1990; Wensel et al., 2004) and that the increase in exercise V̇E is achieved by increasing f more than VT (Agostoni et al., 2002). In these patients the Y-intercept of the V̇E − V̇CO2 relationship remains relatively small and does not increase appreciably with increasing severity of CHF (Buller and Poole-Wilson, 1990; Kleber et al., 2000; Mezzani et al., 2009; Wasserman et al., 1997). It follows that question Q2 regarding the small Y-intercept of the V̇E − V̇CO2 relationship in CHF is again a corollary to Whipp’s law reflecting the relative stability of physiological VD/VT from rest to exercise in this condition.
2.2. Apparent metabolic CO2 load vs. metabolic CO2 flow to the lungs
These observations suggest that in healthy subjects as well as patients with CHF, the V̇E response to moderate exercise is determined not only by V̇CO2 but also by the inherent overhead (in proportion to physiological VD/VT) which V̇E must overcome before CO2 elimination could take place. To account for both these effects collectively, we define a novel CO2 exchange variable called apparent (or real-feel) metabolic CO2 load as:6
| (4) |
Equation (4) represents the overall challenge facing the controller for elimination of metabolic CO2 through the act of breathing. In the ideal case where VD/VT = 0 (100% CO2 exchange efficiency), we have , i.e., the apparent metabolic CO2 load equals the actual metabolic CO2 flow to the lungs; whereas as VD/VT → 1 (0% efficiency), and the apparent metabolic CO2 load becomes prohibitive even though V̇CO2 is finite. For intermediate values of physiological VD/VT, is always > V̇CO2. Put in another way, assuming that the controller cannot distinguish whether an increase in the overall challenge for metabolic CO2 elimination is caused by an increase in V̇CO2 or in physiological VD/VT per se, then represents the apparent ‘metabolic CO2 flow to the lungs’ faced by the controller as though physiological VD/VT = 0, when it is not. The proposed definition of apparent metabolic CO2 load in terms of instead of V̇CO2 rectifies Whipp’s paradox: rather than “knowing” that whenever physiological VD/VT is reduced then V̇E “needs” to increase less per unit V̇CO2 to effect its regulatory function, the controller may actually be totally oblivious to any changes in physiological VD/VT per se and may simply respond to the (subliminally) perceived changes in to effect its regulatory function (Fig. 2a3).
Hence, for healthy subjects and patients with CHF, exercise V̇E (with PI CO2 = 0) is tightly coupled to instead of V̇CO2, with an apparent ventilatory equivalent for CO2 defined herein as:
| (5) |
From Eq. (5), it follows that a tight coupling with constant will ensure that PaCO2 is closely regulated during moderate exercise, no matter any changes in V̇CO2 and/or physiological VD/VT (Fig. 2a2).
2.3. Apparent metabolic CO2 load vs. ‘muscle hypothesis’ and ‘CO2 set point hypothesis’
The controller’s remarkable ability in compensating for both increases and decreases in physiological VD/VT large and small at rest and during exercise in healthy subjects and CHF patients indicates that exercise V̇E is probably coupled to instead of V̇CO2 or the act of exercise per se. In particular, the demonstrated dependence of the V̇E response on changes in physiological VD/VT during exercise (Whipp’s law) suggests that V̇E is not simply driven by putative skeletal muscle ‘ergoreceptors’ (particularly metaboreceptors) which reportedly become hyperactive in CHF (‘muscle hypothesis’) (Grieve et al., 1999; Olson et al., 2010; Piepoli et al., 1996; Piepoli et al., 1999; Scott et al., 2000). The latter studies relied mostly on the technique of regional circulatory occlusion which was applied proximal to the working muscles post-exercise to trap ischemic muscle metabolites in order to delay the decay of V̇E in the recovery period as a means of indirectly inferring metaboreceptor contribution to exercise hyperpnea. This experimental approach (from classical studies of the exercise pressor reflex (Alam and Smirk, 1937)) is fraught with many pitfalls. Indeed, others using a similar approach in CHF patients have reported the lack of such post-exercise stimulation of V̇E and mean arterial pressure, cautioning instead that the occlusion alone could potentially produce a reflex cardioventilatory response via activation of nociceptive pathways (Francis et al., 1999; Middlekauff and Sinoway, 2007). Accumulating evidence in the literature indicates that nociceptive metaboreceptor activation by lactic acid (dissociated into lactate and proton) and other painful anaerobic metabolites (even at trace levels) may contribute importantly to the post-exercise hyperpnea and pressor response dynamics induced by regional circulatory occlusion regardless of whether the pain sensation reaches conscious levels (Appendix A). Post hoc analysis of recent data from exercising healthy humans after group III/V skeletal muscles afferents blockade (Amann et al., 2010, 2011a; Amann et al., 2009; Amann et al., 2011b) reveals that the effects of such afferent feedbacks are short-lived and are not responsible for the late-phase V̇E and cardiovascular responses to sustained exercise (see Appendix B and Fig. S1 in Supplementary Material). Clearly, afferents integration at the controller is not simply an algebraic sum of all individual reflexes. Furthermore, it is far from clear how metaboreceptor and mechanoreceptor activities in selected working muscles with differing response sensitivities (Carrington et al., 2004) might be calibrated so precisely as to compensate for any instantaneous or chronic changes in physiological VD/VT in both healthy subjects and CHF patients in order to maintain PaCO2 homeostasis continually at rest and during exercise in conformance with Whipp’s law. Such robust calibration of the VD/VT -dependent exercise stimulus (if any) would likely occur centrally in the controller instead of peripherally in isolated working muscle receptors if PaCO2 homeostasis is to be maintained regardless of which muscle groups are being activated.
Another possible explanation of the tight coupling of V̇E to instead of V̇CO2 is the hypothesis that [H+]a/PaCO2 are regulated homeostatically around some set point via [H+]a/PaCO2-sensing peripheral chemoreceptors independent of changes in V̇CO2 or VD/VT (Wasserman et al., 2011). However, previous studies have shown that peripheral chemoreceptors serve only to shift the apparent [H+]a/PaCO2 set point and speed up the early-phase exercise ventilatory response dynamics but otherwise are not obligatory for PaCO2 regulation from rest to exercise in the steady state (late phase). For example, PaCO2 is well regulated at rest and during steady-state exercise even after peripheral chemosensitivity is suppressed by hyperoxia (Griffiths et al., 1986; Miyamoto and Niizeki, 1995) or after recovery from bilateral carotid bodies resection (Lugliani et al., 1971; Wasserman et al., 1975). In the latter case, PaCO2 homeostasis is well maintained post-surgery except in patients with pronounced respiratory mechanical limitations as indicated by a significant reduction of FEV1.0 (Honda et al., 1979; Whipp and Ward, 1992). Although increases of central and peripheral chemosensitivities have been reported in CHF patients as a possible mechanism for the augmented exercise hyperpnea (Chua et al., 1996; Narkiewicz et al., 1999; Ponikowski et al., 2001a), the finite magnitudes of such augmented chemosensitivities are still far from those necessary to sustain a tight CO2 set point for the maintenance of [H+]a/PaCO2 homeostasis during moderate exercise in those patients. Indeed, the CO2 set point hypothesis is contradicted by the fact that such presumptive set point during exercise is highly volatile and readily abolished during CO2 breathing in healthy subjects and CHF patients alike as per Comroe’s law.
3. Hypercapnic augmented exercise hyperpnea in dead space loading: effect of virtual airway CO2 load
3.1. Exercise hyperpnea relationship in dead space loading
In CHF, apparent metabolic CO2 load is augmented primarily by increases in parallel (alveolar) VD/VT and secondarily by increases in series (anatomical) VD/VT (see footnote 4). In both healthy subjects and CHF patients, series VD/VT can be exaggerated by dead space loading (tube breathing). The resultant augmentation in relative to V̇CO2 explains the increase in V̇E − V̇CO2 slope that is typical during dead space loading (Poon, 1992b, 2008; Ward and Whipp, 1980; Wood et al., 2011) (Fig. 2b1). Furthermore, since the impact of the dead space load is bound to diminish with increasing V̇E (and hence VT), the Y-intercept of the V̇E − V̇CO2 relationship should also increase, as per Whipp’s law. The interplay between series VD/VT, V̇E and again explains why the V̇E − V̇CO2 relationship under dead space loading is characterized by increases in both the slope and Y-intercept (Fig. 2b1), as demonstrated experimentally in healthy subjects (Poon, 1992a,b, 2008; Ward and Whipp, 1980) and CHF patients (Agostoni et al., 2011). This is in sharp contrast to the V̇E − V̇CO2 relationship in CHF patients breathing freely (without dead space load), where the increase in V̇E − V̇CO2 slope with increasing severity of CHF is without corresponding increases in the Y-intercept (Fig. 2a1). It follows that question Q3 regarding the increases in V̇E − V̇CO2 slope and Y-intercept during dead space loading (Table 3) again can be accounted for at least in part by Whipp’s law (although other factors specific to dead space loading cannot be excluded; see Section 4.1 below).
Indeed, in Ward and Whipp (1980) it is shown that when the V̇E response is corrected for the “wasted ventilation” (V̇D) for varying sizes of the external VD, the corresponding “alveolar ventilation” (V̇E − V̇D) vs. V̇CO2 relationships become closely clustered (Fig. 2b2). It is important to note that although the plots in Fig. 2b2 are mathematically equivalent to that shown in Fig. 2a2, there are fundamental differences between them. Specifically, Fig. 2b2 implies that alveolar ventilation is the ultimate control variable and that V̇CO2 is a prime determinant of this control variable. This notion assumes that the controller necessarily “knows” the values of VD/VT at rest and during exercise (Whipp, 2008) in order to accurately subtract V̇D from V̇E throughout (Fig. 2b3). Similarly, Mitchell (1990) proposed a feedforward controller model for dead space loading in which exercise V̇E was directly driven by V̇CO2 with a gain that was inversely proportional to both the resting PaCO2 level and resting value of the parameter (1 − VD/VT), such that the controller not only must “know” the resting VD/VT value but must “remember” it closely throughout exercise in order to achieve PaCO2 regulation. The assumption of the controller’s rigid adherence to the resting VD/VT value regardless of any breathing pattern-dependent changes in total (physiological and external) VD/VT throughout exercise is at variance with Whipp’s law. In contrast, Fig. 2a2 depicts the control of V̇E relative to , which incorporates the overall challenge imposed by V̇CO2 as well as the adverse effect of the total (series + parallel) VD/VT on pulmonary CO2 elimination, without the controller’s explicit knowledge of the total VD, V̇D or VD/VT per se (Fig. 2a3). As such, the relationship in Fig. 2a2 provides a more accurate representation of the control law in that the VD/VT term (series and/or parallel) is properly attributed to the chemical plant equation instead of the controller equation itself (cf. Fig. 2a3 and 2b3). As elaborated below, the controller may indeed respond differently to changes in series and parallel VD/VT in the chemical plant.
3.2. Ventilatory effects of dead space loading and CHF: differences between series and parallel dead space
In the literature, series and parallel (alveolar) VD are often conflated with one another and their adverse effects on pulmonary gas exchange are represented collectively by the total (series + parallel) VD/VT in an indiscriminate manner. In practice, series VD differs from parallel VD in several respects, such as the decrease in series VD/VT with exercise hyperpnea and increase in alveolar VD/VT with CHF, as noted above. An increase in series VD may actually result in a corresponding decrease in parallel VD by minimizing overall pulmonary ventilation/perfusion heterogeneity (Petrini et al., 1983; Ross and Farhi, 1960). Importantly, although series and parallel VD both impair pulmonary gas exchange by increasing the total VD/VT, only series VD involves rebreathing of CO2 in dead space gas. The rebreathing of CO2 is reminiscent of CO2 breathing, which is subject to Comroe’s law instead of Whipp’s law (Table 2). Arguably, it would be difficult (if not impossible) for the controller to distinguish inspired and rebreathed CO2, since both of them are introduced via the airways. Indeed, CO2 rebreathing is commonly used as a substitute for CO2 breathing for testing the hypercapnic ventilatory response. This observation leads us to surmise that, much like the distinct ventilatory responses to CO2 breathing and increased V̇CO2 during exercise, the controller may respond to increases in series and parallel VD/VT differently if it is somehow also influenced by the CO2 rebreathing process per se that is unique to series VD, independent of the corresponding increase in VD/VT that is common to both.
Indeed, increases in series and parallel VD (with dead space loading and CHF respectively) have been found to result in subtly different ventilatory effects both at rest and during exercise (Johnson, 2001a; Poon, 2001). Specifically, although dead space loading and CHF both cause an increase in VD/VT with a corresponding steepening of the V̇E − V̇CO2 slope, the resultant response remains eucapnic for CHF (Johnson, 2000, 2001b; Wasserman et al., 1997) but becomes hypercapnic for dead space loading—although the resultant elevated PaCO2 remains relatively constant (isocapnic at a hypercapnic level) from rest to moderate exercise in this case (Poon, 1992b, 2008; Ward and Whipp, 1980; Wood et al., 2011). Such ‘hypercapnic regulation’ of exercise hyperpnea under dead space loading reveals a paradoxical dual character of dead space loading: its ventilatory effects resemble those of increased alveolar dead space and CO2 breathing combined. Why does dead space loading induce hypercapnia whereas CHF patients with increased alveolar VD/VT are able to maintain eucapnia at rest and during exercise in conformance with Whipp’s law (Q4, Table 3)? Could the hypercapnic effect of dead space loading be a consequence of Comroe’s law instead?
3.3. Virtual airway CO2 load vs. apparent metabolic CO2 load in dead space loading
Like CO2 breathing, small dead space loads such as anatomical and apparatus VD do not necessarily clog CO2 elimination since the resultant “wasted ventilation” can be compensated for readily by increasing V̇E provided it is not excessive. In long-necked animals such as giraffes and swans, the increased anatomical VD is compensated for by a slow-and-deep breathing pattern such that physiological VD/VT remains relatively unchanged compared with similar-sized animals (Bech and Johansen, 1980; Hugh-Jones et al., 1978; Mitchell and Skinner, 2011). In this case, the increased anatomical VD affects primarily , and Whipp’s law applies as noted above. On the other hand, when the size of the added series VD exceeds the subject’s vital capacity, it becomes physically impossible for the controller to clear the dead space even with maximal respiratory effort, and total CO2 rebreathing inevitably ensues in a manner similar to conventional rebreathing procedures for measuring the ventilatory CO2 sensitivity (Berkenbosch et al., 1989; Duffin, 2011; Read, 1967). In this event CO2 elimination is clogged by mechanical (instead of chemical) limitations of the respiratory apparatus. Furthermore, since CO2 elimination is now completely abolished no matter the V̇E level, PaCO2 must continue to rise indefinitely.
For any sizable dead space load that falls between these extremes, CO2 elimination is not clogged but is impaired by an elevated series VD/VT with resultant increase in , as with elevated parallel VD/VT in CHF. However, because the increase in VD/VT now comes with rebreathing of dead space CO2, the controller may (at the beginning of each inspiration) mistake the dead space load for an airway CO2 load (at PI CO2 ≈ PaCO2) that remains until the dead space is cleared toward late inspiration. This ambiguity may render the controller with an illusion of a virtual PI CO2 (P̂I CO2) with resultant virtual (or illusory) airway CO2 load and corresponding underestimation of given by:
| (6) |
| (7) |
| (8) |
where 0 < r < 1 is the fraction of the total CO2 load [= V̇CO2 /(1 − VD/VT)] facing the controller under dead space loading that is properly attributed to the component and (1 − r) is the balance that is misattributed to the component.
It is important to emphasize that the notion of virtual airway CO2 load (Eq. (7)) is a novel concept to depict the controller’s illusion of CO2 breathing from a phantom CO2-containing external environment, in this case due to the rebreathing of dead space gas.7 Since the latter has a PCO2 level ≈ PaCO2 that necessarily decreases with increasing V̇E on a breath-to-breath basis, such rebreathed CO2 does not really clog CO2 elimination like CO2 breathing with PI CO2 > 0 does (Eq. (3)). Hence strictly speaking, a dead space load should contribute only to instead of , as with increased parallel VD/VT in CHF. However, it is possible that the controller may confuse rebreathed CO2 with inhaled CO2 and treat them alike, since both of them are introduced via the airways. Such an “identity mix-up” is possible provided the respiratory chemosensing mechanism at the controller is responsive to dynamic chemoreceptor signaling mediated by within-breath PaCO2 oscillations rather than (or in addition to) breath-to-breath fluctuations of the mean PaCO2 level, as suggested previously by Yamamoto and others (Band et al., 1980; Collier et al., 2008; Cross et al., 1982; Cunningham et al., 1973; Saunders, 1980; Yamamoto, 1960; Yamamoto, 1962). In other words, the controller is likely “tricked” by the rebreathed CO2 and may mistake it for inhaled CO2 if the controller is “short-sighted” and reacts instinctively to the onrush of rebreathed CO2 during each inspiration rather than “take a long view” to see that such rebreathed CO2 does not really clog the CO2 elimination mechanism on a breath-to-breath basis as inhaled CO2 does.
Hence from Eqs. (6)-(8), Eq. (2) may be rewritten as:
| (9) |
Equation (9) portraits the input–output relationship at the chemical plant as perceived by the controller under dead space loading (where r < 1 with P̂I CO2 > 0 and ), as compared to increased parallel VD/VT in CHF (where r ≈ 1 with , see also Eq. (1)). This model provides a unified framework for predicting how the ventilatory response to dead space loading at rest and during exercise may be influenced by Comore’s law and Whipp’s law (or both) depending on the index r: for r = 1, we have P̂I CO2 = 0 (from Eq. (6)) and Whipp’s law prevails as all of the rebreathed CO2 is properly attributed by the controller to as with increased parallel VD/VT in CHF whereas for r = (1 − VD/VT), Comroe’s law prevails as all of the rebreathed CO2 is misattributed to (Eq. (7)). Between these extremes where (1 − VD/VT) < r < 1, is underestimated by a factor of r with the remainder being misattributed to . The resultant ventilatory response in this case is determined by the differential effects of and . The emergence of the illusory percepts of P̂I CO2 and with corresponding underestimation of for r < 1 provides a plausible explanation of the paradox Q4 (Table 3) regarding the hypercapnic effect of dead space loading as opposed to the eucapnic state of CHF.
4. Influence of within-breath PaCO2 oscillations on respiratory chemosensing
A fundamental premise of Eq. (9) is that the controller’s perception of or (real or virtual) and under varying disturbances of the chemical plant (CO2 breathing or changes in series or parallel VD at rest and during exercise) is influenced by putative dynamic chemoreceptor signaling mediated by within-breath PaCO2 oscillations instead of (or in addition to) breath-to-breath fluctuations of the mean PaCO2 level. To test this hypothesis, we next apply Eq. (9) to three different types of chemical plant disturbances with specific within-breath PaCO2 oscillation profiles which have been reported to produce distinct exercise ventilatory effects: CO2 breathing (with constant PI CO2), late-inspiratory dead space loading (with constant series VD arranged such that rebreathing of dead space CO2 occurs at late inspiration instead of early inspiration) and slug CO2 breathing (with constant that is independent of V̇E).
4.1. Exercise hyperpnea relationship in CO2 breathing
Like dead space loading, CO2 breathing has also been shown to augment both the V̇E − V̇CO2 slope and Y-intercept particularly in young healthy subjects at moderate exercise levels with PaCO2 being held constant at a hypercapnic level (by proportionately decreasing the level of inhaled CO2 with increasing exercise V̇E) (Poon, 1992b; Poon and Greene, 1985). For both CO2 breathing and dead space loading, the resultant augmentation of the V̇E − V̇CO2 slope with increased PaCO2 has been taken to indicate a positive CO2-exercise interaction. However, the increase in V̇E − V̇CO2 slope is significantly greater for dead space loading than CO2 breathing at similar hypercapnic levels indicating a stronger CO2-exercise interaction under dead space loading than CO2 breathing (Poon, 1992b, 2008) (Fig. 3). Furthermore, during CO2 breathing the increase in PaCO2 in the resting state tends to increase further with increasing exercise levels (Clark et al., 1980) unless PI CO2 is decreased proportionately (Poon, 1992b; Poon and Greene, 1985). The lack of PaCO2 regulation spontaneously during exercise under CO2 breathing at fixed PI CO2 again indicates a weaker CO2-exercise interaction in this condition compared with dead space loading, where PaCO2 is regulated from rest to exercise albeit at a hypercapnic level (Poon, 1992b, 2008; Ward and Whipp, 1980; Wood et al., 2011). The greater CO2-exercise interaction observed under dead space loading with increased total VD/VT compared with CO2 breathing is in harmony with classical studies indicating that the resting hypercapnic ventilatory response elicited by tube breathing is higher than that resulting from CO2 breathing in healthy subjects (Fenner et al., 1968; Goode et al., 1969; Masuyama and Honda, 1984) as well as more recent studies indicating that central and peripheral chemosensitivities are enhanced in CHF patients with increased physiological VD/VT (Chua et al., 1996; Narkiewicz et al., 1999; Ponikowski et al., 2001a). Indeed, CO2-exercise interaction is strongest in CHF patients (compared with dead space loading and CO2 breathing) in whom the V̇E − V̇CO2 slope is potentiated without any increase in PaCO2 or in the Y-intercept. These observations beg the question: why is dead space loading (and increased physiological VD/VT in CHF) more effective than CO2 breathing in stimulating V̇E and potentiating the exercise ventilatory response (Q5, Table 3)?
Fig. 3.
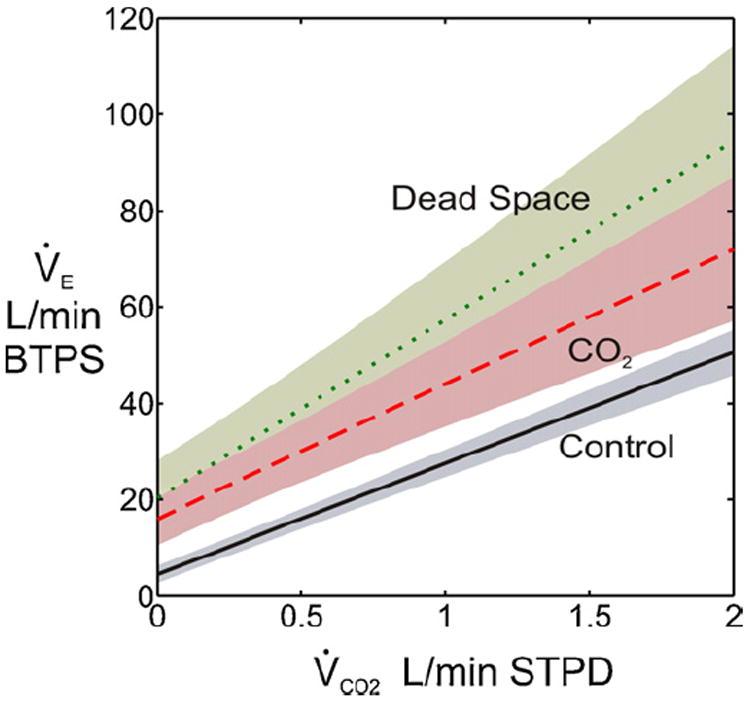
CO2-exercise interaction in healthy subjects. Shadings around plots indicate 95% confidence intervals. Mean PaCO2 levels at rest and during exercise for control, CO2 breathing and dead space loading conditions are approximately 40, 46 and 46 mmHg, respectively. Although CO2 breathing and dead space loading both induce hypercapnia as per Comroe’s law for airway CO2 load (real or virtual), dead space loading causes a greater increase in V̇E − V̇CO2 slope (compared with control) than does CO2 breathing at similar hypercapnic levels. Data adapted from Tables 2 and 3 in Poon (1992b).
Eq. (9) offers some clue for this intriguing paradox. As stated above, for all values of r < 1 Eqs. (6)-(9) predict that dead space loading is predisposed to hypercapnia by virtue of Comroe’s law upon the controller’s illusory perception of P̂I CO2 and and underestimation of . This is in contrast to the predicted eucapnic state in CHF, where r ≈ 1. On the other hand, for any r > (1 − VD/VT) Eqs. (6)-(9) predict and hence the corresponding V̇E − V̇CO2 slope should be greater than that resulting from CO2 breathing at similar PaCO2 levels. In other words, for any resting or exercise V̇CO2 level Eq. (9) predicts that dead space loading is more effective than CO2 breathing in stimulating V̇E because of the increased apparent metabolic CO2 load, whereas the condition of increased alveolar VD/VT in CHF is even more stimulatory because the resultant is greatest. For values of r within the range (1 − VD/VT) < r < 1, therefore, the predictions of Eq. (9) are in excellent agreement with the experimental data shown in Fig. 3 as well as previous studies showing enhanced hypercapnic ventilatory effects of tube breathing (Fenner et al., 1968; Goode et al., 1969; Masuyama and Honda, 1984) and enhanced central and peripheral chemosensitivities in CHF patients (Chua et al., 1996; Narkiewicz et al., 1999; Ponikowski et al., 2001a). Hence, Eq. (9) affords a plausible explanation of the paradox Q5 above that is in harmony with the explanations for Q1–Q4 (Table 3) under the same framework.
The greater CO2-exercise interaction induced by increases in VD/VT than by CO2 breathing is consistent with the predictions of an optimization model of ventilatory control first proposed three decades ago by Poon (Poon, 1983; Poon, 1987a; Poon, 1987b, 1989, 1992a), which is core to the present homeostatic competition theory (Poon, 2009, 2010, 2011; Poon et al., 2007; Tin et al., 2010). In particular, the optimization model predicts that the CO2-exercise interaction during CO2 breathing may be susceptible to respiratory mechanical limitations at higher V̇E levels such that the V̇E − V̇CO2 slope may continually decrease with increasing exercise levels resulting in an increase in the Y-intercept of the linearized V̇E − V̇CO2 relationship, as demonstrated experimentally (Clark et al., 1980; Poon, 1992b; Poon and Greene, 1985). In contrast, the predicted positive CO2-exercise interaction is much better defended against respiratory mechanical limitations when such interaction is induced by increases in physiological VD/VT compared with CO2 breathing. This model prediction is again supported by the small Y-intercept of the V̇E − V̇CO2 relationship seen in CHF patients despite their increased work of breathing and proneness to expiratory flow limitation (Fig. 2a1). Because dead space loading induces hypercapnia with virtual inspired CO2, it follows that increasing respiratory mechanical limitations (in addition to decreasing VD/VT, see Section 3.1 above) with increasing V̇E levels may also contribute to the increased Y-intercept of the V̇E − V̇CO2 relationship seen under this condition as with CO2 breathing (Fig. 3), further accounting for open question Q3 (Table 3).
4.2. Exercise hyperpnea relationship in late-inspiratory dead space loading
The experimental observations of a greater resting hypercapnic ventilatory response and greater V̇E − V̇CO2 slope in dead space loading than in CO2 breathing at similar elevated PaCO2 levels as predicted by Eq. (9) provide further support for the view that within-breath PaCO2 oscillations (rather than or in addition to breath-to-breath fluctuations of mean PaCO2 level) contribute importantly to the controller’s perception of , and in determining the resultant ventilatory response in accordance with Comroe’s law and Whipp’s law. If so, then one would expect that manipulations of the within-breath PaCO2 oscillation profiles of dead space loading at constant mean PaCO2 levels should modulate V̇E. Cunningham et al. (1973) tested this hypothesis by applying a special external dead space load in which rebreathing of dead space CO2 occurred in late inspiration (late-inspiratory dead space loading) instead of early inspiration (as expected for normal tube breathing). Delaying the rebreathing of dead space CO2 from early to late inspiration changed the timing of the PaCO2 oscillation but not its amplitude or mean value. They found that such late-inspiratory dead space loading was less effective than regular tube breathing in stimulating the resting V̇E in healthy subjects under hypoxic conditions. These authors further simulated the effects of these two types of dead space loading by having subjects breathe CO2 only in early or late inspiration in hypoxia. They found that the resting V̇E was stimulated more by the CO2-early than CO2-late maneuver and the difference was similar to those of normal and late-inspiratory dead space loading. Collier et al. (2008) showed similar effects of the CO2-early and CO2-late inspiratory maneuvers in healthy subjects undergoing moderate exercise in hypoxia or normoxia at sea level or after acclimatization at high altitude (5000 m). The difference between the two CO2 timing maneuvers during exercise was particularly pronounced in subjects returning to 5000 m from very high altitude (7100–8848 m). For the subjects at 5000 m such difference appeared to be blunted by supplemental oxygen or the carbonic anhydrase inhibitor acetazolamide. These results were taken to imply that peripheral chemoreceptors played an important role in mediating the chemosensing of the timing-dependent component of the PaCO2 oscillation that modulates V̇E. However, potentiation of the exercise ventilatory response by dead space loading has also been demonstrated when the peripheral chemoreceptors are suppressed under hyperoxic conditions (Poon, 1992b); hence peripheral chemoreceptors are not obligatory for such PaCO2 timing-dependent effect while central chemoreceptor contributions cannot be ruled out.
The distinct effects of regular and late-inspiratory dead space loading on V̇E may be explained within the framework of Eq. (9). As elaborated above, during dead space loading is underestimated by a factor of r < 1 because part of the dead space CO2 that is rebreathed in early inspiration may be mistaken by the controller for inhaled CO2. During late-inspiratory dead space loading, it is possible that is underestimated even further if the rebreathed CO2 at late inspiration is more prone to be mistaken for inhaled CO2 thus rendering it less effective in stimulating V̇E. Presumably, any difference in the controller’s perception of early-and late-inspiratory rebreathed CO2 is probably small since it was discernible only when breathing was stimulated by hypoxia and/or exercise (Collier et al., 2008; Cunningham et al., 1973).
4.3. Exercise hyperpnea relationship in slug CO2 loading
Another classic example of PaCO2 oscillations-induced modulation of V̇E is the paradox of ‘slug CO2 loading’, an experimental paradigm first proposed by Fenn and Craig (1963) in an attempt to simulate metabolic CO2 flow to the lungs by injecting a constant influx of CO2 into the inspired air stream. Fenn and Craig (1963) theorized that the metabolic hyperbola corresponding to such slug CO2 loading with constant CO2 influx independent of V̇E should approximate the metabolic hyperbola under muscular exercise, which is less steep than that corresponding to CO2 breathing at an operating point where these curves intersect one another (Fig. 4a). Based on this theory they conjectured that:8
“It was thought possible [with the injected method] that the respiratory center in “hunting” up and down the pertinent hyperbola would somehow become “aware” of the [decreased] steepness and might be able to come nearer to M than to N…… It may be said that, with inhaled mixtures, the respiratory center “tolerates” the increase of the [PaCO2] from A to C in order to avoid the “effort” of increasing the ventilation from C to N……. [The term CO2 tolerance] has been suggested…… to emphasize the point of view that the increased ventilation due to CO2 inhalation is a balance between the increased respiratory effort required and the “discomfort” of an excessive [PaCO2]…… With the injected method, the “profit” (in terms of lowered [PaCO2]) [is] much greater for an increase in ventilation and the “penalty” (increased [PaCO2]) for decreasing the ventilation [is] much greater than [is] the case with the inhaled method.”
Fig. 4.
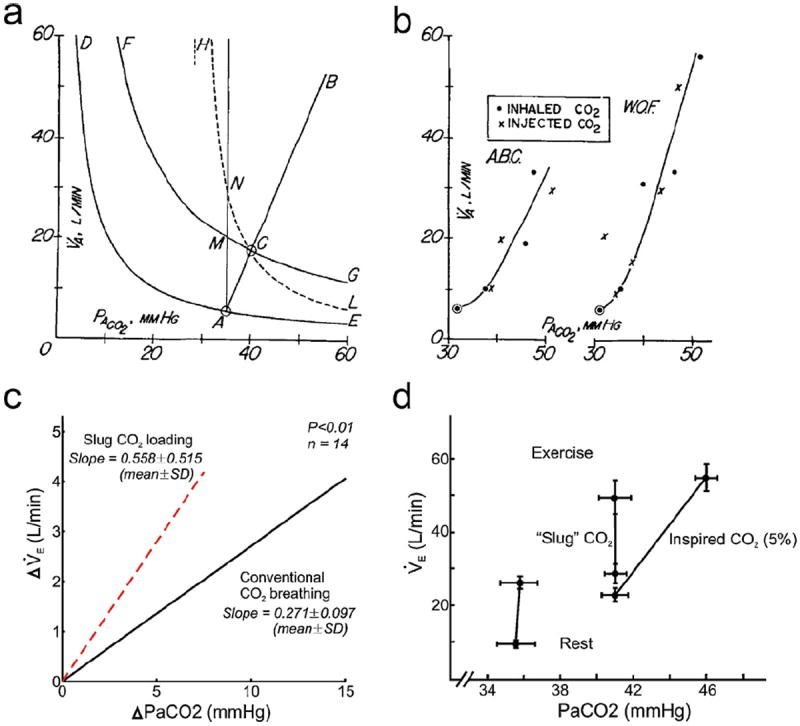
Differential effects of virtual airway CO2 load vs. apparent metabolic CO2 load on ventilatory control in different types of slug CO2 loading. a: Fenn-Craig diagram showing the metabolic hyperbolas for pulmonary CO2 exchange at rest and during exercise, CO2 breathing and (ideal) slug CO2 loading. See text. b: CO2 response curves (or more appropriately, CO2 tolerance curves) are no different under constant-flux slug CO2 loading than CO2 breathing in two subjects. Panels a and b are adapted from Fenn and Craig (1963) with permission. c: CO2 response curve in anesthetized cats is twice steeper in early-inspired slug CO2 loading than in CO2 breathing at constant PI CO2. Adapted from van der Grinten et al. (1992) with permission. d: The mechanism underlying the control of exercise hyperpnea remains intact in early-inspired slug CO2 loading, except that the apparent homeostatic “set point” is shifted to higher PaCO2 levels at rest and during exercise compared with corresponding control values (data points at left). By contrast, CO2 breathing (at 5% level) also induces hypercapnia because of its CO2-clogging effect (Comroe’s law) but homeostatic regulation at the higher PaCO2 level is lost during exercise. Adapted from (Swanson, 1978) with permission.
Fenn and Craig then tested this conjecture in two subjects. Much to their chagrin, however, both subjects exhibited a hypercapnic instead of isocapnic (eucapnic) ventilatory response to the constant-flux slug CO2 load and “increased their alveolar ventilation equally for the same increase in [PaCO2] whether the CO2 was presented as a fixed concentration or a fixed load” (Fig. 4b). Reluctantly, they concluded that their conjecture “described one way in which the respiratory center apparently does not operate” after all (Fenn and Craig, 1963).
This hasty concession proved premature, however. In their seminal work, Fenn and Craig (1963) injected a constant flux of CO2 into the inspired air stream in order to simulate ‘metabolic CO2 flow to the lungs’ via the airways independent of V̇E. Subsequently, van der Grinten et al. (1992) argued that this constant-flux approach may not be effective since the continuous presence of airway CO2 throughout inspiration means that any difference from CO2 breathing (as perceived by the controller) is likely to be small. Instead, they proposed to inject a small bolus of CO2 as early in inspiration as possible as did Swanson (1978), in a manner similar to dead space loading (but with a fixed CO2 slug instead of fixed series VD). With this bolus administration of early-inspired CO2 slug, van der Grinten et al. (1992) showed that the average slope of the CO2 response curves in fourteen anesthetized, spontaneously breathing cats was two times steeper than that resulting from CO2 breathing at constant PI CO2 levels. Thus, although the ventilatory response to an early-inspired CO2 slug was still hypercapnic, the results were closer to the isocapnic exercise hyperpnea response predicted by Fenn and Craig than that predicted by the CO2 response curve (Fig. 4c). Other investigators showed that subjects exposed to such an early-inspired CO2 slug were able to maintain PaCO2 constant albeit at an elevated level from rest to exercise (Mussell et al., 1990; Swanson, 1978). Thus, although the ventilatory response to early-inspired slug CO2 loading was hypercapnic, the mechanism underlying exercise ventilatory control remained intact: the normal eucapnic regulation of exercise hyperpnea was simply shifted to ‘hypercapnic regulation’ at higher but fixed PaCO2 levels as with dead space loading (Fig. 4d).
The distinct ventilatory effects of constant-flux vs. early-inspired slug CO2 loading further underscore the controller’s remarkable responsiveness to even subtle changes in within-breath PaCO2 oscillation profiles. The mechanism underlying these intriguing observations may be understood through a generalized chemical plant equation that extends Eq. (9) to include the effect of slug CO2 loading as follows:
| (10) |
In Eq. (10), is exogenous ‘metabolic CO2 flow to the lungs’ simulated by constant-flux or early-inspired slug CO2 loading; r < 1 is the fraction of the total CO2 load under slug CO2 loading that is properly attributed by the controller to the component, with the remainder being misattributed to the component as virtual airway CO2 load ( , Eq. (7)); and P̂I CO2 is as in Eq. (6). Hence from Eqs. (8) and (10):
| (11) |
In this framework, if all of is properly perceived by the controller as additional ‘metabolic CO2 flow to the lungs’ to be eliminated, then r = 1 and and the ventilatory response to would be similar to isocapnic exercise hyperpnea, as envisioned by Fenn and Craig. However, since comes from the air stream instead of blood stream, it may be misidentified in whole or in part by the controller as an airway CO2 load (with and P̂I CO2 > 0) that potentially clogs the CO2 elimination mechanism in a manner similar to the rebreathing of CO2 in dead space loading. If so, would be underestimated by the controller by a factor of r < 1 as indicated in Eq. (11) and the ventilatory response to would be hypercapnic at a constant elevated PaCO2 level from rest to exercise, as with dead space loading. Indeed, as pointed out by van der Grinten et al. (1992), constant-flux slug CO2 loading as originally employed by Fenn and Craig (1963) may be even more prone to such misidentification than early-inspired slug CO2 loading. In the extreme, if all of is misidentified by the controller as (with corresponding P̂I CO2) then constant-flux slug CO2 loading would be no different than CO2 breathing (with equivalent PI CO2 = P̂I CO2) at any given exercise level (or rest), although ‘hypercapnic regulation’ of PaCO2 from rest to exercise will persist (because the “inspired CO2” is rebreathed and is not real) in a similar manner as illustrated in Fig. 4d for early-inspired slug CO2 loading). These predictions of Eq. (10) are in excellent agreement with the reported ventilatory effects of constant-flux and early-inspired slug CO2 loading, further corroborating the suggested role of within-breath PaCO2 oscillations in modulating V̇E.
5. Concluding remarks
The general framework of respiratory chemosensing presented above rectifies several deep-rooted misconceptions and ill-conceived dogmas and taboos in the field that have long impeded understanding of ventilatory control mechanisms in health and in disease. First, we have shown that the control of V̇E at rest and during exercise is determined not only by the total V̇CO2 to be eliminated but also by the total VD/VT that impairs pulmonary CO2 elimination. The resultant V̇E is coupled to the compound variable , which measures the apparent (real-feel) metabolic CO2 load as perceived by the controller. The constancy of and resultant regulation of PaCO2 is evident in healthy subjects in whom decreases from rest to exercise upon corresponding decreases in anatomical VD/VT (Whipp’s law) and in CHF patients in whom and the V̇E − V̇CO2 slope are augmented in compensation for abnormal increases in alveolar and anatomical VD/VT. The tight coupling of V̇E to compensating for both ventilation-dependent and disease-dependent changes in physiological VD/VT as well as changes in series VD/VT during dead space loading argues against the putative skeletal muscle afferents feedback control of exercise V̇E, a transitory mechanism that appears to die out after the first minutes of exercise. Second, attention has been called to the fact that the classical ‘CO2 response curve’ is not truly a stimulus–response (dose–response) relationship in the Sherringtonian sense as traditionally thought. Instead, it is more appropriately viewed as a ‘CO2 tolerance curve’ (Fenn and Craig, 1963) reflecting the controller’s prudent strategy to tolerate a breakdown of PaCO2 homeostasis with self-imposed “permissive hypercapnia” in order to conserve the work of breathing in the face of severe clogging of V̇E-dependent CO2 elimination (Comroe’s law), as described by the homeostatic competition model (Poon, 2009, 2010, 2011; Poon et al., 2007; Tin et al., 2010). Third, a novel theory of dead space loading has been proposed to highlight the dual character of series VD that distinguishes it from parallel VD. Although series VD does result in an increase in VD/VT thereby augmenting as with parallel VD, it also induces hypercapnia (as with CO2 breathing) through the rebreathing of dead space CO2 thereby creating the illusion of a virtual PI CO2 (P̂I CO2) and virtual with corresponding underestimation of (for r < 1) as perceived by the controller. The subtle difference in the controller’s perception of the relative magnitudes of and under series and parallel VD explains the hypercapnic effect of dead space loading vis-à-vis the eucapnic state of CHF. Last, a novel respiratory chemosensing mechanism at the controller that is responsive to putative drive signals mediated by within-breath PaCO2 oscillations (independent of breath-to-breath fluctuations of the mean PaCO2 level) has been revealed to play an important role in the controller’s perception of and under varying disturbances of the chemical plant (air breathing vs. CO2 breathing, increased alveolar VD in CHF, as well as different types of dead space loading and slug CO2 loading both at rest and during exercise). The demonstrated dependence of the controller’s perception of these chemical plant variables on the corresponding within-breath PaCO2 oscillation profiles provides a unified mechanistic explanation of open questions Q1–Q5 (Table 3) and beyond.
These findings strongly suggest that the chemosensing mechanism at the controller is endowed with cognition and perception capabilities that may be responsive to putative drive signals mediated by within-breath PaCO2 oscillations. The perception process appears rather slow in reaching steady state (phase III) when relying on the proposed chemosensing of PaCO2 oscillations alone. Nonetheless, it may be accelerated by other sensory cues such as central feedforward command at exercise onset (phase I) and the ensuing peripheral chemoreceptor and skeletal muscle afferents feedbacks (phase II) (see Appendix B). In extreme cases such as in congenital patients who lack respiratory chemosensitivity from birth, surrogate sensory cues for exercise such as the wakefulness drive and skeletal muscles feedback may be recruited to play an even more prominent role (Gozal et al., 1996; Paton et al., 1993; Shea et al., 1993). Future studies will explore the neurocircuitry and cellular processes in the controller that are involved in the integration/decoding of central command, central and peripheral chemoreceptor afferents (mediating the mean and oscillatory components of the PaCO2 signal), skeletal muscle afferents and other sensory cues into percepts of and as well as the transformation of these percepts into respiratory motor pattern and resultant ventilatory output in accordance with Whipp’s law and Comroe’s law.
Although the notion of subliminal perception and cognition in cardiorespiratory regulation has been historically eschewed by physiologists and clinicians in favor of Sherringtonian reflex (i.e., chemoreflex, metaboreflex, mechanoreflex, and baroreflex) paradigms, there is no a priori reason to presume that the Cannonian ‘wisdom of the body’ that is richly displayed in these physiological processes (Cannon, 1929; Cannon, 1932; Poon, 2011) should be any less than the brain intelligence that is evident in higher cognitive and sensory/sensorimotor integration processes that reach conscious levels such as temperature sensing, vision, hearing, posture and motor control, touch, pain sensation, etc., all of which are known to exhibit apparent and virtual perceptions of the primary stimuli subject to amplifications/attenuations and distortions by physiological and environmental factors. Indeed, it is well-known that some forms of cardiorespiratory sensations (such as dyspnea or ‘air hunger’) may reach conscious levels in healthy subjects and cardiopulmonary patients and may play an important role in cardiorespiratory control (e.g., (Izumizaki et al., 2011; Jensen et al., 2011)). Therefore, understanding how physiologically and environmentally induced disturbances in the chemical and mechanical plants may modulate the controller’s (and higher centers’) perception of those perturbations through dynamic (rather than mean) respiratory chemosensing and mechanosensing is of utmost fundamental importance in illuminating ventilatory control mechanisms in health and in disease. Such first principles derived from the respiratory system may further inform investigations into other intelligent physiological processes such as those envisioned by Cannon (1929, 1932) as well as other forms of brain intelligence that are traditionally thought to be unique to the higher brain but are extremely difficult to elucidate experimentally because of the complexity of higher brain structures. In particular, an emerging brain intelligence paradigm potentially underlying respiratory motor control that may be common to oculomotor control, skeletomotor control, postural control and other sensory or sensorimotor integration processes in the brain is the notion of ‘internal model’ cognition (Green and Angelaki, 2010; Imamizu and Kawato, 2012; Ito, 2008; Lalazar and Vaadia, 2008; Lisberger, 2009; Poon and Merfeld, 2005; Poon et al., 2007; Tin and Poon, 2005), i.e., the perception of changes in the internal milieu and external environment through adaptive integration of externally elicited afferent inputs and internally generated efference copy (corollary discharge) of motor or mental commands. Interestingly, increased corollary discharge and/or increased respiratory afferent feedback essential for such internal model learning has been implicated in the induction of exertional dyspnea by dead space loading in healthy subjects (Jensen et al., 2011).
The remarkable predictive power of the proposed framework of respiratory chemosensing for decoding and under wide-ranging disturbances of the chemical plant reconciles the limited predictability and mutual inconsistency of the classical chemoreflex model and homeostatic regulation model of ventilatory control that have been the root of the longstanding stalemate in the field. Extension of this framework of respiratory chemosensing to include respiratory mechanosensing mechanisms as provided by the homeostatic competition model (Poon, 2009, 2010, 2011; Poon et al., 2007; Tin et al., 2010) should afford resolution of open questions Q6–Q10 (Table 3) in future.
Supplementary Material
Acknowledgments
We thank Drs. S.A. Ward, K. Wasserman and the late Dr. N.S. Cherniack for generous advice and encouragement and Drs. P.G. Agostoni, M. Amann, T.G. Babb, A.J. Coats, J.A. Dempsey, J. Duffin, B.D. Johnson, M.J.Joyner, H.R. Middlekauff, C.F. Notarius, D.J. Paterson, R. Pellegrino, M.F. Piepoli, H.T. Robertson, N.H. Secher, G.D. Swanson, J.W. Severinghaus, G. Song, D.S. Ward, J.B. West, and C.B. Wolff for valuable comments on the final manuscript. C. Tin was supported by an American Heart Association predoctoral fellowship. This work was supported by NIH grants HL067966 and RR028241.
Appendix A. Nociceptive metaboreceptor modulation of post-exercise hyperpnea and pressor response dynamics
Regional circulatory occlusion as widely employed in muscle metaboreflex studies in the cardiorespiratory physiology literature is also (not coincidentally) an established method of experimental ischemic pain induction at rest and during exercise in the pain literature (Hagenouw et al., 1986; Ray and Carter, 2007; Smith et al., 1966; Smith et al., 1968). Such an occlusion procedure has been shown to stimulate V̇E in a graded manner depending on the level of pain (Borgbjerg et al., 1996). A body of recent evidence indicates that entrapment of lactic acid (in the form of lactate + proton) and other anaerobic metabolites (such as extracellular ATP) in the musculature after intense exercise or ischemic exercise could aggravate exercise-induced muscle pain (Miles and Clarkson, 1994), a complex process that may be triggered by lactic acid and ATP coactivation of acid-sensing ion channels (particularly ASIC subtype 3) (Birdsong et al., 2010; Light et al., 2008; Naves and McCleskey, 2005). The synergistic effect of lactic acid and ATP (and other painful anaerobic metabolites) coactivation of ASICs may explain why even trace levels of lactate production during occlusion are sufficient to induce measurable increases in group III/IV skeletal muscle afferents activity during low levels of induced muscle contraction (Adreani and Kaufman, 1998; Hayes et al., 2006); alternatively, other metaboreceptors/mechanoreceptors independent from lactate might also be involved (Paterson et al., 1990; Vissing et al., 2001). Interestingly, recent evidence indicates that the exercise pressor reflex is associated with increased activity in the periaqueductal gray (Basnayake et al., 2011), a midbrain region that has been identified with integration of the central command for the cardiorespiratory response to exercise (Green et al., 2007) as well as pain information processing and modulation (Depaulis and Bandler, 1991). Possible influences of ischemic pain on the postexercise pressor response were recognized in the classic study by Alam and Smirk (1937) but were dismissed per the subjects’ characterization of the occlusion-provoked sensation as “tiredness or heaviness” and not discomfort or pain, an argument that has since been invoked by many authors. However, activation of nociceptive pathways could stimulate breathing without involving arousal of higher centers (Ward and Karan, 2002). Similarly, activation of skeletal muscle ASIC receptor-mediated nociceptive pathways has been shown to contribute to the pressor reflex during regional circulatory occlusion after muscle contraction in decerebrated cats, without the animals’ conscious perception of pain (McCord et al., 2009).
Post hoc analysis of post-exercise regional circulatory occlusion data reported in the literature reveals the distinct possibility that lactic acid and other painful anaerobic metabolites accumulated within the working musculature (even at trace levels) may indeed play a much more significant role in modulating the cardioventilatory response during post-exercise recovery (with or without conscious sensation of muscle pain) than previously appreciated, a missing link which may underlie the seeming discrepant results from different studies. In (Piepoli et al., 1995) the majority of the healthy subjects (eight out of eleven) reportedly registered “slight discomfort” during regional circulatory occlusion after handgrip exercise, although only four would characterize it as pain. In an attempt to exclude the possibility that occlusion alone could produce any reflex response, the authors found that V̇E was not affected by 4 min of control regional circulatory occlusion of the forearm (at 200 mmHg) at rest without any previous exercise. However, post hoc analysis of the reported data (Piepoli et al., 1995) reveals that the mean V̇E/V̇CO2 over the 4-min control regional circulatory occlusion at rest was indeed 7.5% to 15% higher than the baseline V̇E/V̇CO2 values before handgrip tests in the same subject population (46.9 vs. baseline values of 40.8 for the control handgrip test and 43.6 for the handgrip followed by regional circulatory occlusion test; values calculated from the data in Table 1 of Piepoli et al. (1995)). If so, a conservative estimate of the resultant PaCO2 level during control regional circulatory occlusion would place it lower than the corresponding resting baseline values by as much as 3–6 mmHg on average (although PaCO2 was not reported in that study). Such adverse hyperventilatory effects—if indeed caused by occlusion-induced ischemic muscle lactic acidosis—would likely be exacerbated by increasing exercise intensities and by ischemic exercise particularly in CHF patients who present with exercise intolerance and early onset of lactic acidosis. Furthermore, such occlusion artifact is likely to exert similar confounding influences on the post-exercise pressor response as measured by using this traditional technique.
Evidence favoring this hypothesis can be inferred from the study of Notarius et al. (2001) in which the sympathoneural and pressor responses to post-exercise regional circulatory occlusion were found to be graded by the level and type of exercise and type of subjects in an order that is consistent with increasing proneness to skeletal muscle lactic acidosis. Specifically, the resultant responses were found to be greater at higher levels of exercise and with ischemic than non-ischemic exercise, and greatest for CHF patients with the lowest exercise peak O2 uptake (who were likely predisposed to higher sympathetic activity than patients with higher peak O2 uptake (Notarius et al., 2007)). Such graded sympathoneural response to post-exercise regional circulatory occlusion may account for the discrepant effects reported by other investigators for CHF patients with less severe sympathoexcitation and/or undergoing less intense exercise (Middlekauff et al., 2004; Sterns et al., 1991). In parallel with the graded sympathoneural and pressor responses, the heart rate response during post-exercise regional circulatory occlusion was also graded by increasing exercise intensity in that it was restrained by cardiac parasympathetic reactivation following moderate exercise but not following high-intensity exercise (Fisher et al., 2010). The graded effect on the heart rate response with parasympathetic restraint following moderate- but not high-intensity exercise is consistent with recent data indicating that parasympathetic reactivation is highly impaired after anaerobic exercise (Buchheit et al., 2007).
Appendix B. Nociceptive metaboreceptor modulation of early cardioventilatory dynamics during exercise
Similarly, an important role for muscle lactic acidosis in modulating the ventilatory and pressor response dynamics during brief (~3 min) exercise may also be gleaned from relevant data reported in the literature. In (Amann et al., 2010), partial blockade of group III/IV skeletal muscle afferents with intrathecal injection of the pain reliever fentanyl (an μ-opioid receptor agonist) in healthy subjects had no effect on cardioventilatory variables at rest but significantly attenuated mean arterial pressure and V̇E/V̇CO2 responses and elevated end-tidal PaCO2 response during 3 min of dynamic exercise at varying intensities ranging from moderate to severe (50–325 W). However, in a subsequent study (Amann et al., 2011b) in which healthy subjects performed 3-min mild exercise (15–45 W) that increased V̇CO2 by up to ~5 folds, intrathecal fentanyl had no effect on the resultant V̇E/V̇CO2 and only caused minimal increases in the PaCO2 level during exercise (which were significantly less than the increases in end-tidal PCO2 seen at higher exercise levels reported in (Amann et al., 2010)) even though mean arterial pressure was again significantly depressed by intrathecal fentanyl at each of the work rates. Thus, the suggested influences of group III/IV skeletal muscle afferents on exercise V̇E were graded by increasing V̇CO2. The lack of effect of intrathecal fentanyl on V̇E/V̇CO2 at low work rates is consistent with previous findings that epidural anesthesia attenuated the pressor response but not the V̇E response to dynamic exercise or evoked muscle contractions in humans (Fernandes et al., 1990; Strange et al., 1993). Interestingly, blood lactate levels were found to increase proportionately with increasing V̇CO2 even at moderate exercise intensities (50–150 W) and more markedly at a severe intensity (325 W) (Amann et al., 2010) but not at mild intensities (15–45 W) (Amann et al., 2011b). One may therefore reasonably infer from these data (Amann et al., 2010; Amann et al., 2011b) that the effects of intrathecal fentanyl on exercise V̇E were likely correlated to muscle lactate production, which apparently began to change even at relatively low exercise levels. These observations taken together suggest that lactic acid and other painful anaerobic metabolites could exert appreciable influences on V̇E even during mild-to-moderate “aerobic” exercise in healthy subjects, long before significant systemic lactic acidosis could be discerned that activated the peripheral chemoreceptors at more severe exercise levels. This surprising inference is supported by previous studies which demonstrated that the V̇E response during and after 3 min of moderate dynamic exercise in healthy subjects was inversely related to corresponding intracellular pH within the working musculature (as measured noninvasively by using 31P-magnetic resonance spectroscopy as a surrogate for muscle extracellular fluid pH (Evans et al., 1998)) but not to arterialized pH, regardless of whether regional circulatory occlusion was applied during or after exercise (Oelberg et al., 1998; Systrom et al., 2001).
Although group III/IV skeletal muscle afferents mediating nociceptive metaboreceptor feedback (and possibly also mechanoreceptor feedback) appear to contribute importantly to the increases of V̇E and pressor responses in the first minutes (<3 min) of exercise, it is important to recognize that this does not necessarily imply that such afferent feedback is required for the maintenance of normal exercise hyperpnea in the long term as previously assumed (Amann et al., 2010). Since the work of Dejours (1964) it has been well established that the development of full-blown exercise hyperpnea during constant, mild-to-moderate intensity work typically undergoes three phases: an occasional rapid increase in V̇E at the onset of exercise (phase I), followed by a more gradual time-dependent exponential increase (phase II) toward a final isocapnic steady-state V̇E (phase III). Although many candidate exercise stimuli have been shown to contribute to phase I or phase II development of exercise hyperpnea, none of them has so far been proven obligatory for phase III. For example, classic studies showed that patients who were devoid of peripheral chemosensitivity after bilateral carotid bodies resection (but with central chemosensitivity recovering sufficiently to reestablish eucapnia) might indeed experience hypoventilation during the first minutes (phase II) of constant-load exercise, resulting in a transient overshoot in PaCO2 (Wasserman et al., 1975). Despite this, with prolonged exercise (>8 min) those patients were remarkably able to restore isocapnia in due course with eventual development of normal exercise hyperpnea in the steady state (phase III), albeit belatedly (Wasserman et al., 1975). It is therefore entirely possible that the initial hypoventilation and CO2 retention seen at 3 min (phase II) of exercise in healthy subjects after group III/IV skeletal muscle afferents blockade (Amann et al., 2010) might eventually resolve into normal isocapnic exercise hyperpnea in phase III, had the controller been allowed sufficient time to compensate for the lack of such afferent feedback. Indeed, given that any exerciseinduced muscle pain (whether reaching conscious levels or not) and its effect on V̇E are necessarily transient and may habituate over time (Borgbjerg et al., 1996; Hagenouw et al., 1986; Kato et al., 2001; Poon and Young, 2006), such nociceptive metaboreceptor feedback—while important for the early cardioventilatory dynamics during the first minutes of exercise—would be unlikely to contribute significantly to the late-phase ventilatory and cardiovascular responses during sustained exercise.
Support for this contention can be derived from post hoc analysis of related data with prolonged exercise reported in the literature. In (Amann et al., 2011a), subjects performed constantload high-intensity (318 W) exercise to exhaustion after intrathecal injection of fentanyl or a placebo. As before, fentanyl attenuated the responses in V̇E, V̇E/V̇CO2 and heart rate during the first 5 min of exercise with consequent elevation in end-tidal PCO2. Remarkably, with continuing exercise the impact of fentanyl on these variables diminished progressively toward the end of exercise at exhaustion. Even so, the effects of fentanyl were deemed to persist by comparison of the corresponding response data registered at exhaustion for both conditions (Fig. S1, Supplementary Material). However, because the exercise time to exhaustion was considerably shorter with fentanyl than placebo (6.8 min vs. 8.7 min on average) while all cardioventilatory variables remained nonsteady at exhaustion for both conditions (possibly due to rising systemic lactic acidosis), comparing the fentanyl vs. placebo effects at respective times of exhaustion was improper. On the contrary, Fig. S1 shows that when the data for both conditions were compared simultaneously at the same end point of ~6.8 min after start of exercise (i.e., time of exhaustion under fentanyl) the responses in V̇E, V̇E/V̇CO2, end-tidal PCO2 and heart rate under fentanyl and placebo were virtually identical. (Similar adjusted comparisons may also apply to the mean arterial pressure although only the values at respective times of exhaustion were reported in that study (Amann et al., 2011a)). Thus, the initial adverse effects of muscle afferents blockade on the ventilatory and cardiovascular responses to exercise did not persist as suggested (Amann et al., 2011a) but were completely reversed after ~7 min of exercise. Similar post hoc inference may also be drawn from a related study with healthy subjects undergoing high-intensity variable-load exercise for up to 7.5 min (Amann et al., 2009). In both cases (Amann et al., 2011a; Amann et al., 2009) the contributions of group III/IV skeletal muscle afferents feedback to the V̇E, pressor and heart rate responses were short-lived and were supplanted by other factors after 7–8 min of exercise—possibly including increased central command or increased peripheral chemoreceptor feedback. Since the contribution of the latter to exercise V̇E was again likely limited to the phase II ventilatory dynamics in the first minutes of exercise and should be negligible after ~8 min (Wasserman et al., 1975), increased central commands from the respiratory and cardiovascular controllers remain as the most probable mechanism for the late-phase cardioventilatory response to sustained exercise. Thus central command—in addition to contributing a ‘fight-orflight’ feedforward drive for the early hyperventilatory response during anticipation of exercise or the phase I exercise hyperpnea (Poon et al., 2007)—may likely continually adapt to the changing peripheral chemoreceptor and skeletal muscle afferents feedbacks (and other feedbacks) during phase II until the optimal exercise V̇E response appropriate for is attained in the steady state in phase III.
Footnotes
In this work, the notion of physiological “laws” (Table 2) is adopted in a wide sense to highlight the principles behind empirical relationships that constrain physiological models and are repeatable under ideal, well-defined experimental conditions. A physiological law inferred from a set of experimental observations has explanatory power for the observations of interest and is generalizable to other critical observations derived under similar experimental conditions. This notion is similar to the definition of empirical laws in other branches of life and social sciences (Bowler, 1989; Poon, 1994; Meng et al., 2012). The physiological laws listed in Table 2 are also control laws for the respiratory system under conditions of moderate exercise (Whipp’s law) or CO2 breathing (Comroe’s law).
See West (2012) for a recent review of the physiological legacy of Fenn and co-workers.
The tachypneic breathing pattern (with reduction in VT) in some CHF patients (particularly during exercise; Agostoni et al. (2002)) is likely to cause anatomical (series) and alveolar (parallel) VD to decrease thus mitigating the increase in alveolar VD due to pulmonary ventilation/perfusion mismatch. As a result, physiological (anatomical + alveolar) VD may not change appreciably, as reported by some authors (Woods et al., 2010). Despite this, physiological VD/VT is likely to increase more than predicted by pulmonary ventilation/perfusion mismatch because anatomical VD/VT generally increases with decreases in VT.
It is well established that patients with uncomplicated CHF maintain essentially normal PaCO2 both at rest and during exercise in the face of increased physiological VD/VT (Buller and Poole-Wilson, 1990; Mezzani et al., 2009; Wasserman et al., 1997). In these patients, end-tidal PCO2 may significantly underestimate PaCO2 (Olson et al., 2010; Woods et al., 2010) reflecting an increasingly negative end-tidal-to-arterial PCO2 gradient due to increased alveolar VD/VT, as demonstrated previously in CHF patients (Wasserman et al., 1997) and animal models (Severinghaus and Stupfel, 1957), rather than hyperventilation. Nonetheless, patients with more severe CHF may indeed tend to hyperventilate especially upon exercise likely as a result of increased pulmonary vascular pressures and/or early onset of systemic lactic acidosis (Woods et al., 2010; Sue, 2011; Lorenzi-Filho et al., 2002; Wensel et al., 2005). In this work, we will consider only CHF patients with elevated physiological VD/VT but normal PaCO2 without such complications.
The percept of apparent metabolic CO2 load in respiratory chemosensing is analogous to the percept of ‘apparent temperature’ in temperature sensing, where the perceived outdoor temperature is determined not only by the ambient temperature alone but also other factors such as wind chill and heat index, which tell how the temperature really feels like when the effects of wind speed and humidity, respectively, are taken into account. Another self-evident analogy is found in vision, where an object may appear larger than real when viewed through a magnifying glass.
The illusory percept of virtual airway CO2 load in respiratory chemosensing is analogous to that of optical mirage or virtual reality in visual sensing, where a virtual environment created by optical illusion is perceived by the brain as real.
This avant-garde conjecture conceived by Fenn and Craig as early as in 1963 based on a graphical ‘profit-penalty’ analysis of the ventilatory ‘CO2 tolerance curve’ under CO2 breathing (as opposed to an isocapnic ventilatory response under exercise) was a precursor to the optimization model of ventilatory control (Poon, 1983, 1987b), which provided a more quantitative analysis of this fundamental control law two decades later.
Appendix C. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.resp.2012.12.004.
References
- Adreani CM, Kaufman MP. Effect of arterial occlusion on responses of group III and IV afferents to dynamic exercise. Journal of Applied Physiology. 1998;84:1827–1833. doi: 10.1152/jappl.1998.84.6.1827. [DOI] [PubMed] [Google Scholar]
- Agostoni P, Pellegrino R, Conca C, Rodarte JR, Brusasco V. Exercise hyperpnea in chronic heart failure: relationships to lung stiffness and expiratory flow limitation. Journal of Applied Physiology. 2002;92:1409–1416. doi: 10.1152/japplphysiol.00724.2001. [DOI] [PubMed] [Google Scholar]
- Agostoni P, Apostolo A, Sciomer S. Evolution of the concept of ventilatory limitation during exercise. Combining the pneumologist and cardiologist point of view. Respiratory Physiology and Neurobiology. 2011;179:127–128. doi: 10.1016/j.resp.2011.09.002. [DOI] [PubMed] [Google Scholar]
- Alam M, Smirk FH. Observations in man upon a blood pressure raising reflex arising from the voluntary muscles. Journal of Physiology. 1937;89:372–383. doi: 10.1113/jphysiol.1937.sp003485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amann M, Proctor LT, Sebranek JJ, Pegelow DF, Dempsey JA. Opioid-mediated muscle afferents inhibit central motor drive and limit peripheral muscle fatigue development in humans. Journal of Physiology. 2009;587:271–283. doi: 10.1113/jphysiol.2008.163303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amann M, Blain GM, Proctor LT, Sebranek JJ, Pegelow DF, Dempsey JA. Group III and IV muscle afferents contribute to ventilatory and cardiovascular response to rhythmic exercise in humans. Journal of Applied Physiology. 2010;109:966–976. doi: 10.1152/japplphysiol.00462.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amann M, Blain GM, Proctor LT, Sebranek JJ, Pegelow DF, Dempsey JA. Implications of group III and IV muscle afferents for high-intensity endurance exercise performance in humans. Journal of Physiology. 2011a;589:5299–5309. doi: 10.1113/jphysiol.2011.213769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amann M, Runnels S, Morgan DE, Trinity JD, Fjeldstad AS, Wray DW, Reese VR, Richardson RS. On the contribution of group III and IV muscle afferents to the circulatory response to rhythmic exercise in humans. Journal of Physiology. 2011b;589:3855–3866. doi: 10.1113/jphysiol.2011.209353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthonisen NR, Dhingra S. Ventilatory response to low levels of CO2. Respiration Physiology. 1978;32:335–344. doi: 10.1016/0034-5687(78)90121-4. [DOI] [PubMed] [Google Scholar]
- Asmussen E, Nielsen M. Physiological dead space and alveolar gas pressures at rest and during muscular exercise. Acta Physiologica Scandinavica. 1956;38:1–21. doi: 10.1111/j.1748-1716.1957.tb00169.x. [DOI] [PubMed] [Google Scholar]
- Band DM, Wolff CB, Ward J, Cochrane GM, Prior J. Respiratory oscillations in arterial carbon dioxide tension as a control signal in exercise. Nature. 1980;283:84–85. doi: 10.1038/283084a0. [DOI] [PubMed] [Google Scholar]
- Basnayake SD, Hyam JA, Pereira EA, Schweder PM, Brittain JS, Aziz TZ, Green AL, Paterson DJ. Identifying cardiovascular neurocircuitry involved in the exercise pressor reflex in humans using functional neurosurgery. Journal of Applied Physiology. 2011;110:881–891. doi: 10.1152/japplphysiol.00639.2010. [DOI] [PubMed] [Google Scholar]
- Bech C, Johansen K. Ventilation and gas exchange in the mute swan Cygnus olor. Respiratory Physiology. 1980;39:285–295. doi: 10.1016/0034-5687(80)90060-2. [DOI] [PubMed] [Google Scholar]
- Berkenbosch A, Bovill JG, Dahan A, DeGoede J, Olievier IC. The ventilatory CO2 sensitivities from Read’s rebreathing method and the steady-state method are not equal in man. Journal of Physiology. 1989;411:367–377. doi: 10.1113/jphysiol.1989.sp017578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birdsong WT, Fierro L, Williams FG, Spelta V, Naves LA, Knowles M, Marsh-Haffner J, Adelman JP, Almers W, Elde RP, McCleskey EW. Sensing muscle ischemia: coincident detection of acid and ATP via interplay of two ion channels. Neuron. 2010;68:739–749. doi: 10.1016/j.neuron.2010.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgbjerg FM, Nielsen K, Franks J. Experimental pain stimulates respiration and attenuates morphine-induced respiratory depression: a controlled study in human volunteers. Pain. 1996;64:123–128. doi: 10.1016/0304-3959(95)00088-7. [DOI] [PubMed] [Google Scholar]
- Bowler PJ. The Mendelian revolution: the emergence of hereditarian concepts in modern science and society. Johns Hopkins University Press; Baltimore: 1989. [DOI] [PubMed] [Google Scholar]
- Buchheit M, Laursen PB, Ahmaidi S. Parasympathetic reactivation after repeated sprint exercise. American Journal of Physiology – Heart and Circulatory Physiology. 2007;293:H133–H141. doi: 10.1152/ajpheart.00062.2007. [DOI] [PubMed] [Google Scholar]
- Buller NP, Poole-Wilson PA. Mechanism of the increased ventilatory response to exercise in patients with chronic heart failure. British Heart Journal. 1990;63:281–283. doi: 10.1136/hrt.63.5.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon WB. Organization for physiological homeostasis. Physiological Reviews. 1929;9:399–431. [Google Scholar]
- Cannon WB. The Wisdom of the Body. Norton; New York: 1932. [Google Scholar]
- Carrington CA, Fisher JP, Davies MK, White MJ. Muscle afferent inputs to cardiovascular control during isometric exercise vary with muscle group in patients with chronic heart failure. Clinical Science (London) 2004;107:197–204. doi: 10.1042/CS20040038. [DOI] [PubMed] [Google Scholar]
- Chua TP, Clark AL, Amadi AA, Coats AJ. Relation between chemosensitivity and the ventilatory response to exercise in chronic heart failure. Journal of the American College of Cardiology. 1996;27:650–657. doi: 10.1016/0735-1097(95)00523-4. [DOI] [PubMed] [Google Scholar]
- Chua TP, Ponikowski P, Harrington D, Anker SD, Webb-Peploe K, Clark AL, Poole-Wilson PA, Coats AJ. Clinical correlates and prognostic significance of the ventilatory response to exercise in chronic heart failure. Journal of the American College of Cardiology. 1997;29:1585–1590. doi: 10.1016/s0735-1097(97)00078-8. [DOI] [PubMed] [Google Scholar]
- Clark JM, Sinclair RD, Lenox JB. Chemical and nonchemical components of ventilation during hypercapnic exercise in man. Journal of Applied Physiology. 1980;48:1065–1076. doi: 10.1152/jappl.1980.48.6.1065. [DOI] [PubMed] [Google Scholar]
- Collier DJ, Nickol AH, Milledge JS, van Ruiten HJ, Collier CJ, Swenson ER, Datta A, Wolff CB. Alveolar PCO2 oscillations and ventilation at sea level and at high altitude. Journal of Applied Physiology. 2008;104:404–415. doi: 10.1152/japplphysiol.00166.2007. [DOI] [PubMed] [Google Scholar]
- Comroe JH. Physiology of Respiration, An Introductory Text. Year Book Medical Publishers; Chicago: 1965. [Google Scholar]
- Cross BA, Davey A, Guz A, Katona PG, MacLean M, Murphy K, Semple SJ, Stidwill R. The pH oscillations in arterial blood during exercise; a potential signal for the ventilatory response in the dog. Journal of Physiology. 1982;329:57–73. doi: 10.1113/jphysiol.1982.sp014290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham DJ, Howson MG, Pearson SB. The respiratory effects in man of altering the time profile of alveolar carbon dioxide and oxygen within each respiratory cycle. Journal of Physiology. 1973;234:1–28. doi: 10.1113/jphysiol.1973.sp010331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham DJC, Robbins PA, Wolff CB. Integration of respiratory responses to changes in alveolar partial pressures of CO2 and O2 and in arterial pH. In: Fishman AP, Cherniack NS, Widdicombe JG, editors. Handbook of Physiology, Sect 3: The Respiratory System. American Physiological Society; Bethesda, MD: 1986. pp. 475–528. [Google Scholar]
- Davis JA, Whipp BJ, Wasserman K. The relation of ventilation to metabolic rate during moderate exercise in man. European Journal of Applied Physiology and Occupational Physiology. 1980;44:97–108. doi: 10.1007/BF00421087. [DOI] [PubMed] [Google Scholar]
- Dejours P. Control of respiration in muscular exercise. In: Fenn WO, Rahn H, editors. Handbook of Physiology: Respiration, Section 3. American Physiological Society; Washington, D.C.: 1964. pp. 631–648. [Google Scholar]
- Depaulis A, Bandler R. The Midbrain Periaqueductal Gray Matter: Functional, Anatomical, and Neurochemical Organization. Plenum Press; New York: 1991. North Atlantic Treaty Organization. Scientific Affairs Division. [Google Scholar]
- Duffin J. Role of acid-base balance in the chemoreflex control of breathing. Journal of Applied Physiology. 2005;99:2255–2265. doi: 10.1152/japplphysiol.00640.2005. [DOI] [PubMed] [Google Scholar]
- Duffin J. Measuring the respiratory chemoreflexes in humans. Respiratory Physiology and Neurobiology. 2011;177:71–79. doi: 10.1016/j.resp.2011.04.009. [DOI] [PubMed] [Google Scholar]
- Evans AB, Tsai LW, Oelberg DA, Kazemi H, Systrom DM. Skeletal muscle ECF pH error signal for exercise ventilatory control. Journal of Applied Physiology. 1998;84:90–96. doi: 10.1152/jappl.1998.84.1.90. [DOI] [PubMed] [Google Scholar]
- Fenn WO, Craig AB., Jr Effect of CO2 on respiration using a new method of administering CO2. Journal of Applied Physiology. 1963;18:1023–1024. doi: 10.1152/jappl.1963.18.5.1023. [DOI] [PubMed] [Google Scholar]
- Fenner A, Jansson EH, Avery ME. Enhancement of the ventilatory response to carbon dioxide by tube breathing. Respiration Physiology. 1968;4:91–100. doi: 10.1016/0034-5687(68)90010-8. [DOI] [PubMed] [Google Scholar]
- Fernandes A, Galbo H, Kjaer M, Mitchell JH, Secher NH, Thomas SN. Cardiovascular and ventilatory responses to dynamic exercise during epidural anaesthesia in man. Journal of Physiology. 1990;420:281–293. doi: 10.1113/jphysiol.1990.sp017912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher JP, Seifert T, Hartwich D, Young CN, Secher NH, Fadel PJ. Autonomic control of heart rate by metabolically sensitive skeletal muscle afferents in humans. Journal of Physiology. 2010;588:1117–1127. doi: 10.1113/jphysiol.2009.185470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fordyce WE, Knuth SL, Bartlett D., Jr Ventilatory responses to low levels of CO2 inhalation in the cat. Respiration Physiology. 1984;55:81–94. doi: 10.1016/0034-5687(84)90118-x. [DOI] [PubMed] [Google Scholar]
- Francis N, Cohen-Solal A, Logeart D. Peripheral muscle ergoreceptors and ventilatory response during exercise recovery in heart failure. American Journal of Physiology. 1999;276:H913–H917. doi: 10.1152/ajpheart.1999.276.3.H913. [DOI] [PubMed] [Google Scholar]
- Goode RC, Brown EB, Jr, Howson MG, Cunningham DJ. Respiratory effects of breathing down a tube. Respiration Physiology. 1969;6:343–359. doi: 10.1016/0034-5687(69)90033-4. [DOI] [PubMed] [Google Scholar]
- Gozal D, Marcus CL, Ward SL, Keens TG. Ventilatory responses to passive leg motion in children with congenital central hypoventilation syndrome. American Journal of Respiratory and Critical Care Medicine. 1996;153:761–768. doi: 10.1164/ajrccm.153.2.8564130. [DOI] [PubMed] [Google Scholar]
- Green AM, Angelaki DE. Internal models and neural computation in the vestibular system. Experimental Brain Research. 2010;200:197–222. doi: 10.1007/s00221-009-2054-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green AL, Wang S, Purvis S, Owen SL, Bain PG, Stein JF, Guz A, Aziz TZ, Paterson DJ. Identifying cardiorespiratory neurocircuitry involved in central command during exercise in humans. Journal of Physiology. 2007;578:605–612. doi: 10.1113/jphysiol.2006.122549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieve DA, Clark AL, McCann GP, Hillis WS. The ergoreflex in patients with chronic stable heart failure. International Journal of Cardiology. 1999;68:157–164. doi: 10.1016/s0167-5273(98)00349-0. [DOI] [PubMed] [Google Scholar]
- Griffiths TL, Henson LC, Whipp BJ. Influence of inspired oxygen concentration on the dynamics of the exercise hyperpnoea in man. Journal of Physiology. 1986;380:387–403. doi: 10.1113/jphysiol.1986.sp016292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grodins FS, Gray JS, Schroeder KR, Norins AL, Jones RW. Respiratory responses to CO2 inhalation; a theoretical study of a nonlinear biological regulator. Journal of Applied Physiology. 1954;7:283–308. doi: 10.1152/jappl.1954.7.3.283. [DOI] [PubMed] [Google Scholar]
- Hagenouw RR, Bridenbaugh PO, van Egmond J, Stuebing R. Tourniquet pain: a volunteer study. Anesthesia and Analgesia. 1986;65:1175–1180. [PubMed] [Google Scholar]
- Hayes SG, Kindig AE, Kaufman MP. Cyclooxygenase blockade attenuates responses of group III and IV muscle afferents to dynamic exercise in cats. American Journal of Physiology – Heart and Circulatory Physiology. 2006;290:H2239–H2246. doi: 10.1152/ajpheart.01274.2005. [DOI] [PubMed] [Google Scholar]
- Honda Y, Myojo S, Hasegawa S, Hasegawa T, Severinghaus JW. Decreased exercise hyperpnea in patients with bilateral carotid chemoreceptor resection. Journal of Applied Physiology. 1979;46:908–912. doi: 10.1152/jappl.1979.46.5.908. [DOI] [PubMed] [Google Scholar]
- Hugh-Jones P, Barter CE, Hime JM, Rusbridge MM. Dead space and tidal volume of the giraffe compared with some other mammals. Respiration Physiology. 1978;35:53–58. doi: 10.1016/0034-5687(78)90040-3. [DOI] [PubMed] [Google Scholar]
- Imamizu H, Kawato M. Cerebellar internal models: implications for the dexterous use of tools. Cerebellum. 2012;11:325–335. doi: 10.1007/s12311-010-0241-2. [DOI] [PubMed] [Google Scholar]
- Ito M. Control of mental activities by internal models in the cerebellum. Nature Reviews Neuroscience. 2008;9:304–313. doi: 10.1038/nrn2332. [DOI] [PubMed] [Google Scholar]
- Izumizaki M, Masaoka Y, Homma I. Coupling of dyspnea perception and tachypneic breathing during hypercapnia. Respiratory Physiology and Neurobiology. 2011;179:276–286. doi: 10.1016/j.resp.2011.09.007. [DOI] [PubMed] [Google Scholar]
- Jensen D, O’Donnell DE, Li R, Luo YM. Effects of dead space loading on neuro-muscular and neuro-ventilatory coupling of the respiratory system during exercise in healthy adults: Implications for dyspnea and exercise tolerance. Respiratory Physiology and Neurobiology. 2011;179:219–226. doi: 10.1016/j.resp.2011.08.009. [DOI] [PubMed] [Google Scholar]
- Johnson RL., Jr Gas exchange efficiency in congestive heart failure. Circulation. 2000;101:2774–2776. doi: 10.1161/01.cir.101.24.2774. [DOI] [PubMed] [Google Scholar]
- Johnson RL. Possible mechanism of augmented exercise hyperpnea in congestive heart failure - Response. Circulation. 2001a;104:E131–E131. [PubMed] [Google Scholar]
- Johnson RL., Jr Gas exchange efficiency in congestive heart failure II. Circulation. 2001b;103:916–918. doi: 10.1161/01.cir.103.7.916. [DOI] [PubMed] [Google Scholar]
- Jones NL. Normal values for pulmonary gas exchange during exercise. American Review of Respiratory Disease. 1984;129:S44–S46. doi: 10.1164/arrd.1984.129.2P2.S44. [DOI] [PubMed] [Google Scholar]
- Kato Y, Kowalski CJ, Stohler CS. Habituation of the early pain-specific respiratory response in sustained pain. Pain. 2001;91:57–63. doi: 10.1016/s0304-3959(00)00419-x. [DOI] [PubMed] [Google Scholar]
- Kleber FX, Vietzke G, Wernecke KD, Bauer U, Opitz C, Wensel R, Sperfeld A, Glaser S. Impairment of ventilatory efficiency in heart failure: prognostic impact. Circulation. 2000;101:2803–2809. doi: 10.1161/01.cir.101.24.2803. [DOI] [PubMed] [Google Scholar]
- Lalazar H, Vaadia E. Neural basis of sensorimotor learning: modifying internal models. Current Opinion in Neurobiology. 2008;18:573–581. doi: 10.1016/j.conb.2008.11.003. [DOI] [PubMed] [Google Scholar]
- Lamara N, Whipp BJ, Ward SA. Physiological inferences from intra-breath measurement of pulmonary gas exchange. Proceedings of Annual International Conference of IEEE/EMBS IEEE; New Orleans. 1988. pp. 825–826. [Google Scholar]
- Light AR, Hughen RW, Zhang J, Rainier J, Liu Z, Lee J. Dorsal root ganglion neurons innervating skeletal muscle respond to physiological combinations of protons, ATP, and lactate mediated by ASIC, P2X, and TRPV1. Journal of Neurophysiology. 2008;100:1184–1201. doi: 10.1152/jn.01344.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisberger SG. Internal models of eye movement in the floccular complex of the monkey cerebellum. Neuroscience. 2009;162:763–776. doi: 10.1016/j.neuroscience.2009.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzi-Filho G, Azevedo ER, Parker JD, Bradley TD. Relationship of carbon dioxide tension in arterial blood to pulmonary wedge pressure in heart failure. European Respiratory Journal. 2002;19:37–40. doi: 10.1183/09031936.02.00214502. [DOI] [PubMed] [Google Scholar]
- Lugliani R, Whipp BJ, Seard C, Wasserman K. Effect of bilateral carotid-body resection on ventilatory control at rest and during exercise in man. New England Journal of Medicine. 1971;285:1105–1111. doi: 10.1056/NEJM197111112852002. [DOI] [PubMed] [Google Scholar]
- Masuyama H, Honda Y. Differences in overall ‘gain’ of CO2-feedback system between dead space and CO2 ventilations in man. Bulletin Europeen de Physiopathologie Respiratoire. 1984;20:501–506. [PubMed] [Google Scholar]
- McCord JL, Tsuchimochi H, Kaufman MP. Acid-sensing ion channels contribute to the metaboreceptor component of the exercise pressor reflex. American Journal of Physiology – Heart and Circulatory Physiology. 2009;297:H443–H449. doi: 10.1152/ajpheart.00328.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Y, Tanaka S, Poon C-S. Comment on Universality in the evolution of orientation columns in the visual cortex. Science. 2012;336:413. doi: 10.1126/science.1205737. [DOI] [PubMed] [Google Scholar]
- Mezzani A, Agostoni P, Cohen-Solal A, Corra U, Jegier A, Kouidi E, Mazic S, Meurin P, Piepoli M, Simon A, Laethem CV, Vanhees L. Standards for the use of cardiopulmonary exercise testing for the functional evaluation of cardiac patients: a report from the Exercise Physiology Section of the European Association for Cardiovascular Prevention and Rehabilitation. European Journal of Cardiovascular Prevention and Rehabilitation. 2009;16:249–267. doi: 10.1097/HJR.0b013e32832914c8. [DOI] [PubMed] [Google Scholar]
- Middlekauff HR, Sinoway LI. Increased mechanoreceptor stimulation explains the exaggerated exercise pressor reflex seen in heart failure. Journal of Applied Physiology. 2007;102:492–494. doi: 10.1152/japplphysiol.00994.2006. discussion 496. [DOI] [PubMed] [Google Scholar]
- Middlekauff HR, Chiu J, Hamilton MA, Fonarow GC, Maclellan WR, Hage A, Moriguchi J, Patel J. Muscle mechanoreceptor sensitivity in heart failure. American Journal of Physiology – Heart and Circulatory Physiology. 2004;287:H1937–H1943. doi: 10.1152/ajpheart.00330.2004. [DOI] [PubMed] [Google Scholar]
- Miles MP, Clarkson PM. Exercise-induced muscle pain, soreness, and cramps. Journal of Sports Medicine and Physical Fitness. 1994;34:203–216. [PubMed] [Google Scholar]
- Mitchell GS. Ventilatory control during exercise with increased respiratory dead space in goats. Journal of Applied Physiology. 1990;69:718–727. doi: 10.1152/jappl.1990.69.2.718. [DOI] [PubMed] [Google Scholar]
- Mitchell G, Skinner JD. Lung volumes in giraffes, Giraffa camelopardalis. Comparative Biochemistry and Physiology. Part A: Molecular and Integrative Physiology. 2011;158:72–78. doi: 10.1016/j.cbpa.2010.09.003. [DOI] [PubMed] [Google Scholar]
- Miyamoto Y, Niizeki K. Ventilatory responses during incremental exercise in men under hyperoxic conditions. Japanese Journal of Physiology. 1995;45:59–68. doi: 10.2170/jjphysiol.45.59. [DOI] [PubMed] [Google Scholar]
- Mussell MJ, Paulev PE, Miyamoto Y, Nakazono Y, Sugawara T. A constant flux of carbon dioxide injected into the airways mimics metabolic carbon dioxide in exercise. Japanese Journal of Physiology. 1990;40:877–891. doi: 10.2170/jjphysiol.40.877. [DOI] [PubMed] [Google Scholar]
- Narkiewicz K, Pesek CA, van de Borne PJ, Kato M, Somers VK. Enhanced sympathetic and ventilatory responses to central chemoreflex activation in heart failure. Circulation. 1999;100:262–267. doi: 10.1161/01.cir.100.3.262. [DOI] [PubMed] [Google Scholar]
- Naves LA, McCleskey EW. An acid-sensing ion channel that detects ischemic pain. Brazilian Journal of Medical and Biological Research. 2005;38:1561–1569. doi: 10.1590/s0100-879x2005001100001. [DOI] [PubMed] [Google Scholar]
- Neder JA, Nery LE, Peres C, Whipp BJ. Reference values for dynamic responses to incremental cycle ergometry in males and females aged 20 to 80. American journal of respiratory and critical care medicine. 2001;164:1481–1486. doi: 10.1164/ajrccm.164.8.2103007. [DOI] [PubMed] [Google Scholar]
- Notarius CF, Atchison DJ, Floras JS. Impact of heart failure and exercise capacity on sympathetic response to handgrip exercise. American Journal of Physiology – Heart and Circulatory Physiology. 2001;280:H969–H976. doi: 10.1152/ajpheart.2001.280.3.H969. [DOI] [PubMed] [Google Scholar]
- Notarius CF, Spaak J, Morris BL, Floras JS. Comparison of muscle sympathetic activity in ischemic and nonischemic heart failure. Journal of Cardiac Failure. 2007;13:470–475. doi: 10.1016/j.cardfail.2007.03.014. [DOI] [PubMed] [Google Scholar]
- Oelberg DA, Evans AB, Hrovat MI, Pappagianopoulos PP, Patz S, Systrom DM. Skeletal muscle chemoreflex and pHi in exercise ventilatory control. Journal of Applied Physiology. 1998;84:676–682. doi: 10.1152/jappl.1998.84.2.676. [DOI] [PubMed] [Google Scholar]
- Olson TP, Joyner MJ, Johnson BD. Influence of locomotor muscle metaboreceptor stimulation on the ventilatory response to exercise in heart failure. Circulation: Heart Failure. 2010;3:212–219. doi: 10.1161/CIRCHEARTFAILURE.109.879684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P, De Filippis F, Fraioli F, Cinquanta A, Valli G, Laveneziana P, Vaccaro F, Martolini D, Palange P. Cardiopulmonary exercise testing (CPET) in pulmonary emphysema. Respiratory Physiology and Neurobiology. 2011;179:167–173. doi: 10.1016/j.resp.2011.07.013. [DOI] [PubMed] [Google Scholar]
- Paterson DJ, Friedland JS, Bascom DA, Clement ID, Cunningham DA, Painter R, Robbins PA. Changes in arterial K+ and ventilation during exercise in normal subjects and subjects with McArdle’s syndrome. Journal of Physiology. 1990;429:339–348. doi: 10.1113/jphysiol.1990.sp018260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paton JY, Swaminathan S, Sargent CW, Hawksworth A, Keens TG. Ventilatory response to exercise in children with congenital central hypoventilation syndrome. American Review of Respiratory Disease. 1993;147:1185–1191. doi: 10.1164/ajrccm/147.5.1185. [DOI] [PubMed] [Google Scholar]
- Petrini MF, Robertson HT, Hlastala MP. Interaction of series and parallel dead space in the lung. Respiration Physiology. 1983;54:121–136. doi: 10.1016/0034-5687(83)90118-4. [DOI] [PubMed] [Google Scholar]
- Piepoli M, Clark AL, Coats AJ. Muscle metaboreceptors in hemodynamic, autonomic, and ventilatory responses to exercise in men. American Journal of Physiology. 1995;269:H1428–H1436. doi: 10.1152/ajpheart.1995.269.4.H1428. [DOI] [PubMed] [Google Scholar]
- Piepoli M, Clark AL, Volterrani M, Adamopoulos S, Sleight P, Coats AJ. Contribution of muscle afferents to the hemodynamic, autonomic, and ventilatory responses to exercise in patients with chronic heart failure: effects of physical training. Circulation. 1996;93:940–952. doi: 10.1161/01.cir.93.5.940. [DOI] [PubMed] [Google Scholar]
- Piepoli M, Ponikowski P, Clark AL, Banasiak W, Capucci A, Coats AJ. A neural link to explain the “muscle hypothesis” of exercise intolerance in chronic heart failure. American Heart Journal. 1999;137:1050–1056. doi: 10.1016/s0002-8703(99)70361-3. [DOI] [PubMed] [Google Scholar]
- Ponikowski P, Chua TP, Anker SD, Francis DP, Doehner W, Banasiak W, Poole-Wilson PA, Piepoli MF, Coats AJ. Peripheral chemoreceptor hypersensitivity: an ominous sign in patients with chronic heart failure. Circulation. 2001a;104:544–549. doi: 10.1161/hc3101.093699. [DOI] [PubMed] [Google Scholar]
- Ponikowski P, Francis DP, Piepoli MF, Davies LC, Chua TP, Davos CH, Florea V, Banasiak W, Poole-Wilson PA, Coats AJ, Anker SD. Enhanced ventilatory response to exercise in patients with chronic heart failure and preserved exercise tolerance: marker of abnormal cardiorespiratory reflex control and predictor of poor prognosis. Circulation. 2001b;103:967–972. doi: 10.1161/01.cir.103.7.967. [DOI] [PubMed] [Google Scholar]
- Poon C-S. Optimal control of ventilation in hypoxia, hypercapnia and exercise. In: Whipp BJ, Wiberg DM, Bellville JW, Ward SA, editors. Modelling and Control of Breathing. Elsevier Biomedical; New York: 1983. pp. 189–196. [Google Scholar]
- Poon C-S. Optimal control of ventilation in hypercapnia and exercise: an extended model. In: Benchetrit G, Baconnier P, Demongeot J, editors. Concept and Formalizations in the Control of Breathing. Manchester University Press; Manchester, UK: 1987a. pp. 119–131. [Google Scholar]
- Poon C-S. Ventilatory control in hypercapnia and exercise: optimization hypothesis. Journal of Applied Physiology. 1987b;62:2447–2459. doi: 10.1152/jappl.1987.62.6.2447. [DOI] [PubMed] [Google Scholar]
- Poon C-S. Optimal regulation of ventilation during exercise. In: Khoo MC, editor. Modeling and Parameter Estimation in Respiratoy Control. Plenum; New York: 1989. pp. 25–37. [Google Scholar]
- Poon C-S. Introduction: Optimization hypothesis in the control of breathing. In: Honda Y, Miyamoto Y, Konno K, Widdicombe JG, editors. Control of Breathing and its Modeling Perspective. Plenum; New York, NY: 1992a. pp. 371–384. [Google Scholar]
- Poon CS. Potentiation of exercise ventilatory response by airway CO2 and dead space loading. Journal of Applied Physiology. 1992b;73:591–595. doi: 10.1152/jappl.1992.73.2.591. [DOI] [PubMed] [Google Scholar]
- Poon C-S. Quantitative social science. Nature. 1994;368:297–298. [Google Scholar]
- Poon CS. Possible mechanism of augmented exercise hyperpnea in congestive heart failure. Circulation. 2001;104:E131. [PubMed] [Google Scholar]
- Poon CS. The classic potentiation of exercise ventilatory response by increased dead space in humans is more than short-term modulation. Journal of Applied Physiology. 2008;105:390. doi: 10.1152/japplphysiol.90543.2008. [DOI] [PubMed] [Google Scholar]
- Poon CS. Optimal interaction of respiratory and thermal regulation at rest and during exercise: role of a serotonin-gated spinoparabrachial thermoafferent pathway. Respiratory Physiology and Neurobiology. 2009;169:234–242. doi: 10.1016/j.resp.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon CS. Homeostatic competition: evidence of a serotonin-gated spinoparabrachial pathway for respiratory and thermoregulatory interaction. Advances in Experimental Medicine and Biology. 2010;669:61–65. doi: 10.1007/978-1-4419-5692-7_13. [DOI] [PubMed] [Google Scholar]
- Poon CS. Evolving paradigms in H+ control of breathing: from homeostatic regulation to homeostatic competition. Respiratory Physiology and Neurobiology. 2011;179:122–126. doi: 10.1016/j.resp.2011.08.002. [DOI] [PubMed] [Google Scholar]
- Poon C-S. Hypercapnic augmented exercise ventilation in dead space loading: effect of virtual (illusory) airway CO2 load in respiratory chemosensing, Experimental Biology. Boston: Massachusetts, USA; 2013a. [Google Scholar]
- Poon C-S. Experimental Biology 2013. Boston: Massachusetts, USA; 2013b. Isocapnic augmented exercise ventilation in congestive heart failure: effect of apparent (real-feel) metabolic CO2 load in respiratory chemosensing. [Google Scholar]
- Poon CS, Greene JG. Control of exercise hyperpnea during hypercapnia in humans. Journal of Applied Physiology. 1985;59:792–797. doi: 10.1152/jappl.1985.59.3.792. [DOI] [PubMed] [Google Scholar]
- Poon C-S, Merfeld D. Internal models: the state of the art. Journal of Neural Engineering. 2005;2 http://dx.doi.org/10.1088/1741-2552/1082/1083/E1001. [Google Scholar]
- Poon CS, Young DL. Nonassociative learning as gated neural integrator and differentiator in stimulus-response pathways. Behav Brain Funct. 2006;2:29. doi: 10.1186/1744-9081-2-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon CS, Tin C, Yu Y. Homeostasis of exercise hyperpnea and optimal sensorimotor integration: the internal model paradigm. Respiratory Physiology and Neurobiology. 2007;159:1–13. doi: 10.1016/j.resp.2007.02.020. discussion 14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray CA, Carter JR. Central modulation of exercise-induced muscle pain in humans. Journal of Physiology. 2007;585:287–294. doi: 10.1113/jphysiol.2007.140509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read DJ. A clinical method for assessing the ventilatory response to carbon dioxide. Australasian Annals of Medicine. 1967;16:20–32. doi: 10.1111/imj.1967.16.1.20. [DOI] [PubMed] [Google Scholar]
- Reischl P, Stavert DM. Arterial CO2 response to low levels of inspired CO2 in awake beagle dogs. Journal of Applied Physiology. 1982;52:672–676. doi: 10.1152/jappl.1982.52.3.672. [DOI] [PubMed] [Google Scholar]
- Robertson HT. Comprehensive Physiology. John Wiley & Sons, Inc; 2011. Gas exchange consequences of left heart failure. [DOI] [PubMed] [Google Scholar]
- Ross BB, Farhi LE. Dead-space ventilation as a determinant in the ventilation-perfusion concept. Journal of Applied Physiology. 1960;15:363–371. doi: 10.1152/jappl.1960.15.3.363. [DOI] [PubMed] [Google Scholar]
- Saunders KB. Oscillations of arterial CO2 tension in a respiratory model: some implications for the control of breathing in exercise. Journal of Theoretical Biology. 1980;84:163–179. doi: 10.1016/s0022-5193(80)81042-3. [DOI] [PubMed] [Google Scholar]
- Scott AC, Francis DP, Davies LC, Ponikowski P, Coats AJ, Piepoli MF. Contribution of skeletal muscle ‘ergoreceptors’ in the human leg to respiratory control in chronic heart failure. Journal of Physiology. 2000;529(Pt (3)):863–870. doi: 10.1111/j.1469-7793.2000.00863.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severinghaus JW, Stupfel M. Alveolar dead space as an index of distribution of blood flow in pulmonary capillaries. Journal of Applied Physiology. 1957;10:335–348. doi: 10.1152/jappl.1957.10.3.335. [DOI] [PubMed] [Google Scholar]
- Shea SA, Andres LP, Shannon DC, Banzett RB. Ventilatory responses to exercise in humans lacking ventilatory chemosensitivity. Journal of Physiology. 1993;468:623–640. doi: 10.1113/jphysiol.1993.sp019792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GM, Egbert LD, Markowitz RA, Mosteller F, Beecher HK. An experimental pain method sensitive to morphine in man: the submaximum effort tourniquet technique. Journal of Pharmacology and Experimental Therapeutics. 1966;154:324–332. [PubMed] [Google Scholar]
- Smith GM, Lowenstein E, Hubbard JH, Beecher HK. Experimental pain produced by the submaximum effort tourniquet technique: further evidence of validity. Journal of Pharmacology and Experimental Therapeutics. 1968;163:468–474. [PubMed] [Google Scholar]
- Sterns DA, Ettinger SM, Gray KS, Whisler SK, Mosher TJ, Smith MB, Sinoway LI. Skeletal muscle metaboreceptor exercise responses are attenuated in heart failure. Circulation. 1991;84:2034–2039. doi: 10.1161/01.cir.84.5.2034. [DOI] [PubMed] [Google Scholar]
- Strange S, Secher NH, Pawelczyk JA, Karpakka J, Christensen NJ, Mitchell JH, Saltin B. Neural control of cardiovascular responses and of ventilation during dynamic exercise in man. Journal of Physiology. 1993;470:693–704. doi: 10.1113/jphysiol.1993.sp019883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sue DY. Excess ventilation during exercise and prognosis in chronic heart failure. American Journal of Respiratory and Critical Care Medicine. 2011 doi: 10.1164/rccm.201006-0965CI. [DOI] [PubMed] [Google Scholar]
- Sullivan MJ, Higginbotham MB, Cobb FR. Increased exercise ventilation in patients with chronic heart failure: intact ventilatory control despite hemodynamic and pulmonary abnormalities. Circulation. 1988;77:552–559. doi: 10.1161/01.cir.77.3.552. [DOI] [PubMed] [Google Scholar]
- Sun XG, Hansen JE, Garatachea N, Storer TW, Wasserman K. Ventilatory efficiency during exercise in healthy subjects. American journal of respiratory and critical care medicine. 2002;166:1443–1448. doi: 10.1164/rccm.2202033. [DOI] [PubMed] [Google Scholar]
- Swanson GD. The exercise hyperpnea dilemma. Chest. 1978;73:277–279. doi: 10.1378/chest.73.2.277. [DOI] [PubMed] [Google Scholar]
- Systrom DM, Hrovat M, Oelberg D, Kazemi H. Skeletal muscle chemoreflex in exercise ventilatory control. Advances in Experimental Medicine and Biology. 2001;499:343–348. doi: 10.1007/978-1-4615-1375-9_55. [DOI] [PubMed] [Google Scholar]
- Tin C, Poon CS. Internal models in sensorimotor integration: perspectives from adaptive control theory. Journal of Neural Engineering. 2005;2:S147–S163. doi: 10.1088/1741-2560/2/3/S01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tin C, Wasserman K, Cherniack NS, Poon CS. Paradoxical potentiation of exercise hyperpnea in congestive heart failure contradicts Sherrington chemoreflex model and supports a respiratory optimization model. Advances in Experimental Medicine and Biology. 2010;669:69–72. doi: 10.1007/978-1-4419-5692-7_15. [DOI] [PubMed] [Google Scholar]
- van der Grinten CP, Schoute E, de Vries WR, Luijendijk SC. Conventional versus slug CO2 loading and the control of breathing in anaesthetized cats. Journal of Physiology. 1992;445:487–498. doi: 10.1113/jphysiol.1992.sp018935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissing J, MacLean DA, Vissing SF, Sander M, Saltin B, Haller RG. The exercise metaboreflex is maintained in the absence of muscle acidosis: insights from muscle microdialysis in humans with McArdle’s disease. Journal of Physiology. 2001;537:641–649. doi: 10.1111/j.1469-7793.2001.00641.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward DS, Karan S. Effects of pain and arousal on the control of breathing. J Anesth. 2002;16:216–221. doi: 10.1007/s005400200028. [DOI] [PubMed] [Google Scholar]
- Ward SA, Whipp BJ. Ventilatory control during exercise with increased external dead space. Journal of Applied Physiology. 1980;48:225–231. doi: 10.1152/jappl.1980.48.2.225. [DOI] [PubMed] [Google Scholar]
- Wasserman K. Breathing during exercise. New England Journal of Medicine. 1978;298:780–785. doi: 10.1056/NEJM197804062981408. [DOI] [PubMed] [Google Scholar]
- Wasserman K, Van Kessel AL, Burton GG. Interaction of physiological mechanisms during exercise. Journal of Applied Physiology. 1967;22:71–85. doi: 10.1152/jappl.1967.22.1.71. [DOI] [PubMed] [Google Scholar]
- Wasserman K, Whipp BJ, Koyal SN, Cleary MG. Effect of carotid body resection on ventilatory and acid-base control during exercise. Journal of Applied Physiology. 1975;39:354–358. doi: 10.1152/jappl.1975.39.3.354. [DOI] [PubMed] [Google Scholar]
- Wasserman K, Whipp B, Casaburi R, Beaver W, Brown H. CO2 flow to the lungs and ventilatory control. In: Dempsey J, Reed C, editors. Muscular Exercise and the Lung. University of Wisconsin Press; Madison, WI: 1977. pp. 105–135. [Google Scholar]
- Wasserman K, Zhang YY, Gitt A, Belardinelli R, Koike A, Lubarsky L, Agostoni PG. Lung function and exercise gas exchange in chronic heart failure. Circulation. 1997;96:2221–2227. doi: 10.1161/01.cir.96.7.2221. [DOI] [PubMed] [Google Scholar]
- Wasserman K, Hansen JE, Sue DY, Stringer WW, Whipp BJ. Principles of Exercise Testing and Interpretation. Lippincott Williams & Wilkins; Philadelphia, PA: 2005. [Google Scholar]
- Wasserman K, Beaver WL, Sun XG, Stringer WW. Arterial H+ regulation during exercise in humans. Respiratory Physiology and Neurobiology. 2011;178:191–195. doi: 10.1016/j.resp.2011.05.018. [DOI] [PubMed] [Google Scholar]
- Wensel R, Georgiadou P, Francis DP, Bayne S, Scott AC, Genth-Zotz S, Anker SD, Coats AJ, Piepoli MF. Differential contribution of dead space ventilation and low arterial pCO2 to exercise hyperpnea in patients with chronic heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. American Journal of Cardiology. 2004;93:318–323. doi: 10.1016/j.amjcard.2003.10.011. [DOI] [PubMed] [Google Scholar]
- Wensel R, Francis DP, Georgiadou P, Scott A, Genth-Zotz S, Anker SD, Coats AJ, Piepoli MF. Exercise hyperventilation in chronic heart failure is not caused by systemic lactic acidosis. European Journal of Heart Failure. 2005;7:1105–1111. doi: 10.1016/j.ejheart.2004.12.005. [DOI] [PubMed] [Google Scholar]
- West JB. The physiological legacy of the Fenn, Rahn, and Otis school. Journal of Physiology – Lung Cellular and Molecular Physiology. 2012;303:L845–L851. doi: 10.1152/ajplung.00322.2012. [DOI] [PubMed] [Google Scholar]
- Whipp BJ. Control of the exercise hyperpnea: the unanswered question. Advances in Experimental Medicine and Biology. 2008;605:16–21. doi: 10.1007/978-0-387-73693-8_3. [DOI] [PubMed] [Google Scholar]
- Whipp BJ, Ward SA. The coupling of ventilation to pulmonary gas exchange during exercise. In: Whipp BJ, Wasserman K, editors. Pulmonary Physiology and Pathophysiology of Exercise. Marcel Dekker; New York: 1991. pp. 271–307. [Google Scholar]
- Whipp BJ, Ward SA. Physiologic changes following bilateral carotid-body resection in patients with chronic obstructive pulmonary disease. Chest. 1992;101:656–661. doi: 10.1378/chest.101.3.656. [DOI] [PubMed] [Google Scholar]
- Whipp BJ, Wasserman K. Alveolar-arterial gas tension differences during graded exercise. Journal of Applied Physiology. 1969;27:361–365. doi: 10.1152/jappl.1969.27.3.361. [DOI] [PubMed] [Google Scholar]
- Wood HE, Mitchell GS, Babb TG. Short-term modulation of the exercise ventilatory response in younger and older women. Respiratory Physiology and Neurobiology. 2011;179:235–247. doi: 10.1016/j.resp.2011.08.011. [DOI] [PubMed] [Google Scholar]
- Woods PR, Olson TP, Frantz RP, Johnson BD. Causes of breathing inefficiency during exercise in heart failure. Journal of Cardiac Failure. 2010;16:835–842. doi: 10.1016/j.cardfail.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto WS. Mathematical analysis of the time course of alveolar CO2. Journal of Applied Physiology. 1960;15:215–219. doi: 10.1152/jappl.1960.15.2.215. [DOI] [PubMed] [Google Scholar]
- Yamamoto WS. Transmission of information by the arterial blood stream with particular reference to carbon dioxide. Biophysical Journal. 1962;2:143–159. doi: 10.1016/s0006-3495(62)86846-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.