

Abstract
Tumor antigen–encoding mRNA for dendritic cell (DC)-based vaccination has gained increasing popularity in recent years. Within this context, two main strategies have entered the clinical trial stage: the use of mRNA for ex vivo antigen loading of DCs and the direct application of mRNA as a source of antigen for DCs in vivo. DCs transfected with mRNA-encoding Wilms' tumor 1 (WT1) protein have shown promising clinical results. Using a stepwise approach, we re-engineered a WT1 cDNA-carrying transcription vector to improve the translational characteristics and immunogenicity of the transcribed mRNA. Different modifications were performed: (i) the WT1 sequence was flanked by the lysosomal targeting sequence of dendritic cell lysosomal-associated membrane protein to enhance cytoplasmic expression; (ii) the nuclear localization sequence (NLS) of WT1 was deleted to promote shuttling from the nucleus to the cytoplasm; (iii) the WT1 DNA sequence was optimized in silico to improve translational efficiency; and (iv) this WT1 sequence was cloned into an optimized RNA transcription vector. DCs electroporated with this optimized mRNA showed an improved ability to stimulate WT1-specific T-cell immunity. Furthermore, in a murine model, we were able to show the safety, immunogenicity, and therapeutic activity of this optimized mRNA. This work is relevant for the future development of improved mRNA-based vaccine strategies K.
Introduction
The use of mRNA-based vaccines in immunotherapy for cancer holds great promise. mRNA is an attractive candidate because it does not integrate into the genome of the host, it is safe, and easy to produce. The interest in using mRNA as a means to load dendritic cells (DCs) for the development of anticancer vaccines began to rise in the late 1990s, thanks to the pioneering work of E. Gilboa and colleagues at the Duke University.1,2,3 Nowadays, both ex vivo antigen loading4,5,6 and the direct administration of tumor-associated antigen mRNA to load DCs in vivo7,8,9 are under investigation. The success of both strategies depends on the uptake of the mRNA by the DCs in an efficient way and on the maturation status of the DCs when presenting antigenic peptides to immune cells. Several parameters of these mRNA-based cancer vaccines can be optimized, including the maturation stimuli, the choice of target antigen, and the presentation of antigenic peptide. We have shown that DCs matured by electroporation with TriMix mRNA (TriMix-DC) encoding CD40Ligand, CD70, and a constitutively active Toll-like receptor 4 (ca TLR4) are efficient T-cell stimulators.10,11 In 2009, a National Cancer Institute's expert panel ranked and selected the most relevant tumor antigens for immunotherapeutic targeting.12 Antigen importance was determined using predefined and preweighted objective criteria, the most important being proof of therapeutic efficacy, immunogenicity and tumor specificity (representing together >60% of the relative weight). Although none of the evaluated antigens fulfilled all criteria of the ideal cancer antigen, the Wilms' tumor 1 antigen (WT1) was ranked at position one in this list and can therefore be regarded as a highly suitable antigen. In the current study, we set out to optimize the presentation of WT1-derived antigenic peptides through the design of an optimized WT1 mRNA construct.
WT1 is a zinc-finger transcription factor that is encoded by the WT1 gene located on chromosome 11p13.13,14,15 It has a temporal and spatial expression pattern during the development of the human urogenital system. After birth, normal tissue distribution of WT1 is restricted to the urogenital system, the central nervous system, and hematopoietic tissues.14,16,17,18 By contrast, WT1 is aberrantly overexpressed in a wide range of malignancies and therefore considered as a nearly universal tumor-associated antigen. These characteristics, as well as its established role in the malignant process, render WT1 an attractive target for immunotherapeutic intervention.16,19,20
Several WT1 peptide vaccination trials have been performed in patients with hematological and solid malignancies,21 providing proof-of-concept that in vivo targeting of WT1 can elicit selective and WT1-specific antitumor immune responses.22,23 Nevertheless, peptide vaccination has some limitations, in particular its human leukocyte antigen (HLA) restriction and the need for prior identification of immunogenic epitopes.
To circumvent these limitations, DCs electroporated with WT1-encoding mRNA have been used, allowing the introduction of the full-length tumor antigen into the DC (multiepitope vaccination), which is desirable to target a broad range of epitopes.24,25,26,27 Recently, Van Tendeloo et al. were able to demonstrate the immunogenicity and clinical efficacy of WT1 mRNA-electroporated DC vaccinations in the postremission setting of acute myeloid leukemia, providing strong support for the further development and implementation of this immunotherapeutic approach.24,26 Besides the introduction of the entire tumor antigen into the DCs through mRNA electroporation, an additional advantage of this strategy is that the antigen of interest can be genetically modified.25 In this study, we aimed to design a novel WT1 mRNA construct with improved translational characteristics and enhanced immunogenicity through a stepwise molecular engineering approach: (i) linking of the WT1 sequence with the lysosomal targeting signal of dendritic cell lysosomal-associated membrane protein (DC-LAMP);28,29,30 (ii) removal of the nuclear localization sequence (NLS) region from the WT1 sequence; (iii) in silico gene optimization for optimal codon usage and G/C content and for removal of factors known to affect translational efficacy (e.g. splice sites)31,32; and (iv) subcloning of the entire sequence into a previously described RNA transcription vector with enhanced translational properties.33
Results
Stepwise engineering of the WT1 mRNA constructs
Several molecular modifications were made to our original WT1 mRNA-encoding pGEM vector,34 further referred to as “wild-type WT1” mRNA. First, we removed the antigen's natural 5′- and 3′-untranslated regions (UTRs), and we flanked the open reading frame of WT1 at its 5′-end by a signal peptide (sig) to allow translocation to the endoplasmic reticulum (ER) and at its 3′-end by the transmembrane and cytoplasmic domains of DC-LAMP (CD208) to mediate transport of WT1 to lysosomal compartments (pGEM/ΔUT/sig-WT1-DC-LAMP vector, coding for “WT1-DC-L” mRNA). To improve the cytoplasmic expression of WT1, a sequence containing the NLS of zinc-finger domain 135 was deleted resulting in the pGEM/ΔUT/sig-WT1-NLS-DC-LAMP vector (encoding “WT1-sh-DC-L” mRNA). Next, the insert of the latter vector was optimized in silico, resulting in the pGEM/ΔUT/sig-WT1-DC-LAMP OPT vector (encoding “WT1-sh-DC-L-OPT” mRNA). As a last modification step, we subcloned this optimized WT1 sequence into another vector, the pST1 vector. This RNA transcription vector can augment and prolong the antigen expression as shown by Holtkamp et al.33 The in vitro synthesized mRNA derived from the pST1 plasmid as template contains two consecutive 3′-UTRs of the β-globin and a free-ending poly(A)-tail of 120 adenosine residues. Figure 1a provides a schematic representation of the different WT1-encoding vectors and their characteristics. The quality, size, and concentration of the different in vitro transcribed mRNA constructs were analyzed with the Agilent 2100 Bioanalyzer (Figure 1b,c). All mRNA preparations derived from the different WT1-encoding constructs were of high quality, without impurities or mRNA degradation.
Figure 1.
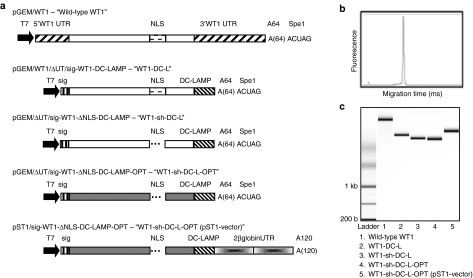
Schematic of the different WT1-encoding vectors and quality control of the in vitro transcribed mRNA. (a) The T7 promoter, 5′-UTR, 3′-UTR, human β-globin UTR, poly(A)-tail (A64 or A120), the NLS, sig, and the lysosomal targeting sequence (DC-LAMP) are shown. All pGEM constructs have an overhang of nucleotides derived from the vector backbone, which remains as 3′ attachment to the poly(A)-tail after linearization with SpeI. The pST1 vector has a free-ending poly(A)-tail after linearization with SapI. The original WT1 sequence is depicted in white and the optimized WT1 sequence in gray. (b) Electropherogram of the wild-type WT1 mRNA, representative of all mRNA samples, and (c) gel-like image of mRNA derived from the different WT1 constructs. NLS, nuclear localization sequence; sig, signal peptide; UTR, untranslated regions; WT1, Wilms' tumor 1.
WT1 expression in K562 cells and human DCs
In a first set of experiments, K562 cells were electroporated with the different WT1-encoding mRNA constructs and analyzed for WT1 protein expression kinetics by flow cytometry. As shown in Figure 2a, all WT1 mRNA constructs yielded a maximal WT1 expression pattern 4-hour postelectroporation with a rapid decline over time. WT1 expression was still detectable 24-hour postelectroporation but returned to baseline levels 48-hour postelectroporation with the exception of the “WT1-sh-DC-L-OPT” mRNA transcribed from the pST1 vector (Figure 2a). Similar results were obtained with DCs coelectroporated with TriMix mRNA combined with the WT1-encoding mRNA molecules (data not shown), showing that the improvement of protein expression by these modifications is not cell type dependent and is also applicable to DCs. However, in comparison with K562 cells, the overall WT1 expression was lower in DCs (Figure 2b).
Figure 2.

Expression and presentation of WT1 protein after electroporation with mRNA derived from the different WT1-encoding vectors. (a) Intracellular staining of WT1 in K562 cells. K562 cells were electroporated with the indicated WT1 mRNA constructs and analyzed for WT1 expression by intracellular staining 4-, 24-, and 48-hour postelectroporation (open histograms). As a control for nonspecific WT1 immunoreactivity, parallel electroporations were performed with a control mRNA (pGEM/NEF-DC-LAMP), which yielded similar expression levels as the isotype controls (gray-filled histograms). This figure is representative of three independent experiments. (b) Comparison of WT1 ΔMFI (MFI of positive population substracted by the basal WT1 epxression) values of the total K562 population and the total immature DC population after electroporation with WT1-sh-DC-L-OPT mRNA encoded by the pGEM vector. Data are presented as mean ± SEM of three independent experiments (**P = 0.0081). (c) Immunocytochemical detection of WT1 in TriMix-matured, WT1 mRNA-electroporated DCs. Immunocytochemistry for WT1 was performed on DCs fixated 24 hours after coelectroporation with WT1-encoding mRNA or control mRNA. Immunocytochemical staining patterns are shown from one representative experiment out of five (upper panel). Expression data were scored according to the staining intensity (middle panel) and according to the percentage of WT1-positive cells (lower panel; data are expressed as mean ± SEM of five independent experiments). (d) TriMix-DCs were coelectroporated with the indicated WT1 mRNA constructs or with the control mRNA. Twenty-four hours later, electroporated DCs were cocultured with the WT1-specific T-cell clone for 20 hours. IFN-γ release during this coculture was measured. Results are shown as mean ± SEM of three independent experiments (*P < 0.05). (e) Human HLA-A2+ TriMix-DCs were loaded with the WT1 antigen either by electroporation with the pST1-derived WT1-sh-DC-L-OPT mRNA or by pulsing with the HLA-A2-restricted WT1126–134 peptide. Negative controls included TriMix-DCs electroporated with the control mRNA specified above or pulsed with the HLA-A2-restricted Melan-A26-35 peptide. Four, 24, or 48 hours after antigen loading, cells were used to stimulate a human HLA-A2-restricted WT1126-134-specific CD8+ T-cell clone. IFN-γ release was measured by ELISA after a 20-hour coculture. DC, dendritic cell; ELISA, enzyme-linked immunosorbent assay; IFN-γ, interferon-γ MFI, mean fluorescence intensity; SEM, standard error of the mean; WT1, Wilms' tumor 1.
Localization of the WT1 protein in human DCs
Immunocytochemistry for WT1 was performed to further investigate the impact of the above-mentioned mRNA modifications on the protein expression level and subcellular localization 24 hours after coelectroporation of DCs with TriMix and WT1-encoding mRNA (Figure 2c). A control mRNA was included in the DC experiments as a control for nonspecific WT1 immunoreactivity. In contrast to TriMix-DCs electroporated with control mRNA, low but detectable WT1 protein expression was observed in ~20% of the cells following electroporation with wild-type WT1 mRNA. Localization of WT1 immunoreactivity was almost exclusively restricted to the nucleus. Addition of the lysosomal targeting sequence of DC-LAMP to the original WT1 mRNA construct (WT1-DC-L) in addition to the removal of the natural UTRs of the WT1 mRNA resulted in a substantial increase in protein expression, as demonstrated by the enhanced WT1 immunoreactivity in ~50% of the electroporated DCs (% positive cells “WT1-DC-L” versus “wild-type WT1” P = 0.004). Further improvement in translational efficiency was observed after removal of the NLS (WT1-sh-DC-L), resulting in even higher WT1 immunopositivity in 61.6 ± 4.1% of the cells (% positive cells “WT1-sh-DC-L” versus “WT1-DC-L” P = 0.02). Notably, deletion of the NLS clearly favored the cytoplasmic localization of WT1. The in silico optimization step (WT1-sh-DC-L-OPT) did not result in a further improvement of the translational characteristics of WT1-sh-DC-L mRNA, as demonstrated by the identical WT1-immunostaining pattern and the comparable degree of WT1 protein expression (% positive cells “WT1-sh-DC-L-OPT” versus “WT1-sh-DC-L” P = 0.53). Immunocytochemical analysis of pST1-derived mRNA-electroporated DCs revealed a strong cytoplasmic expression of WT1 in nearly all cells (% positive cells “WT1-sh-DC-L-OPT” (pST1 vector) versus “WT1-sh-DC-L-OPT” (pGEM vector); P = 0.0002).
Antigen-specific T-cell stimulatory capacity of human DCs electroporated with the different WT1-encoding mRNA constructs
To compare their functional capacity, TriMix-DCs were coelectroporated with the different WT1-encoding mRNA constructs and cocultured at various time points postelectroporation (4, 24, and 48 hours) with a human HLA-A2-WT1126–134-restricted CD8+ T-cell clone. Antigen-specific release of interferon-γ (IFN-γ) was measured after 20 hours of coculture. A summary of four independent antigen presentation assays is shown, where WT1-specific T-cells were stimulated with DCs 24 hours after electroporation (Figure 2d). A trend to an increased IFN-γ secretion was observed after every modification made to the WT1 sequence, except for the in silico optimization step. The antigen-presenting capacity of WT1 mRNA-electroporated DCs rapidly declined over time, and IFN-γ secretion was reduced by >50% in cocultures initiated 24-hour postelectroporation with DCs electroporated with pGEM-derived WT1 mRNA (data not shown). Nevertheless, at this time point, all optimized mRNAs resulted in higher IFN-γ secretion as compared with the maximum IFN-γ level observed with the wild-type mRNA (data not shown). We observed that subcloning the WT1 sequence into the pST1 vector significantly increased WT1 antigen presentation, as shown by the high IFN-γ release. Indeed, whereas the IFN-γ release of the T cells stimulated with DCs electroporated with mRNA derived from the pGEM vectors had disappeared 48 hours after electroporation, DCs electroporated with the pST1-derived mRNA still exhibited stimulatory potential (data not shown). Compared with peptide-pulsed DCs, the stimulatory capacity of DCs electroporated with pST1-derived mRNA remained higher at 24 and 48 hours after electroporation, although they were comparable at 4-hour postelectroporation or postpeptide loading (Figure 2e).
In vivo CTL activation after immunization with WT1 mRNA
To further illustrate the superiority of the pST1-derived WT1-sh-DC-L-OPT mRNA, we analyzed its functionality in an in vivo mouse model. This WT1-encoding mRNA was selected for further in vivo immunizations, given its demonstrated superiority over the other constructs during the in vitro experiments. As a reference mRNA, the wild-type WT1 mRNA was used. Both constructs are of direct relevance from a translational clinical research perspective. The wild-type WT1 mRNA has already been used in clinical trials24,36, and the WT1-sh-DC-L-OPT mRNA derived from the pST1 vector is currently being used in a clinical trial using mRNA-electroporated DCs for patients with solid malignancies (NCT01291420).
C57BL/6 mice were immunized via intranodal injection with TriMix mRNA together with the optimized WT1 mRNA or the wild-type WT1 mRNA, as described before.7 Five days after immunization, the in vivo WT1-specific cytotoxic T-lymphocyte (CTL) response was assessed in the spleen, the injected and noninjected lymph node, and the blood. We observed specific lysis of peptide-loaded target cells after immunization with both the optimized WT1 mRNA and wild-type WT1 mRNA in each compartment. Importantly, the lysis observed after immunization with the optimized WT1-encoding mRNA was significantly increased when compared with the lysis observed on immunization with the wild-type WT1 mRNA (Figure 3a; P = 0.0002).
Figure 3.

Functionality of the WT1 protein encoded by the optimized vector in an in vivo mouse model. (a) In vivo CTL assay after immunization with wild-type WT1 mRNA or mRNA encoded by the optimized vector. In vivo cytolysis of the target cells was assessed ex vivo by flow cytometry analysis of CFSElo or CFSEhi cells in the spleen, injected and noninjected lymph nodes, and the peripheral blood. Data shown are from the blood and are representative for the other compartments. This figure is representative of three independent experiments. Data are presented as the percentage of specific lysis. (b) C57BL/6 mice (n = 7) were inoculated with 5 × 105 C1498-WT1 cells s.c. 7 days later, and mice were immunized intranodally with control, wild-type WT1+TriMix, or optimized WT1+TriMix mRNA. Tumor volume was assessed (**P < 0.01; ***P < 0.001). WT1, Wilms' tumor 1.
Therapeutic antitumor immunity by intranodal WT1 mRNA immunization
C57BL/6 mice were inoculated subcutaneously with C1498-WT1 leukemia cells. On day 7, in the presence of a palpable tumor, mice were immunized intranodally with control, wild-type WT1, or optimized WT1 mRNA in combination with TriMix mRNA. Mice from the control group showed rapid tumor growth (Figure 3b). Tumor volume of the optimized WT1 mRNA-immunized mice was significantly better than the wild-type WT1 (P < 0.01) and control group (P < 0.001). WT1 mRNA-immunized mice inoculated with C1498 leukemia cells (not expressing WT1) could not reject the tumor (data not shown).
Toxicity of WT1 mRNA immunization
To investigate the autotoxicity against organs expressing WT1 under physiological conditions, such as the kidney (Figure 4a), WT1 mRNA-immunized mice were screened for proteinuria twice a week for 24 day. In addition, histopathologic studies of the kidney, lung, and liver were performed 35 days after intranodal immunization. Neither the tissues examined nor the urinary analysis showed signs of autotoxicity (Figure 4b,c).
Figure 4.
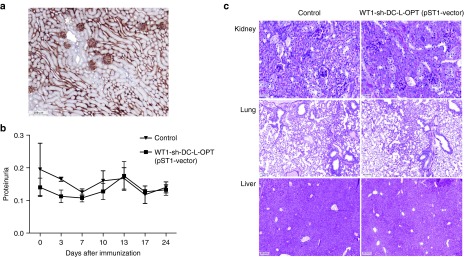
Autotoxicity-related side effects after WT1 immunization. (a) WT1 expression in the kidney under physiological conditions. (b) No proteinuria was observed in mice immunized with the optimized WT1 mRNA, indicating no damage to the kidney (n = 8 mice/group). (c) No pathological changes, such as lymphocyte infiltration or tissue destruction and repair, were observed in the kidney, lung, or liver (negative control) of WT1 mRNA-immunized mice. WT1, Wilms' tumor 1.
Discussion
Recently, WT1 has been selected by a National Cancer Institute expert panel as a very promising antigen candidate, which deserves the highest priority for implementation as a target antigen for cancer immunotherapy.12 WT1 functions as a transcription factor, explaining its predominant nuclear localization.37 This was confirmed by our results, demonstrating nuclear WT1 staining in DCs electroporated with mRNA encoding the full-length (wild-type) WT1 protein (Figure 2c). As could be expected from their nuclear localization and low WT1 expression level, these cells had an overall weak capacity to stimulate WT1-specific T-cell immunity (Figure 2d). Although the wild-type WT1 mRNA has already been used in clinical studies with some positive results,24,36,38 we hypothesized that the localization, expression level, and presentation of the WT1 antigen could be further enhanced by competent molecular design of the tumor antigen–encoding sequence.
In line with previous work from our group,28,29 we introduced a first modification of the wild-type WT1 mRNA by linking it to the lysosomal targeting sequence of DC-LAMP. In this study, we observed that the addition of the DC-LAMP signal redirects WT1 to the cytosol and dramatically improves the translational efficacy, resulting in an enhanced presentation of the antigen to CD8+ T-cells (Figure 2d and data not shown). This corroborates our previous work24,25 and of Sahin et al.39 showing that the coupling of antigens to the trafficking sequences of endosomal/lysosomal proteins augments the MHC I antigen presentation efficiency. Two possible reasons can account for this. First, peptides from mRNAs without lysosomal targeting sequence, such as DC-LAMP, are generated during the pioneer round of translation, the major source of antigenic peptides for the MHC class I pathway.40,41,42 Peptides from the WT1-DC-L mRNA, which includes the DC-LAMP sequence, are also generated via the pioneer round of translation for MHC I presentation, but in addition enter the lysosomal and endosomal MHC class II compartments. Via this route, cross-presentation of antigenic peptides by the MHC class I molecules can occur, and this in turn results in an overall superior CD8+ T-cell stimulation capacity. Another possible explanation has been described in a recent study by Kreiter et al.,39 showing that an increased MHC class I presentation is mainly mediated by the additional signal peptide at the N-terminal. Their hypothesis is that antigens fused to a signal peptide undergo translocation to the ER where misfolded proteins undergo rapid translocation to the cytosol. ER-associated degradation occurs by the proteasome in the vicinity of the ER. This results in rapid translocation of the peptides by TAP back into the ER, eventually leading to an increased MHC class I presentation. The first modification step also involved the deletion of the natural 5′- and 3′-UTRs of WT1. Although we have not investigated the effect of the deletion of these UTRs separately, we argue that an isolated removal of the UTRs would not have a negative impact on the processing and the presentation of the antigen for several reasons. First, the pGEM vector without UTRs leads to a longer and stronger protein expression after mRNA electroporation compared with a similar vector containing the 5′- and 3′-UTRs of the Xenopus laevis β-globin gene (unpublished data). Second, the expression of a transcription factor, such as WT1, is partly regulated by the stability of its mRNA. This can be controlled by its own UTRs as these sequences may therefore promote the degradation of the mRNA more rapidly.43
As stated before, the WT1 protein is predominantly located in the nucleus. Transport of a transcription factor from the cytoplasm to the nucleus is usually driven by NLS present in the zinc-finger domains. We hypothesized that deleting the NLS of zinc-finger domain 1 would retain WT1 in the cytoplasm, resulting in an enhanced delivery of WT1 protein to the antigen-processing compartments.16,35 Moreover, this would prevent the presence of two trafficking signals with opposite cellular destinations, being the NLS for translocation to the nucleus and the DC-LAMP signal for rerouting to the cytosolic lysosomal compartments. Although, an NLS has been described in zinc-finger domain 3 of WT144 after the start of our study, we could show that the nuclear expression after electroporation with the WT1-sh-DC-L mRNA was strongly diminished. Our results indicate that deletion of one WT1-derived NLS indeed led to a higher cytoplasmic expression level of WT1 and a subsequent gain in antigen processing and presentation (Figure 2c,d). It would be interesting to investigate the added influence of deleting the NLS in zinc-finger domain 3 in the future. A downside to this modification is that 57 amino acids (aa292-348) are deleted, resulting in a possible loss of WT1 epitopes. Several epitopes have been located in this region.20,45 Nevertheless, since only a relatively small portion of the total WT1 sequence is deleted (about 10%), we believe that the improvement in antigen expression and presentation observed after deletion of the NLS region outweighs the potential loss of immunogenic epitopes.
In general, protein expression may be limited by the short half-life of the mRNA. Furthermore, translation may be hampered by several characteristics of the mRNA (including rare codon usage, low G/C-content or presence of negatively cis-acting motifs). In silico optimization can lead to an increase in protein expression, demonstrating the reliability and general validity of this approach.31,32 Our observations also reveal some increased expression as seen with CD40Ligand and ca TLR4 (unpublished data). Nevertheless, for some genes such as CD70, sequence modification appears not to have a major impact on transgene expression (unpublished data). A similar observation was made in the present study for WT1. Indeed, we consistently found that the WT1-sh-DC-L-OPT mRNA construct was not superior to the WT1-sh-DC-L construct in terms of translational efficacy and T-cell stimulatory capacity, indicating that in silico sequence optimization is not useful for all genes.
In a final step, we subcloned the optimized WT1-sh-DC-L-OPT sequence into the pST1 vector containing two repeats of the 3′-UTR of human β-globin and a free-ending poly-A tail of 120 adenosine residues.33,43 These features are known to enhance the translation efficiency and stability of the mRNA. We were able to confirm that the use of pST1 vector–transcribed mRNA results in a higher and longer-lasting WT1 antigen expression and presentation by DCs in vitro as compared with the pGEM vector. In parallel, we observed that DCs electroporated with WT1 mRNA transcribed from the pST1 vector are superior to peptide-pulsed DCs for antigen-specific T-cell stimulation. This can be ascribed to the prolongation of the half-life of pST1-derived mRNA, resulting in increased cumulative expression levels of the antigen that enables de novo generation and presentation of peptides over an extended period of time. Consequently, a high density of peptides will be presented for a longer time, which is essential for optimal T-cell stimulation. This is in clear contrast with peptide-pulsed DCs where peptides are loaded exogenously onto the MHC class I molecules, which is known to be a very dynamic process with a short half-life.46
The superior T-cell stimulatory capacity of the optimized vector was confirmed in vivo. Intranodal injection of mRNA results in the uptake of RNA molecules by local DCs that are able to translate and present the antigen to the regional T-cells.7,47 Our in vivo results demonstrated that mice immunized with the optimized WT1 mRNA together with TriMix mRNA had a higher WT1-specific lytic capacity than mice immunized with wild-type WT1 mRNA and TriMix. Moreover, we observed strong antitumor effects against WT1-overexpressing leukemia cells after immunization with the optimized WT1 mRNA, without overt signs of autoimmune toxicity. The lack of overt autoimmune toxicity indicates that the normal level of WT1 expression by the few WT1-expressing tissues may be inadequate for recognition by WT1-specific CTLs.
Altogether, our findings show that the modification of an antigen-encoding mRNA template harbors multiple options to optimize its antigen presentation efficacy. This study proves that molecular optimization of a strong immunogenic tumor-associated antigen, such as WT1, can result in a high surface density of MHC/peptide complexes for an extended period of time. As a result, sufficiently high stimulatory signals can lead to the activation of sufficient numbers of T-cells to elicit significant cytotoxic impact without inducing autotoxicity. In conclusion, these data have important implications for the future design of mRNA-based cancer vaccine strategies targeting WT1.
Materials and methods
Mice. Six-to 12-week-old, female C57BL/6 mice were purchased from Harlan (Horst, The Netherlands). All the animals were maintained and treated according to the European guidelines for animal experimentation. All experiments were approved by the Ethical Committee for the use of laboratory animals of the Vrije Universiteit Brussel (CEP n°10-214-2).
Cells and media. Human DCs were generated as previously described.48 Cells were cultured in RPMI1640 medium (Lonza, Verviers, Belgium) supplemented with 5% fetal bovine serum (Harlan, Sera-Lab, West-Sussex, UK), 100 U/ml penicillin, and 2 mmol/l L-glutamine (PS/L-GLU; Lonza). Antigen presentation assays were performed in stimulation medium consisting of Iscove's modified Dulbecco's medium (Gibco) supplemented with 1% heat-inactivated human AB serum (PAA Laboratories, Linz, Austria), PS/L-GLU, 1 mmol/l sodium pyruvate, nonessential amino acids, 0.24 mmol/l L-asparagine, and 0.55 mmol/l L-arginine (all from Lonza). C1498 is a WT1-negative leukemia cell line of C57BL/6 origin. C1498-WT1 was established by transfection of murine WT1 cDNA into C1498 cells. C1498 and C1498-WT1 cells were cultured in Iscove's modified Dulbecco's medium (Lonza) supplemented with 10% fetal bovine serum and PS/L-GLU. WT1 expression of C1498-WT1 was maintained by culture in 0.5-mg/ml G418 (Alexis Biochemicals, Belgium). Both cell lines are a kind gift from HE Kohrt (CCSR, Stanford University, Stanford, CA) and were tested by polymerase chain reaction for WT1 expression.
Genetic constructs. The cloning of the CD40Ligand-, CD70-, and ca TLR4-encoding plasmids has been described.7,10 In this study, the TriMix genes (human and mouse analogs) were optimized in silico to improve their performance (GENEART, Regensburg, Germany) and subcloned into the pST1 transcription vector (kindly provided by U. Sahin, Johannes-Gutenberg University, Mainz, Germany).49 pGEM/tNGFR encoding a truncated form of the nerve growth factor receptor (containing the extracellular and transmembrane fragments) used for in vivo experiments, and the pGEM/NEF-DC-LAMP constructs used for in vitro experiments were described previously.50,51
The pGEM/WT1 plasmid containing the full-length WT1 cDNA flanked by its natural 5′- and 3′-UTRs and a poly(A)-tail of 64 adenines has been described before.38 The pGEM/ΔUT/sig-WT1-DC-LAMP plasmid was generated by cloning the WT1 cDNA in-frame as a BglII–BglII fragment between the sig and the lysosomal targeting sequence of DC-LAMP in the pGEM/ΔUT/sig-WT1-DC-LAMP vector without UTRs.28 To obtain the pGEM/ΔUT/sig-WT1-NLS-DC-LAMP vector, a 171 bp (aa292-348) long sequence containing the NLS of zinc-finger domain 1 was removed from the WT1 sequence.16,35 In the next step, in silico multiparameter sequence optimization of the sig-WT1-NLS-DC-LAMP sequence was performed using GENEART technology (sig-WT1-NLS-DC-LAMP-OPT). Finally, this optimized sequence was cloned into the pGEM and pST1 vectors. The latter contains a T7 promoter, 2-serial human 3′-β-globin UTRs, and a poly(A)-tail of 120 adenines.
In vitro transcription of mRNA. Before the in vitro mRNA synthesis, the pGEM and pST1 plasmids were linearized with SpeI and SapI, respectively. The in vitro transcription and mRNA quality control were performed as described before.28 The quality, size, and concentration of the different in vitro transcribed mRNA constructs were analyzed with the Agilent 2100 Bioanalyzer (Diegem, Belgium).
Electroporation of DCs and K562. Four to five million cells were coelectroporated with WT1-encoding mRNA (20 µg) and TriMix mRNA (5µg of each component) to allow antigen loading and DC maturation, respectively. Electroporation was performed as described before.11 Immediately after electroporation, the cells were transferred to culture medium and incubated at 37 °C in a humidified 5% CO2 atmosphere.
Synthetic peptides and peptide pulsing. Synthetic peptides corresponding to the HLA-A2-restricted epitopes of WT1 (aa126-134; RMFPNAPYL) and Melan-A (aa26-35; ELAGIGILTV) were purchased from Eurogentec (Seraing, Belgium). HLA-A2+ DCs (5 × 105 cells/ml) were loaded with 5 µg/ml peptide for 2 h. Subsequently, cells were washed and resuspended to a final density of 2 × 105 cells/ml.
Flow cytometry. To analyze intracellular WT1 expression, cells were fixed and permeabilized using the BD Cytofix/Cytoperm plus kit and stained intracellularly with an anti-WT1 monoclonal antibody (clone 6F-H2; Dako Cytomation, Carpinteria, CA). An immunoglobulin G isotype-matched PE-labeled anti-mouse antibody was used as secondary Ab (Becton & Dickinson, Erembodegem, Belgium). Nonreactive isotype-matched antibody (eBioscience, Vienna, Austria) was used as control. Data acquisition was performed on a FACSCanto flow cytometer (BD, Erembodegem, Belgium) and analyzed using FlowJo software.
Immunochemistry and histological examination. Immunocytochemistry of electroporated DCs was performed on Saccomanno-fixed (Yvsolab, Turnhout, Belgium), paraffin-embedded cell-block sections (5 µm) using an anti-WT1 monoclonal antibody (clone 6F-H2; Dako, Heverlee, Belgium) and the EnVisionTM FLEX detection system (Dako). Slides were examined quantitatively (percentage of WT1-positive cells) and qualitatively (signal intensity as compared with a WT1-positive control specimen) for expression of WT1 by light microscopy.
For histological examination, mice were killed by cervical dislocation at d35 postimmunization; kidney, lung, and liver were immediately prelevated for formalin fixation and paraffin embedding. Five-micrometer sections were cut and stained with periodic acid-Schiff and analyzed by light microscopy.
WT1-specific T-cell clone and in vitro antigen presentation assay. A human WT1-specific T-cell clone (B3) recognizing the HLA-A2-restricted WT1126–134 epitope was used. Clone B3 was expanded in vitro using the rapid expansion protocol as previously described.52
To investigate the T-cell stimulatory capacity of human DCs, 2 × 104 electroporated and/or peptide-pulsed HLA-A2+ DCs were cocultured with 5 × 103 WT1-specific B3 T-cells. Cocultures were started 4, 24, or 48 hours after electroporation. After 20 hours of coculture, supernatant was analyzed for the presence of IFN-γ by ELISA using commercially available antibodies (Thermo Scientific, Doornik, Belgium).
In vivo CTL induction. Mice were immunized intranodally with either control (truncated nerve growth factor receptor) mRNA, wild-type WT1 mRNA in combination with TriMix mRNA, or pST1-derived WT1-sh-DC-L-OPT mRNA in combination with TriMix mRNA. To that end, 10 µg of each mRNA molecule was resuspended in a volume of 10 μl 0.8 Hartman solution (Baxter, Braine-l'Alleud, Belgium) and injected into the inguinal lymph node as described before.7
Five days later, spleen cells isolated from naive syngeneic mice were pulsed with an overlapping pool of WT1 peptides spanning the entire sequence (JPT, Innovative Peptide Solutions, Berlin, Germany) at 1 µmol/l for 2 hours, after which they were labeled with 10 µmol/l carboxyfluoresceinsuccinimidyl ester (CFSE) (CFSEhi) (Life Technologies, Merelbeke, Belgium). These cells were mixed at a 1:1 ratio with unpulsed spleen cells labeled with 0.5 µmol/l CFSE (CFSElo). Ten million- to twenty million-mixed CFSE-labeled cells were injected via the tail vein. Sixteen hours later, specific lysis of target cells was analyzed in spleen, peripheral blood, and (injected and noninjected) lymph nodes by flow cytometry. The percentage of killing was calculated as described in ref. 7.
Tumor rejection experiments. To assess the therapeutic efficacy of intranodal WT1 immunizations, mice were inoculated with 5 × 105 C1498-WT1 cells on day 0 and immunized with a control (truncated nerve growth factor receptor) mRNA, the wild-type WT1, or the optimized WT1 mRNA in combination with TriMix mRNA 7 days later, as specified above. Mice were killed when the tumor reached a diameter of >2,500 mm.3
Urine collection. All animals were allowed ad libitum drinking water and free access to standard chow. During the course of the experiment, mice were housed in metabolic cages at different time points for the collection of 24-hour urine samples. Proteinuria was expressed as total urinary protein over creatinine.53
Statistical analysis. Data were analyzed with GraphPad Prism 5 software using a Student's t-test for human experiments or ANOVA with Bonferroni post hoc for in vivo experiments. Findings were considered statistically significant when P values were >0.05.
Acknowledgments
The authors wish to thank Petra Roman, Elsy Vaeremans, Xavier Debaere, Gwenny De Metter, Chiraz Mahmoud and Siegrid Pauwels for their excellent technical assistance.
TriMix-DCs are the topic of a patent application in the EU and are patented in the United States with the patent n° 8,476,419. AB and KT are inventors of this application. None of the authors involved in this study receive any form of support or remuneration related to this platform. This work was supported by grants to KT from the Interuniversity Attraction Poles Program—Belgian State—Belgian Science Policy, the Belgian Foundation against Cancer, the Flemish League against Cancer, an Integrated Project and Network of Excellence sponsored by the EU, and the Research Foundation—Flanders (FWO). VVT received grants from the Research Foundation—Flanders (# G.0370.08 and G.0082.08), the Belgian Foundation against Cancer, the Flemish League against Cancer, the National Cancer Plan 29 of the Belgian government, the Flemish Agency for Innovation by Science and Technology (IWT), and the Methusalem financement program of the Flemish government and from the Interuniversity Attraction Pole financement program (IAP #P6/41) of the Belgian government. SA is a former PhD fellow of the Research Foundation—Flanders (FWO) and, currently, holds an Emmanuel van der Schueren of the Flemish League against Cancer; SA also received personal financial support from the Belgian public utility foundation VOCATIO. KB and AB are postdoctoral fellows of the Research Foundation—Flanders (FWO).
References
- Boczkowski D, Nair SK, Snyder D, Gilboa E. Dendritic cells pulsed with RNA are potent antigen-presenting cells in vitro and in vivo. J Exp Med. 1996;184:465–472. doi: 10.1084/jem.184.2.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair SK, Boczkowski D, Morse M, Cumming RI, Lyerly HK, Gilboa E. Induction of primary carcinoembryonic antigen (CEA)-specific cytotoxic T lymphocytes in vitro using human dendritic cells transfected with RNA. Nat Biotechnol. 1998;16:364–369. doi: 10.1038/nbt0498-364. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Senovilla L, Vacchelli E, Eggermont A, Fridman WH, Galon J, et al. Trial watch: Dendritic cell-based interventions for cancer therapy. Oncoimmunology. 2012;1:1111–1134. doi: 10.4161/onci.21494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nuffel AM, Benteyn D, Wilgenhof S, Corthals J, Heirman C, Neyns B, et al. Intravenous and intradermal TriMix-dendritic cell therapy results in a broad T-cell response and durable tumor response in a chemorefractory stage IV-M1c melanoma patient. Cancer Immunol Immunother. 2012;61:1033–1043. doi: 10.1007/s00262-011-1176-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilgenhof S, Van Nuffel AM, Corthals J, Heirman C, Tuyaerts S, Benteyn D, et al. Therapeutic vaccination with an autologous mRNA electroporated dendritic cell vaccine in patients with advanced melanoma. J Immunother. 2011;34:448–456. doi: 10.1097/CJI.0b013e31821dcb31. [DOI] [PubMed] [Google Scholar]
- Benteyn D, Van Nuffel AM, Wilgenhof S, Corthals J, Heirman C, Neyns B, et al. Characterization of CD8+ T-cell responses in the peripheral blood and skin injection sites of melanoma patients treated with mRNA electroporated autologous dendritic cells (TriMixDC-MEL). Biomed Res Int. 2013;2013:976383. doi: 10.1155/2013/976383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lint S, Goyvaerts C, Maenhout S, Goethals L, Disy A, Benteyn D, et al. Preclinical evaluation of TriMix and antigen mRNA-based antitumor therapy. Cancer Res. 2012;72:1661–1671. doi: 10.1158/0008-5472.CAN-11-2957. [DOI] [PubMed] [Google Scholar]
- Weide B, Pascolo S, Scheel B, Derhovanessian E, Pflugfelder A, Eigentler TK, et al. Direct injection of protamine-protected mRNA: results of a phase ½ vaccination trial in metastatic melanoma patients. J Immunother. 2009;32:498–507. doi: 10.1097/CJI.0b013e3181a00068. [DOI] [PubMed] [Google Scholar]
- Weide B, Carralot JP, Reese A, Scheel B, Eigentler TK, Hoerr I, et al. Results of the first phase I/II clinical vaccination trial with direct injection of mRNA. J Immunother. 2008;31:180–188. doi: 10.1097/CJI.0b013e31815ce501. [DOI] [PubMed] [Google Scholar]
- Bonehill A, Tuyaerts S, Van Nuffel AM, Heirman C, Bos TJ, Fostier K, et al. Enhancing the T-cell stimulatory capacity of human dendritic cells by co-electroporation with CD40L, CD70 and constitutively active TLR4 encoding mRNA. Mol Ther. 2008;16:1170–1180. doi: 10.1038/mt.2008.77. [DOI] [PubMed] [Google Scholar]
- Bonehill A, Van Nuffel AM, Corthals J, Tuyaerts S, Heirman C, François V, et al. Single-step antigen loading and activation of dendritic cells by mRNA electroporation for the purpose of therapeutic vaccination in melanoma patients. Clin Cancer Res. 2009;15:3366–3375. doi: 10.1158/1078-0432.CCR-08-2982. [DOI] [PubMed] [Google Scholar]
- Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, et al. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res. 2009;15:5323–5337. doi: 10.1158/1078-0432.CCR-09-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohenstein P, Hastie ND. The many facets of the Wilms' tumour gene, WT1. Hum Mol Genet. 2006;15 Spec No 2:R196–R201. doi: 10.1093/hmg/ddl196. [DOI] [PubMed] [Google Scholar]
- Wilsher M, Cheerala B. WT1 as a complementary marker of malignant melanoma: an immunohistochemical study of whole sections. Histopathology. 2007;51:605–610. doi: 10.1111/j.1365-2559.2007.02843.x. [DOI] [PubMed] [Google Scholar]
- Wagner N, Panelos J, Massi D, Wagner KD. The Wilms' tumor suppressor WT1 is associated with melanoma proliferation. Pflugers Arch. 2008;455:839–847. doi: 10.1007/s00424-007-0340-1. [DOI] [PubMed] [Google Scholar]
- Yang L, Han Y, Suarez Saiz F, Saurez Saiz F, Minden MD. A tumor suppressor and oncogene: the WT1 story. Leukemia. 2007;21:868–876. doi: 10.1038/sj.leu.2404624. [DOI] [PubMed] [Google Scholar]
- Mundlos S, Pelletier J, Darveau A, Bachmann M, Winterpacht A, Zabel B. Nuclear localization of the protein encoded by the Wilms' tumor gene WT1 in embryonic and adult tissues. Development. 1993;119:1329–1341. doi: 10.1242/dev.119.4.1329. [DOI] [PubMed] [Google Scholar]
- Sugiyama H. WT1 (Wilms' tumor gene 1): biology and cancer immunotherapy. Jpn J Clin Oncol. 2010;40:377–387. doi: 10.1093/jjco/hyp194. [DOI] [PubMed] [Google Scholar]
- Oka Y, Tsuboi A, Taguchi T, Osaki T, Kyo T, Nakajima H, et al. Induction of WT1 (Wilms' tumor gene)-specific cytotoxic T lymphocytes by WT1 peptide vaccine and the resultant cancer regression. Proc Natl Acad Sci USA. 2004;101:13885–13890. doi: 10.1073/pnas.0405884101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anguille S, Van Tendeloo VF, Berneman ZN. Leukemia-associated antigens and their relevance to the immunotherapy of acute myeloid leukemia. Leukemia. 2012;26:2186–2196. doi: 10.1038/leu.2012.145. [DOI] [PubMed] [Google Scholar]
- Van Driessche A, Berneman ZN, Van Tendeloo VF. Active specific immunotherapy targeting the Wilms' tumor protein 1 (WT1) for patients with hematological malignancies and solid tumors: lessons from early clinical trials. Oncologist. 2012;17:250–259. doi: 10.1634/theoncologist.2011-0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka Y, Elisseeva OA, Tsuboi A, Ogawa H, Tamaki H, Li H, et al. Human cytotoxic T-lymphocyte responses specific for peptides of the wild-type Wilms' tumor gene (WT1) product. Immunogenetics. 2000;51:99–107. doi: 10.1007/s002510050018. [DOI] [PubMed] [Google Scholar]
- Gao L, Bellantuono I, Elsässer A, Marley SB, Gordon MY, Goldman JM, et al. Selective elimination of leukemic CD34(+) progenitor cells by cytotoxic T lymphocytes specific for WT1. Blood. 2000;95:2198–2203. [PubMed] [Google Scholar]
- Van Tendeloo VF, Van de Velde A, Van Driessche A, Cools N, Anguille S, Ladell K, et al. Induction of complete and molecular remissions in acute myeloid leukemia by Wilms' tumor 1 antigen-targeted dendritic cell vaccination. Proc Natl Acad Sci USA. 2010;107:13824–13829. doi: 10.1073/pnas.1008051107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits EL, Anguille S, Cools N, Berneman ZN, Van Tendeloo VF. Dendritic cell-based cancer gene therapy. Hum Gene Ther. 2009;20:1106–1118. doi: 10.1089/hum.2009.145. [DOI] [PubMed] [Google Scholar]
- Anguille S, Lion E, Smits E, Berneman ZN, van Tendeloo VF. Dendritic cell vaccine therapy for acute myeloid leukemia: questions and answers. Hum Vaccin. 2011;7:579–584. doi: 10.4161/hv.7.5.14652. [DOI] [PubMed] [Google Scholar]
- Van Nuffel AM, Benteyn D, Wilgenhof S, Corthals J, Heirman C, Neyns B, et al. Intravenous and intradermal TriMix-dendritic cell therapy results in a broad T-cell response and durable tumor response in a chemorefractory stage IV-M1c melanoma patient. Cancer Immunol Immunother. 2012;61:1033–1043. doi: 10.1007/s00262-011-1176-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonehill A, Heirman C, Tuyaerts S, Michiels A, Breckpot K, Brasseur F, et al. Messenger RNA-electroporated dendritic cells presenting MAGE-A3 simultaneously in HLA class I and class II molecules. J Immunol. 2004;172:6649–6657. doi: 10.4049/jimmunol.172.11.6649. [DOI] [PubMed] [Google Scholar]
- De Keersmaecker B, Heirman C, Allard S, Bonehill A, Corthals J, Thielemans K, et al. Lumenal part of the DC-LAMP protein is not required for induction of antigen-specific T cell responses by means of antigen-DC-LAMP messenger RNA-electroporated dendritic cells. Hum Gene Ther. 2010;21:479–485. doi: 10.1089/hum.2009.080. [DOI] [PubMed] [Google Scholar]
- Van Nuffel AM, Benteyn D, Wilgenhof S, Pierret L, Corthals J, Heirman C, et al. Dendritic cells loaded with mRNA encoding full-length tumor antigens prime CD4+ and CD8+ T cells in melanoma patients. Mol Ther. 2012;20:1063–1074. doi: 10.1038/mt.2012.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fath S, Bauer AP, Liss M, Spriestersbach A, Maertens B, Hahn P, et al. Multiparameter RNA and codon optimization: a standardized tool to assess and enhance autologous mammalian gene expression. PLoS ONE. 2011;6:e17596. doi: 10.1371/journal.pone.0017596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turksma AW, Bontkes HJ, Ruizendaal JJ, van den Heuvel H, Scholten KB, Santegoets SJ, et al. Increased cytotoxic capacity of tumor antigen specific human T cells after in vitro stimulation with IL21 producing dendritic cells. Hum Immunol. 2013;74:506–513. doi: 10.1016/j.humimm.2013.01.014. [DOI] [PubMed] [Google Scholar]
- Holtkamp S, Kreiter S, Selmi A, Simon P, Koslowski M, Huber C, et al. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood. 2006;108:4009–4017. doi: 10.1182/blood-2006-04-015024. [DOI] [PubMed] [Google Scholar]
- Van Driessche A, Van de Velde AL, Nijs G, Braeckman T, Stein B, De Vries JM, et al. Clinical-grade manufacturing of autologous mature mRNA-electroporated dendritic cells and safety testing in acute myeloid leukemia patients in a phase I dose-escalation clinical trial. Cytotherapy. 2009;11:653–668. doi: 10.1080/14653240902960411. [DOI] [PubMed] [Google Scholar]
- Bruening W, Moffett P, Chia S, Heinrich G, Pelletier J. Identification of nuclear localization signals within the zinc fingers of the WT1 tumor suppressor gene product. FEBS Lett. 1996;393:41–47. doi: 10.1016/0014-5793(96)00853-8. [DOI] [PubMed] [Google Scholar]
- Coosemans A, Wölfl M, Berneman ZN, Van Tendeloo V, Vergote I, Amant F, et al. Immunological response after therapeutic vaccination with WT1 mRNA-loaded dendritic cells in end-stage endometrial carcinoma. Anticancer Res. 2010;30:3709–3714. [PubMed] [Google Scholar]
- Niksic M, Slight J, Sanford JR, Caceres JF, Hastie ND. The Wilms' tumour protein (WT1) shuttles between nucleus and cytoplasm and is present in functional polysomes. Hum Mol Genet. 2004;13:463–471. doi: 10.1093/hmg/ddh040. [DOI] [PubMed] [Google Scholar]
- Van Driessche A, Van de Velde AL, Nijs G, Braeckman T, Stein B, De Vries JM, et al. Clinical-grade manufacturing of autologous mature mRNA-electroporated dendritic cells and safety testing in acute myeloid leukemia patients in a phase I dose-escalation clinical trial. Cytotherapy. 2009;11:653–668. doi: 10.1080/14653240902960411. [DOI] [PubMed] [Google Scholar]
- Kreiter S, Selmi A, Diken M, Sebastian M, Osterloh P, Schild H, et al. Increased antigen presentation efficiency by coupling antigens to MHC class I trafficking signals. J Immunol. 2008;180:309–318. doi: 10.4049/jimmunol.180.1.309. [DOI] [PubMed] [Google Scholar]
- Rock KL, Farfán-Arribas DJ, Shen L. Proteases in MHC class I presentation and cross-presentation. J Immunol. 2010;184:9–15. doi: 10.4049/jimmunol.0903399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apcher S, Manoury B, Fåhraeus R. The role of mRNA translation in direct MHC class I antigen presentation. Curr Opin Immunol. 2012;24:71–76. doi: 10.1016/j.coi.2012.01.007. [DOI] [PubMed] [Google Scholar]
- Apcher S, Daskalogianni C, Lejeune F, Manoury B, Imhoos G, Heslop L, et al. Major source of antigenic peptides for the MHC class I pathway is produced during the pioneer round of mRNA translation. Proc Natl Acad Sci USA. 2011;108:11572–11577. doi: 10.1073/pnas.1104104108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn AN, Diken M, Kreiter S, Vallazza B, Türeci Ö, Sahin U. Determinants of intracellular RNA pharmacokinetics: Implications for RNA-based immunotherapeutics. RNA Biol. 2011;8:35–43. doi: 10.4161/rna.8.1.13767. [DOI] [PubMed] [Google Scholar]
- Depping R, Schindler SG, Jacobi C, Kirschner KM, Scholz H. Nuclear transport of Wilms' tumour protein Wt1 involves importins a and ß. Cell Physiol Biochem. 2012;29:223–232. doi: 10.1159/000337603. [DOI] [PubMed] [Google Scholar]
- Van der Bruggen PS, Vigneron N, Van den Eynde B.Peptide database at < http://www.cancerimmunity.org/peptidedatabase/Tcellepitopes.htm >
- Reits E, Griekspoor A, Neijssen J, Groothuis T, Jalink K, van Veelen P, et al. Peptide diffusion, protection, and degradation in nuclear and cytoplasmic compartments before antigen presentation by MHC class I. Immunity. 2003;18:97–108. doi: 10.1016/s1074-7613(02)00511-3. [DOI] [PubMed] [Google Scholar]
- Kreiter S, Selmi A, Diken M, Koslowski M, Britten CM, Huber C, et al. Intranodal vaccination with naked antigen-encoding RNA elicits potent prophylactic and therapeutic antitumoral immunity. Cancer Res. 2010;70:9031–9040. doi: 10.1158/0008-5472.CAN-10-0699. [DOI] [PubMed] [Google Scholar]
- Tuyaerts S, Noppe SM, Corthals J, Breckpot K, Heirman C, De Greef C, et al. Generation of large numbers of dendritic cells in a closed system using Cell Factories. J Immunol Methods. 2002;264:135–151. doi: 10.1016/s0022-1759(02)00099-6. [DOI] [PubMed] [Google Scholar]
- Holtkamp S, Kreiter S, Selmi A, Simon P, Koslowski M, Huber C, et al. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood. 2006;108:4009–4017. doi: 10.1182/blood-2006-04-015024. [DOI] [PubMed] [Google Scholar]
- Michiels A, Tuyaerts S, Bonehill A, Corthals J, Breckpot K, Heirman C, et al. Electroporation of immature and mature dendritic cells: implications for dendritic cell-based vaccines. Gene Ther. 2005;12:772–782. doi: 10.1038/sj.gt.3302471. [DOI] [PubMed] [Google Scholar]
- Allard SD, Pletinckx K, Breckpot K, Heirman C, Bonehill A, Michiels A, et al. Functional T-cell responses generated by dendritic cells expressing the early HIV-1 proteins Tat, Rev and Nef. Vaccine. 2008;26:3735–3741. doi: 10.1016/j.vaccine.2008.04.077. [DOI] [PubMed] [Google Scholar]
- Ho WY, Nguyen HN, Wolfl M, Kuball J, Greenberg PD. In vitro methods for generating CD8+ T-cell clones for immunotherapy from the naïve repertoire. J Immunol Methods. 2006;310:40–52. doi: 10.1016/j.jim.2005.11.023. [DOI] [PubMed] [Google Scholar]
- Van Beneden K, Geers C, Pauwels M, Mannaerts I, Wissing KM, Van den Branden C, et al. Comparison of trichostatin A and valproic acid treatment regimens in a mouse model of kidney fibrosis. Toxicol Appl Pharmacol. 2013;271:276–284. doi: 10.1016/j.taap.2013.05.013. [DOI] [PubMed] [Google Scholar]