

ABSTRACT
The proteins that form vertebrate gap junctions, the connexins, are highly regulated and have short (<2 hour) half-lives. Phosphorylation of connexin43 (Cx43) affects gap junction assembly, channel gating and turnover. After finding dramatic effects on gap junctions with Akt inhibitors, we created an antibody specific for Cx43 phosphorylated on S373, a potential Akt substrate. We found S373 phosphorylation in cells and skin or heart almost exclusively in larger gap-junctional structures that increased dramatically after wounding or hypoxia. We were able to mechanistically show that Akt-dependent phosphorylation of S373 increases gap junction size and communication by completely eliminating the interaction between Cx43 and ZO-1. Thus, phosphorylation on S373 acts as a molecular ‘switch’ to rapidly increase gap-junctional communication, potentially leading to initiation of activation and migration of keratinocytes or ischemic injury response in the skin and the heart, respectively.
KEY WORDS: Gap junctions, Connexin43, Akt, Wounding, Hypoxia
INTRODUCTION
Gap-junction-mediated intercellular communication allows the passage of ions and small metabolites between adjacent cells (Laird, 2010; Scemes et al., 2007; Söhl and Willecke, 2004). Gap junctions are composed of integral membrane proteins from the connexin gene family and are critically important in regulating development, coordinated contraction of excitable cells, tissue homeostasis, controlled cell growth and differentiation (Scemes et al., 2007; Söhl and Willecke, 2004). Mutations in connexin proteins have been linked to many diseases, including hereditary deafness (Bergoffen et al., 1993; Kelsell et al., 1997; Laird, 2006) and oculodentodigital dysplasia, a disease caused by connexin43 (Cx43) mutations that can lead to syndactyly, atrioseptal defects and arrhythmias (Paznekas et al., 2003).
Cx43, which is the most ubiquitous connexin, can be phosphorylated at 12 or more sites, with some sites apparently increasing gap junction assembly whereas others inhibit formation of gap junctions, or decrease channel open time (Solan and Lampe, 2009). Given the unusually short half-life (<2 hours) of Cx43 in cell culture and tissue, Cx43 trafficking and turnover also appear to influence gap-junctional communication. There are some specific events that can regulate gap junction assembly and turnover. Phosphorylation at S365 and S325, S328 or S330 positively regulates gap junction assembly (Cooper and Lampe, 2002; Lampe et al., 2006; Solan et al., 2007). Gap junction size is negatively regulated by interaction with ZO-1 because a peptide mimetic of the Cx43 ZO-1 binding domain (termed α-CT) can compete out the endogenous ZO-1?–Cx43 interaction (Hunter et al., 2005). α-CT can also reduce remodeling of gap junctions and induced arrhythmia following ventricular injury (O'Quinn et al., 2011). ZO-1 small interfering RNA experiments also verified that loss of ZO-1 function increases the rate of aggregation of connexons into gap junctions (Rhett et al., 2011).
We have previously shown that ubiquitin-mediated activation of Akt is responsible for increased gap-junctional communication and stability following proteasomal inhibition (Dunn et al., 2012). Here, we show that Akt activity is necessary for phosphorylation of Cx43 at S373 and this phosphorylation event causes an increase in gap junction size and communication levels by limiting the interaction of ZO-1 with Cx43. Furthermore, we show that Akt rapidly becomes activated in response to scratch-wound injury or hypoxia, and leads to Cx43 phosphorylation at S373. This phosphorylation event increases in cardiac tissue in response to hypoxia and in skin in response to wounding, leading to larger junctions. Thus, gap junction size and activity is dynamically regulated by phosphorylation at S373 and is highly responsive to injury, both in vitro and in vivo.
RESULTS
Cx43 phosphorylation on S373
Previously, we used phosphotryptic peptide/mass spectrometry analysis to show that the C-terminal region of Cx43 (A371–I382), containing S372 and S373, was phosphorylated in cells (Cooper and Lampe, 2002). More recently, we showed that proteasomal inhibition increased gap junction stability through activation of Akt (Dunn et al., 2012). Because S373 has been described as a possible Akt kinase substrate (Park et al., 2007), we decided to create a phosphospecific antibody that reacts with Cx43 when it is phosphorylated at S373 (pS373). To examine Cx43 phosphorylation in cells, we used an MDCK cell line that was null for Cx43 in the parental line (Jordan et al., 1999), which upon stable transfection expresses Cx43 at nominal levels (MDCK43) and routinely forms functional gap junctions (Dunn et al., 2012). We found that untreated MDCK43 cells showed low levels of pS373, but inhibition of the proteasome with MG132 caused a 3.2±0.4-fold increase (significantly different, P = 0.007; n = 6) in the pS373 signal, primarily in a single band with reduced SDS-PAGE mobility (Fig. 1A). Like many phosphorylated proteins, Cx43 normally migrates as multiple bands in SDS-PAGE owing to phosphorylation, and the pS373 band overlaid exactly with a major Cx43 phosphoisoform (Fig. 1A, arrow) that was increased by addition of MG132. Cell lysates from MG132-treated cells also showed increased overall phosphorylation of Akt substrates, as shown in cell lysates that were blotted with the phospho-(Ser/Thr) Akt substrate antibody, including a minor band that aligned with the same Cx43 and pS373 signal noted above (Fig. 1A). Furthermore, the pS373 antibody was quite specific for Cx43 because the only major band was eliminated if the peptide that was used to create the antibody was preincubated with the antibody prior to blotting (pS373/pep). These results show that phosphorylation of Cx43 on S373, a potential Akt site, is increased by treatment with MG132.
Fig. 1.

Cx43 is phosphorylated on Serine 373 in gap junctions. (A) Immunoblot detection of Cx43 in MDCK43 cell lysates using antibodies against the N-terminus (Cx43), Cx43 phosphorylated at S373 (pS373) and phosphorylated Akt ‘consensus’ sites (Akt sub). Arrows indicate the S373-phosphorylated isoform of Cx43. The fastest migrating Cx43 isoform is marked P0 and the multiple phosphoisoforms are bracketed with a P. (B–D) Cx43 immunofluorescence in MDCK43 cells using the pS373 antibody (B) and total Cx43 (C). The overlay (D) shows that the pS373 antibody signal is almost exclusively gap junctional; DAPI in blue. Scale bar: 25 µm. (E) High magnification immunofluorescence of a series of gap junctions of different size with total Cx43 (green), pS373 (red) and overlay. (F) Histogram showing size distribution of pS373 (red) and total Cx43 (green) signals. Scale bar: 2 µm.
When our pS373 antibody was used to examine its immunolocalization within MDCK43 cells, the pS373 signal overlaid with the signal from a total Cx43 antibody but only within larger, punctate, apparent gap-junctional structures (Fig. 1B–D). A higher-resolution image of a cell–cell interface (Fig. 1E) indicated that the pS373 signal was localized primarily within the central regions of the larger punctate total Cx43 staining. Quantification of the size distribution of pS373 and total Cx43 signal at over 4000 discrete fluorescent objects confirmed that the pS373 signal was shifted to larger objects (Fig. 1F). Note that light microscopy does not have the resolution to determine whether these larger objects are actually larger junctions or more tightly packed assemblages of junctions. In either case, the increase in fluorescence does reflect an apparent increase in area and/or density of junctional Cx43, and we generically denote this hereafter as an increase in gap junction size.
Proteasomal inhibition increases S373 phosphorylation, gap junction size and gap-junctional communication
To further examine the functions of S373 phosphorylation and Akt activity on gap junctions, we stably transfected the parental MDCK cells with Cx43 containing mutations at S373 to mimic (i.e. S373D) or block (i.e. S373A) phosphorylation at this site in order to examine its effect on gap junction size compared with the wild type in the presence of MG132 or the specific Akt1 inhibitor MK2206. In western immunoblots (Fig. 2A), MDCK43 cells showed a large increase in pS373 levels in the presence of MG132 that was completely eliminated when MK2206 was added (Fig. 2). Neither S373A nor S373D mutants showed a response to MK2206 treatment, but both showed a reduced increase in the slower-migrating total Cx43 isoform compared with the control, in response to MG132. As expected, neither lysate from the mutant cell lines showed a positive signal for the S373 phosphospecific antibody, again proving the high specificity of the antibody. When Cx43 was examined by immunofluorescence (Fig. 2B), striking differences were observed in these cells. Cells expressing wild-type Cx43 increased the level of signal in gap junctions in response to MG132 and decreased them in response to MK2206. The role of pS373 was clearly apparent because cells expressing S373A lost characteristic punctate gap-junctional staining whereas S373D cells had large concentrations of gap-junctional signal, and these distributions did not change appreciably in either cell line in response to drug treatment (Fig. 2B). Quantification of gap junction size expressed as a volume distribution of fluorescent objects (minimum gap junction/object size defined as 0.005 µm3) shows that expression in S373D cells essentially mimicked MG132 treatment, whereas S373A cells showed a distribution similar to that observed with MK2206 treatment (Fig. 2C). Specifically, MDCK43 S373D cells had a mean object size that was 1.6-times larger than control cells whereas treatment with MG132 caused a 1.7-fold increase. MDCK43 S373A cells had a mean object size that was 1.6-times smaller than control cells and MK2206 treatment caused a 3.6-times decrease compared with control cells. Chi-square analysis of these volume distributions comparing MG132 treatment or S373A and S373D substitution each individually to the wild-type MDCK43 untreated control cells are all statistically different (P≤0.003). Comparing the number of objects labeled by total Cx43 (9562) to that for pS373 (589) antibodies on the same images reinforced the idea that S373 phosphorylation only occurs on a subset of large gap junctions. From these results, we conclude that phosphorylation at S373 via Akt controls gap junction size.
Fig. 2.
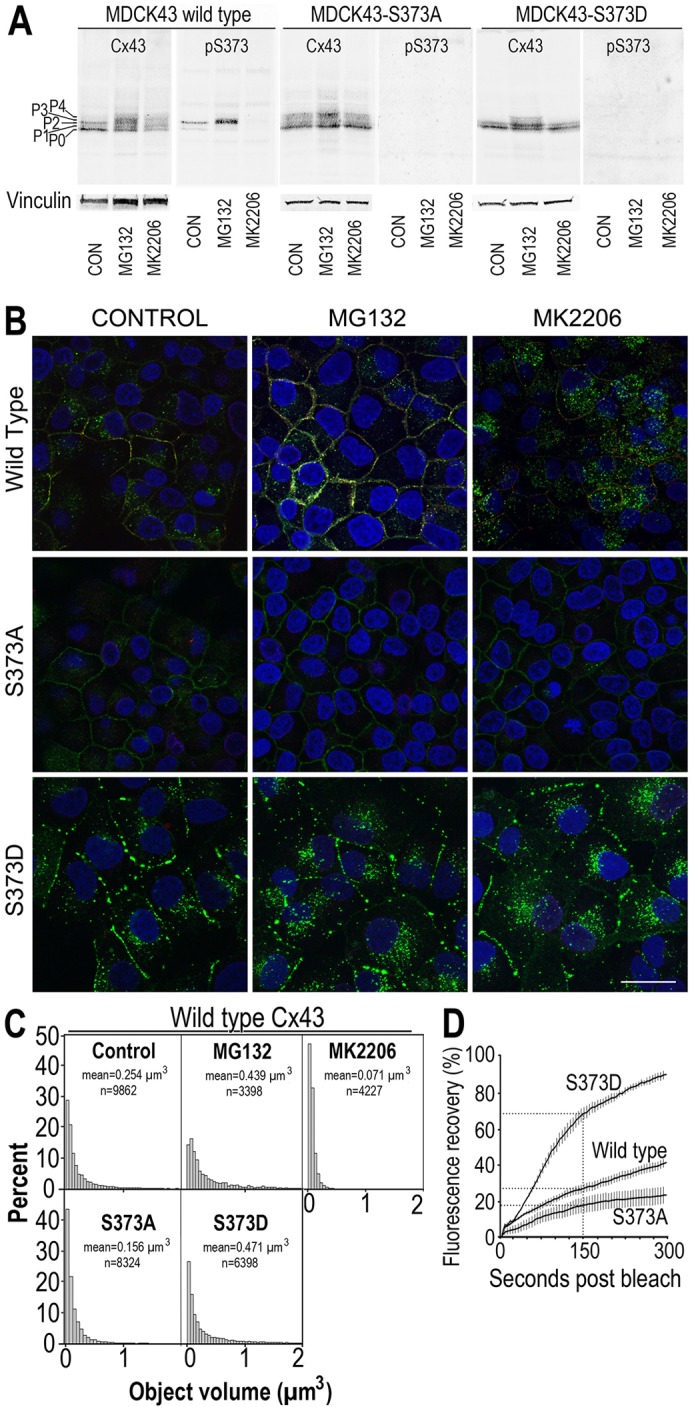
Cx43 is phosphorylated at S373 in large gap-junctional structures and mutation of S373 to A or D can inhibit or promote gap junction formation. MDCK43, MDCK43 S373A and MDCK43 S373D cells were untreated or treated with MG132 or MK2206. (A) Immunoblot analysis of total Cx43 and pS373. Cx43 isoforms are marked with P0 and P (phosphorylated). (B) Immunofluorescence analysis with staining for total Cx43 (green), pS373 (red) and DAPI (blue); overlay images are shown. Scale bar: 25 µm. (C) Distribution of fluorescent object size after MG132 or MK2206 treatment or mutation of S373 to A or D. (D) Gap-junctional communication is increased in MDCK43 S373D and decreased in MDCK43 S373A cells compared with control MDCK43 cells expressing wild-type Cx43. Dotted lines indicate percentage recovery at 150 seconds post bleach. Data are means ± s.e.m.
Next, we determined whether these mutations affected gap-junctional communication. Thus, we performed fluorescence recovery after photobleaching (FRAP) by loading MDCK43 cells with Calcein-AM, bleaching a single cell and quantifying fluorescence recovery that occurs by gap-junctional transfer of calcein from its neighbors, so called ‘GAP-FRAP’ (Yum et al., 2007). Examination of the time to recovery at 150 seconds post bleach (Fig. 2D) showed significantly different results with MDCK cells expressing Cx43 through wild-type, S373D and S373A constructs with 26.9%, 68.5% and 17.4% recovery, respectively (S373D versus wild type, P<0.001 and S373A versus wild type, P = 0.03 or S373D, P<0.001). Although the wild-type Cx43-expressing cells had ∼30% more Cx43 relative to vinculin by immunoblotting than the S373A and S373D transfectants, the latter two expressed very similar amounts of Cx43, but dye-transfer rates were very different. These results indicate that the larger gap junctions observed in S373D cells are functional and result in more gap-junctional communication than wild-type-expressing cells whereas those containing S373A had the smallest gap junctions and had the least functionality.
Interaction between ZO-1 and Cx43 is eliminated by S373 phosphorylation
Given results from the Gourdie lab concerning the influence of ZO-1 on accretion of Cx43 into larger gap-junctional structures (Hunter et al., 2005; Rhett et al., 2011), we set out to test whether the interaction of ZO-1 with Cx43 might be influenced by phosphorylation at S373. We used MDCK cells expressing wild- type (WT), S365/369/373A (S3A) or S373D versions of Cx43 and examined their ability to pull down endogenous ZO-1 in the presence and absence of MG132. In Fig. 3A, we show that MG132 treatment decreased the amount of ZO-1–Cx43 interaction by ∼40% in the WT cells. Conversely, cells expressing the S3A mutant showed the highest level of interaction, whether or not the cells were treated with MG132, indicating that prevention of phosphorylation at these residues allowed maximal interaction with or without drug. In MDCK43 S373D cells, we detected only 3.8% of the level of coprecipitated ZO-1 signal compared with that in S373A cells, with or without MG132 (significantly different P<0.005; n = 7). Thus, maintaining S373 in the non-phosphorylated state was sufficient to allow Cx43–ZO-1 interaction whereas the phospho-mimetic essentially eliminated this interaction.
Fig. 3.
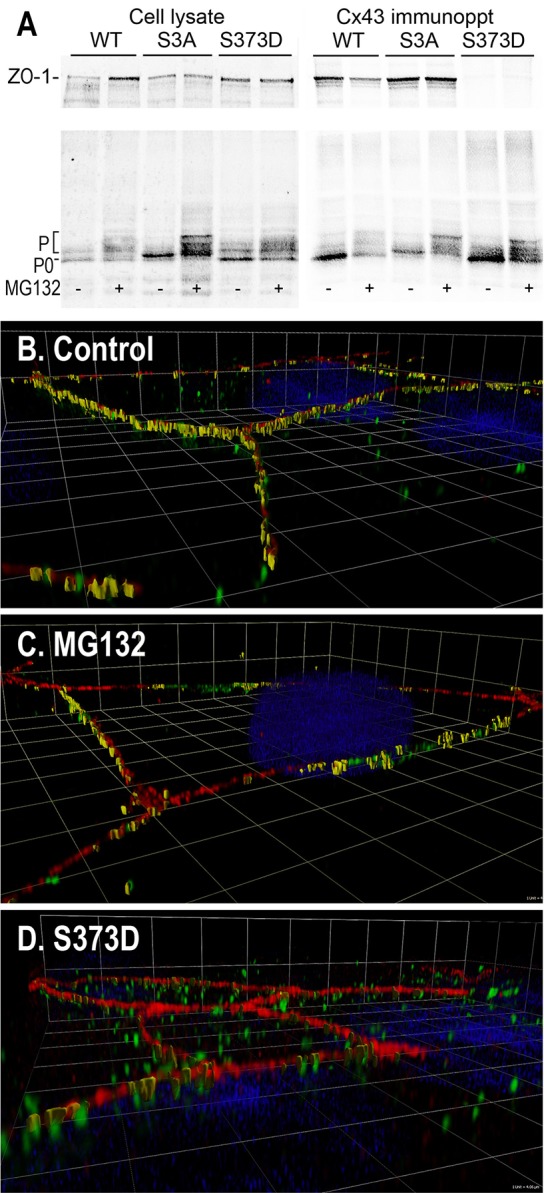
Interaction of ZO-1 and Cx43 is regulated by S373 phosphorylation. (A) Coimmunoprecipitation of ZO-1 with Cx43 antibody from MDCK43 (WT), MDCK43 cells with Cx43 mutant at S365/369/373 (S3A) and MDCK43-S373D (S373D) cells treated (+) or not untreated (−) with MG132. Cx43 isoforms are marked with P0 and P (phosphorylated). (B) Tilted edge-on volume-rendered immunofluorescence colocalization of Cx43 and ZO-1 in MDCK43 (B), MDCK43 treated with MG132 (C) and MDCK43 S373D (D) cells. Cx43 is shown in green and ZO-1 in red (DAPI in blue). Grid lines are 4.06 µm apart.
We then examined the interaction of ZO-1 and Cx43 by immunofluorescence (Fig. 3B). To examine this interaction at higher resolution we used a DeltaVision OMX® super-resolution 3D structured illumination system that yields a twofold improvement in resolution. Angled edge-on volume rendered views are shown to illustrate the amount of colocalization in the z-direction of the cell–cell interface with Cx43 shown in green and ZO-1 in red (Fig. 3B). Untreated MDCK43 cells showed significant amounts of colocalization (yellow) and fairly extensive intermixing of Cx43 and ZO-1 within the same narrow z-plane of cell–cell interface. When quantified, we found that 6.8±0.9% of the ZO-1 signal overlaid with Cx43. Note that the interaction is expressed in terms of percentage of ZO-1 overlaid with Cx43 because the Cx43 present in cytoplasmic membranes was represented to varying degrees using this technology. MG132 treatment (Fig. 3C) significantly (P = 0.0007, t-test; n = 6 full images of >1000 total identified objects each) reduced the amount of ZO-1 overlaid ∼10-times to 0.6±0.2% and in contrast to the intermixed expression pattern in the control, long stretches of just ZO-1 or Cx43 staining occurred independent of each other while still remaining in the same general z-plane. S373D cells (Fig. 3D) also had ∼10-times less extensive colocalization (0.7±0.2%) than control (P = 0.0007; n = 6 full images of >1000 total identified objects each), but in contrast to both of the other cell types, ZO-1 and Cx43 staining was totally independent even in the z-direction. The MDCK43 S373A cells lacked punctate structures typical of gap junctions and were not well represented using super-resolution techniques (i.e. only very low levels of staining were apparent) so they are not shown here.
Treatment with 12-O-tetradecanoylphorbol-13-acetate (TPA) transiently increases S373 phosphorylation and gap junction size
When cell lines are treated with phorbol esters (e.g. TPA), there can be a dramatic and dynamic rearrangement of gap junctions, with many cell types showing a transient increase in Cx43 immunolocalization to apparent gap-junctional structures followed by loss of this staining (e.g. Sirnes et al., 2008). Because our results showed that pS373 levels could readily control gap junction size, we decided to study pS373 levels in TPA-treated MDCK43 cells. Examination of the pS373 signal through a time course of TPA treatment by immunoblotting showed a dramatic increase in signal after 5 minutes, followed by a gradual decrease up to 45 minutes (Fig. 4A). Immunofluorescence similarly showed a dramatic increase in pS373 signal at apparent gap junction structures with both total Cx43 (green) and pS373 (red) antibodies after 5 minutes of TPA treatment (Fig. 4B). At 45 minutes, the pS373 signal was essentially gone and much more of the total Cx43 signal was intracellular. Because MAPK activation has been proposed to be involved in relocalization of Cx43 from the plasma membrane to intracellular membranes in response to phorbol esters and growth factors (e.g. Leithe and Rivedal, 2004; Ruch et al., 2001; Sirnes et al., 2008) and phosphorylation at S279 or S282 has been proposed to be involved in association with NEDD4 and turnover of Cx43 (Leykauf et al., 2006), we decided to examine the levels of phosphorylated S279 and/or S282 (pS279/282) during this TPA treatment time course. We found that the level of pS279/282 peaked at 15–30 minutes (Fig. 4A), consistent with a role in the relocalization of Cx43 to cytoplasmic membranes during this time period.
Fig. 4.
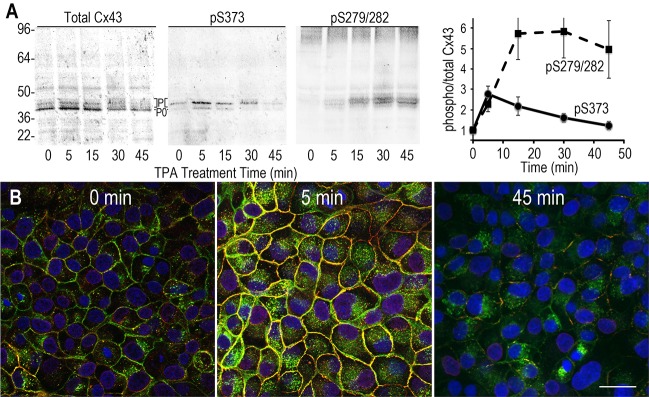
TPA treatment leads to rapid phosphorylation of Cx43 at S373 and slower phosphorylation at S279/282. (A) Immunoblot analysis of a TPA treatment time course for total Cx43, Cx43 phosphorylated on S373 (pS373) and S279/282 (pS279/282). Cx43 isoforms are marked with P0 and P (phosphorylated). Data in graph on right are means ± s.e.m. of five blots. (B) Immunofluorescence analysis with staining for total Cx43 (green), pS373 (red) and DAPI (blue). Overlay images are shown. Scale bar: 25 µm.
Ischemia or scratch wounding leads to increased Cx43 phosphorylation on S373
We decided to examine other biologically important events that involved Cx43 relocalization, including ischemia and wounding. Specifically, we examined MDCK43 cells scratched with a pipette tip or incubated in 1% O2. Scratch-wounded MDCK43 cells showed a statistically significant (P<0.004; n = 5) and rapid 34±3% increase in pS373 signal detectable on Cx43 at 1 minute with a rapid return to pre-scratch levels by 15 minutes (Fig. 5A). Examination of the pS373 signal by immunofluorescence indicated that the level of Cx43 in gap junctions was much higher at 5 minutes than in the unscratched control (Fig. 5B). We also examined the activation of Akt using the ratio of phosphoAkt to total Akt levels (phosphorylated S473 and S308 over total Akt) resulting from scratch wounding and found an increase in phosphorylation/activation in the 1–15 minute range, with a peak at 5 minutes (Fig. 5A).
Fig. 5.

Scratch wounding or incubation of MDCK43 cells at 1% O2 cells increases phosphorylation of Cx43 on S373. (A) Immunoblot analysis of wounded cells over time post scratch showing increases in pS373 on Cx43 and pS308 and pS473 on Akt. The change in ratio of Akt signal from pS308 and pS473 over total Akt shows a maximum at 5 minutes. (B) Immunofluorescence of Cx43 phosphorylation at S373 (red) and total Cx43 (green) at 0 and 5 minutes post scratch with DAPI (blue). Scratched area is indicated by arrow. (C) Immunoblot analysis of cells incubated over time show increases in pS373, pS308 and pS473. Cx43 isoforms are marked with P0 and P (phosphorylated). The change in ratio of Akt signal from pS308 and pS473 over total Akt shows a maximum at 5 minutes. Data are means ± s.e.m. (D) Immunofluorescence of Cx43 phosphorylation at S373 (red) and total Cx43 (green) at 0 and 5 minutes after reduction to 1% O2. DAPI is blue. Scale bar: 25 µm.
Examination of S373 phosphorylation at 1% O2 by immunoblotting showed a statistically significant (P = 0.029; n = 7) and rapid 81±29% increase in pS373 within 5 minutes, which gradually tapered off over 30–60 minutes (Fig. 5C). Similarly, phosphoAkt levels at S308 and S473 showed increases at 1 minute that peaked at 5 minutes before returning to very low levels at 60 minutes. Note that the levels of Akt phosphorylation at S473 were much higher than at S308, implying that hypoxia triggers more PI3K than PDK activity. Immunofluorescence of cells incubated at 1% O2 for 5 minutes showed a dramatic increase in the level of Cx43 phosphorylation at S373 (Fig. 5D).
Cx43 phosphorylation on S373 increases during cardiac ischemia or skin wounding
We sought to determine whether S373 phosphorylation occurred in tissue and whether it might be dynamically regulated. Cardiac ischemia leads to loss of specific Cx43 localization to the intercalated disc region and gain of Cx43 at the lateral edges of the myocytes in a process called remodeling or lateralization (Beardslee et al., 2000; Ek-Vitorin et al., 2006; Schulz et al., 2003). When whole mouse hearts were exposed to no-flow ischemia for 30 minutes, the level of pS373 was increased 3.6±0.4-times over control levels (P = 0.001, n = 6) when assayed by immunoblotting (Fig. 6A). Immunofluorescence showed sharp and intense pS373 and total Cx43 signals entirely at the ends of myocytes in the region of the intercalated discs under control conditions (Fig. 6B). However, after 30 minutes of ischemia, the total and pS373 Cx43 staining was more diffuse and present at both the intercalated discs and lateral edges, with more intense pS373 staining in larger puncta. Thus, these results in tissue appear to be consistent with the MDCK43 cell response to hypoxia with phosphorylation on S373 increasing during hypoxia (shown by immunoblotting) particularly in larger gap junctions (shown by immunofluorescence).
Fig. 6.
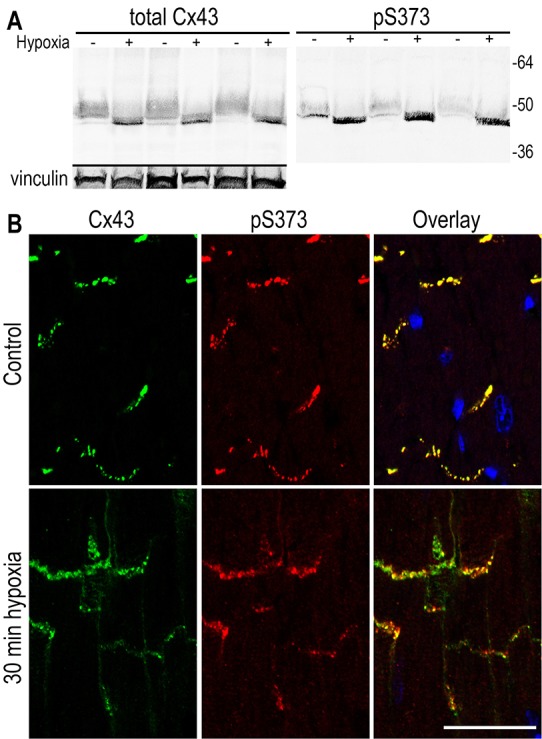
Cx43 is phosphorylated on S373 in murine cardiac tissue particularly after 30 minutes of hypoxia. (A) Immunoblot analysis probing for total Cx43 or pS373 in three pairs of heart tissues that had been either untreated or rendered hypoxic for 30 minutes. (B) Immunofluorescence analysis with staining of control or 30 minute hypoxic heart tissue for total Cx43 (green), pS373 (red) and DAPI (blue). Overlay images are shown. Scale bar: 25 µm.
We then sought to determine whether there were changes in the level of S373 phosphorylation in human skin explants in response to epidermal wounding. Unwounded and excisional wounds fixed at 30 minutes and 24 hours post wounding were immunostained (Fig. 7) for pS373 (red) and total Cx43 (green). We observed extensive staining for both Cx43 and pS373 in punctate gap junctional structures in the unwounded and the 30 minute wounded skin (arrowhead denotes edge of wound) but saw a loss of both signals in the wound by 24 hours. We measured the integrated density values for each signal and found that the phosphorylated S373 to total Cx43 ratio significantly (P = 0.003; n = 9) increased 43±11% in the epidermal tissue proximal to the wound margin relative to unwounded control skin. The ratio in the wounds after 24 hours was slightly but not significantly higher (P = 0.2; n = 9) than control levels.
Fig. 7.
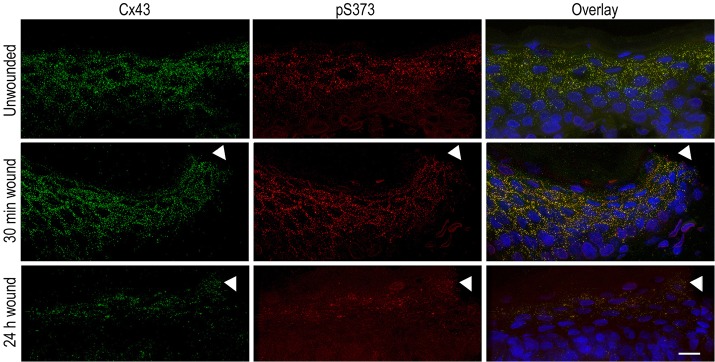
Phosphorylation of Cx43 on S373 is increased in human skin 30 minutes post wounding. Unwounded, 30 minutes and 24 hours post-wound are shown. Total Cx43 (green), pS373 (red) and their overlay with DAPI (blue) are shown. The site of the wound is marked with an arrowhead. Scale bar: 25 µm.
DISCUSSION
Results from this study allow us to conclude that: (1) Cx43 is phosphorylated at S373 in cells and tissues; (2) S373 phosphorylation is dependent on and correlates with Akt activation; (3) S373 phosphorylation is found in larger gap junctions; (4) S373 phosphorylation makes larger junctions in response to reagents such as TPA, hypoxia and scratch wounding; (5) Phosphorylation at S373 eliminates Cx43 interaction with ZO-1, thereby allowing gap junctions to grow in size; (6) Akt is phosphorylated on S308 and S473 in response to scratch wounding or hypoxia; (7) Cx43 is phosphorylated at S373 at low levels in cardiac tissue that increase dramatically during hypoxia; and (8) Cx43 is phosphorylated in human skin and the phosphorylation level increases following wounding, consistent with the requirement for cell–cell communication before cell activation and migration to repair the wound (Richards et al., 2004).
A few specific phosphorylation events have been correlated with turnover of Cx43 (e.g. S279/282), but the mechanistic explanation of how specific Cx43 phosphorylation events actually cause changes in gap-junctional properties is lacking. We and others had previously shown that S372 and/or S373 of Cx43 are phosphorylated (Cooper and Lampe, 2002; Yogo et al., 2006) potentially directly by Akt (Dunn et al., 2012; Park et al., 2007). Therefore, we created an antibody that can detect Cx43 when it is phosphorylated on S373 and mutant constructs that eliminate or mimic S373 phosphorylation. These reagents showed that phosphorylation at S373 regulates gap junction size and is necessary and sufficient to eliminate the interaction of Cx43 with ZO-1 under our experimental conditions.
Here, we present many lines of evidence that prove phosphorylation at S373 is a simple molecular switch that can control the interaction of Cx43 with ZO-1. Mutation of S373 to alanine can eliminate the formation of large junctions, whereas mutation to aspartate to mimic phosphorylation results in larger gap junctions that are refractory to drugs that inhibit Akt and eliminate large junctions. Furthermore, these mutations lead to an increase in or elimination of interaction with ZO-1. The fact that the data are so definitive makes the molecular switch model viable and mechanistically explains the consequences of S373 phosphorylation. In terms of a specific mode of action, different potential connexin phosphorylation events have been structurally modeled in vitro. Specific to S373, Chen and colleagues showed that a phosphopeptide representing the last 12 amino acids of Cx43 bound in vitro to a R193A mutant construct encoding the PDZ2 domain of ZO-1 with a Kd of 7.2 µM and the same peptide substituted with glutamic acid at the S373 equivalent position had several fold lower affinity, whereas other substitutions had less of an effect (Chen et al., 2008). Furthermore, modeling predictions of the interaction indicated that phosphorylation at S373 would significantly weaken the interaction of the end of Cx43 with the ZO-1 PDZ2 domain (Xiao et al., 2011). These calculations are consistent with our coimmunoprecipitation and coimmunolabeling results in cells and indicate that phosphorylation at S373 changes the binding affinity of ZO-1.
Knockdown of ZO-1 and hence reduced interaction with Cx43 results in larger junctions and increased gap-junctional communication (Rhett et al., 2011) as does treatment with proteasomal inhibitors (e.g. Laing et al., 1997). These results raise the question as to whether interaction with ZO-1 affects the assembly process and allows larger junctions to form or whether it blocks their disassembly/turnover. Given that cells expressing S373A mutant Cx43 did not show typical punctate gap-junction-like immunofluorescence staining and exhibited very limited dye transfer, our results would be more consistent with the idea proposed by Rhett and co-workers (Rhett et al., 2011) that ZO-1 interaction inhibits gap junction assembly from connexons. This would imply that Akt phosphorylation of Cx43 occurs, displacing or preventing the ZO-1 interaction, which is consistent with previous reports indicating that Akt (Dunn et al., 2012; Park et al., 2007) and ZO-1 (Hunter et al., 2005) interact with Cx43 at the outer edge of gap junctions. Because our data indicate that the pS373 signal is more concentrated in the central regions of the gap junction and S373 phosphorylation is quite rapid in response to hypoxia, wounding or growth factor treatment, this could imply some significant diffusion of Cx43 within the outer, ‘perinexus’ region of the gap junction (Rhett et al., 2011) when S373 is not phosphorylated, and concentration and stabilization in the middle when it is phosphorylated.
Our data indicated that TPA treatment first leads to increased S373 phosphorylation and increased gap junction size prior to dramatic gap junction turnover. Because imaging studies have shown that larger gap junctions can be turned over by removal of ‘older’ channels from the center of the gap junction plaque (Gaietta et al., 2002; Gumpert et al., 2008), one hypothesis is that phosphorylation on S373 leads to larger gap junctions in order to increase the likelihood of removal by this mechanism. We are currently testing this hypothesis by examining the response of our cell lines expressing mutant Cx43 to TPA treatment. However, these S373D cells contain larger gap junctions and have excellent intercellular communication, therefore it seems unlikely that S373 phosphorylation necessarily and directly leads to gap junction turnover. Our preferred model would involve subsequent Cx43 phosphorylation at S279, S282 and/or S368, consistent with their reported roles in gap junction disassembly (Johnson et al., 2013; Lampe, 1994).
There are several reports that have examined Akt phosphorylation during ischemia or hypoxia. Most involve treatment for longer periods than we used, which show a reduction in Akt phosphorylation (Majmundar et al., 2012); however, shorter-term experiments have shown an increase with a return to basal levels or lower within 2 hours (Mockridge et al., 2000) similar to our results. There are also a few reports of changes in Akt phosphorylation following wounding (Pankow et al., 2006), but few mechanistic conclusions have been drawn. Because gap junctions can control migration and proliferation, we propose that gap junctions might be a major effector of wound-dependent Akt signaling.
Our immunostaining of heart and skin tissues and their response to injury is temporally consistent with the results we obtained in tissue-cultured cells. S373 phosphorylation increased during cardiac ischemia, but the signal primarily remained in larger gap junctions located at the intercalated disc and lateral edges. Akt activation has been shown to preserve cardiac function and limit injury after cardiac ischemia (Matsui et al., 2001; Oshima et al., 2008). One hypothesis consistent with our data would be that hypoxic injury causes activation of PI3K and mTorc2, leading to Akt phosphorylation on S473 and activation and translocation to the membrane. Then, Cx43 phosphorylation at S373 occurs to transiently maximize communication and maintain larger junctions at the intercalated disc. This explanation is consistent with the observations of O'Quinn and colleagues (O'Quinn et al., 2011), who found that treatment of injured hearts with a cell-permeable peptide termed α-CT, which that mimics the C-terminal tail of Cx43 and competes out the interaction of Cx43 with cellular ZO-1 (i.e. similar effect to that of S373 phosphorylation) led to decreased remodeling and arrhythmia. An alternative explanation could be that the increased phosphorylation at S373 is a precursor to Cx43 degradation and/or lateralization. To investigate this further, we aim to develop a more sophisticated ischemia model appropriate for additional treatment, including reperfusion injury.
Upon excisional epidermal wounding, we observed an increase in the amount of phosphorylated S373 at 30 minutes. Although not well defined functionally at this point, Akt has been shown to be activated and important for wound repair (Squarize et al., 2010). Previously, we have shown that gap-junctional communication within the first 6 hours post wounding is crucial for migration of primary human keratinocytes to fill the wounded region (Richards et al., 2004), so S373 phosphorylation might occur to increase communication in this time frame. At 24 hours and longer, several groups have shown that total Cx43 is reduced near the wound margin and that this event promotes more rapid healing (Coutinho et al., 2003; Goliger and Paul, 1995; Lampe et al., 1998), consistent with our results. The Cx43 C-terminal mimetic peptide α-CT also accelerated wound closure (Moore et al., 2013), implying a key regulatory role in the S373 phosphorylation event we observed.
In summary, we have shown that Akt phosphorylation of Cx43 at S373 eliminates ZO-1 binding (the ‘switch’), which allows increased gap junction size and communication. These events are regulated in vivo because S373 phosphorylation increases in myocytes during hypoxia and in keratinocytes in response to wounding. Thus, our data indicate that cells and tissues use S373 phosphorylation to maintain intercellular communication during crucial stages of their response to injury.
MATERIALS AND METHODS
Antibodies, cDNA constructs and other reagents
All general chemicals, unless otherwise noted, were purchased from Fisher Scientific. TPA was purchased from Sigma Chemical (St Louis, MO) and used at 75 nM final concentration. MK2206 and MG132 were purchased from Selleck (Houston, TX) and EMD Biosciences (Gibbstown, NJ), respectively, and both used at 10 µM final concentration. Mouse anti-Cx43 antibodies Cx43IF1 (against a peptide representing amino acids 360–382) and Cx43NT1 (against amino acids 1–20) were prepared at the Fred Hutchinson Cancer Research Center Hybridoma Development Facility (Seattle, WA) and have been described previously (Cooper and Lampe, 2002; Lampe et al., 2006; Sosinsky et al., 2007). We made rabbit anti-pS373 Cx43 antibody by custom commercial preparation (ProSci Inc., Poway, CA; 13 week schedule) against a synthetic peptide that was phosphorylated at a position equivalent to S373, i.e. acetyl-PASS(p)RPRD and linked through a C-terminal cysteine to maleimide-activated KLH (Thermo Scientific, Rockford, IL), and phosphospecific antibody was affinity purified essentially identically to our previously published method (Lampe et al., 2006). Phospho-Akt (Thr308, #2965), Phospho-Akt (Ser473, #4060), and Akt pan (#2920) antibodies were purchased from Cell Signaling (Danvers, MA). Cx43 cDNA with S373A, S373D and S365/369/373A mutations were made using the GeneTailor site-directed mutagenesis system (Invitrogen, Carlsbad, CA) on full-length Cx43 cloned into the mammalian expression vector pIREShyg (Clontech, Mountain View, CA).
Cell line maintenance and transfection
MDCK cells were cultured in DMEM (Mediatech, Pittsburgh PA) supplemented with 10% FCS (Atlanta Biologicals, Lawrenceville, GA) and antibiotics in a humidified 5% CO2 environment. For the 1% O2 experiments, we used a NAPCO Series 8000WJ equipped with regulated nitrogen displacement and oxygen sensor. Wild-type and mutant Cx43 were cloned into the pIRESHyg vector and electroporated into a MDCK cell line lacking Cx43 expression (Jordan et al., 1999) with Nucleofector apparatus (Amaxa Biosystems, Gaithersburg, MD), and cell lines expressing similar levels of Cx43 were dilution cloned and selected using cloning rings. MG132 and MK2206 were added to cells for 3 hours except where indicated. MDCK43 cells were scratch wounded as previously described (Márquez-Rosado et al., 2012; Richards et al., 2004). Briefly, cells were cultured to confluence on 10 cm plates, and the medium was replaced with Opti-MEM I (Invitrogen) reduced serum medium 24 hours before scratching with a 200 ml pipette tip, and the remaining cells were incubated for the time indicated.
Immunoblotting
Cells or cardiac tissue were lysed in sample buffer containing 50 mM NaF, 500 µM Na3VO4, 2 mM PMSF and 1× Complete protease inhibitors (Roche Molecular Biochemicals, Alameda, CA) and cellular proteins were separated by SDS-PAGE (10% polyacrylamide). After electrophoresis, protein was transferred to nitrocellulose, the membrane was blocked, and antibodies were incubated as previously indicated (Lampe et al., 2006). IRDye800 donkey anti-rabbit (Rockland Immunochemicals, Gilbertsville, PA) and Alexa Fluor 680 anti-mouse (Molecular Probes, Inc., Eugene, OR) secondary antibodies were used to simultaneously visualize the phosphospecific and NT1 antibodies, respectively, and were directly quantified using the Li-Cor Biosciences Odyssey infrared imaging system and associated software (Lincoln, NE).
Immunofluorescence, immunohistochemistry and coimmunoprecipitation
Cultured cells were washed twice in PBS, and fixed in cold methanol and acetone (50∶50) for 1 minute followed by a 1 hour block in 1% BSA in PBS. Formalin-fixed cardiac tissue was paraffin embedded and sectioned (4 µm) as previously described (King and Lampe, 2004). Cells or tissue were incubated with a mouse anti-Cx43 antibody (Cx43IF1) or rabbit anti-Cx43 in blocking solution for 1 hour. Following several PBS washes, the cell cultures and skin were incubated with Alexa-Fluor-568-conjugated goat anti-rabbit antibody and/or Alexa-Fluor-488-conjugated goat anti-mouse antibody for 30–60 minutes and counterstained with DAPI (Molecular Probes), followed by several washes in PBS. Heart sections were incubated with Alexa-Fluor-568-conjugated goat anti-rabbit antibody and Alexa-Fluor-647-conjugated goat anti-mouse antibody to avoid the auto-fluorescence associated with paraffin embedded tissue. The coverslips were mounted onto slides with DABCO anti-fade medium [25 mg/ml of 1,4-Diazobicyclo-(2,2,2)octane (Sigma) diluted in 90% glycerol and 10% PBS, pH 8.6] and viewed with a Zeiss LSM 510 laser-scanning fluorescence microscope (cultured cells and heart) or an Applied Precision DeltaVision Elite (Issaquaw WA) system (skin). The image analysis software package Volocity version 6.1 (PerkinElmer, Waltham MA) was used to determine the size of gap junctions in the cell lines. We defined a minimum size object for a gap junction of 0.005 µm3. Using Volocity measurement view, an object volume measurement protocol was built to determine volume, and the results were graphed using Minitab software.
For ZO-1 and Cx43 colocalization studies, cells were grown on #1.5 square coverslips (Fisher Scientific) to 90% confluence and fixed, immunostained and mounted as described above and a DeltaVision OMX system (Applied Precision, Issaquah, WA) was used for imaging the cells. Images were deconvolved using a constrained iterative algorithm (softWoRx 4.1.2; Applied Precision), and Volocity was used to determine the volume of objects for each channel, with an additional task module used to calculate volume of overlap.
Cardiac tissue was collected and ischemic conditions were generated as previously described (Ek-Vitorin et al., 2006). Hearts at the 0 or 30 minute time points were longitudinally bisected and either sonicated in Laemmli sample buffer (for immunoblot analysis) or fixed overnight at 4°C in 10% formalin (for immunohistochemistry). Animal studies were conducted in compliance with the US Department of Health and Human Services Guide for the Care and Use of Laboratory Animals and approved by the FHCRC Institutional Review Board.
Human foreskin tissue was collected under an Institutional Review Board approved protocol and maintained in DMEM and 2× penicillin-streptomycin. For the wounding experiments, a 5 mm seamless disposable biopsy punch (Robbins Instruments, Chatham, NJ) was used to remove a round section of epidermal tissue. The tissue was then incubated at 37°C floating in KBM medium (Lonza). After 30 minutes, the tissue was bisected through the wound, mounted in OCT, frozen, and 10 µm sections were cut on a cryostat. After sections were cut, they were placed under vacuum overnight. Prior to staining, they were fixed with cold 50∶50 methanol and acetone, washed three times in PBS and blocked with 1% BSA. The sections were labeled and processed for Cx43 and pS373 immunofluorescence as described above. A DeltaVision Elite system using an Olympus IX71 microscope was used to collect images at the indicated wavelengths using 0.2 µm optical sections that were subsequently deconvolved (softWoRx 4.1.2) and examined using ImageJ (Schneider et al., 2012).
Coimmunoprecipitation experiments were performed as described previously (Cooper and Lampe, 2002; Lampe et al., 2006; Solan et al., 2007). Briefly, cells were lysed in immunoprecipitation buffer (0.5% deoxycholate, 0.5% Triton X-100, 100 mM NaCl, 10 mM EDTA, 50 mM NaF, 500 µM Na3VO4, 2 mM PMSF, and 1× Complete Protease inhibitor, 50 mM Tris-HCl, pH 7.2) and pre-cleared. Lysates were incubated with IF1 Cx43 antibody for 30 minutes followed by the addition of Protein-G beads for 2.5 hours, washed three times with cold immunoprecipitation assay buffer, and eluted with sample buffer prior to SDS-PAGE and immunoblotting, as described above.
FRAP measurement of gap-junctional communication
FRAP experiments were carried out using an Ultraview Vox (PerkinElmer) on a Nikon Eclipse Ti platform. Cells were grown on a 35/23 mm glass bottom FluoroDish (World Precision Instruments, Inc., Sarasota, FL) to 85–95% confluence, washed in PBS, and incubated with Calcein-AM (0.5 µM; Invitrogen) in Opti-MEM (Invitrogen) for 30 minutes. The cells were then washed three times with PBS and maintained in Opti-MEM for the remainder of the experiment. Using a 60× objective and the 488 nm laser line, individual cells were selected for photobleaching. The interactive software for the microscope (Volocity) was used to identify the selected cells to bleach and to input the design of the FRAP experiment. We captured two images per second for 10 seconds at baseline followed by photobleaching of individual cells at 4% laser excitation. Recovery of fluorescence was followed for 300 seconds with images captured every 5 seconds using a Hamamatsu C9100-13 camera and 100 ms exposure time. The integrated intensity of each selected cell in each image was exported to Excel where the fluorescence intensity signal of selected cells in the last image prior to photobleaching was normalized to 100% and the first image following photobleaching was normalized to 0%. Recovery was calculated based on the fluorescence intensity in the photobleached cell at each time point relative to the fluorescence intensity of the same cell at the same region before the bleaching and expressed as percentage recovery.
Statistics
Except where specifically indicated otherwise, results are reported as mean ± s.e.m. with significance expressed as the P-value calculated using a standard two-sided Student's t-test.
Acknowledgments
We thank Leanna Ferrand at GE Healthcare for her assistance with the DeltaVision OMX imaging.
Footnotes
Competing interests
The authors declare no competing financial interests.
Author contributions
C.A.D. performed the majority of experiments. P.D.L. developed the phosphospecific antibody, led the project and jointly wrote the manuscript and analysed the data with C.A.D.
Funding
This work was funded by the National Institutes of Health [grant number R01GM055632]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Deposited in PMC for release after 12 months.
References
- Beardslee M. A., Lerner D. L., Tadros P. N., Laing J. G., Beyer E. C., Yamada K. A., Kléber A. G., Schuessler R. B., Saffitz J. E. (2000). Dephosphorylation and intracellular redistribution of ventricular connexin43 during electrical uncoupling induced by ischemia. Circ. Res. 87, 656–662 10.1161/01.RES.87.8.656 [DOI] [PubMed] [Google Scholar]
- Bergoffen J., Scherer S. S., Wang S., Scott M. O., Bone L. J., Paul D. L., Chen K., Lensch M. W., Chance P. F., Fischbeck K. H. (1993). Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science 262, 2039–2042 10.1126/science.8266101 [DOI] [PubMed] [Google Scholar]
- Chen J., Pan L., Wei Z., Zhao Y., Zhang M. (2008). Domain-swapped dimerization of ZO-1 PDZ2 generates specific and regulatory connexin43-binding sites. EMBO J. 27, 2113–2123 10.1038/emboj.2008.138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper C. D., Lampe P. D. (2002). Casein kinase 1 regulates connexin-43 gap junction assembly. J. Biol. Chem. 277, 44962–44968 10.1074/jbc.M209427200 [DOI] [PubMed] [Google Scholar]
- Coutinho P., Qiu C., Frank S., Tamber K., Becker D. (2003). Dynamic changes in connexin expression correlate with key events in the wound healing process. Cell Biol. Int. 27, 525–541 10.1016/S1065--6995(03)00077--5 [DOI] [PubMed] [Google Scholar]
- Dunn C. A., Su V., Lau A. F., Lampe P. D. (2012). Activation of Akt, not connexin 43 protein ubiquitination, regulates gap junction stability. J. Biol. Chem. 287, 2600–2607 10.1074/jbc.M111.276261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ek-Vitorin J. F., King T. J., Heyman N. S., Lampe P. D., Burt J. M. (2006). Selectivity of connexin 43 channels is regulated through protein kinase C-dependent phosphorylation. Circ. Res. 98, 1498–1505 10.1161/01.RES.0000227572.45891.2c [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaietta G., Deerinck T. J., Adams S. R., Bouwer J., Tour O., Laird D. W., Sosinsky G. E., Tsien R. Y., Ellisman M. H. (2002). Multicolor and electron microscopic imaging of connexin trafficking. Science 296, 503–507 10.1126/science.1068793 [DOI] [PubMed] [Google Scholar]
- Goliger J. A., Paul D. L. (1995). Wounding alters epidermal connexin expression and gap junction-mediated intercellular communication. Mol. Biol. Cell 6, 1491–1501 10.1091/mbc.6.11.1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumpert A. M., Varco J. S., Baker S. M., Piehl M., Falk M. M. (2008). Double-membrane gap junction internalization requires the clathrin-mediated endocytic machinery. FEBS Lett. 582, 2887–2892 10.1016/j.febslet.2008.07.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter A. W., Barker R. J., Zhu C., Gourdie R. G. (2005). Zonula occludens-1 alters connexin43 gap junction size and organization by influencing channel accretion. Mol. Biol. Cell 16, 5686–5698 10.1091/mbc.E05--08--0737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K. E., Mitra S., Katoch P., Kelsey L. S., Johnson K. R., Mehta P. P. (2013). Phosphorylation on Ser-279 and Ser-282 of connexin43 regulates endocytosis and gap junction assembly in pancreatic cancer cells. Mol. Biol. Cell 24, 715–733 10.1091/mbc.E12--07--0537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan K., Solan J. L., Dominguez M., Sia M., Hand A., Lampe P. D., Laird D. W. (1999). Trafficking, assembly, and function of a connexin43-green fluorescent protein chimera in live mammalian cells. Mol. Biol. Cell 10, 2033–2050 10.1091/mbc.10.6.2033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelsell D. P., Dunlop J., Stevens H. P., Lench N. J., Liang J. N., Parry G., Mueller R. F., Leigh I. M. (1997). Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature 387, 80–83 10.1038/387080a0 [DOI] [PubMed] [Google Scholar]
- King T. J., Lampe P. D. (2004). The gap junction protein connexin32 is a mouse lung tumor suppressor. Cancer Res. 64, 7191–7196 10.1158/0008--5472.CAN--04--0624 [DOI] [PubMed] [Google Scholar]
- Laing J. G., Tadros P. N., Westphale E. M., Beyer E. C. (1997). Degradation of connexin43 gap junctions involves both the proteasome and the lysosome. Exp. Cell Res. 236, 482–492 10.1006/excr.1997.3747 [DOI] [PubMed] [Google Scholar]
- Laird D. W. (2006). Life cycle of connexins in health and disease. Biochem. J. 394, 527–543 10.1042/BJ20051922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird D. W. (2010). The gap junction proteome and its relationship to disease. Trends Cell Biol. 20, 92–101 10.1016/j.tcb.2009.11.001 [DOI] [PubMed] [Google Scholar]
- Lampe P. D. (1994). Analyzing phorbol ester effects on gap junctional communication: a dramatic inhibition of assembly. J. Cell Biol. 127, 1895–1905 10.1083/jcb.127.6.1895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampe P. D., Nguyen B. P., Gil S., Usui M., Olerud J., Takada Y., Carter W. G. (1998). Cellular interaction of integrin α3β1 with laminin 5 promotes gap junctional communication. J. Cell Biol. 143, 1735–1747 10.1083/jcb.143.6.1735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampe P. D., Cooper C. D., King T. J., Burt J. M. (2006). Analysis of Connexin43 phosphorylated at S325, S328 and S330 in normoxic and ischemic heart. J. Cell Sci. 119, 3435–3442 10.1242/jcs.03089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leithe E., Rivedal E. (2004). Epidermal growth factor regulates ubiquitination, internalization and proteasome-dependent degradation of connexin43. J. Cell Sci. 117, 1211–1220 10.1242/jcs.00951 [DOI] [PubMed] [Google Scholar]
- Leykauf K., Salek M., Bomke J., Frech M., Lehmann W. D., Dürst M., Alonso A. (2006). Ubiquitin protein ligase Nedd4 binds to connexin43 by a phosphorylation-modulated process. J. Cell Sci. 119, 3634–3642 10.1242/jcs.03149 [DOI] [PubMed] [Google Scholar]
- Majmundar A. J., Skuli N., Mesquita R. C., Kim M. N., Yodh A. G., Nguyen-McCarty M., Simon M. C. (2012). O(2) regulates skeletal muscle progenitor differentiation through phosphatidylinositol 3-kinase/AKT signaling. Mol. Cell. Biol. 32, 36–49 10.1128/MCB.05857--11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Márquez-Rosado L., Singh D., Rincón-Arano H., Solan J. L., Lampe P. D. (2012). CASK (LIN2) interacts with Cx43 in wounded skin and their coexpression affects cell migration. J. Cell Sci. 125, 695–702 10.1242/jcs.084400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T., Tao J., del Monte F., Lee K. H., Li L., Picard M., Force T. L., Franke T. F., Hajjar R. J., Rosenzweig A. (2001). Akt activation preserves cardiac function and prevents injury after transient cardiac ischemia in vivo. Circulation 104, 330–335 10.1161/01.CIR.104.3.330 [DOI] [PubMed] [Google Scholar]
- Mockridge J. W., Marber M. S., Heads R. J. (2000). Activation of Akt during simulated ischemia/reperfusion in cardiac myocytes. Biochem. Biophys. Res. Commun. 270, 947–952 10.1006/bbrc.2000.2522 [DOI] [PubMed] [Google Scholar]
- Moore K., Bryant Z. J., Ghatnekar G., Singh U. P., Gourdie R. G., Potts J. D. (2013). A synthetic connexin 43 mimetic peptide augments corneal wound healing. Exp. Eye Res. 115, 178–188 10.1016/j.exer.2013.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Quinn M. P., Palatinus J. A., Harris B. S., Hewett K. W., Gourdie R. G. (2011). A peptide mimetic of the connexin43 carboxyl terminus reduces gap junction remodeling and induced arrhythmia following ventricular injury. Circ. Res. 108, 704–715 10.1161/CIRCRESAHA.110.235747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima Y., Ouchi N., Sato K., Izumiya Y., Pimentel D. R., Walsh K. (2008). Follistatin-like 1 is an Akt-regulated cardioprotective factor that is secreted by the heart. Circulation 117, 3099–3108 10.1161/CIRCULATIONAHA.108.767673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankow S., Bamberger C., Klippel A., Werner S. (2006). Regulation of epidermal homeostasis and repair by phosphoinositide 3-kinase. J. Cell Sci. 119, 4033–4046 10.1242/jcs.03175 [DOI] [PubMed] [Google Scholar]
- Park D. J., Wallick C. J., Martyn K. D., Lau A. F., Jin C., Warn-Cramer B. J. (2007). Akt phosphorylates Connexin43 on Ser373, a “mode-1” binding site for 14-3-3. Cell Commun. Adhes. 14, 211–226 10.1080/15419060701755958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paznekas W. A., Boyadjiev S. A., Shapiro R. E., Daniels O., Wollnik B., Keegan C. E., Innis J. W., Dinulos M. B., Christian C., Hannibal M. C. et al. (2003). Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. Am. J. Hum. Genet. 72, 408–418 10.1086/346090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhett J. M., Jourdan J., Gourdie R. G. (2011). Connexin 43 connexon to gap junction transition is regulated by zonula occludens-1. Mol. Biol. Cell 22, 1516–1528 10.1091/mbc.E10--06--0548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards T. S., Dunn C. A., Carter W. G., Usui M. L., Olerud J. E., Lampe P. D. (2004). Protein kinase C spatially and temporally regulates gap junctional communication during human wound repair via phosphorylation of connexin43 on serine368. J. Cell Biol. 167, 555–562 10.1083/jcb.200404142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruch R. J., Trosko J. E., Madhukar B. V. (2001). Inhibition of connexin43 gap junctional intercellular communication by TPA requires ERK activation. J. Cell. Biochem. 83, 163–169 10.1002/jcb.1227 [DOI] [PubMed] [Google Scholar]
- Scemes E., Suadicani S. O., Dahl G., Spray D. C. (2007). Connexin and pannexin mediated cell-cell communication. Neuron Glia Biol. 3, 199–208 10.1017/S1740925X08000069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C. A., Rasband W. S., Eliceiri K. W. (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz R., Gres P., Skyschally A., Duschin A., Belosjorow S., Konietzka I., Heusch G. (2003). Ischemic preconditioning preserves connexin 43 phosphorylation during sustained ischemia in pig hearts in vivo. FASEB J. 17, 1355–1357 [DOI] [PubMed] [Google Scholar]
- Sirnes S., Leithe E., Rivedal E. (2008). The detergent resistance of Connexin43 is lost upon TPA or EGF treatment and is an early step in gap junction endocytosis. Biochem. Biophys. Res. Commun. 373, 597–601 10.1016/j.bbrc.2008.06.095 [DOI] [PubMed] [Google Scholar]
- Söhl G., Willecke K. (2004). Gap junctions and the connexin protein family. Cardiovasc. Res. 62, 228–232 10.1016/j.cardiores.2003.11.013 [DOI] [PubMed] [Google Scholar]
- Solan J. L., Lampe P. D. (2009). Connexin43 phosphorylation: structural changes and biological effects. Biochem. J. 419, 261–272 10.1042/BJ20082319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solan J. L., Marquez-Rosado L., Sorgen P. L., Thornton P. J., Gafken P. R., Lampe P. D. (2007). Phosphorylation at S365 is a gatekeeper event that changes the structure of Cx43 and prevents down-regulation by PKC. J. Cell Biol. 179, 1301–1309 10.1083/jcb.200707060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosinsky G. E., Solan J. L., Gaietta G. M., Ngan L., Lee G. J., Mackey M. R., Lampe P. D. (2007). The C-terminus of connexin43 adopts different conformations in the Golgi and gap junction as detected with structure-specific antibodies. Biochem. J. 408, 375–385 10.1042/BJ20070550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squarize C. H., Castilho R. M., Bugge T. H., Gutkind J. S. (2010). Accelerated wound healing by mTOR activation in genetically defined mouse models. PLoS ONE 5, e10643 10.1371/journal.pone.0010643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao F., Weng J., Fan K., Wang W. (2011). Detailed regulatory mechanism of the interaction between ZO-1 PDZ2 and connexin43 revealed by MD simulations. PLoS ONE 6, e21527 10.1371/journal.pone.0021527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yogo K., Ogawa T., Akiyama M., Ishida-Kitagawa N., Sasada H., Sato E., Takeya T. (2006). PKA implicated in the phosphorylation of Cx43 induced by stimulation with FSH in rat granulosa cells. J. Reprod. Dev. 52, 321–328 10.1262/jrd.17107 [DOI] [PubMed] [Google Scholar]
- Yum S. W., Zhang J., Valiunas V., Kanaporis G., Brink P. R., White T. W., Scherer S. S. (2007). Human connexin26 and connexin30 form functional heteromeric and heterotypic channels. Am. J. Physiol. 293, C1032–C1048 10.1152/ajpcell.00011.2007 [DOI] [PubMed] [Google Scholar]