

Abstract
Antimicrobial photodynamic therapy (PDT) is used for the eradication of pathogenic microbial cells and involves the light excitation of dyes in the presence of O2, yielding reactive oxygen species including the hydroxyl radical (•OH) and singlet oxygen (1O2). In order to chemically enhance PDT by the formation of longer-lived radical species, we asked whether thiocyanate (SCN−) could potentiate the methylene blue (MB) and light-mediated killing of the gram-positive Staphylococcus aureus and the gram-negative Escherichia coli. SCN− enhanced PDT (10 μM MB, 5J/cm2 660 nm hv) killing in a concentration-dependent manner of S. aureus by 2.5 log10 to a maximum of 4.2 log10 at 10 mM (P < 0.001) and increased killing of E. coli by 3.6 log10 to a maximum of 5.0 log10 at 10 mM (P < 0.01). We determined that SCN− rapidly depleted O2 from an irradiated MB system, reacting exclusively with 1O2, without quenching the MB excited triplet state. SCN− reacted with 1O2, producing a sulfur trioxide radical anion (a sulfur-centered radical demonstrated by EPR spin trapping). We found that MB-PDT of SCN− in solution produced both sulfite and cyanide anions, and that addition of each of these salts separately enhanced MB-PDT killing of bacteria. We were unable to detect EPR signals of •OH, which, together with kinetic data, strongly suggests that MB, known to produce •OH and 1O2, may, under the conditions used, preferentially form 1O2.
Keywords: Thiocyanate, Antimicrobial photodynamic inactivation, Sulfite, Cyanide, ESR spin trapping, Gram-positive bacteria, Gram-negative bacteria
Introduction
The increase in disease caused by antimicrobial-resistant microorganisms is a major medical concern of the twenty-first century [1,2]. In the United States, antimicrobial-resistant infections cost an excess of $55 billion and result in a collective 8 million additional hospital days annually [3]. In an effort to reduce morbidity, mortality, and economic losses associated with resistant pathogens, there is currently a global search for alternative antibacterial techniques that are able not only to kill antimicrobial-resistant isolates but also do not cause resistance to said alternative therapeutics.
Photodynamic therapy (PDT) was discovered in 1900 and has since emerged for the treatment of neoplastic conditions [4]. Because PDT can destroy antibiotic-resistant bacteria and selectively kill pathogens over host cells, PDT is considered a promising treatment for localized infections [5]. PDT involves the visible light excitation of a photosensitizer (PS) in the presence of O2. When light excites a PS, the PS reaches an excited electronic singlet state and may convert to a long-lived excited triplet state which reacts with ground (triplet) state O2 in two different ways [6]. In the type I photochemical reaction an excited PS molecule transfers an electron to an appropriate acceptor molecule such as ground state O2. Type I photochemistry forms partially reduced oxygen species, including hydroxyl radicals (•OH), superoxide radical anions (O2•−), and hydrogen peroxide (H2O2). Alternatively, in the type II photochemical reaction, an energy-transfer reaction occurs between the excited PS and the ground state O2, forming excited singlet oxygen (1O2). Both oxygen radicals and 1O2 cause acute oxidative stress, resulting in destruction of target cells. Because of the high reactivity and thus short lifetimes of these reactive oxygen species, oxidative damage by PDT typically occurs in the immediate proximity of PS localization following excitation [7]. In the case of antimicrobial PDT, where dyes localize externally on the surface of bacteria, oxidative damage is limited to the outer portion of the bacterial cell envelope [8].
Recently, we reported the paradoxical potentiation of methylene blue-mediated antimicrobial PDT by azide (N3−) [9]—paradoxical as N3− physically quenches 1O2, lessening 1O2 killing of bacteria [10]. We concluded that oxygen radicals or the excited PS itself can directly abstract an electron from N3−, forming azidyl radicals (N3•) which are less reactive, but more selective, oxidizing agents, because they are longer lived than •OH. This longer lifetime may be responsible for enhanced PDT killing as N3• may diffuse deeply into bacteria and then wreak havoc while the more reactive •OH is consumed at the cell wall. Because N3− is a pseudohalide—a collection of atoms that collectively behaves like halides—we subsequently investigated alkali halide salts for potentiation of PDT and observed enhanced antimicrobial activity due to the formation of iodine and bromine radicals (manuscript in preparation). These findings are in accordance with the observation that KBr and KI react with •OH [11].
Pseudohalides besides N3−, such as thiocyanate (SCN−), also function as •OH scavengers [11]. Lassiter et al. augmented lactoferrin-H2O2 (a •OH generator) killing of Streptococcus mutans via application of SCN− [12]. SCN− and •OH react through the following mechanisms:
| (1) |
| (2) |
| (3) |
One-electron oxidation of SCN− by •OH results in the formation of the thiocyanate radical (•SCN) [Eqs. (1) and (2)]. •SCN reacts with excess SCN−, forming the dirhodane radical anion ((SCN)2•−) [Eq. (3)] [13,14]. Even though the SCN− reaction with radicals provides precedent to analyze the effect of SCN− on antimicrobial PDT, 1O2, which is also formed, is particularly thiophilic, reacting with sulfur species affording sulfoxides, sulfones, and sulfonic acids [15]. Due to the known reactivity of SCN− with •OH and sulfur with 1O2, we sought to investigate whether SCN− enhanced antibacterial PDT and to study the chemical mechanism of any potentiation.
Materials and methods
Chemicals and reagents
Methylene blue (MB), NaSCN, KSCN, FeSO4, FeCl3, KCN, Na2SO3, and malachite green were purchased from Sigma-Aldrich (St. Louis, MO). MB stock solutions were prepared in dH2O and stored at 4 °C in the dark for no more than 24 h prior to use. NaSCN and KSCN solutions were prepared in dH2O as required immediately before experimentation. The EPR oxygen-sensitive spin probe 4-protio-3-carbamoyl-2,2,5,5-tetraperdeuteromethyl-3-pyrrolin-1-yloxy (mHCTPO) was a generous gift from Prof. H. J. Halpern (University of Chicago, Chicago, IL) [16]. The spin-trap 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) was purchased from Sigma-Aldrich.
Bacterial strains and culture conditions
Escherichia coli K12 (ATCC 33780) and Staphylococcus aureus (NCTC 8325) were chosen as representative gram-negative and gram-positive bacteria, respectively. Bacteria were routinely suspended in brain heart infusion (BHI) broth (Becton, Dickinson, and Company, Franklin Lakes, NJ) and grown overnight, aerobically in a 37 °C shaker incubator (New Brunswick Scientific, Edison, NJ) at 130 rpm. Cells were removed at an optical density at 600 nm (OD600) of 1.0, approximately equivalent to 108 colony forming units (CFU) per milliliter as determined by spectrophotometry (Thermo Scientific, Waltham, MA).
Photodynamic inactivation
E. coli and S. aureus were harvested at an OD600 of 1.0, centrifuged at 12,000 rpm for 3 min (24,148 g), and resuspended in phosphate-buffered saline (PBS) to arrest microbial growth. For photodynamic inactivation E. coli and S. aureus were incubated with MB for 5 min in the dark at final concentrations of 10 μM. PBS suspensions of bacteria exposed to MB were then illuminated with 5 J cm−2 of 660 ± 15nm (100–200 mW) light using a Lumacare LC-122 lamp (Lumacare, Newport Beach, CA) with the appropriate band pass filter [17].
SCN− combination studies
Cellular toxicity of SCN− was first determined by preparing bacteria in PBS, as described above and exposing them to KSCN at final concentrations of 10 μM, 100 μM, 1 mM, and 10 mM. PDT SCN− combination was accomplished by first incubating PBS suspensions of bacteria with SCN− for 5 min and then exposing these cultures to MB for 5 min. Samples were then irradiated as described above, serially diluted (6 tenfold dilutions), and plated on BHI agar and colonies were counted, following the methods of Jett et al. [18]. PDT killing is reported as the log10 reduction in cell viability relative to control values. Cellular killing is graphed as the mean of three separate experiments. Error bars are standard deviation from the mean.
PDT dependence on oxygen
PDT was performed as described above in the presence of 10 mM KSCN. One-milliliter aliquots were transferred to quartz cuvettes (Model 32Q10, Starna Cells Inc, Atascadero, CA) containing a magnetic stirrer and were sealed with a rubber septum in the dark. Cuvettes allowed O2-free samples to be irradiated without exposure to ambient air. The septum was pierced with a hollow needle connected to a N2/Ar line and samples were then bubbled with 75% N2/25% Ar gas for 10 min. Samples were irradiated with 5 or 10 J cm−2 of 660 ± 15 nm hv (150 mW/cm2).
Photosensitized O2 consumption measurements
Time-dependent changes in O2 concentration were determined by electron paramagnetic resonance (EPR) oximetry using mHCTPO at concentration 100 μM as an oxygen (3O2)-sensitive spin probe [16,19]. Samples with and without NaSCN were irradiated in EPR quartz flat cells in the resonant cavity with 540–740 nm (70 mW/cm2) light derived from a 300 W high pressure compact arc xenon lamp (Cermax, PE300CE–13FM/Module300W, Perkin-Elmer) equipped with a water filter, heat reflecting hot mirror, cutoff filter blocking light below 390 nm, and long-pass filter transmitting light above 540 nm. Concentrations of MB and NaSCN were 25 μM and 10 mM, respectively. O2 consumption measurements were performed in PBS (H2O and D2O for comparison). EPR samples were run using a microwave power 1.06 mW, modulation amplitude 0.006 mT, scan width 0.3 mT, and scan time 5.2 s. EPR measurements were carried out using a Bruker EMX–AA EPR spectrometer (Bruker BioSpin, Germany).
Time-resolved spectroscopic detection of 1O2
The efficiency of SCN− to quench 1O2 was determined directly by measuring 1O2 lifetime in deuterium oxide (D2O) with and without NaSCN. Time-resolved luminescence of 1O2 was measured at 1270 nm. Solutions of MB in 1-cm fluorescence cuvette (QA-1000, HelmaOptik) were excited with 532 nm microjoule pulses (750 ps duration) generated by a microchip Nd:YAG laser (Pulselas-P-1064-FC, Alphalas GmbH, Goettingen, Germany) operating with 2–10 kHz repetition rate. To filter first and third harmonics of laser radiation, 50-cm water filter and dichroic mirrors (BK7series, Eksma Optics, Vilnius, Lithuania) were used. Near-infrared luminescence was measured perpendicularly to the excitation beam in a photon counting mode [20] using a thermoelectric cooled NIR PMT module (Model H10330-45, Hamamatsu, Japan) equipped with 1100-nm cutoff filter and additional selected narrow-band filters (NB series, NDC Infrared Engineering LTD, Maldon, UK). A computer-mounted PCI-board multichannel scaler was used (NanoHarp 250, PicoQuant GmbH, Berlin, Germany) and data collection was synchronized with laser pulses using an ultrafast photodiode (UGP-300-SP, Alphalas GmbH, Goettingen, Germany) as a trigger. First-order luminescence decay fitting by the Levenberg-Marquardt algorithm was performed by custom-written software.
Detection of free radicals
EPR spin-trapping was employed using DMPO as a spin trap at concentrations of 10 mM or 50 mM [21,22]. The formation of DMPO spin adducts with radicals accompanying the photosensitized oxidation of SCN− was initiated by irradiation of appropriate samples, as described above. The concentration of MB was 25 μM and that of NaSCN was 10 mM. EPR samples were run using microwave power 10.6 mW, modulation amplitude 0.05 mT, center field 339.0 mT, scan width 8 mT, and scan time 84 s. To establish anaerobic conditions, samples were bubbled with argon for at least 30 min and vacuum transferred into an EPR flat cell that was prepurged with argon. Simulations of EPR spectra were performed using a WinSIM (Version 0.98) program. EPR measurements were carried out using the same spectrometer as described above.
Cyanide assay
The Prussian Blue (PB) assay was used in order to determine the amount of CN− produced from the MB-PDT-mediated oxidation of SCN−. This assay is based on the formation of a blue colored Fe4[Fe(CN)6]3 complex, PB, produced from CN− in the presence of Fe2+ and Fe3+ which can be detected with a spectrophotometer at 700 nm [1]. A calibration curve was constructed from absorbance values of standard concentrations of CN− with Fe (II) sulfate and Fe (III) chloride in acidic conditions. A sample of 20 μM MB and 10 mM KSCN was then vigorously stirred and illuminated using a Lumacare LC-122 lamp and the appropriate filter of 635 nm light with fluences of 0–320 J/cm2. During illumination, an aliquot of 100 μl of sample was removed and added to the reagent mixture (Fe2+/Fe3+/H+) and the absorption was measured at 700 nm using the SpectraMax M5 plate reader (Molecular Devices, Sunnyvale, CA). Data points are plotted as the mean of three separate experiments. Errors bars are SD.
Sulfite assay
The concentration of sulfite that was released from MB-PDT-mediated decomposition of KSCN was measured using UV-visible spectrophotometry in the presence of malachite green dye. The idea behind this assay is that sulfite (SO3−) ion can reduce the malachite green and reduce its absorbance [2].
A calibration curve was plotted of the absorbance change at 617 nm. The calibration curve was shown to be linear with the range SO3− concentration from 50 μM to 1.0 mM in the presence of malachite green and MB. A mixture of 10 mM KSCN and 20 μM MB was vigorously stirred and then illuminated with a Lumacare LC-122 lamp and the appropriate filter of 660 nm light with fluence of 0–320 J/cm2. During illumination, an aliquot of 100 μl of sample was added to a 100-μl solution of 5 μM malachite green. The absorption change value was measured in 617 nm using the SpectraMax M5 plate reader (Molecular Devices, Sunnyvale, CA). Data points are plotted as the mean of three separate experiments.
Potentiation of MB-antibacterial PDI by addition of sulfite and cyanide and mixtures of both
In order to test whether addition of KCN or Na2SO3 could increase the MB-mediated PDI killing of bacteria we carried PDI studies with S. aureus and with E. coli as described above (10 μM MB and 1–7 J/cm2 of 660 nm light) but with either addition or not of sodium sulfite (10 μM) or potassium cyanide (1 mM). Furthermore at the suggestion of a reviewer we tested the mixture of equimolar concentrations (50 μM) of KCN or Na2SO3 as a potentiator of PDI (10 μM MB+1–7 J/cm2 of 660 nm light).
Statistical analyses
Statistical analyses were performed for photodynamic inactivation studies. One-way analysis of variance (one-way ANOVA) and the Tukey post hoc test were used to compare survival fractions of PDT+SCN− with PDT alone. Analysis was performed using Microsoft Excel Statistical Package (Microsoft, Redmond, Washington). Results were considered statistically significant when P < 0.05.
Results
Photodynamic inactivation in the presence of SCN−
SCN− was capable of increasing MB photodynamic inactivation of S. aureus (10 μM MB+5 J/cm2 660 nm light) in a concentration-dependent manner, reaching a maximum increase in killing of 2.5 log10 (from 1.4 log10 without KSCN to 4.2 log10 at 10 mM KCSN) (P < 0.01, Fig. 1A). E. coli killing by MB-PDT was also potentiated by KSCN in a concentration-dependent manner (from 1.4 log10 without KSCN to 5.0 log10 at 10 mM KCSN) (P < 0.01, Fig. 1B). The concentration dependence was statistically significant because the killing at 10 mM was significantly different from that at 10 μM (P < 0.05) for both bacterial strains. Slight yet negligible (> 1 log10) dark toxicity was noted in S. aureus (Fig. 1A) and slight yet negligible stimulatory effects were observed in E. coli when cells were exposed to MB in the dark (Fig. 1B). SCN− alone was nontoxic at all concentrations for both S. aureus and E. coli. In order the confirm that the potentiation of killing was strictly dependent on photochemical action we carried out studies where bacteria were exposed to a fixed concentration of MB (10 μM) and KCSN (10 mM) and increasing fluences of light were delivered. Fig. 1C shows that the degree of potentiation of killing increases as more light is delivered in the case of S. aureus, and Fig. 1D shows the same phenomenon in the case of E. coli.
Fig. 1.
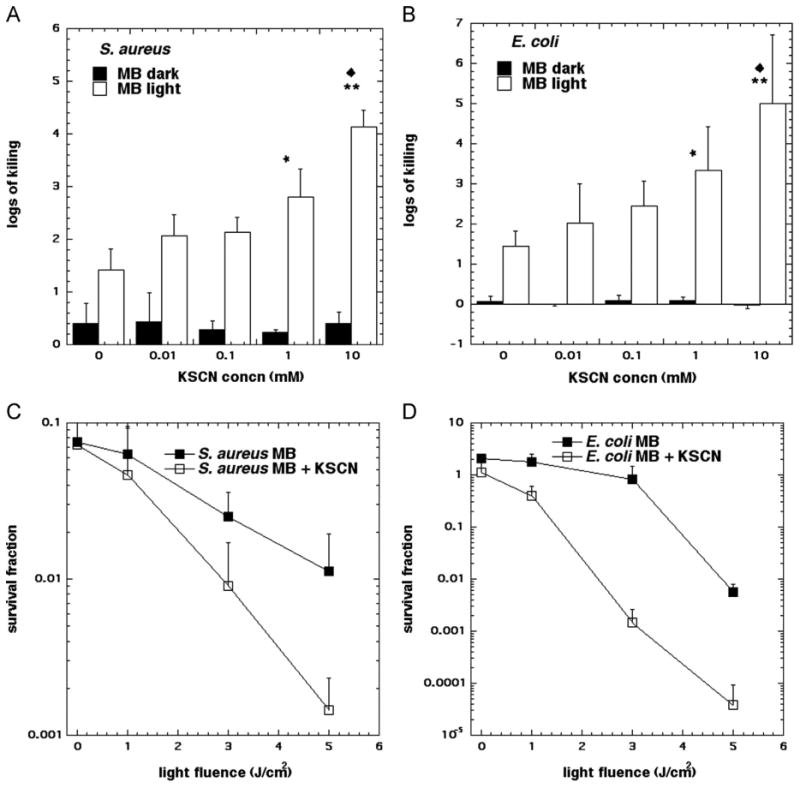
Concentration-dependent SCN− potentiation of antimicrobial PDT killing. (A) S. aureus, (B) E. coli. Cells were first incubated with NaSCN of the appropriate concentration for 10 min in the presence of 10 μM MB for both E. coli and S. aureus and then 5 J cm−2 of 660 nm light was delivered. Light-dose-dependent killing of (C) S. aureus and (D) E. coli in the presence of 10 μM MB and 10 mM NaSCN as above. Values are means of triplicate determinations from separate experiments, and bars are SD. Significance was determined by one-way ANOVA+Tukey post hoc analyses. (A and B) P values are * < 0.05; ** < 0.01 vs no NaSCN. (C and D) P values are * < 0.05; ** < 0.01 vs no NaSCN. ◆ is P < 0.05 vs 0.01 mM NaSCN.
EPR oximetry of the PDT-SCN− system
Irradiation of EPR samples containing 25 μM MB and 10 mM NaSCN in PBS with 12.6 J cm−2 of 540–740 nm light induced rapid depletion of dissolved O2, as determined by mHCTPO. Compared to controls, NaSCN increased the rate of O2 photo-consumption by 500-fold to 5.7 μM s−1 (Fig. 2A). To test if 1O2 was implicated in the observed PDT potentiation, 5 mM NaN3 was added to MB-SCN− samples [23]. This resulted in a 17-fold reduction in O2 photo-consumption in the MB-SCN− system (Fig. 2A). To further determine if 1O2 was involved in the MB-SCN− interaction, EPR was carried out in deuterated PBS, as 1O2 is known to have an extended lifetime in D2O [24]. Solvent deuteration increased the rate of O2 photo-consumption by NaSCN almost 40-fold (Fig. 2A).
Fig. 2.
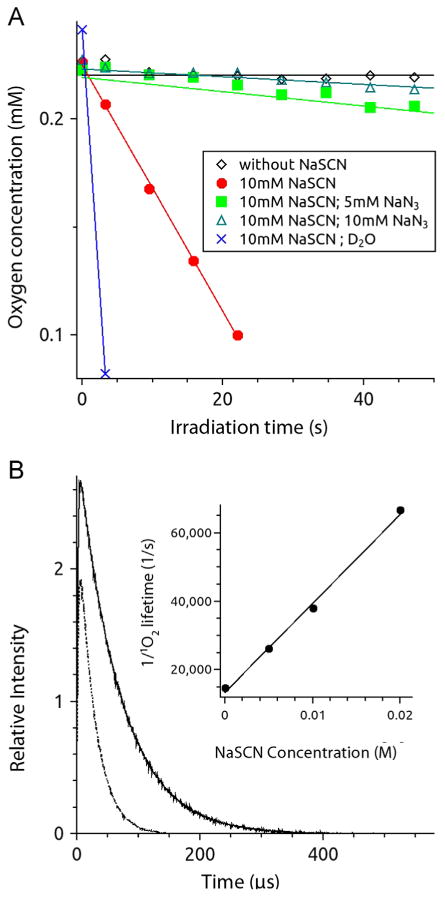
The effect of NaSCN on O2 photo-consumption and 1O2 lifetime. (A) Effects of NaSCN on MB and light on O2 photo-consumption. Samples contained 100 μM mHCTPO, 25 μM MB, were in the PBS pH 7.4 and were irradiated with 540–740 nm (70 mW/cm2) light. Open diamonds, MB in PBS/H2O; filled circles, sample in PBS/H2O with 10 mM NaSCN; filled squares, sample in PBS/H2O with 10 mM NaSCN and 5 mM NaN3; open triangles, sample in PBS/H2O with 10 mM NaSCN and 10 mM NaN3; exes, sample in PBS/D2O with 10 mM NaSCN. (B) Kinetics of 1O2 phosphorescence. Sample contained 50 μM MB in D2O without (solid line) and with (dotted line) 10 mM NaSCN and was excited with 532-nm laser pulses. Total acquisition time was 30 s. Inset: the inverse of singlet oxygen lifetime as a function of sodium thiocyanate concentration.
Time-resolved detection of 1O2 lifetime
Time-resolved detection of 1O2 luminescence at 1270 nm was used to analyze the effect of NaSCN on the lifetime of 1O2. Fig. 2B shows representative data obtained in D2O used as a solvent with or without 10 mM NaSCN. NaSCN shortened the 1O2 lifetime by a factor of ≈ 2.7 (Fig. 2B). SCN− interacted with 1O2 in a concentration-dependent manner (Fig. 2B, inset); the biomolecular rate constant for SCN− quenching of 1O2 was moderate (2.6 × 106 M −1 s −1), indicating that the lifetime of 1O2 is preferentially determined by its interaction with solvent molecules.
Interaction of excited MB with SCN−
Absorbance changes between 360 and 500 nm, with a maximum around 420 nm, could be assigned to the absorption band of the lowest triplet excited state of MB [25]. We monitored the lowest triplet state of MB at 420 nm via laser flash photolysis in the presence and absence of SCN− to determine if excited MB interacts with SCN−. The MB triplet state lifetime is of the order of 10−5 s and strongly depends on O2. Transients (Fig. S1A) represent changes in absorbance of MB induced by pulse photo-excitation of MB molecules, detected at different wavelengths after selected time delays. In air-saturated samples, the lifetime of the transient was shortened to below 1 μs (insets in Figs. S1A and S1B). Fig. S1B shows corresponding transients obtained in the presence of 10 mM NaSCN without O2. It is evident that NaSCN failed to quench the MB excited triplet state, as no spectral modifications in the presence of NaSCN were noted.
Effect of O2 on SCN− potentiation of MB-PDT
PDT with SCN− was performed in the absence of O2 by bubbling samples with N2 for 30 min prior to irradiation (Fig. 3). It is evident that O2 is necessary for SCN− potentiation of PDT, as O2-free irradiated MB samples were incapable of killing either S. aureus (Fig. 3A) or E. coli (Fig. 3B) even in the presence of SCN−. This confirms that SCN− is incapable of quenching the excited triplet state of MB and that SCN− -mediated PDT strongly relies on the presence of O2.
Fig. 3.
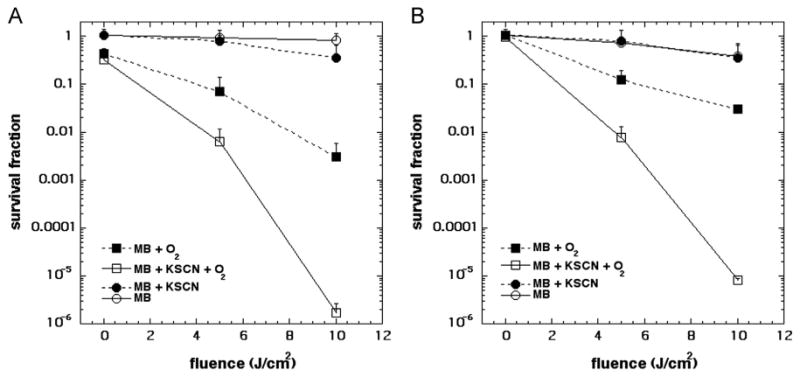
Effect of O2 on KSCN potentiation of MB-PDT. (A) S. aureus was incubated with 20 μM MB and 10 mM KSCN and (B) E. coli was incubated with 200 μM MB and 10 mM KSCN in the dark. One-milliliter aliquots were irradiated with 660±15 nm light in air or in nitrogen. Values are means of triplicate determinations from separate experiments, and bars are SD.
EPR spin trapping of radical sulfur species
To elucidate the product of the 1O2-SCN− reaction EPR spin trapping with DMPO spin trap was employed. Irradiation of samples containing 25 μM MB, 10 mM DMPO, and 10 mM NaSCN in H2O led to a time-dependent accumulation of a spin adduct with the corresponding hyperfine splittings of aN=1.46mT and aH=1.62mT (Fig. 4A, compare to simulation Fig. 4E). No photochemical generation of SCN−-derived radical species occurred in the absence of O2 (Fig. 4B). The radical DMPO adduct detected in our MB-SCN− system was sufficiently different from the DMPO• SCN adduct (whose hyperfine splitting constants are aN=1.50 mT, aH=1.65mT) [26]. Solvent deuteration had a dramatic effect on the accumulation of the spin adduct, increasing the rate of formation by almost two orders of magnitude and EPR signal intensity was 12-fold more intense than that formed in H2O-containg samples (Fig. 4C). Moreover, addition of 5 mM NaN3 to samples dissolved in D2O decreased the rate of photo-formation of the spin adduct by almost 200-fold and significantly decreased the intensity of its EPR signal (Fig. 4D, Fig. S2). D2O exchange and addition of NaN3 to D2O and the respective increase and decrease in EPR signal of the DMPO spin trap suggested that the detected radical species formation is dependent on 1O2. Additionally, in the MB-NaSCN samples with NaN3, the observable EPR signal consisted of a mixture of two spectra—the DMPO-N3, and a residual DMPO spin adduct with the radical discussed above (compare to control DMPO spin adduct in Figs. 4A, C).
Fig. 4.
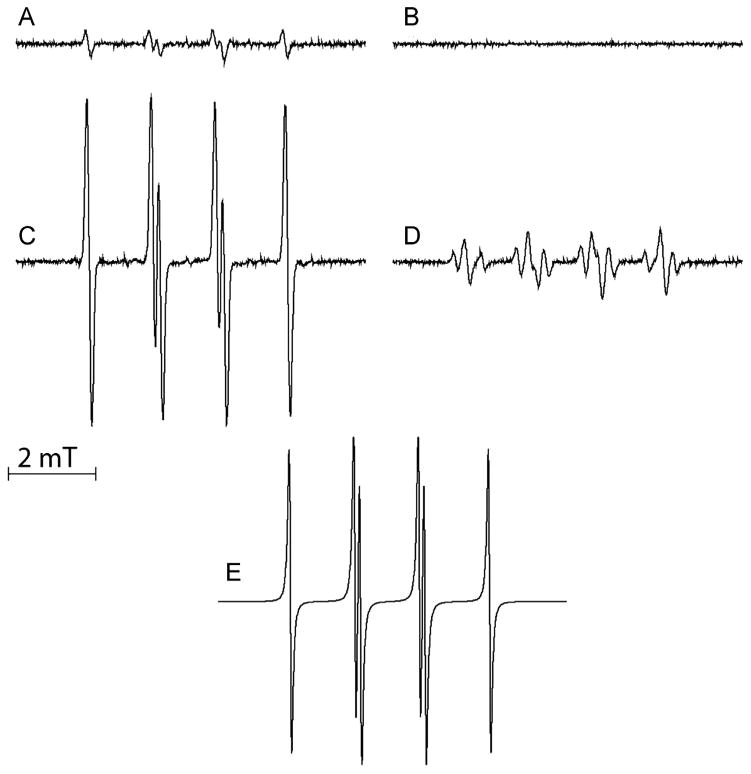
MB-photosensitized formation of DMPO spin adducts. Samples contained 10 mM DMPO (except sample in B, which contained 50 mM DMPO), 25 μM MB, 10 mM NaSCN and were air-saturated except sample B which was O2 free, after saturation with argon. Spectra A–D were recorded after irradiation of samples with light (540–740 nm, 70 mW/cm2) for 180 s (12.6 J). (A) Sample in H2O, (B) in H2O in O2-free system, (C) sample in D2O, (D) sample in D2O with 5 mM NaN3. (E) Simulation of the EPR signal of DMPO spin adduct.
DMPO spin-trap identification of the sulfur trioxide radical anion
The hyperfine constants of the spin adduct detected in the DMPO spin-trapping experiments are characteristic of the sulfur trioxide radical anion (SO3•−), a biologically relevant radical sulfur species. It has been reported that both photolysis of bisulfite and peroxidase oxidation of sulfite form SO3•−, with DMPO spin adduct splitting constants of aN= 1.47 mT and aH= 1.60 mT [27–29]. To confirm our EPR adduct identification, we compared the DMPO radical adduct of our irradiated MB-SCN− system with a control photolysis of bisulfite (Fig. 5). Overlap of a photolyzed sodium sulfite (Fig. 5A, red) with photosensitized MB in the presence of SCN− (Fig. 5A, blue) underscores negligible differences between the control and our experimental adduct. The reaction governing the formation of the detected spin adduct and the structure of that spin adduct are specified in Fig. 5B.
Fig. 5.
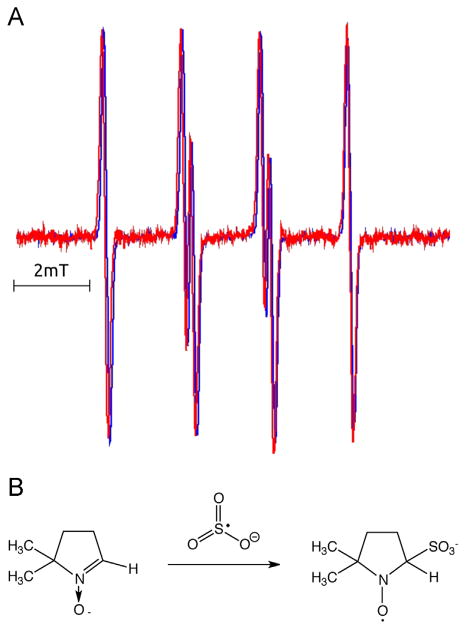
Comparison of EPR spin adducts of the MB-SCN− system and photolyzed Na2SO3. (A) EPR spin adducts generated by photolysis of 100 mM Na2SO3 in H2O with pH adjusted to 7.5, in the presence of 50 mM DMPO (red spectrum), or by oxidation of 10 mM NaSCN photosensitized by 25 μM MB photosensitized in PBS/D2O in the presence of 10 mM DMPO (blue spectrum). Deaerated solution of sulfite was irradiated for 800 s with light (240 kJ), derived from an ozone-free 300 W 300–770 nm hv high pressure compact-arc xenon lamp (Cermax, PE300CE–13FM/Module300W, Perkin-Elmer), equipped with a 5-cm water filter; while in photosensitized reaction aerated sample in PBS/D2O was irradiated with orange light (540–740 nm). Red spectrum was multiplied by a factor of 2.8 to obtain comparable amplitude of the EPR signals. (B) Formation and structure of the DMPO•-SO3− spin adduct by interaction with SO3•−.
Detection of cyanide as a product of 1O2 oxidation of SCN−
The Prussian Blue CN− detection assay was used to determine if CN− is ejected from an SCN-derived intermediate and if it is conserved or oxidized. As the applied light was increased from 0 to 80 J cm−2, the Prussian Blue complex concentration increased (Fig. 6A). Formation of the Prussian Blue complex from MB and KSCN in the dark or from KSCN and light with no MB was negligible, even after a 180-min incubation, confirming the specificity of the assay (data not shown). CN− generation was proportional to the light-mediated oxidative reactions occurring during PDT.
Fig. 6.
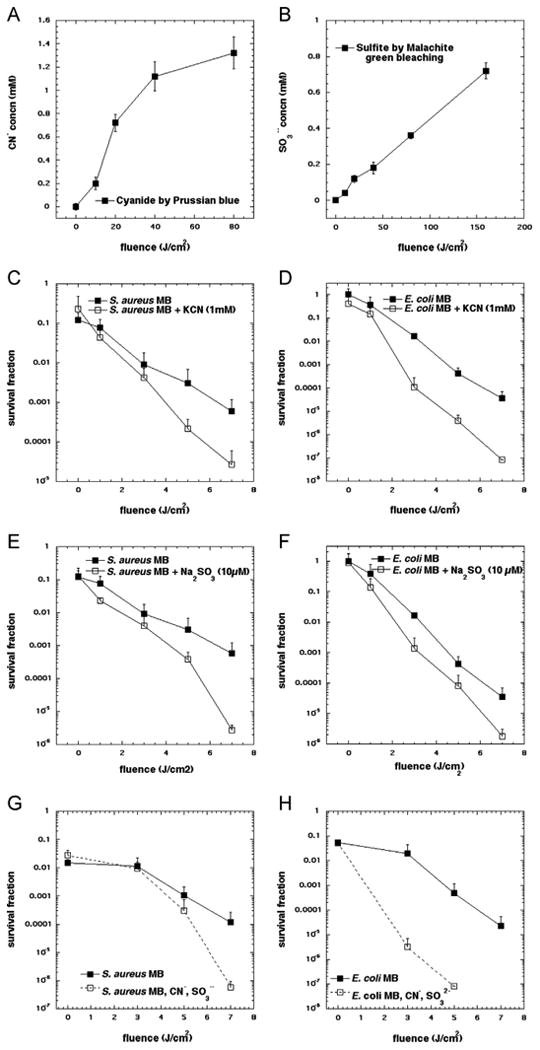
(A) Cyanide formation as a function of light dose in the MB-SCN− system. Absorption of 700 nm corresponds to the Prussian Blue complex Fe4[Fe(CN)6]3. A sample of 20 μM MB and 10 mM KSCN was irradiated with variable amounts of 660 nm hv and aliquots of 100 μl of sample were removed and added to the reagent mixture (Fe2+/Fe3+/H+) and the absorption was measured at 700 nm. (B) Sulfite formation as a function of light dose in the MB-SCN− system. Malachite green is reduced by sulfite and shift in absorbance at 617 nm may be detected and recorded. A 10 mM KSCN and 20 μM MB were used and irradiated with variable amounts of 660 nm hv in the presence of 2.5 μM malachite green. (C and D) Potentiation of antimicrobial PDT killing mediated by 10 μM MB and 1–7 J/cm2 of 660 nm light by addition of 1 mM KCN for (C) S. aureus, and (D) E. coli. (E and F) Potentiation of antimicrobial PDT killing mediated by 10 μM MB and 1–7 J/cm2 of 660 nm light by addition of 10 μM Na2SO3 for (E) S. aureus, and (F) E. coli. (G and H) Potentiation of antimicrobial PDT killing mediated by 10 μM MB and 1–7 J/cm2 of 660 nm light by addition of an equimolar mixture of 50 μM KCN and Na2SO3 for (G) S. aureus, and (H) E. coli. Points represent the mean from three separate experiments and error bars represent SD from the mean.
Detection of sulfite as a product of 1O2 oxidation of SCN−
Since the proposed mechanism of sulfur trioxide radical anion called for the intermediate production of sulfite anion we used a malachite green bleaching assay that was able to measure the formation of sulfite in the presence of MB. Fig. 6B shows a good light dose-dependent generation of sulfite anion, albeit at higher doses than cyanide (Fig. 6A).
Potentiation of MB-mediated PDI of bacteria by addition of sulfite or cyanide
Since our proposed mechanism of increased bacterial killing called for the generation of sulfur trioxide radical anion formed from sulfite produced from oxidation of SCN−, it was very relevant to test if addition of sulfite itself to MB-PDT could also potentiate killing. As shown in Figs. 6E and F using 10 μM SO32- we found an additional 2.5 logs of killing with S. aureus (Fig. 6C) and 1.5 logs of killing of E. coli in Fig. 6E. It should be noted that the dependence of this effect on the sulfite concentration was complex and at higher sulfite concentrations disappeared. This biphasic effect will be the subject of further study.
We also asked if the synergistic bacterial killing effects of SCN- were due to the formation of CN− either in the dark or in a photochemical reaction caused by light. We first measured the minimum inhibitory concentration (MIC) of CN− for both bacterial species (Fig. S3). CN− in the dark was likely not responsible for the enhanced killing as the MICCN− for S. aureus was 5 mM, while the MICCN- for E. coli was 2 mM. We then went on to determine if there was any potentiation of MB-PDT by addition of CN-. Somewhat to our surprise we found that addition of 1 mM CN- gave about 1 log more killing of S. aureus in a light dose-dependent manner (Fig. 6C) and an even greater increase of killing (2 logs) against E. coli (Fig. 6D). The mechanism for the potentiation of MB-PDT by cyanide will be the subject of further investigation. Because both cyanide and sulfite are proposed to be generated simultaneously as products of 1O2 oxidation of SCN- it was of interest to test a mixture of CN- and SO32- as potentiators, which we did at an equimolar concentration of 50 μM. Figs. 6G and H show that while there was little difference in the potentiation of killing with either salt alone for S. aureus (compare Fig. 6G with Figs. 6C and E), there was clearly increased killing of E. coli with MB-PDT using the salt mixture over either salt alone (compare Fig. 6H with Figs. 6D and F).
Discussion
Photodynamic inactivation in the presence of SCN−
The lactoperoxidase, SCN−, H2O2 system has been of biological interest due to the formation of an unstable sulfur-centered radical and a stable, toxic SCN− oxidation product, resulting in short- and long-term antibacterial effects [30]. Consequently, we sought to investigate if similar effects could be noted during PDT. Both S. aureus and E. coli showed a high degree of synergistic bacterial killing when MB-PDT was combined with SCN-. Somewhat surprisingly the effect was greater for E. coli than for S. aureus, since gram-negative bacteria are less permeable than their gram-positive counterparts due to the presence of a lipopolysaccharide layer in addition to a peptidoglycan cell wall and cell membrane and this difference makes gram-negative species less susceptible to PDT [5].
The potentiation of killing depended on the concentration of SCN− at the concentrations tested (Fig. 1). Two previous investigations on the SCN− potentiation of lactoferrin-H2O2 and H2O2 antibacterial systems by Lassiter et al. and Michot et al. [12,31] found concentration independence with respect to SCN−. Lassiter et al. attributed the concentration independence to the formation of a reactive radical species (e.g., •SCN) from SCN− at the cell surface. Moreover, Milligan et al. noted that increasing SCN− concentration in the presence of •OH attenuated •SCN radical damage of DNA by disturbing the •SCN, SCN−/(SCN)2•− equilibrium [Eq. (3)], whereby (SCN)2•− is less damaging than •SCN [13].
1O2 is key for PDT-SCN− synergy
After observing a pronounced PDT antibacterial effect in the presence of SCN−, we sought to identify the mechanism of potentiation. EPR oximetry indicated that O2 photo-consumption was enhanced in the presence of SCN−. Adding NaN3, a 1O2 physical quenching agent, reduced O2 photo-consumption, and exchange of H2O for D2O, known to extend the 1O2 lifetime, enhanced O2 photo-consumption (Fig. 2A). To confirm that 1O2 was in fact consumed during this reaction, we compared luminescence emission at 1270 nm in our PDT system with and without the presence of SCN−: the marked decrease in phosphorescence at 1270 nm in the presence of SCN− strongly suggested that 1O2 is being consumed. These two findings indicate that SCN− quenching of 1O2 was predominately chemical in nature and may lead to the formation of a distinct product.
To verify that such a reaction was taking place between 1O2 and SCN−, rather than some transient species that is undetectable by EPR oximetry/1O2 emission, we analyzed the effect of physical and bactericidal effects of the PDT-SCN− system in the absence of O2. Our previous investigations indicated that the pseudohalide N3− is capable of quenching the excited triplet state of MB (9). On the contrary, SCN− did not react with excited states of MB, substantiating the notion that a SCN− 1O2 interaction is responsible for synergistic potentiation of PDT (Fig. S1). The necessity of O2 for the antibacterial effect was confirmed by performing PDT in the absence of O2. Unlike N3−, which can quench the excited triplet state of MB and thus lead to photosensitized bacterial killing in the absence of O2 [9], no such effect was observed for SCN− (Fig. 3). The SCN− effects on O2 photo-consumption and 1O2 1270 nm luminescence in addition to the lack of effects of SCN− on the lifetime of the excited MB triplet state and the absence of PDT in the absence of O2 are all highly indicative of a discreet interaction between SCN− and 1O2. This conclusion was supported by the observation that, contrary to N3−, which under O2-free conditions still interacts with the photoexcited MB molecules forming N3 [9], no photochemical generation of SCN− -derived radical species occurred in MB-SCN− samples in the absence of O2 (Fig. 4B).
DMPO spin-trapping identification of SO3•−
EPR data indicated that neither •SCN nor (SCN)2• were formed in the photosensitized MB-SCN− system. A one-electron oxidation of SCN− by 1O2 is not thermodynamically feasible, as evident by the one-electron reduction potentials of 1O2/O2•− and •SCN/SCN− which have been reported to be +0.65 and +1.62 V, respectively [32,33]. This is confirmed by our DMPO radical adducts, which could not be identified as DMPO•-OH or DMPO•-SCN, suggesting that oxygen-centered radicals are not involved in the SCN− enhancement of PDT. While MB mediates both type I and type II photochemical reactions, it would appear that in our MB-SCN− system, type II photochemistry is significantly favored [34]. If •OH were formed, •SCN would undoubtedly be formed (and detected) as evident by the high rate constant of 1.4 × 1010 M−1 s−1 for reaction 1 which is approximately four orders of magnitude greater than our reported rate constant for the interaction of 1O2 and SCN− (2.6 × 106 M−1 s−1) (14). This was supported by our use of a DMPO spin-trapping system: DMPO is a fairly promiscuous spin trap, known to react with •OH [34]. If •OH were formed, it would react with SCN−, precluding a 1O2 reaction with SCN−.
It appears that in the presence of SCN−, MB predominantly undergoes type II photochemical reactions. Because SCN− appears to silence any type I reactions and favor type II reactions, and N3− appears to do the opposite, this suggests that the photochemistry of MB is surprisingly variable. The fact that •SCN is not formed in the oxidation of SCN− is noteworthy seeing as most (if not all) previous investigations of interactions of SCN− in biological environments identified •SCN as the radical sulfur species responsible [35,36]. An additional study by Løvaas identified the carbon-centered radical methanimidate(thioperoxidate)O-acid (−OSC• = N−) as the active radical species of the lactoperoxidase, SCN−, H2O2 system [30]. Nevertheless, our hyperfine splitting constants for the DMPO•-SO3− adduct of aN=1.46 mT and aH=1.62 mT are almost identical to those of DMPO•-SO3− as determined by Zamora and Villamena, where aN= 1.45 mT and aH= 1.61 mT, thus corroborating our interpretation of EPR spectra [37]. To our knowledge, this is the first reported case of the oxidation of SCN− to SO3•−. We are confident that SO3•− is being formed in situ and is directly attacking DMPO rather than an alternative mechanism consisting of sulfite nucleophilic attack of DMPO followed by subsequent oxidation by a background oxidant (the Forrester-Hepburn pathway for radical adduct formation), as two recent reports have demonstrated that the DMPO•-SO3- adduct cannot be formed by the Forrester-Hepburn pathway or an inverted spin-trapping pathway (reverse order of the Forester-Hepburn pathway) [37–40].
Formation of SO3•− from SCN− is by no means a straightforward process. First, the cyanide (CN−) component of SCN− must be detached from sulfur; whether it was preserved or chemically modified needed to be established. Moreover, single addition of 1O2 to the electron-rich sulfur of SCN− only accounts for two of the three oxygen atoms found in SO3•−. We postulated that the CN− of an SCN−-derived intermediate was replaced with an equivalent of water, explaining the seemingly elusive third oxygen atom and the apparent loss of CN−. This was confirmed by the Prussian Blue assay, wherein CN− production was produced during irradiation of a MB sample.
Our PDT-SCN− system generating the unstable SO3•− and stable, toxic CN− mirrors that of the lactoperoxidase, SCN−, H2O2 system in that an unstable sulfur-centered radical and a stable, toxic SCN− oxidation product resulting in short- and long-term antibacterial effects [30]. We consequently asked whether SCN− enhancement of PDT killing could also be explained by the production of CN−, which is known to competitively bind with chelated iron of heme in cytochromes, thus disrupting electron transport [41]. In terms of bacterial susceptibility to CN−, E. coli is known to have CN− assimilatory pathways, which serve as protection from CN−, and is capable of growing on media containing CN− [42]. This could explain the aforementioned susceptibility differences to the PDT-SCN− system. Our MICCN− tests confirmed that this was not the case (Fig. S3) and that the antibacterial effects observed in this study were likely not due to dark toxicity of CN−. However, the finding that addition of cyanide can also potentiate MB-PDT bacterial killing does suggest that CN− may subsequently be oxidized to an unknown reactive species that can further damage bacteria. Further study will be needed to establish the role of released cyanide in the potentiation. The proposed mechanisms do call for the intermediacy of sulfite anion (SO32−), and we were able to confirm this release from the MB/SCN−/light reaction with an assay for sulfite using reduction of malachite green dye. We were also able to show that addition of sodium sulfite to MB-PDT of bacteria was able to potentiate killing, albeit within a fairly narrow range of concentration of sulfite (10–100 μM). When the concentration of sulfite was raised to levels of 1–10 mM the bacterial killing was inhibited rather than potentiated. This biphasic concentration response has previously been reported with compounds that can act as radical generators at low concentrations, but can act as quenching antioxidants at high concentrations. For instance ascorbic acid (vitamin C) potentiated PDT killing of cancer cells mediated by a functionalized C84 fullerene and UVA light at concentrations of 3–30 μM, but quenched killing at higher concentrations [43].
We propose a possible mechanism for SO3•− formation, involving the ejection of CN− (Fig. 7). The electron-rich and thus nucleophilic sulfur atom of SCN−, 1, may attack 1O2, 2, yielding a persulfoxide intermediate, or 1O2 may undergo a cyclo-addition with sulfur yielding a thiadioxirane intermediate. Both of these mechanisms have been previously reported, and may yield the key cyanosulfonyl-like species, 3 [15,44]. Attack of 3 by water with concomitant or subsequent ejection of the CN− followed by deprotonation yields the bisulfite anion, 4. Oxidation of 4 (from excited state MB or other type I species) with concomitant or subsequent deprotonation may then yield SO3•−, 5. We determined that 1O2 does not oxidize 4 but that excited state MB oxidizes 4 in the absence of O2, forming SO3•− (Fig. S4), confirming formation of SO3•− via a bisulfite intermediate.
Fig. 7.
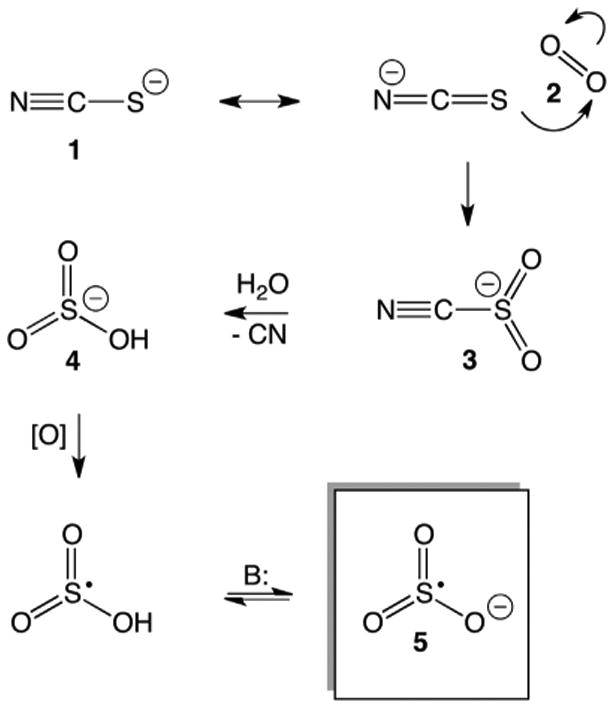
Proposed mechanism of SO3•− formation. SCN−, 1; 1O2, 2; cyanosulfonyl-intermediate, 3; bisulfite, 4; SO3•−, 5.
Even though the one-electron reduction potential of SO3•−/SO32− is relatively low (+0.8 V), SO3•− can induce oxidative damage to different cellular constituents, particularly if its intrinsic lifetime, determined by recombination and/or disproportionation, is sufficiently long [45]. SO3•− is a sulfur-centered radical, as confirmed by theoretical and empirical investigations, and is one of the few radical sulfur species encountered in cells. Moreover cells are incapable of reducing sulfonic acid adducts between SO3•− and biomolecules [37,46]. This is exemplified by the fact that a naturally occurring sulfonic acid, taurine, which is synthesized by oxidation of cysteine sulfinic acid is considered redox inactive. Moreover, SO3•− may exert antibacterial effects due to its being a strong prooxidant. SO3•− reacts with ground state O2, forming the persulfoxyl free radical (−O3SOO•)[Eq. (4)]:
| (4) |
This species −O3SOO• may then react with sulfite to form the sulfate free radical [Eq. (5)][29,47]: The sulfate free radical is an extremely strong oxidizing species which, in the presence of chloride, exists in equilibrium with the chlorine radical [Eq. (6)] [48,49]:
| (5) |
| (6) |
Moreover, SO3•− may react with O2 to form the sulfur pentoxide radical anion, an oxygen-centered radical. Hydroperoxysulfuryl radicals may also be formed from SO3•−; all of these species are presumed to be deleterious to cell homeostasis [47]. Additionally, the prospect of indirectly forming chlorine radicals from −O3SOO• is interesting in that the chlorine radical has not been thoroughly evaluated as an antimicrobial agent due to the difficulty of production.
Conclusion
The last universal common ancestor evolved on earth in the presence of ROS some 3.5 billion years ago; consequently, all forms of life have highly complex cellular machineries for quenching or sequestering oxygen–centered radicals [50]. This is underscored by the ubiquity and high conservation of superoxide dismutases and catalases. Seeing as the cytotoxicity of PDT is due to oxygen-centered radicals, we sought to redirect the oxidizing power of PDT in a nonoxygen inorganic radical form, so as to bypass microbial mechanisms of ROS tolerance. Though we did achieve antimicrobial PDT potentiation via in situ formation of SO3•−, to our surprise this was mediated through 1O2 and not oxygen-centered radicals.
The findings reported in this study have several important implications for antimicrobial PDT as well as photochemistry. Though pseudohalide–enhanced PDT has been demonstrated with N3−, N3− is quite toxic; on the other hand, SCN− is the least toxic of the pseudohalides and evidence from this report suggests that SCN− is capable of achieving PDT-enhancement comparable to that of N3−. Secondly, in contrast to N3−, which physically quenches 1O2 exclusively permitting type I photochemistry-mediated cytotoxicity, SCN− reacts with 1O2 to form sulfite, which is oxidized to SO3•−. While this in and of itself is interesting, no oxygen–centered radicals were detected. Though the mechanism of this type I sequestration has yet to be elucidated, our observations suggest that SCN− exclusively permits type II photochemistry. This phenomenon should be explored in greater depth so as to allow the photochemistry community to potentially exploit a tool that coerces PS to undergo exclusive type II photochemistry.
Finally, this study is the first to report sulfur–centered radical antimicrobial PDT. We propose that other sulfur–containing polyatomic anions should be explored for enhancement of antimicrobial PDT. Sulfite is of particular interest, seeing as it is an intermediate in the chemical mechanism of SCN− enhancement of PDT. Two questions thus emerge with regard to sulfite-enhanced PDT: (A) does sulfite abrogate oxygen-centered radical formation like SCN− and (B) can a sulfite/O2/sulfate/chloride cocktail be engineered to allow for in situ production of chlorine radicals [Eqs. (4), (5), and (6)]? If such a system could be achieved, we envisage that it would be highly antimicrobial and an achievement seeing as the oxidation potential of chloride is such that no ROS produced by PDT can generate chlorine radicals. Though this is highly speculative, the results of this study suggest that MB-mediated PDT may be used to generate chlorine radicals provided competing reactions have significantly lower rate constants.
Supplementary Material
Acknowledgments
We thank Michael Davies of the Heart Research Institute in Australia and Nicholas J. Turro of Columbia University in New York for invaluable discussion on the mechanism of SO3•− formation. Research conducted by Tyler G. St. Denis is supported by the Columbia University I. I. Rabi Science Research Fellowship. Research conducted by the Tadeusz Sarna Laboratory in Poland is supported by National Science Centre (2011/03/B/NZ1/00007). Research conducted in the Hamblin Laboratory is supported by NIH (R01A1050875).
Abbreviations
- DMPO
5,5-dimethyl-1-pyrroline-N-oxide
- MB
methylene blue
- mHCTPO
4-protio-3-carbamoyl-2,2,5,5-tetraperdeuteromethyl-3-pyrrolin-1-yloxy
- PB
Prussian Blue
- PBS
phosphate-buffered saline
- PDT
photodynamic therapy
- PS
photosensitizer
Footnotes
Appendix A. Supplementary Information: Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.freeradbiomed..2013.08.1623.
References
- 1.Canton R, Akova M, Carmeli Y, Giske CG, Glupczynski Y, Gniadkowski M, Livermore DM, Miriagou V, Naas T, Rossolini GM, Samuelsen O, Seifert H, Woodford N, Nordmann P. Rapid evolution and spread of carbapenemases among Enterobacteriaceae in Europe. Clin Microbiol Infect. 2012;18:413–431. doi: 10.1111/j.1469-0691.2012.03821.x. [DOI] [PubMed] [Google Scholar]
- 2.Nordmann P, Poirel L, Toleman MA, Walsh TR. Does broad-spectrum beta-lactam resistance due to NDM-1 herald the end of the antibiotic era for treatment of infections caused by gram-negative bacteria? J Antimicrob Chemother. 2011;66:689–692. doi: 10.1093/jac/dkq520. [DOI] [PubMed] [Google Scholar]
- 3.Bush K, Courvalin P, Dantas G, Davies J, Eisenstein B, Huovinen P, Jacoby GA, Kishony R, Kreiswirth BN, Kutter E, Lerner SA, Levy S, Lewis K, Lomovskaya O, Miller JH, Mobashery S, Piddock LJ, Projan S, Thomas JR, Tomasz A, Tulkens PM, Walsh TR, Watson JD, Witkowski J, Witte W, Wright G, Yeh P, Zgurskaya HI. Tackling antibiotic resistance. Nat Rev Microbiol. 2011;9:894–896. doi: 10.1038/nrmicro2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Raab O. Ueber die Wirkung fluorescirender Stoffe auf Infusorien. Z Biol. 1900;39:524–546. [Google Scholar]
- 5.Dai T, Huang YY, Hamblin MR. Photodynamic therapy for localized infections—state of the art. Photodiagnosis Photodyn Ther. 2009;6:170–188. doi: 10.1016/j.pdpdt.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Foote CS. Definition of type I and type II photosensitized oxidation. Photochem Photobiol. 1991;54:659. doi: 10.1111/j.1751-1097.1991.tb02071.x. [DOI] [PubMed] [Google Scholar]
- 7.Castano AP, Demidova TN, Hamblin MR. Mechanisms in photodynamic therapy: part one, photosensitizers, photochemistry and cellular localization. Photodiagnosis Photodyn Ther. 2004;1:279–293. doi: 10.1016/S1572-1000(05)00007-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hamblin MR, O'Donnell DA, Murthy N, Rajagopalan K, Michaud N, Sherwood ME, Hasan T. Polycationic photosensitizer conjugates: effects of chain length and gram classification on the photodynamic inactivation of bacteria. J Antimicrob Chemother. 2002;49:941–951. doi: 10.1093/jac/dkf053. [DOI] [PubMed] [Google Scholar]
- 9.Huang L, St Denis TG, Xuan Y, Tanaka M, Huang YY, Zadlo A, Sama T, Hamblin MR. Paradoxical potentiation of methylene blue-mediated antimicrobial photodynamic inactivation by sodium azide: role of ambient oxygen and azide radicals. Free Radic Biol Med. 2012 doi: 10.1016/j.freeradbiomed.2012.09.006. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tavares A, Dias SR, Carvalho CM, Faustino MA, Tome JP, Neves MG, Tome AC, Cavaleiro JA, Cunha A, Gomes NC, Alves E, Almeida A. Mechanisms of photodynamic inactivation of a gram-negative recombinant bioluminescent bacterium by cationic porphyrins. Photochem Photobiol Sci. 2011;10:1659–1669. doi: 10.1039/c1pp05097d. [DOI] [PubMed] [Google Scholar]
- 11.Malešič J, Kolar J, Strlič M, Polanc S. The influence of halide and pseudohalide antioxidants in Fenton-like reaction systems. Acta Chim Slov. 2006;53:450–456. [Google Scholar]
- 12.Lassiter MO, Newsome AL, Sams LD, Arnold RR. Characterization of lactoferrin interaction with Streptococcus mutans. J Dent Res. 1987;66:480–485. doi: 10.1177/00220345870660021601. [DOI] [PubMed] [Google Scholar]
- 13.Milligan JR, Aguilera JA, Paglinawan RA, Ward JF. Mechanism of DNA damage by thiocyanate radicals. Int J Radiat Biol. 2000;76:1305–1314. doi: 10.1080/09553000050151574. [DOI] [PubMed] [Google Scholar]
- 14.Herrmann H, Hoffmann D, Schaefer T, Brauer P, Tilgner A. Tropospheric aqueous-phase free-radical chemistry: radical sources, spectra, reaction kinetics and prediction tools. Chemphyschem. 2010;11:3796–3822. doi: 10.1002/cphc.201000533. [DOI] [PubMed] [Google Scholar]
- 15.Clennan EL. Persulfoxide: key intermediate in reactions of singlet oxygen with sulfides. Acc Chem Res. 2001;34:875–884. doi: 10.1021/ar0100879. [DOI] [PubMed] [Google Scholar]
- 16.Rozanowska M, Jarvis-Evans J, Korytowski W, Boulton ME, Burke JM, Sarna T. Blue light-induced reactivity of retinal age pigment. In vitro generation of oxygen-reactive species. J Biol Chem. 1995;270:18825–18830. doi: 10.1074/jbc.270.32.18825. [DOI] [PubMed] [Google Scholar]
- 17.Lillie RD, Conn HJ, Commission BS. H J Conn's Biological stains: a handbook on the nature and uses of the dyes employed in the biological laboratory. Williams & Wilkins; 1977. [Google Scholar]
- 18.Jett BD, Hatter KL, Huycke MM, Gilmore MS. Simplified agar plate method for quantifying viable bacteria. Biotechniques. 1997;23:648–650. doi: 10.2144/97234bm22. [DOI] [PubMed] [Google Scholar]
- 19.Halpern HJ, Peril M, Nguyen TD, Spencer DP, Teicher BA, Lin YJ, Bowman MK. Selective isotopic labeling of a nitroxide spin label to enhance sensitivity for T2 oxymetry. J Magn Res. 1990;90:40–51. [Google Scholar]
- 20.Jimenez-Banzo A, Ragas X, Kapusta P, Nonell S. Time-resolved methods in biophysics. 7. Photon counting vs. analog time-resolved singlet oxygen phosphorescence detection. Photochem Photobiol Sci. 2008;7:1003–1010. doi: 10.1039/b804333g. [DOI] [PubMed] [Google Scholar]
- 21.Vakrat-Haglili Y, Weiner L, Brumfeld V, Brandis A, Salomon Y, McLlroy B, Wilson BC, Pawlak A, Rozanowska M, Sarna T, Scherz A. The microenvironment effect on the generation of reactive oxygen species by Pd-bacteriopheophorbide. J Am Chem Soc. 2005;127:6487–6497. doi: 10.1021/ja046210j. [DOI] [PubMed] [Google Scholar]
- 22.Mroz P, Pawlak A, Satti M, Lee H, Wharton T, Gali H, Sarna T, Hamblin MR. Functionalized fullerenes mediate photodynamic killing of cancer cells: type I versus type II photochemical mechanism. Free Radic Biol Med. 2007;43:711–719. doi: 10.1016/j.freeradbiomed.2007.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mashiko S, Suzuki N, Koga S, Nakano M, Goto T, Ashino T, Mizumoto I, Inaba H. Measurement of rate constants for quenching singlet oxygen with a Cypridina luciferin analog (2-methyl-6-[p-methoxyphenyl]-3,7-dihydroimidazo [1,2-a]pyrazin-3-one) and sodium azide. J Biolumin Chemilumin. 1991;6:69–72. doi: 10.1002/bio.1170060203. [DOI] [PubMed] [Google Scholar]
- 24.Rodgers MAJ, Snowden PT. Lifetime of oxygen (O2(1Δg)) in liquid water as determined by time-resolved infrared luminescence measurements. J Am Chem Soc. 1982;104:5541–5543. [Google Scholar]
- 25.Danziger RM, Bar-Eli KH, Weiss K. The laser photolysis of methylene blue. J Phys Chem. 1967;71:2633–2640. [Google Scholar]
- 26.Adak S, Mazumdar A, Banerjee RK. Low catalytic turnover of horseradish peroxidase in thiocyanate oxidation. Evidence for concurrent inactivation by cyanide generated through one-electron oxidation of thiocyanate. J Biol Chem. 1997;272:11049–11056. doi: 10.1074/jbc.272.17.11049. [DOI] [PubMed] [Google Scholar]
- 27.Ranguelova K, Chatterjee S, Ehrenshaft M, Ramirez DC, Summers FA, Kadiiska MB, Mason RP. Protein radical formation resulting from eosinophil peroxidase-catalyzed oxidation of sulfite. J Biol Chem. 2010;285:24195–24205. doi: 10.1074/jbc.M109.069054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chignell CF, Kalyanaraman B, Sik RH, Mason RP. Spectroscopic studies of cutaneous photosensitizer agents. II. Spin trapping of photolysis products from sulfanilamide and 4-aminobenzoic acid using 5,5-dimethyl-1-pyrroline-1-oxide. Photochem Photobiol. 1981;34:147–156. [Google Scholar]
- 29.Mottley C, Mason RP. Sulfate anion free radical formation by the peroxidation of (Bi)sulfite and its reaction with hydroxyl radical scavengers. Arch Biochem Biophys. 1988;267:681–689. doi: 10.1016/0003-9861(88)90077-x. [DOI] [PubMed] [Google Scholar]
- 30.Lovaas E. Free radical generation and coupled thiol oxidation by lactoperoxidase/SCN-/H2O2. Free Radic Biol Med. 1992;13:187–195. doi: 10.1016/0891-5849(92)90014-8. [DOI] [PubMed] [Google Scholar]
- 31.Michot JL, Osty J, Nunez J. Regulatory effects of iodide and thiocyanate on tyrosine oxidation catalyzed by thyroid peroxidase. Eur J Biochem. 1980;107:297–301. doi: 10.1111/j.1432-1033.1980.tb06029.x. [DOI] [PubMed] [Google Scholar]
- 32.Koppenol WH. Reactions involving singlet oxygen and the superoxide anion. Nature. 1976;262:420–421. doi: 10.1038/262420a0. [DOI] [PubMed] [Google Scholar]
- 33.Alfassi Z, Harriman A, Huie R, Mosseri S, Neta P. The redox potential of the azide/azidyl couple. J Phys Chem. 1987;91:2120–2122. [Google Scholar]
- 34.Buettner GR, Doherty TP, Bannister TD. Hydrogen peroxide and hydroxyl radical formation by methylene blue in the presence of ascorbic acid. Radiat Environ Biophys. 1984;23:235–243. doi: 10.1007/BF01407595. [DOI] [PubMed] [Google Scholar]
- 35.Modi S, Behere DV, Mitra S. Horseradish peroxidase catalyzed oxidation of thiocyanate by hydrogen peroxide: comparison with lactoperoxidasecatalysed oxidation and role of distal histidine. Biochim Biophys Acta. 1991;1080:45–50. doi: 10.1016/0167-4838(91)90110-l. [DOI] [PubMed] [Google Scholar]
- 36.Exner M, Hermann M, Hofbauer R, Hartmann B, Kapiotis S, Gmeiner B. Thiocyanate catalyzes myeloperoxidase-initiated lipid oxidation in LDL. Free Radic Biol Med. 2004;37:146–155. doi: 10.1016/j.freeradbiomed.2004.04.039. [DOI] [PubMed] [Google Scholar]
- 37.Zamora PL, Villamena FA. Theoretical and experimental studies of the spin trapping of inorganic radicals by 5,5-dimethyl-1-pyrroline N-oxide (DMPO). 3. Sulfur dioxide, sulfite, and sulfate radical anions. J Phys Chem A. 2012;116:7210–7218. doi: 10.1021/jp3039169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ranguelova K, Mason RP. New insights into the detection of sulfur trioxide anion radical by spin trapping: radical trapping versus nucleophilic addition. Free Radic Biol Med. 2009;47:128–134. doi: 10.1016/j.freeradbiomed.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ranguelova K, Mason RP. The fidelity of spin trapping with DMPO in biological systems. Magn Reson Chem. 2011;49:152–158. doi: 10.1002/mrc.2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leinisch F, Ranguelova K, DeRose EF, Jiang J, Mason RP. Evaluation of the Forrester-Hepburn mechanism as an artifact source in ESR spin-trapping. Chem Res Toxicol. 2011;24:2217–2226. doi: 10.1021/tx2003323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bryan LE, Kwan S. Roles of ribosomal binding, membrane potential, and electron transport in bacterial uptake of streptomycin and gentamicin. Anti-microb Agents Chemother. 1983;23:835–845. doi: 10.1128/aac.23.6.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dzombak DA, Ghosh RS, Wong-Chong GM. Cyanide in water and soil: chemistry, risk, and management. CRC/Taylor & Francis; 2006. [Google Scholar]
- 43.Sperandio FF, Sharma SK, Wang M, Jeon S, Huang YY, Dai T, Nayka S, de Sousa SC, Chiang LY, Hamblin MR. Photoinduced electron-transfer mechanisms for radical-enhanced photodynamic therapy mediated by water-soluble decacationic C70 and C84O2 Fullerene derivatives. Nanomedicine. 2013;9:570–579. doi: 10.1016/j.nano.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Watanabe Y, Kuriki N, Ishiguro K, Sawaki Y. Persulfoxide and thiadioxirane intermediates in the reaction of sulfides and singlet oxygen. J Am Chem Soc. 1991;113:2677–2682. [Google Scholar]
- 45.Huie RE. Fossil fuels utilization. American Chemical Society; 1986. Chemical kinetics of intermediates in the autoxidation of SO2; pp. 284–292. [Google Scholar]
- 46.Gruhlke MC, Slusarenko AJ. The biology of reactive sulfur species (RSS) Plant Physiol Biochem. 2012 doi: 10.1016/j.plaphy.2012.03.016. [DOI] [PubMed] [Google Scholar]
- 47.Giles GI, Jacob C. Reactive sulfur species: an emerging concept in oxidative stress. Biol Chem. 2002;383:375–388. doi: 10.1515/BC.2002.042. [DOI] [PubMed] [Google Scholar]
- 48.Buxton GV, Bydder M, Arthur Salmon G. The reactivity of chlorine atoms in aqueous solution Part II. The equilibrium SO4-+Cl-ClNsbd+SO42. Phys Chem Chem Phys. 1999;1:269–273. [Google Scholar]
- 49.Yu XY, Bao ZC, Barker JR. Free radical reactions involving Cl(dot), Cl2-(dot), and SO4-(dot) in the 248 nm photolysis of aqueous solutions containing S2O82- and Cl- J Phys Chem A. 2003;108:295–308. [Google Scholar]
- 50.Slesak I, Slesak H, Kruk J. Oxygen and hydrogen peroxide in the early evolution of life on earth: in silico comparative analysis of biochemical pathways. Astrobiology. 2012;12:775–784. doi: 10.1089/ast.2011.0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.