

Abstract
Class I PI 3-kinases are heterodimeric proteins with distinct catalytic (p110) and regulatory (p85) subunits. The minimal fragment of p85 capable of regulating p110 activity (p85ni) is the N-terminal SH2 domain linked to the iSH2 coiled-coil domain. We used cysteine mutagenesis and 14C-NEM labeling to show that the p110-binding site in the iSH2 domain includes two regions: residues 482-484 and 532-541. These regions are adjacent to each other in the three dimensional structural model of the iSH2 domain, and define a coherent binding site. We then used spin labeling and EPR spectroscopy to demonstrate that the conformation of the iSH2 domain is unaffected by binding to the N-terminal fragment of p110 (residues 1-108), and or by phosphopeptide binding to p85ni/p110(1-108) heterodimers. Finally, we show that the cSH2 domain cannot substitute for the nSH2 domain with regard to inhibition of p110. These data support a model in which the iSH2 domain is a rigid tether for p110, and regulation of p85/p110 is mediated by nSH2-p110 contacts.
Keywords: PI 3-kinase, p85, p110, phosphoinositides, SH2 domain, coiled-coil, iSH2 domain
Introduction
PI 3-kinases are critical regulators of cell survival, motility, and proliferation [1, 2]. Class IA PI 3-kinases, which produce PI[3,4,5] P3 in intact cells, are heterodimeric proteins with distinct catalytic (p110) and regulatory (p85) subunits. Class IA PI 3-kinases are obligate heterodimers in mammalian cells, because the p110 catalytic subunits are unstable as monomers at 37oC [3]. Thus, mice lacking p85 are also deficient for expression of p110 [4]. The p85 regulatory subunit stabilizes p110 by binding to a structurally uncharacterized N-terminal domain, p110(1-108) [3, 5-7].
In addition to stabilizing p110, p85 binding maintains p110 in a low activity state [3]. Given that p85 and p110 do not dissociate, subsequent activation of p85/p110 dimers involves a conformational change that disinhibits p110. We previously showed that the minimal fragment of p85 required for regulation of p110 consists of a coiled-coil region, the iSH2 domain, linked at its amino terminus to a single SH2 domain (the nSH2 domain) [8]. The nSH2-iSH2 fragment (hereafter referred to as p85ni) inhibits p110, and p85ni/p110 complexes are activated when YXXM phosphopeptides bind to the SH2 domain of p85ni [9]. In contrast, isolated nSH2 domains do not bind p110 or affect its activity. Isolated iSH2 domains are sufficient to bind to p110 [5-7, 10], but this binding does not affect p110 activity [8].
How does phosphopeptide binding to the nSH2 domain lead to disinhibition of p110? Presumably, nSH2 domain occupancy causes conformational changes that are somehow transmitted to p110. To address the mechanism of p110 disinhibition, we previously used site specific spin labeling and EPR spectroscopy to study the iSH2 domain, a 100 Å long coiled-coil consisting of two anti-parallel helices linked to SH2 domains at its extreme N- and C-terminal ends [5, 11]. We showed that phosphopeptide binding to the nSH2 domain of p85ni had no effect on the conformation of the iSH2 domain [11]. These data are in contrast to studies suggesting that the iSH2 domain is a modular molecular switch [12].
However, it is possible that the presence of p110 alters the conformation of the iSH2 domain, and renders it conformationally responsive to phosphopeptide-nSH2 domain binding. This consideration motivates the current study, in which we use a biochemical footprinting approach to define the p110 binding site in the iSH2 domain. We then use spin labeling and EPR spectroscopy to show that the iSH2 domain fails to undergo major conformational changes upon p110(1-108) binding, or upon phosphopeptide binding to p85ni/p110(1-108) complexes. These studies strongly support a model in which iSH2 domain is a static structure that provides a conformationally rigid tether for p110. The iSH2 domain does not act as a conformational switch during phosphopeptide activation of p85/p110 dimers, which must therefore involve regulatory interactions between the nSH2 domain and p110.
Materials and Methods
Mutagenesis and preparation of recombinant proteins
Mutants of the p85ni fragment of human p85 (residues 321-600) were produced by standard 4-primer PCR techniques. All mutants were confirmed by sequencing, and were >95% pure by SDS-PAGE and Coomasie staining. All p85ni mutants were demonstrated to bind to recombinant p110 in vitro, and to inhibit the activity of recombinant p110 to the same extent as wild-type p85ni, as previously described [11]. Recombinant myc-p110 was produced in baculovirus-infected Sf-9 cells as previously described [3].
Preparation of MBP-p110(1-108) fusion protein
The N-terminal fragment of human p110α (residues 1-108) was cloned into pMAL-C2 (New England Biolabs). Due to the low efficiency of Factor Xa cleavage, a thrombin site was engineered into pMAL-C2 such that the MBP tag could be efficiently removed by thrombin digestion. Expression and affinity-purification of MBP-p110(1-108) fusion protein was performed as described by the manufacturer. After affinity-purification, the sample was dialyzed against PBS (pH 7.4), concentrated and further purified by chromatography on Superdex-75 (Pharmacia). Protein eluting at the molecular weight corresponding to monomeric MBP-p110(1-108) was collected for subsequent studies.
Binding studies
GST-p85ni or GST-p85i (125-160 ng) was immobilized on glutathione-Sepharose beads, and incubated for 1h at 4°C with various concentrations of MBP-p110(1-108) and a constant amount of recombinant myc-p110 produced in baculovirus-infected Sf-9 cells. The beads were washed and protein were eluted and analyzed by SDS-PAGE followed by blotting with anti-myc antibodies and visualization using ECL reagent (Amersham).
NEM Labeling
p85ni was mixed with either MBP or MBP-p110(1-108) at a molar ratio of 1:20 in PBS (pH 7.4), and incubated overnight at 4°C. The MBP tag was removed by thrombin digestion, and the protein mixture was incubated for 15 min with 14C-NEM (34mci/mmol; 0.5mM final concentration) at room temperature. The alkylation reaction was terminated by addition of 15 mM DTT, and the samples were separated by reducing SDS-PAGE. 14C incorporation into p85ni was quantitated using a Phosphorimager (Molecular Dynamics). Images of the 35S-labeled gels were obtained by autoradiography followed by scanning using an Epson 3200 flatbed scanner; identical settings were used for each scan. Figures were assembled using Adobe Photoshop, using identical contrast and brightness settings for all scanned images.
EPR spectroscopy
Spin-labeling, phosphopeptide stimulation and EPR spectroscopy of p85ni mutants has been previously described [11]. Briefly, purified p85ni mutants were spin labeled with (1-oxyl-2,2,5,5-tetramethylpyrolinyl-3-methyl)-methanethiosulfonate (MTSSL; Toronto Research, Toronto). After extensive dialysis, the labeled samples were incubated with MBP-p110 or MBP at 4°C overnight, and treated with thrombin as above. Where indicated, samples were also incubated with a phosphorylated YXXM peptide derived from the PDGFβ receptor. Continuous wave EPR measurements were carried out on a Varian E112 X-Band Spectrometer. For studies at 300K the power was 2 mW with 1kHz modulation frequency and 1.6G modulation amplitude. Double labeled samples contained 40% sucrose. The sample volume was 10 to 30 μl contained in sealed 50 μl melting point capillaries.
p110 activity studies
Recombinant myc-p110 was incubated in the absence or presence of recombinant fragments of p85 for 1h at 4°C. The samples were then assayed for lipid kinase activity towards phosphatidylinositol as described [8]. The data are the mean −/+ SEM from three separate experiments.
Results
In vitro binding of p85ni to p110(1-108)
The p110 binding site in the iSH2 domain was determined using a biochemical footprinting approach, in which we measured the ability of p110(1-108) to block 14C-NEM labeling of p85ni mutants containing cysteine at various positions in the iSH2 domain. To determine binding conditions appropriate for analysis of p85-p110 interactions, we incubated immobilized GST-p85ni with a constant amount of recombinant N-myc-p110α plus various concentrations of MBP-p110(1-108) for 1h at 4°C. The immobilized GST-p85ni was washed, and bound myc-p110 was measured by blotting with anti-myc antibodies. Saturation of p85ni was observed at a MBP-p110(1-108):p85ni ratio of approximately 20:1 (Fig. 1A). This ratio was used in subsequent footprinting experiments. Similar results were obtained with GST-p85i (Fig 1B), which bound with slightly lower affinity than GST-p85ni. Gel filtration of p85ni/MBP-p110(1-108) complexes showed a mass of approximately 90kDa, consistent with a 1:1 stoichiometry (data not shown).
Figure 1. Binding of MBP-p110(1-108) to immobilized fragments of p85.
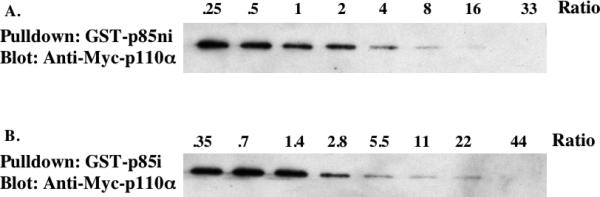
GST-p85ni (A) or GST-p85i (B) were bound to glutathione-Sepharose, and incubated with a constant amount of recombinant N-myc-p110α plus various concentrations of purified MBP-p110(1-108). The beads were washed and proteins were eluted and analyzed by SDS-PAGE and western blotting with anti-myc antibody. The ratios refer to the amount of MBP-p110(1-108) relative to GST-p85ni or GST-p85i.
NEM labeling of p85ni mutants
Footprinting experiments were conducted by incubation of p85ni mutants in the presence of MBP alone, or MBP-p110(1-108), followed by digestion with thrombin to remove the MPB tag. This procedure was necessary because the p110(1-108) fragment is stable as an MBP-fusion or as a dimer with p85ni, but precipitates as a monomer in solution. Removal of the MBP tag eliminates potential reductions in NEM labeling at a given site due to steric hindrance by MBP. Cleavage was approximately 80-90% complete for each mutant, based on autoradiography of cleaved MBP-p110(1-108) (data not shown).
Data from a typical experiment is shown in Figure 2. The figure shows examples of four types of labeling results. The cysteine-less C498S mutant was used as the basis for all the additional cysteine mutants; we have previously shown that removal of the sole endogenous cysteine in p85ni does not affect its function [11]. As expected, this mutant was not labeled by 14C-NEM (Fig. 2A). In the C498S/E496C mutant, 14C-NEM labeling was unaffected by the presence of p110(1-108) (Fig. 2B). In a few cases, for example the C498S/E547C mutant, labeling was enhanced by p110(1-108) (Fig. 2C). Finally, labeling of the C498S/T482C and C498S/I535C mutants was significantly attenuated by p110(1-108) (Fig. 2D and 2E).
Figure 2. 14C-NEM labeling of p85ni mutants.
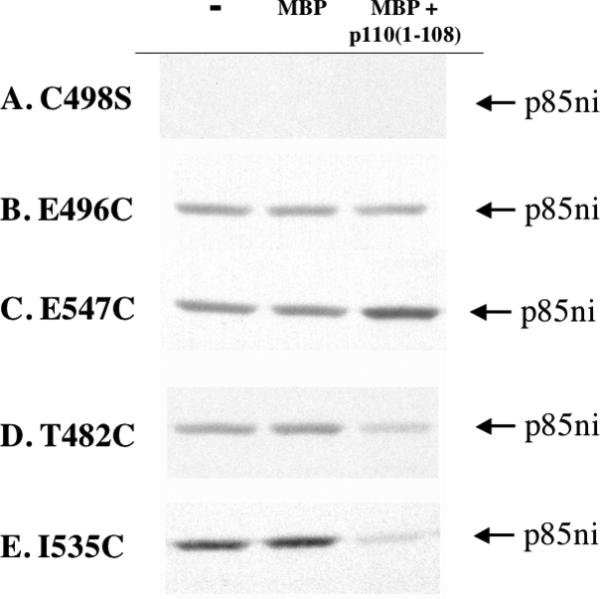
p85ni mutants were incubated in the absence or presence of MBP or MBP-p110(1-108) (20X molar excess) overnight at 4°C. The MBP tag was removed by thrombin digestion, and the protein mixture was incubated for 15 min with 14C-NEM at room temperature. The reaction was terminated by addition of 15 mM DTT, and the samples were analyzed by SDS-PAGE and autoradiography. A. The C498S mutant is not labeled by 14C-NEM. B. Labeling of the E496C mutant is unchanged by p110(1-108) binding. C. Labeling of E547C is increased by p110(1-108) binding. D. Labeling of the T482C is decreased by p110(1-108) binding. E. Labeling of I535C is decreased by p110(1-108) binding. All images were from the same experiment.
The overall results from the analysis of p110(1-108) binding to the iSH2 domain are shown in Table 1 and Figure 3A. While 14C-NEM-labeling of most sites was minimally affected by p110(1-108) binding, substantial decreases in labeling were detected between residues 482-484 in the upper helix of the coiled-coil, and residues 532-541 in the lower helix. Within these two regions, statistically significant decreases were observed at residues C482 (−63.9 +/− 7%, p < .001), and C484 (−28.8 +/− 12. %, p = .057) in the upper helix, and at C532 (−73.2 +/− 7.5%, p = .066), C535 (−84.6 +/− 4.3 %, p < .001), C539 (−17.5 +/− 5%, p = .075), and C541 (−58.2 +/− .5%, p < .001) in the lower helix. In the three dimensional structural model of the iSH2 domain, these regions of the upper and lower helices are adjacent, consistent with a single binding site (Figure 3B).
Table 1.
NEM labeling data is presented as % change in the presence of MBP-p110(1-108) versus MBP. p values are calculated using a students t test.
| Residue | % Change | SEM | p value | Replicates |
|---|---|---|---|---|
| 449 | 6.6 | 8.0 | 0.451 | 6 |
| 454 | −2.6 | 7.4 | 0.739 | 6 |
| 461 | 15.9 | 6.6 | 0.045 | 9 |
| 468 | 7.4 | 15.8 | 0.661 | 6 |
| 475 | 66.5 | 22.4 | 0.031 | 6 |
| 477 | −15.5 | 8.2 | 0.109 | 7 |
| 481 | −17.2 | 8.2 | 0.170 | 3 |
| 482 | −63.9 | 7.3 | <.0001 | 8 |
| 484 | −28.8 | 12.7 | 0.057 | 8 |
| 485 | −21.0 | 13.5 | 0.259 | 3 |
| 489 | −4.1 | 9.6 | 0.682 | 7 |
| 492 | 29.0 | 28.1 | 0.412 | 3 |
| 495 | 16.8 | 7.3 | 0.148 | 3 |
| 496 | −8.6 | 6.9 | 0.249 | 7 |
| 498 | −8.4 | 7.1 | 0.284 | 10 |
| 503 | 11.6 | 11.6 | 0.348 | 9 |
| 516 | 5.0 | 15.2 | 0.751 | 7 |
| 521 | −24.1 | 13.5 | 0.107 | 10 |
| 532 | −73.2 | 7.6 | 0.066 | 2 |
| 533 | −0.5 | 13.2 | 0.971 | 4 |
| 535 | −84.6 | 4.3 | <0.001 | 9 |
| 536 | −1.9 | 9.8 | .866 | 3 |
| 559 | −17.5 | 5.1 | 0.075 | 5 |
| 540 | 32.0 | 12.8 | 0.129 | 5 |
| 541 | −58.2 | 0.5 | <0.001 | 3 |
| 544 | −1.0 | 0.5 | 0.298 | 2 |
| 547 | 33.7 | 10.2 | 0.021 | 6 |
| 548 | 2.9 | 6.6 | 0.701 | 3 |
| 549 | 21.7 | 20.3 | 0.364 | 4 |
| 561 | −3.7 | 10.5 | 0.739 | 5 |
| 568 | 4.7 | 15.0 | 0.770 | 5 |
| 574 | 0.8 | 8.0 | 0.919 | 9 |
| 575 | 13.1 | 4.9 | 0.116 | 3 |
| 582 | 15.1 | 11.5 | 0.294 | 8 |
Figure 3. Mapping of the p110 binding site in p85ni.

A. 14C-labeling of p85ni mutants was quantitated by Phosphorimager scanning. The data from each experiment is expressed as the difference in labeling in the presence of MBP alone versus MBP-p110(1-108), as a percentage of the labeling with MBP alone. The data are pooled from 3-10 separate experiments, depending on the mutant (see Table 1). B. Structural model of the p110 binding site in the iSH2 domain of p85.
Conformational changes in p85ni upon p110(1-108) binding
To determine whether the conformation of the iSH2 domain changed upon binding to p110(1-108), we used EPR spectroscopy to measure the distance between pairs of spin-labeled cysteine residues in the iSH2 domain. We chose one pair of cysteines at 489/549, close to the p110 binding site, and a second pair at 461/577, which lies between the p110 binding site and the nSH2 domain (Figure 4A). The mutants were labeled with the spin probe MTSSL and incubated without or with MBP-p110(1-108), followed by cleavage of the MBP moiety with thrombin. The distances between the probes were then measured as previously described [11]. As shown in Figure 4B, no significant changes in the spectra were observed upon binding of either labeled mutant to p110(1-108). EPR spectra are highly sensitive to changes in the distance between spin labels, which for these mutants is dependent on the extended helical coiled-coil structure of the iSH2 domain [11]. These data therefore show that the binding of p110(1-108) to p85ni does not cause a significant conformational rearrangement of the iSH2 domain.
Figure 4. EPR spectra of p85ni mutants in the absence or presence of MBP-p110(1-108) and phosphopeptide detect minimal conformational changes.
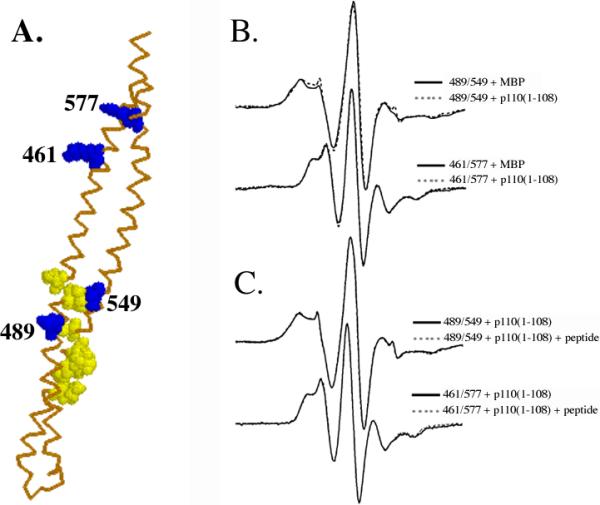
A. Homology model of the iSH2 domain [11], with the positions of cysteine mutants labeled with the spin-probe MTSSL shown in blue, and the p110-binding region shown in yellow. B. Top: 489C/549C double spin-labeled p85ni in the presence of MBP alone (solid line) and upon addition of MBP-p110(1-108) (dashed lines) at 300K. Bottom: 461C/577C double spin-labeled p85ni in the presence of MBP alone (solid line) and upon addition of MBP-p110(1-108) (dashed lines) at 300K. C. Top: Spin-labeled p85ni 489C/549C with MBP-p110(1-108) in the absence (solid line) or presence (dashed line) of PDGF phosphopeptide. Bottom: spin labeled p85ni 461C/577C with MBP-p110(1-108) in the absence (solid line) or presence (dashed line) of PDGF phosphopeptide. No significant changes (corresponding to changes in spin label separation and therefore coiled-coil structure) are observed upon binding of either MBP-p110(1-108) or phosphopeptide.
We have previously shown that the distances between pairs of probes in the iSH2 domain were unaffected by phosphopeptide binding to the nSH2 domain in isolated p85ni [11]. However, it is possible that conformational coupling between the nSH2 and iSH2 domains might only occur in the presence of p110. We therefore repeated these experiments, using the 489/549 and 461/577 mutants described above bound to p110(1-108). As shown in Figure 4C, addition of YXXM phosphopeptide caused no change in the spectrum of either spin-labeled p85ni/p110(1-108) complex. Thus, even when bound to the N-terminal fragment from p110, the iSH2 domain remains conformationally rigid.
The cSH2 domain is not required for regulation of p110
Recent studies on an oncogenic mutant of p85 have attributed its phenotype to the loss of inhibitory residues in the distal iSH2 and cSH2 domains [12]. In contrast, our previous studies have shown that the nSH2 domain is required for regulation of p110, which is inhibited by binding to p85ni but unaffected by binding to either the iSH2 domain or an iSH2-cSH2 fragment (p85ic) [8]. This latter fragment contains the putative inhibitory domains, yet does not inhibit p110.
While our data suggest that the nSH2 is specifically required for regulation of p110, inhibition of p110 could be explained by the N-terminal location of the nSH2 domain, rather than by the existence of specific phosphopeptide-regulated nSH2-p110 contacts. To test this possibility, we measured the activity of p110 complexed with p85ni, p85ic, and a construct in which the cSH2 domain was swapped with the nSH2 domain (p85ci). As previously described, p110 is inhibited by p85ni, but was minimally affected by the iSH2 domain (Fig. 5). While both the p85ic and the p85ci constructs bound to p110 (data not show), the effect of these constructs on p110 activity was similar to that seen in with the iSH2 domain alone (Fig. 5). These data show that the nSH2 domain is specifically required for inhibition of p110.
Figure 5. Regulation of p110 specifically requires the nSH2 domain.
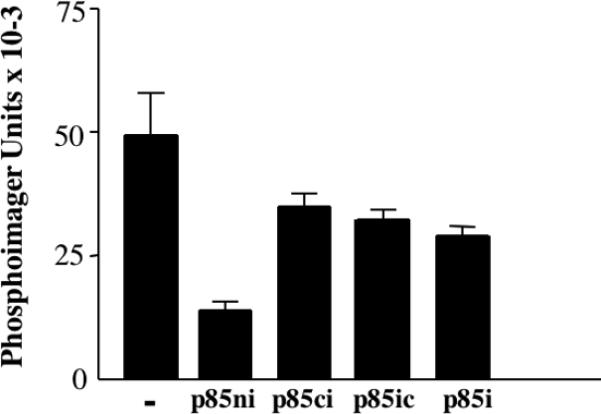
Recombinant p110, produced in baculovirus-infected Sf-9 cells, was incubated in the absence or presence of equal amounts of p85ni, p85i, p85ic or p85ci, for 1h at 4°C. Lipid kinase activity was then determined as previously described [8].
Discussion
The p85ni fragment of p85 is sufficient to mimic phosphopeptide regulation of intact p85/p110: p85ni inhibits p110, and p85ni/p110 dimers are activated by phosphopeptide [3]. p85 and p110 are obligate dimers, due to the heat lability of p110 monomers [3]. Therefore, during activation of p85/p110 dimers, a conformational change initiated by nSH2 domain binding to phosphopeptide is transferred to p110. Given the fact that the iSH2 domain is necessary and sufficient for p85 binding to p110 [5, 6, 10], the iSH2 domain might seem to be likely candidate for the conformational switch linking the nSH2 domain to p110. Indeed, murine and human oncogenic mutations have been described at the distal end of the iSH2 domain [13-15], and the iSH2 domain has been proposed to act as a molecular switch in vivo [12]. In contrast, we have previously shown that the iSH2 coiled-coil is unaffected by phosphopeptide binding to the nSH2 domain [11]. However, our initial study did not examine the possibility that the conformation of the iSH2 domain is altered by p110 binding, such that the iSH2 domain becomes conformationally responsive to nSH2 domain occupancy. We have now defined the p110 binding site in the iSH2 domain. We have furthermore shown that EPR probes near this site, or between this site and the nSH2 domain, show no measurable changes in probe-probe separation upon phosphopeptide stimulation. These data effectively rule out the iSH2 domain as a conformational switch involved in the regulation of p110 activity by phosphopeptides. In addition, we show that the nSH2 domain is specifically required for inhibition of p110 by p85, and cannot be supplanted by the cSH2 domain. These data point to interactions between the nSH2 and p110 as critical for regulation of p85/p110 dimers.
Our mapping experiments have defined two segments of the iSH2 domain that interact with p110. Binding to p110(108) caused decreases in 14C-NEM labeling in residues 482-484 in the N-terminal helix, and residues 532-541 in the C-terminal helix. Viewed in the context of our previously described model of the iSH2 domain [11], and given the 1:1 stoichiometry of the p85ni-p110(1-108) complex (data not shown), these two domains define a single binding site that encompasses both helices of the coiled-coil (Figure 3B). Our studies are consistent in part with a previous study by Dhand et al., which identified residues 478-492 in the upper helix as a potential binding site for p110 [5]. However, this study argued that whereas residues in the N-terminal helix were involved in direct p110 binding, corresponding residues in the C-terminal helix were required for overall structural integrity but were not involved in p110 binding. In contrast, we see significant reductions in the labeling of residues in the C-terminal helix by p110(1-108), suggesting that these residues are likely to be at the site of iSH2-p110 contact.
Statistically significant increases in 14C-NEM-labeling were observed at residues C461 (15.8 +/− 7%, p = .045) , C475 (66.5 +/− 22 %, p = 0.031) and residue C547 (33.7 +/− 10%, p = 0.021). While these increases suggest some conformational change induced by p110 binding, the EPR data shows that the magnitude of this change is small. As demonstrated previously [11], the spin label side chain assumes a distribution of orientations, which may render the EPR spectrum somewhat less sensitive than NEM labeling to small changes in side chain orientation or accessibility induced by p110 binding.
We have not directly tested the necessity of the 477-485 and 521-535 regions for p85-p110 binding. In the mutants used in this study, introduction of single or double cysteine residues does not interfere with p110 binding (data not shown). Given the large binding surface detected by the footprinting experiment, which involves multiple residues in both helices, this is not surprising. In addition, it should also be noted that residues buried in the p110-iSH2 interface may not in fact be the same residues whose interactions with complementary residues in p110 create a high affinity binding site. Deletion of the two domains would be expected to disrupt binding, as was shown with the 478-492 deletion [5]. However, these experiments would be difficult to interpret, given the likelihood of nonspecific disruption of the iSH2 coiled-coil.
In this paper, we have shown that (a) the iSH2 domain is conformationally rigid and unaffected by phosphopeptide or p110 binding, and (b) the nSH2 domain is specifically required for inhibition of p110, and cannot be supplanted by the cSH2 domain. We also note that previous NMR and crystal structures of isolated nSH2 domains have detected conformational changes upon phosphopeptide binding, particularly in the BG loop involved in phosphopeptide binding [16, 17], although the magnitude of these changes has been controversial [18-20]. Our data, combined with these previous studies, make the nSH2 domain a reasonable candidate for a conformational switch involved in phosphopeptide regulation of p85/p110 dimers.
In summary, this work is the first biochemical mapping study of the p85-p110 binding interface. We have detected two segments of the iSH2 domain primary structure, in both the upper and lower helices, that are implicated in p110 binding. Modeling studies suggest that these two regions lie physically close to each other, and are likely to form a single binding site involving both helices. We cannot detect significant conformational changes in the iSH2 domain, either upon p110 binding or upon subsequent stimulation of p85ni-p110(1-108) dimers with tyrosine phosphopeptides. We conclude that the p110 binding site is composed of a conformationally inflexible region of the iSH2 domain, containing contributions from both helices of the coiled-coil. Regulation of p110 specifically requires the nSH2 domain, as p85ic and p85ci constructs bind but do not inhibit p110. Conformational changes involved in phosphopeptide-mediated activation of p85/p110 dimers presumably occur at the nSH2-p110 interface, and do not involve the iSH2 domain.
Acknowledgements
We would like to thank Drs. Mark Girvin and S.C. Shekar (Albert Einstein College of Medicine) for helpful discussion and critical reading of the manuscript. This work was supported by NIH grant GM55692 (J.M.B.) and GM60609 (to G.J.G.). E.A.-S. is supported by the Medical Scientist Training Program at Albert Einstein College of Medicine.
Abbreviations
- DTT
dithiothreitol
- EPR
Electron Paramagnetic Resonance
- GST
Glutathione-S-transferase
- iSH2
Inter-SH2
- MBP
Maltose binding protein
- MTSSL
(1-oxyl-2,2,5,5-tetramethylpyrolinyl-3-methyl)-methanethiosulfonate
- NEM
N-Ethyl-maleimide
- PBS
phosphate buffered saline
- PI 3-kinase
Phosphoinositide 3-kinase
- PI(3)P
phosphatidylinositol –3-phosphate
- PI(3,4)P2
phosphatidylinositol –3,4-bisphosphate
- PI(3,4,5)P3
phosphatidylinositol –3,4,5-trisphosphate
- SH2
Src-homology 2
- SH3
Src-homology 3
REFERENCES
- 1.Fruman DA, Meyers RE, Cantley LC. Phosphoinositide kinases. Annu.Rev.Biochem. 1998;67:481–507. doi: 10.1146/annurev.biochem.67.1.481. [DOI] [PubMed] [Google Scholar]
- 2.Vanhaesebroeck B, Leevers SJ, Ahmadi K, Timms J, Katso R, Driscoll PC, Woscholski R, Parker PJ, Waterfield MD. Synthesis and function of 3-phosphorylated inositol lipids. Annu Rev Biochem. 2001;70:535–602. doi: 10.1146/annurev.biochem.70.1.535. [DOI] [PubMed] [Google Scholar]
- 3.Yu J, Zhang Y, McIlroy J, Rordorf-Nikolic T, Orr GA, Backer JM. Regulation of the p85/p110 phosphatidylinositol 3′-kinase: Stabilization and inhibition of the p110-alpha catalytic subunit by the p85 regulatory subunit. Mol.Cell.Biol. 1998;18:1379–1387. doi: 10.1128/mcb.18.3.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fruman DA, Mauvais-Jarvis F, Pollard DA, Yballe CM, Brazil D, Bronson RT, Kahn CR, Cantley LC. Hypoglycaemia, liver necrosis and perinatal death in mice lacking all isoforms of phosphoinositide 3-kinase p85 alpha. Nat Genet. 2000;26:379–382. doi: 10.1038/81715. [DOI] [PubMed] [Google Scholar]
- 5.Dhand R, Hara K, Hiles I, Bax B, Gout I, Panayotou G, Fry MJ, Yonezawa K, Kasuga M, Waterfield MD. PI 3-kinase: Structural and functional analysis of intersubunit interactions. EMBO J. 1994;13:511–521. doi: 10.1002/j.1460-2075.1994.tb06289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klippel A, Escobedo JA, Hirano M, Williams LT. The interaction of small domains between the subunits of phosphatidylinositol 3-kinase determines enzyme activity. Mol Cell Biol. 1994;14:2675–2685. doi: 10.1128/mcb.14.4.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu P, Mondino A, Skolnik EY, Schlessinger J. Cloning of a novel, ubiquitously expressed human phosphatidylinositol 3-kinase and identification of its binding site on p85. Mol.Cell.Biol. 1993;13:7677–7688. doi: 10.1128/mcb.13.12.7677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu JH, Wjasow C, Backer JM. Regulation of the p85/p110α Phosphatidylinositol 3′-kinase - Distinct roles for the N-terminal and C-terminal SH2 domains. J.Biol.Chem. 1998;273:30199–30203. doi: 10.1074/jbc.273.46.30199. [DOI] [PubMed] [Google Scholar]
- 9.Rordorf-Nikolic T, Van Horn DJ, Chen D, White MF, Backer JM. Regulation of phosphatidylinositol 3′-kinase by tyrosyl phosphoproteins. Full activation requires occupancy of both SH2 domains in the 85-kDa regulatory subunit. J.Biol.Chem. 1995;270:3662–3666. doi: 10.1074/jbc.270.8.3662. [DOI] [PubMed] [Google Scholar]
- 10.Holt KH, Olson AL, Moye-Rowley WS, Pessin JE. Phosphatidylinositol 3-kinase activation is mediated by high-affinity interactions between distinct domains within the p110 and p85 subunits. Mol.Cell.Biol. 1994;14:42–49. doi: 10.1128/mcb.14.1.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fu Z, Aronoff-Spencer E, Backer JM, Gerfen GJ. The structure of the inter-SH2 domain of class IA phosphoinositide 3-kinase determined by site-directed spin labeling EPR and homology modeling. Proc Natl Acad Sci U S A. 2003;100:3275–3280. doi: 10.1073/pnas.0535975100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan TO, Rodeck U, Chan AM, Kimmelman AC, Rittenhouse SE, Panayotou G, Tsichlis PN. Small GTPases and tyrosine kinases coregulate a molecular switch in the phosphoinositide 3-kinase regulatory subunit. Cancer Cell. 2002;1:181–191. doi: 10.1016/s1535-6108(02)00033-8. [DOI] [PubMed] [Google Scholar]
- 13.Janssen JWG, Schleithoff L, Bartram CR, Schulz AS. An oncogenic fusion product of the phosphatidylinositol 3-kinase p85β subunit and HUMORF8, a putative deubiquitinating enzyme. Oncogene. 1998;16:1767–1772. doi: 10.1038/sj.onc.1201695. [DOI] [PubMed] [Google Scholar]
- 14.Philp AJ, Campbell IG, Leet C, Vincan E, Rockman SP, Whitehead RH, Thomas RJ, Phillips WA. The phosphatidylinositol 3′-kinase p85alpha gene is an oncogene in human ovarian and colon tumors. Cancer Res. 2001;61:7426–7429. [PubMed] [Google Scholar]
- 15.Jimenez C, Jones DR, Rodríguez-Viciana P, Gonzalez-García A, Leonardo E, Wennström S, Von Kobbe C, Toran JL, Borlado LR, Calvo V, Copin SG, Albar JP, Gaspar ML, Diez E, Marcos MAR, Downward J, Martinez C, Mérida I, Carrera AC. Identification and characterization of a new oncogene derived from the regulatory subunit of phosphoinositide 3-kinase. EMBO J. 1998;17:743–753. doi: 10.1093/emboj/17.3.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shoelson SE, Sivaraja M, Williams KP, Hu P, Schlessinger J, Weiss MA. Specific phosphopeptide binding regulates a conformational change in the PI 3-kinase SH2 domain associated with enzyme activation. Embo J. 1993;12:795–802. doi: 10.1002/j.1460-2075.1993.tb05714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nolte RT, Eck MJ, Schlessinger J, Shoelson SE, Harrison SC. Crystal structure of the PI 3-kinase p85 amino-terminal SH2 domain and its phosphopeptide complexes. Nat Struct Biol. 1996;3:364–374. doi: 10.1038/nsb0496-364. [DOI] [PubMed] [Google Scholar]
- 18.Weber T, Schaffhausen B, Liu Y, Gunther UL. NMR structure of the N-SH2 of the p85 subunit of phosphoinositide 3-kinase complexed to a doubly phosphorylated peptide reveals a second phosphotyrosine binding site. Biochemistry. 2000;39:15860–15869. doi: 10.1021/bi001474d. [DOI] [PubMed] [Google Scholar]
- 19.O'Brien R, Rugman P, Renzoni D, Layton M, Handa R, Hilyard K, Waterfield MD, Driscoll PC, Ladbury JE. Alternative modes of binding of proteins with tandem SH2 domains. Protein Sci. 2000;9:570–579. doi: 10.1110/ps.9.3.570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hensmann M, Booker GW, Panayotou G, Boyd J, Linacre J, Waterfield M, Campbell ID. Phosphopeptide binding to the N-terminal SH2 domain of the p85 alpha subunit of PI 3′-kinase: a heteronuclear NMR study. Protein Sci. 1994;3:1020–1030. doi: 10.1002/pro.5560030704. [DOI] [PMC free article] [PubMed] [Google Scholar]