

Abstract
Adoptive transfer of antigen-specific T cells is a compelling tool to treat cancer. To overcome issues of immune tolerance which limits the endogenous adaptive immune response to tumor-associated antigens, robust systems for the genetic modification and characterization of T cells expressing chimeric antigen receptors (CARs) to redirect specificity have been produced. Refinements with regards to persistence and trafficking of the genetically modified T cells are underway to help improve the potency of genetically modified T cells. Clinical trials utilizing this technology demonstrate feasibility, and increasingly, antitumor activity, paving the way for multi-center trials to establish the efficacy of this novel T-cell therapy.
2. Introduction
Allogeneic hematopoietic stem-cell transplantation (HSCT) cures a substantial portion of patients with hematological malignancies who are refractory to conventional chemotherapy, and underscores the powerful therapeutic effect of the T-cell immune response in controlling advanced disease. Polyclonal (non-targeted) T-cell therapy in the form of donor lymphocyte infusion (DLI) following HSCT has been used to effectively treat relapse of slow-growing malignancies in a subset of patients (1-7). However, disease relapse and graft-versus-host-disease (GVHD) following HSCT and DLI illustrate the two most significant limitations of non-directed cellular therapy, namely, immune evasion of the tumor leading to relapse, and on-target effects in which donor-derived T cells target major or minor histocompatibility antigens leading to GVHD.
To achieve remission, infused T cells must recognize and eliminate tumor cells that have arisen in the immunocompetent host and that have evolved a range of passive and active immune evasion strategies to avoid immunemediated destruction. Passive evasion strategies include the emergence of tumor escape variants that have lost the targeted tumor-associated antigen (TAA) such as described in a report by Vago and colleagues(8). In 5 of 17 patients who relapsed with acute myeloid leukemia following haploidentical HSCT, they identified antigen-loss variants of the original leukemic cells in which a region of chromosome 6 encoding the mismatched human leukocyte antigen (HLA) haplotype was deleted, with consequent loss of the tumor target for the donor T cells (8). Active evasion strategies are exemplified by the ability of tumors to adversely modulate the tumor microenvironment that impair T-cell effector functions, such as through secretion of TGFbeta (9).
The antigenic similarities of many tumors limit immune-mediated recognition and clearance by T cells. Many TAA expressed in the tumor microenvironment are self-antigens and endogenous T cells are tolerant due to the lack of their recognition of, or activation by, TAA. Investigators have used genetic tools to overcome the limitation of immune tolerance by genetically modifying T cells to express transgenic T-cell receptor (TCR) alpha and beta chains that recognize TAA in context of human leukocyte antigen (HLA), or by expressing a single-chain chimeric antigen receptor (CAR) to redirect T-cell specificity to a TAA expressed on the cell surface independent of HLA(10, 11). In this review, we focus on the design and implementation of CARs.
3. The CAR structure
The prototypical CAR uses a mouse monoclonal antibody (mAb) that docks with a designated cell-surface TAA triggering desired T-cell activation and effector functions. The specificity of a CAR is achieved by its exodomain which is typically derived from the antigen-binding motif from a mAb that links VH with VL sequences to construct a single-chain fragment variable (scFv) region. In the event that the TAA is itself a receptor, exodomains of CARs have also been fashioned from ligands or peptides (e.g. cytokines) to redirect specificity to receptors (e.g. cytokine receptors), such as the IL-13Ralpha2–specific “zetakine” (12). The exodomain is completed by the inclusion of a flexible (hinge), such as from CD8α or immunoglobulin(13, 14) and is expressed on the T-cell surface via a transmembrane domain. Upon binding TAA, the CAR activates T cells via an endodomain which typically includes cytoplasmic domains from CD3 or high-affinity receptor FcεRI (15-17). The docking of CAR to TAA ideally provides the genetically modified T cell with a fully-competent activation signal, minimally defined as CAR-dependent killing, proliferation, and cytokine production. Specific effector functions can be engineered by the design of CARs, such as the inclusion of more than one chimeric activation domain. Thus, iterative modifications to the CAR have resulted in first-, second-, and third-generation CARs designed with one, two, or three signaling motifs within an endodomain (Figure 1) that include cytoplasmic signaling motifs derived from CD28, CD134, CD137, Lck, ICOS, and DAP10(14, 18-20).
Figure 1. Descriptions of prototypical CARs and genetically modified T cells from which they are expressed.
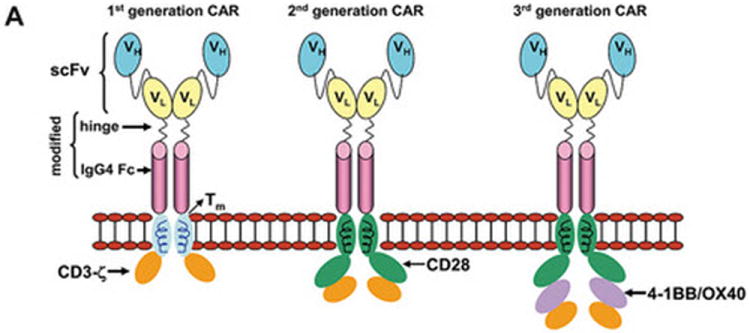
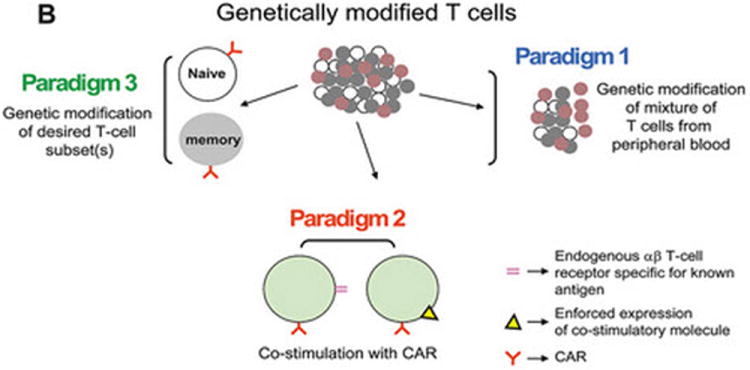
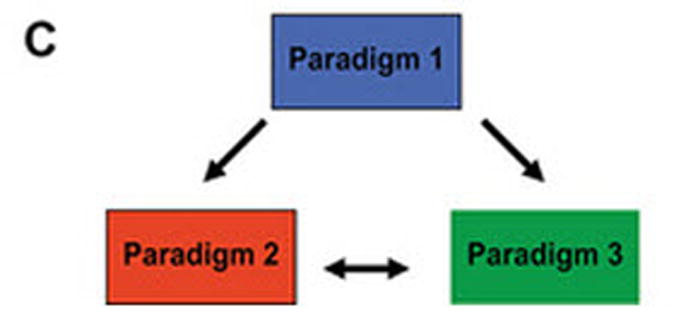
(A) Dimerized CARs demonstrating the extracellular scFv (VH linked to VL) via a linker) region, linked to a flexible hinge and Fc region (for example, from IgG4) fused to intracellular signaling motifs via a transmembrane domain. The 1st generation CAR is shown as activating T cells through an endodomain composed of only CD3-ζ. The 2nd generation CAR activates T cells through chimeric CD3-ζ and CD28. The 3rd generation CAR activates T cells through three signaling motifs, e.g., CD3-ζ with CD28 and CD134 or CD137. The modular structure of the extracellular and intracellular domains can be readily altered to achieve fully competent CAR-dependent signaling. (B) Paralleling the design changes to CAR is an understanding that the type of T cell into which the CAR is expressed can impact the therapeutic potential of adoptive immunotherapy. Paradigm 1 refers to the collection of (naïve, memory, effector) T cells in peripheral blood which can be genetically modified to express just the CAR without further manipulation. Paradigm 2 generates CAR+ T cells that can signal with other desired receptor(s) such as endogenous αβ TCR or introduced co-stimulatory molecule(s). Paradigm 3 expresses the CAR in desired T-cell subsets such as naïve or central memory. (C) The three proposed paradigms are not mutually exclusive of each other as T cells from peripheral blood can be used as cellular templates for Paradigms 2 and 3. Clinical trials are needed to determine whether the CAR design or the type of T cell from which the CAR is expressed, or both, will result in superior persistence and anti-tumor response.
Implicit in the design of CARs is the desire by investigators to improve the survival of adoptively transferred T cells, as their persistence correlates with their therapeutic potential. While the optimal CAR design remains to be determined, results from early clinical trials appear to indicate that 1st generation technology, in which a CAR signals solely through immunoreceptor tyrosine-based activation motif (ITAM) domains on CD3-zeta, is unlikely to sustain the in vivo persistence of T cells in most patients (21-25). Second-generation CARs, which have signaling domains in addition to CD3-zeta coupled to other co-stimulatory molecules, have improved T-cell effector functioning(21, 26, 27). For this reason, most clinical trials infusing CAR+ T cells at this point are using the 2nd generation CAR design. Indeed, Kochenderfer and colleagues treated a patient with advanced follicular lymphoma with fludarabine and cyclophosphamide preconditioning followed by infusion of autologous T cells modified via gamma retrovirus transduction to express a second generation CAR that recognized CD19 (28). Significant regression of the lymphoma was noted, which could be attributed to the chemotherapy and/or T cell infusion. Importantly, B-cell precursors were selectively eliminated from the patient’s bone marrow and absent in the peripheral blood for at least 39 weeks following infusion of the modified T cell suggesting prolonged activity and efficacy of the modified T cell, rather than chemotherapy. Third-generation CARs that include a combination of co-stimulatory endodomains (e.g. CD28 and CD137) (14) are being tested in a limited number of clinical trials. Newer strategies to enhance T cell signaling following ligation of the CAR with the TAA (beyond the current 2nd generation design) will likely need rigorous safeguards to prevent excessive toxicity as these T cells may be capable of synchronous and supra-physiologic signaling which could lead to adverse events (29).
To improve the targeting of TAA, the avidity of the genetically modified T cell for TAA may be altered by changes to the scFv to improve functional affinity based on selecting high-affinity binding variants from phage arrays(30, 31). The approach to developing CAR+ T cells with a calibrated increase in functional affinity may be necessary to enable genetically modified T cells to target tumors with low levels of TAA expression or perhaps to target a cell-surface molecule in the presence of soluble antigen(32, 33). Other modifications to the scFv include reducing potential immunogenicity by using humanized scFv regions, for example to target carcinoembryonic antigen (CEA) (34) and ERBB2 (14). It is anticipated that these humanized CARs may avoid immune-mediated recognition leading to elimination of the genetically modified T cells. However, a benefit for using humanized scFv regions is yet to be established in the clinical setting.
4. Approaches to genetic modification of T cells to express CAR
Approaches to the genetic manipulation of T cells for the introduction of CAR transgene have mostly relied on transduction using recombinant retrovirus. As an alternative, we and others are investigating the clinical potential of non-viral approaches to gene transfer. The different approaches to the expression of transgenes are summarized in two recent reviews by June and Jena(35, 36). Recombinant retroviral systems can efficiently and stably genetically modify populations of T cells with the inherent goal to shorten their in vitro time to production and release, since prolonged time in culture can lead to terminal differentiation and replication senescence (37-39) (40-42). Despite the theoretical risk for insertional mutagenesis there has been no documented genotoxicity or tumor induction attributed to T cells genetically modified with retrovirus. However, for many investigators recombinant clinical grade retroviruses are often cumbersome and expensive to manufacture requiring specialized facilities and personnel skilled in current good manufacturing practice (cGMP). Nevertheless, the retroviral transduction systems have been most extensively studied and validated in the clinical setting.
As an alternative to transduction, electroporation has been adapted as an approach to the nonviral gene transfer of DNA plasmids to generate CAR+ T cells (24, 43, 44). Early clinical data demonstrates the feasibility and safety of infusing autologous CAR+ T cells in patients with lymphoma and such T cells have been attributed to have an antitumor effect (22, 23). The electrotransfer and integration of naked plasmid DNA into T cells is considered inefficient because it depends on illegitimate recombination for stable genomic insertion of nonviral sequences. As a result, lengthy in vitro culturing times were required to select for stably transfected T cells, leading to senescence of some of the T cells and decreased efficacy (45, 46). The efficiency of integration can be greatly improved leading to shortened time in tissue culture using transposon and transposase systems such as derived from Sleeping Beauty (SB) (43, 47, 48) and piggyBac (49, 50) to stably introduce CAR from electrotransferred DNA plasmids (44, 49, 51-55). The electroporation of T cells in compliance with cGMP with clinical-grade DNA plasmids is less costly compared with retrovirus systems. We have shown that the SB system can be used to introduce CAR and other transgenes into primary human T cells with approximately 60-fold improved integration efficiency, compared with electrotransfer of DNA transposon plasmid without transposase (47). After electroporation, T cells can be rapidly expanded in a CAR-dependent manner by recursive culture on γ-irradiated artificial antigen-presenting cells (aAPC) achieving clinically sufficient numbers of cells for infusion within 3 to 4 weeks after electroporation.
5. Improved persistence of CAR+ T cells
Limited survival of infused genetically modified T cells has been noted in many of the initial Phase I trials described in Table 1. While these trials were not powered for efficacy, the lack of long-term persistence of the infused T cells likely contributes to the lack of major clinical responses reported from these studies. The reasons for poor persistence are (i) incomplete T-cell activation through the CAR, (ii) diminished proliferative capacity of the T-cell sub-population into which CAR was inserted, (iii) unfavorable environment into which T cells are infused and/or home to, and (iv) immune response by the recipient leading to clearance of infused T cells. As previously mentioned, the CAR design has been modified to improve the ability of T cells to undergo a fully competent activation signal to ultimately improve efficacy (Figure 1).
Table 1.
Completed clinical trials with CAR+ T cells
| 1st Author, Year | Type of T cell | CAR construct | Cell Dose (T cells/m2) | Cancer/No. Pt. | Serious Adverse Events | Persistence | Anti-tumor response |
|---|---|---|---|---|---|---|---|
| Park, 2007 (24) | OKT3 activated T cells | CE7R-1st gen. CAR plasmid with HyTK, nonviral transduction | 108 - 109 | Neuro-blastoma/ 6 | None | 1-42 days | 1/6 PR |
| Kershaw, 2008 (65) | OKT3/allo-antigen activated T cells | α-folate receptor, 1st gen. CAR, retroviral vector | 3 × 109 - 5 × 1010 (OKT3) 4 × 109-1.69 × 1011 (alloantigen) | Ovarian/ 14 | None | Up to 3 wks (OKT3)12 mo. in one patient (alloantigen) | None reported |
| Till, 2008 (23) | OKT3 activated T cells | CD20, 1st gen, CAR, nonviral transduction | 108 - 3.3 × 109 | CD20+ NHL/7 | None | 1-3 wks (clones) 5-9 wks (lines+ IL2) | 5 eval: 4 SD, 1 PR |
| Pule, 2008 (25) | OKT3 activated T cells and EBV-specific CTLs | GD2-CAR retroviral vector | 2 × 107 - 2 × 108 of each product | Neuro-blastoma/ 11 | None | Up to 3 wks for activated T cells; up to 6 mo. CTLs | 8 eval: 4 response with 1 CR |
| Lamers, 2010 (64) | G250-1st gen. CAR, retroviral vector | 0.38 - 2.13 × 109 | Renal/ 11 | Liver | Up to 53 days | None reported | |
| Kochen-derfer, 2010 (28) | CD19, 2nd gen. CAR, retroviral vector | 4 × 108 in 2 divided doses | Follicular/1 | Absent Ig | 27 wks | PR |
Additionally, there is increasing awareness that the type of T cell into which the CAR is expressed impacts the ability of the T cell to proliferate and survive after adoptive transfer. Initially, pools of T cells directly obtained from peripheral blood were genetically modified to express CAR (Figure 1, Paradigm 1). However, sub-sets of T cells may be selected for improved persistence and thus improved therapeutic effect (Figure 2, Paradigm 2). These T cells signal through an additional receptor, for example, via an endogenous alpha-beta TCR with specificity for known antigen (e.g. viral antigen or allo-antigen)(25, 56, 57) or via enforced expression of a co-stimulatory molecule such as CD80 and CD137L (58). Triggering such TCRs in vivo can lead to enhanced T-cell proliferation thereby improving antitumor effect delivered by the introduced CAR. This was recently demonstrated in a report by Pule and colleagues describing patients infused with autologous bispecific T cells that recognized EBV-specific antigens via the endogenous alpha-betaTCR and the disialoganglioside antigen GD2 on neuroblastoma cells via introduced CAR. The EBV-specific CAR+ T cells had prolonged persistence in vivo compared with CAR+ T cells for which the specificity of the TCR was not known (25). Moreover, the CAR can be expressed on naive and memory T cells that were pre-selected prior to genetic modification in order to generate a desired population of T cells with an enhanced ability to persist after adoptive transfer (Figure 1, Paradigm 3)(59, 60). A debate remains regarding the preferred T-cell phenotype. For example, infusion of central memory T cells persisted to a greater extent than more differentiated T cells in the non-human primate model (61), whereas infusion of naive T cells were more long lived in a murine model (59).
Another strategy to improve the survival of T cells is to deplete lymphocytes in the recipient prior to the infusion of the genetically modified T cells. The efficacy of this strategy is demonstrated by the adoptive transfer of ex vivo expanded tumor-infiltrating lymphocytes (TIL) after iatrogenic lymphodepletion in patients with advanced melanoma (62). Sustained persistence of genetically modified T cells has also been observed when patients received lymphodepleting chemotherapy prior to T-cell infusion (28) (23). Infused T cells may proliferate more efficiently in the lymphopenic host through homeostatic mechanisms mediated by the removal of regulatory and suppressor cells, and increased availability of cytokines, while immune responses that develop against the CAR may be attenuated.
Clinical data reveals that the recipient can recognize immunogenic transgenes which might lead to immune-mediated clearance of infused genetically modified T cells. As initially reported by Lamers and colleagues in 2006 (63), and recently updated (64), 11 patients with metastatic renal cell carcinoma (RCC) received multiple doses (without prior lymphodepletion) of autologous T cells modified via retrovirus transduction to express a 1st generation CAR specific for carbonic anhydrase IX (CAIX), that is over-expressed on RCC. Humoral and cellular anti-CAIX-CAR T-cell immune responses were noted that were associated with limited persistence of the transferred T cells (64). Similarly, development of anti-CAR humoral immunity was thought to curtail persistence of CAR+ T cells in the study reported by Kershaw and colleagues, in which 14 patients with ovarian cancer received autologous T cells modified via retroviral transduction to express a 1st generation CAR specific for the ovarian cancer-associated antigen α-folate receptor (65). Patients received a single dose of T cells with or without IL-2, without prior lymphodepletion (65). Immune responses to the CAR may be avoided using a humanized CAR (66) (31). However, the vector itself may be a target for an immune response as two patients developed immunity directed against the retroviral vector epitopes in the study reported by Lamers and colleagues (64). Immune responses to epitopes encoded by the viral vector may be avoided by using non-viral, electroporation techniques to introduce DNA plasmids to express CAR.
The ability to genetically modify T cells with different extracellular receptors and intracellular signaling domains also offers an opportunity to engineer for improved persistence. Investigators have enforced expression of cytokines that signal through the common γ-cytokine receptor, to replace the dependence of T cells on exogenous cytokines for survival. For example, animal experiments (67) as well as clinical experience (68) have shown that long-lived T cells are associated with expression of the IL-7Rα chain (CD127), but genetically modified and propagated T cells tend to down-regulate this cytokine receptor. Therefore, to enhance the ability of T cells to respond to the pro-survival cytokine IL-7, investigators have enforced the expression of IL-7Rα to demonstrate the improved survival of genetically modified EBV-specific T cells in an animal model (69). A novel membrane-bound variant of IL-7 when co-expressed on the cell surface improved persistence of CAR+ T cells (Hurton ASH 2009). Furthermore, T-cell over-expression of receptors for IL-2 and IL-15, as well as enforced expression of cytokines for secretion, have improved persistence of T cells in vitro (70) (71) (72-74) (75). However, when this approach was tested in a clinical trial infusing TIL genetically modified to constitutively secrete recombinant IL-2, the persistence of the adoptively transferred T cells was not improved compared with genetically unmodified TIL (76). The reasons are likely multifactorial, but the authors concluded that extensive manipulation and prolonged culture of the T cells, resulting in shortened telomere length, likely contributed significantly to the truncated persistence of the modified T cells. Cytokine receptors have also been modified in T cells to improve their ability for effector functioning within a tumor-suppressive environment. For example, investigators have introduced a dominant-negative receptor for TGFβ receptor to enable genetically modified T cells to resist the suppressive effects of this cytokine (77). Recognizing that the tumor microenvironment contains regulatory T cells, the CAR signaling motif has been adapted to resist the suppressive effects of these cells by the expression of chimeric CD28 (78).
6. Improved trafficking of CAR+ T cells
To effectively penetrate the tumor, genetically modified T cells must home to the sites of malignancy. Migration may be compromised by the loss of desired chemokine receptors during genetic modification and passage ex vivo, or may result from the selection of T cells that are inherently unable to localize to certain tissues. Panels of tissue-specific homing receptors which are typically composed of integrins, chemokines, and chemokine receptors are associated with T-cell migration to anatomic sites of malignancy. Therefore, flow cytometry can be used to describe the potential migration of the T cells that will be infused (79, 80). Whether specific subsets of T cells expressing the desired endogenous homing receptors can be genetically modified to express CAR is unclear. As a result, investigators are manipulating the homing potential of T cells through the enforced expression of chemokine receptors such as CCR4 (81).
7. Non-invasive imaging of CAR+ T cells
The ability to genetically modify T cells also provides investigators with a platform to express other transgenes such as those that permit noninvasive imaging by positron emission tomography (PET). The ability to visualize trafficking and localization of the modified T cells would provide significant biological information. One imaging transgene coexpressed with CAR is thymidine kinase (TK) (and associated TK mutants) from herpes simplex virus-1 (HSV-1) (82) which can be used to enzymatically trap radioactive substrates within the cytoplasm to image the trafficking of T cells by PET (83, 84). Furthermore, expression of TK also renders CAR+ T cells sensitive to conditional ablation using ganciclovir (85, 86). The potential immunogenicity of using viral-derived TK may be overcome by enforcing the expression of mitochondrial human TK in the cytoplasm (87). Recently, another naturally occurring human gene has been adapted for PET. Deoxycytidine kinase (dCK) has been imaged with designer PET probes and dCK itself has been redesigned for efficient uptake of PET probes which are currently in use (88, 89).
8. Safety of CAR+ T cells
While the safety profile of adoptive transfer of non-modified ex vivo propagated autologous T cells is established (90), there are several potential concerns with the use of engineered lymphocytes. Firstly, toxicity attributable to undesired, on-target effects of the transgene has been observed (63, 64, 91). Significant hepatic toxicity necessitating discontinuation of treatment was seen in one patient, and dose reduction in 2 patients were required after infusion of T cells engineered to express CAR specific for CAIX. While CAIX is over-expressed on RCC cells it is also expressed, to a lesser extent, on epithelial cells lining the digestive tract, including liver bile ducts. Biopsy of the liver in one of these patients indicated infiltration by modified T cells around bile ducts expressing CAIX resulting in cholangitis (63, 64). Furthermore, evidence it appears that low-level expression of ERBB2 on normal lung cells may have been associated with the sudden death of a patient who received autologous HER2-specific T cells expressing a 3rd generation CAR (29). The elimination of normal B cells in the study reported by Kochenderfer and colleagues (28) is another example of an undesired, on-target toxicity. However, this toxicity was expected as CD19-specific T cells can not distinguish between CD19 expressed on malignant B cells versus normal B cells. In this situation, the risk of loss of normal B-cell function is offset by the potential benefit from an antitumor effect and that patients can tolerate B-cell lymphopenia. (We note that the report by Brentjens describing the death of a patient after infusing CD19-specific CAR+ T cells was not attributed to the genetically modified T cells (92).)
Secondly, there is concern for genotoxicity attributable to the vector. This remains a theoretical concern for genetically modified T cells, in contrast to hematopoietic progenitor cells (93). The stable expression of CAR currently requires the introduction of a promoter and the transgene which raises the possibility of insertional mutagenesis (94). To date there have been no genotoxic events attributed to genetically modified T cells that have been transduced by recombinant virus or electroporation (54, 95). The risk for insertional mutagenesis may be alleviated by electrotransfer of in vitro transcribed mRNA coding for a CAR. RNA-based electroporation mediates transient expression (96), but as technologies improve to synchronously electroporate large numbers of cells, it may be possible to overcome the expected loss of CAR expression from transient transfection by repeatedly electroporating and administering CAR+ T cells(97).
The third area of safety concern relates to the type of T cell being infused. For example, will the genetically modified T cells develop autonomous proliferation independent of binding TAA? Or, in the allogeneic setting, will the donor-derived T cells target major or minor histocompatibility antigens that could lead to GVHD? Co-expression of a conditional suicide gene, such as TK or dimerizable caspase, may prevent long-term toxicity (74), however, administration of the appropriate “suicide substrate” may not impact the pace of a potential CAR-mediated clinical toxicity arising in the acute setting. Currently, as infused CAR+ T cells are not (yet) long-lived, there does not appear the need to co-express a conditional suicide gene for CAR+ T cells. As T-cell subsets are isolated for genetic modification and as CARs and other co-stimulatory molecules are refined to provide supra-physiologic signaling, the decision to include a conditional suicide gene may then be warranted.
9. Future directions of CARs
The initial results of early phase trials using current CAR technology demonstrate the feasibility and therapeutic potential of genetically modified T cells. The technology will continue to improve, and future directions will likely include combination therapies. For example, CAR+ T cells may benefit from concomitant therapy with therapeutic monoclonal antibodies (98) or immunocytokine support (99). As the technology becomes more complex, involving multiple genes, the regulatory oversight for the production and release of the T cells and the safety of the recipient becomes more complex. Most pilot trials, including those accruing patients to receive cell and gene therapy, enroll research participants with advanced disease and it is not unexpected for a subset of these medically fragile patients to unfortunately expire during the trial. While it is incumbent on the clinical team to safeguard patient well-being, it is also important for regulatory bodies to monitor gene therapy trials in such a way as to maintain safety, but not hinder progress. The technology to manufacture CAR+ T cells has now reached a point of “mass-production” so that many investigators can readily participate in the design and implementation of clinical trials. Future clinical, multicenter trials will require streamlining the current regulatory processes governing T-cell manufacturing so that studies powered for efficacy can be efficiently conducted to definitively establish the therapeutic potential of CAR+ T cells.
Acknowledgments
Support from: Cancer Center Core Grant (CA16672); PO1 (CA100265); RO1 (CA124782, CA120956, CA141303); R33 (CA116127); DOD PR064229; The Alliance for Cancer Gene Therapy; The Alex Lemonade Stand Foundation; Burroughs Wellcome Fund; The Harry T. Mangurian, Jr., Foundation, The Gillson Longenbaugh Foundation; Institute of Personalized Cancer Therapy; The Leukemia and Lymphoma Society; The Lymphoma Research Foundation; The Miller Foundation; Mr. and Mrs. Joe H. Scales; The National Foundation for Cancer Research; The Pediatric Cancer Research Foundation; The National Marrow Donor Program; The William Lawrence and Blanche Hughes Children’s Foundation.
References
- 1.Kolb HJ. Graft-versus-leukemia effects of transplantation and donor lymphocytes. Blood. 2008;112(12):4371–83. doi: 10.1182/blood-2008-03-077974. [DOI] [PubMed] [Google Scholar]
- 2.Cullis JO, Jiang YZ, Schwarer AP, Hughes TP, Barrett AJ, Goldman JM. Donor leukocyte infusions for chronic myeloid leukemia in relapse after allogeneic bone marrow transplantation. Blood. 1992;79(5):1379–81. [PubMed] [Google Scholar]
- 3.Drobyski WR, Keever CA, Roth MS, Koethe S, Hanson G, McFadden P, Gottschall JL, Ash RC, van Tuinen P, Horowitz MM, et al. Salvage immunotherapy using donor leukocyte infusions as treatment for relapsed chronic myelogenous leukemia after allogeneic bone marrow transplantation: efficacy and toxicity of a defined T-cell dose. Blood. 1993;82(8):2310–8. [PubMed] [Google Scholar]
- 4.Ferster A, Bujan W, Mouraux T, Devalck C, Heimann P, Sariban E. Complete remission following donor leukocyte infusion in ALL relapsing after haploidentical bone marrow transplantation. Bone Marrow Transplant. 1994;14(2):331–2. [PubMed] [Google Scholar]
- 5.Kolb HJ, Mittermuller J, Clemm C, Holler E, Ledderose G, Brehm G, Heim M, Wilmanns W. Donor leukocyte transfusions for treatment of recurrent chronic myelogenous leukemia in marrow transplant patients. Blood. 1990;76(12):2462–5. [PubMed] [Google Scholar]
- 6.Lokhorst HM, Schattenberg A, Cornelissen JJ, Thomas LL, Verdonck LF. Donor leukocyte infusions are effective in relapsed multiple myeloma after allogeneic bone marrow transplantation. Blood. 1997;90(10):4206–11. [PubMed] [Google Scholar]
- 7.Pati AR, Godder K, Lamb L, Gee A, Henslee-Downey PJ. Immunotherapy with donor leukocyte infusions for patients with relapsed acute myeloid leukemia following partially mismatched related donor bone marrow transplantation. Bone Marrow Transplant. 1995;15(6):979–81. [PubMed] [Google Scholar]
- 8.Vago L, Perna SK, Zanussi M, Mazzi B, Barlassina C, Stanghellini MT, Perrelli NF, Cosentino C, Torri F, Angius A, Forno B, Casucci M, Bernardi M, Peccatori J, Corti C, Bondanza A, Ferrari M, Rossini S, Roncarolo MG, Bordignon C, Bonini C, Ciceri F, Fleischhauer K. Loss of mismatched HLA in leukemia after stem-cell transplantation. N Engl J Med. 2009;361(5):478–88. doi: 10.1056/NEJMoa0811036. [DOI] [PubMed] [Google Scholar]
- 9.Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005;5(4):263–74. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
- 10.Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A. 1993;90(2):720–4. doi: 10.1073/pnas.90.2.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8(4):299–308. doi: 10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kahlon KS, Brown C, Cooper LJ, Raubitschek A, Forman SJ, Jensen MC. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res. 2004;64(24):9160–6. doi: 10.1158/0008-5472.CAN-04-0454. [DOI] [PubMed] [Google Scholar]
- 13.Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL, Campana D. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 2004;18(4):676–84. doi: 10.1038/sj.leu.2403302. [DOI] [PubMed] [Google Scholar]
- 14.Zhao Y, Wang QJ, Yang S, Kochenderfer JN, Zheng Z, Zhong X, Sadelain M, Eshhar Z, Rosenberg SA, Morgan RA. A herceptin-based chimeric antigen receptor with modified signaling domains leads to enhanced survival of transduced T lymphocytes and antitumor activity. J Immunol. 2009;183(9):5563–74. doi: 10.4049/jimmunol.0900447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Letourneur F, Klausner RD. T-cell and basophil activation through the cytoplasmic tail of T-cell-receptor zeta family proteins. Proc Natl Acad Sci U S A. 1991;88(20):8905–9. doi: 10.1073/pnas.88.20.8905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yun CO, Nolan KF, Beecham EJ, Reisfeld RA, Junghans RP. Targeting of T lymphocytes to melanoma cells through chimeric anti-GD3 immunoglobulin T-cell receptors. Neoplasia. 2000;2(5):449–59. doi: 10.1038/sj.neo.7900108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abken H, Hombach A, Heuser C. Immune response manipulation: recombinant immunoreceptors endow T-cells with predefined specificity. Curr Pharm Des. 2003;9(24):1992–2001. doi: 10.2174/1381612033454289. [DOI] [PubMed] [Google Scholar]
- 18.Wang J, Jensen M, Lin Y, Sui X, Chen E, Lindgren CG, Till B, Raubitschek A, Forman SJ, Qian X, James S, Greenberg P, Riddell S, Press OW. Optimizing adoptive polyclonal T cell immunotherapy of lymphomas, using a chimeric T cell receptor possessing CD28 and CD137 costimulatory domains. Hum Gene Ther. 2007;18(8):712–25. doi: 10.1089/hum.2007.028. [DOI] [PubMed] [Google Scholar]
- 19.Yvon E, Del Vecchio M, Savoldo B, Hoyos V, Dutour A, Anichini A, Dotti G, Brenner MK. Immunotherapy of metastatic melanoma using genetically engineered GD2-specific T cells. Clin Cancer Res. 2009;15(18):5852–60. doi: 10.1158/1078-0432.CCR-08-3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sadelain M, Brentjens R, Riviere I. The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol. 2009;21(2):215–23. doi: 10.1016/j.coi.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kowolik CM, Topp MS, Gonzalez S, Pfeiffer T, Olivares S, Gonzalez N, Smith DD, Forman SJ, Jensen MC, Cooper LJ. CD28 costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Res. 2006;66(22):10995–1004. doi: 10.1158/0008-5472.CAN-06-0160. [DOI] [PubMed] [Google Scholar]
- 22.Jensen MC, Popplewell L, Cooper LJ, DiGiusto D, Kalos M, Ostberg JR, Forman SJ. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant. 16(9):1245–56. doi: 10.1016/j.bbmt.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Till BG, Jensen MC, Wang J, Chen EY, Wood BL, Greisman HA, Qian X, James SE, Raubitschek A, Forman SJ, Gopal AK, Pagel JM, Lindgren CG, Greenberg PD, Riddell SR, Press OW. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood. 2008;112(6):2261–71. doi: 10.1182/blood-2007-12-128843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park JR, Digiusto DL, Slovak M, Wright C, Naranjo A, Wagner J, Meechoovet HB, Bautista C, Chang WC, Ostberg JR, Jensen MC. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther. 2007;15(4):825–33. doi: 10.1038/sj.mt.6300104. [DOI] [PubMed] [Google Scholar]
- 25.Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, Huls MH, Liu E, Gee AP, Mei Z, Yvon E, Weiss HL, Liu H, Rooney CM, Heslop HE, Brenner MK. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14(11):1264–70. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Finney HM, Lawson AD, Bebbington CR, Weir AN. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J Immunol. 1998;161(6):2791–7. [PubMed] [Google Scholar]
- 27.Hombach A, Wieczarkowiecz A, Marquardt T, Heuser C, Usai L, Pohl C, Seliger B, Abken H. Tumor-specific T cell activation by recombinant immunoreceptors: CD3 zeta signaling and CD28 costimulation are simultaneously required for efficient IL-2 secretion and can be integrated into one combined CD28/CD3 zeta signaling receptor molecule. J Immunol. 2001;167(11):6123–31. doi: 10.4049/jimmunol.167.11.6123. [DOI] [PubMed] [Google Scholar]
- 28.Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, Maric I, Raffeld M, Nathan DA, Lanier BJ, Morgan RA, Rosenberg SA. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically-engineered to recognize CD19. Blood. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 18(4):843–51. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pameijer CR, Navanjo A, Meechoovet B, Wagner JR, Aguilar B, Wright CL, Chang WC, Brown CE, Jensen MC. Conversion of a tumor-binding peptide identified by phage display to a functional chimeric T cell antigen receptor. Cancer Gene Ther. 2007;14(1):91–7. doi: 10.1038/sj.cgt.7700993. [DOI] [PubMed] [Google Scholar]
- 31.Willemsen RA, Debets R, Hart E, Hoogenboom HR, Bolhuis RL, Chames P. A phage display selected fab fragment with MHC class I-restricted specificity for MAGE-A1 allows for retargeting of primary human T lymphocytes. Gene Ther. 2001;8(21):1601–8. doi: 10.1038/sj.gt.3301570. [DOI] [PubMed] [Google Scholar]
- 32.Chmielewski M, Hombach A, Heuser C, Adams GP, Abken H. T cell activation by antibody-like immunoreceptors: increase in affinity of the single-chain fragment domain above threshold does not increase T cell activation against antigen-positive target cells but decreases selectivity. J Immunol. 2004;173(12):7647–53. doi: 10.4049/jimmunol.173.12.7647. [DOI] [PubMed] [Google Scholar]
- 33.Vera J, Savoldo B, Vigouroux S, Biagi E, Pule M, Rossig C, Wu J, Heslop HE, Rooney CM, Brenner MK, Dotti G. T lymphocytes redirected against the kappa light chain of human immunoglobulin efficiently kill mature B lymphocyte-derived malignant cells. Blood. 2006;108(12):3890–7. doi: 10.1182/blood-2006-04-017061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hombach A, Schneider C, Sent D, Koch D, Willemsen RA, Diehl V, Kruis W, Bolhuis RL, Pohl C, Abken H. An entirely humanized CD3 zeta-chain signaling receptor that directs peripheral blood t cells to specific lysis of carcinoembryonic antigen-positive tumor cells. Int J Cancer. 2000;88(1):115–20. doi: 10.1002/1097-0215(20001001). [DOI] [PubMed] [Google Scholar]
- 35.June CH, Blazar BR, Riley JL. Engineering lymphocyte subsets: tools, trials and tribulations. Nat Rev Immunol. 2009;9(10):704–16. doi: 10.1038/nri2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jena B, Dotti G, Cooper LJ. Redirecting T-cell specificity by introducing a tumor-specific chimeric antigen receptor. Blood. 116(7):1035–44. doi: 10.1182/blood-2010-01-043737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rosenberg SA, Aebersold P, Cornetta K, Kasid A, Morgan RA, Moen R, Karson EM, Lotze MT, Yang JC, Topalian SL, et al. Gene transfer into humans--immunotherapy of patients with advanced melanoma, using tumor-infiltrating lymphocytes modified by retroviral gene transduction. N Engl J Med. 1990;323(9):570–8. doi: 10.1056/NEJM199008303230904. [DOI] [PubMed] [Google Scholar]
- 38.Rischer M, Pscherer S, Duwe S, Vormoor J, Jurgens H, Rossig C. Human gammadelta T cells as mediators of chimaeric-receptor redirected anti-tumour immunity. Br J Haematol. 2004;126(4):583–92. doi: 10.1111/j.1365-2141.2004.05077.x. [DOI] [PubMed] [Google Scholar]
- 39.Turatti F, Figini M, Alberti P, Willemsen RA, Canevari S, Mezzanzanica D. Highly efficient redirected anti-tumor activity of human lymphocytes transduced with a completely human chimeric immune receptor. J Gene Med. 2005;7(2):158–70. doi: 10.1002/jgm.647. [DOI] [PubMed] [Google Scholar]
- 40.Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, Samanta M, Lakhal M, Gloss B, Danet-Desnoyers G, Campana D, Riley JL, Grupp SA, June CH. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17(8):1453–64. doi: 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272(5259):263–7. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 42.Varela-Rohena A, Carpenito C, Perez EE, Richardson M, Parry RV, Milone M, Scholler J, Hao X, Mexas A, Carroll RG, June CH, Riley JL. Genetic engineering of T cells for adoptive immunotherapy. Immunol Res. 2008;42(1-3):166–81. doi: 10.1007/s12026-008-8057-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Geurts AM, Yang Y, Clark KJ, Liu G, Cui Z, Dupuy AJ, Bell JB, Largaespada DA, Hackett PB. Gene transfer into genomes of human cells by the sleeping beauty transposon system. Mol Ther. 2003;8(1):108–17. doi: 10.1016/s1525-0016(03)00099-6. [DOI] [PubMed] [Google Scholar]
- 44.Huang X, Guo H, Kang J, Choi S, Zhou TC, Tammana S, Lees CJ, Li ZZ, Milone M, Levine BL, Tolar J, June CH, Scott McIvor R, Wagner JE, Blazar BR, Zhou X. Sleeping Beauty transposon-mediated engineering of human primary T cells for therapy of CD19+ lymphoid malignancies. Mol Ther. 2008;16(3):580–9. doi: 10.1038/sj.mt.6300404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jensen MC, Clarke P, Tan G, Wright C, Chung-Chang W, Clark TN, Zhang F, Slovak ML, Wu AM, Forman SJ, Raubitschek A. Human T lymphocyte genetic modification with naked DNA. Mol Ther. 2000;1(1):49–55. doi: 10.1006/mthe.1999.0012. [DOI] [PubMed] [Google Scholar]
- 46.Cooper LJ, Topp MS, Serrano LM, Gonzalez S, Chang WC, Naranjo A, Wright C, Popplewell L, Raubitschek A, Forman SJ, Jensen MC. T-cell clones can be rendered specific for CD19: toward the selective augmentation of the graft-versus-B-lineage leukemia effect. Blood. 2003;101(4):1637–44. doi: 10.1182/blood-2002-07-1989. [DOI] [PubMed] [Google Scholar]
- 47.Singh H, Manuri PR, Olivares S, Dara N, Dawson MJ, Huls H, Hackett PB, Kohn DB, Shpall EJ, Champlin RE, Cooper LJ. Redirecting specificity of T-cell populations for CD19 using the Sleeping Beauty system. Cancer Res. 2008;68(8):2961–71. doi: 10.1158/0008-5472.CAN-07-5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xue X, Huang X, Nodland SE, Mates L, Ma L, Izsvak Z, Ivics Z, LeBien TW, McIvor RS, Wagner JE, Zhou X. Stable gene transfer and expression in cord blood-derived CD34+ hematopoietic stem and progenitor cells by a hyperactive Sleeping Beauty transposon system. Blood. 2009;114(7):1319–30. doi: 10.1182/blood-2009-03-210005. [DOI] [PubMed] [Google Scholar]
- 49.Manuri PV, Wilson MH, Maiti SN, Mi T, Singh H, Olivares S, Dawson MJ, Huls H, Lee DA, Rao PH, Kaminski JM, Nakazawa Y, Gottschalk S, Kebriaei P, Shpall EJ, Champlin RE, Cooper LJ. piggyBac transposon/transposase system to generate CD19-specific T cells for the treatment of B-lineage malignancies. Hum Gene Ther. 21(4):427–37. doi: 10.1089/hum.2009.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nakazawa Y, Huye LE, Dotti G, Foster AE, Vera JF, Manuri PR, June CH, Rooney CM, Wilson MH. Optimization of the PiggyBac transposon system for the sustained genetic modification of human T lymphocytes. J Immunother. 2009;32(8):826–36. doi: 10.1097/CJI.0b013e3181ad762b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hackett PB, Ekker SC, Largaespada DA, McIvor RS. Sleeping beauty transposon-mediated gene therapy for prolonged expression. Adv Genet. 2005;54:189–232. doi: 10.1016/S0065-2660(05)54009-4. [DOI] [PubMed] [Google Scholar]
- 52.Ivics Z, Hackett PB, Plasterk RH, Izsvak Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell. 1997;91(4):501–10. doi: 10.1016/s0092-8674(00)80436-5. [DOI] [PubMed] [Google Scholar]
- 53.Izsvak Z, Chuah MK, Vandendriessche T, Ivics Z. Efficient stable gene transfer into human cells by the Sleeping Beauty transposon vectors. Methods. 2009;49(3):287–97. doi: 10.1016/j.ymeth.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 54.Hackett PB, Largaespada DA, Cooper LJ. A transposon and transposase system for human application. Mol Ther. 18(4):674–83. doi: 10.1038/mt.2010.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Izsvak Z, Hackett PB, Cooper LJ, Ivics Z. Translating Sleeping Beauty transposition into cellular therapies: victories and challenges. Bioessays. 32(9):756–67. doi: 10.1002/bies.201000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rossig C, Bollard CM, Nuchtern JG, Rooney CM, Brenner MK. Epstein-Barr virus-specific human T lymphocytes expressing antitumor chimeric T-cell receptors: potential for improved immunotherapy. Blood. 2002;99(6):2009–16. doi: 10.1182/blood.v99.6.2009. [DOI] [PubMed] [Google Scholar]
- 57.Cooper LJ, Al-Kadhimi Z, Serrano LM, Pfeiffer T, Olivares S, Castro A, Chang WC, Gonzalez S, Smith D, Forman SJ, Jensen MC. Enhanced antilymphoma efficacy of CD19-redirected influenza MP1-specific CTLs by cotransfer of T cells modified to present influenza MP1. Blood. 2005;105(4):1622–31. doi: 10.1182/blood-2004-03-1208. [DOI] [PubMed] [Google Scholar]
- 58.Stephan MT, Ponomarev V, Brentjens RJ, Chang AH, Dobrenkov KV, Heller G, Sadelain M. T cell-encoded CD80 and 4-1BBL induce auto- and transcostimulation, resulting in potent tumor rejection. Nat Med. 2007;13(12):1440–9. doi: 10.1038/nm1676. [DOI] [PubMed] [Google Scholar]
- 59.Hinrichs CS, Borman ZA, Cassard L, Gattinoni L, Spolski R, Yu Z, Sanchez-Perez L, Muranski P, Kern SJ, Logun C, Palmer DC, Ji Y, Reger RN, Leonard WJ, Danner RL, Rosenberg SA, Restifo NP. Adoptively transferred effector cells derived from naive rather than central memory CD8+ T cells mediate superior antitumor immunity. Proc Natl Acad Sci U S A. 2009;106(41):17469–74. doi: 10.1073/pnas.0907448106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest. 2008;118(1):294–305. doi: 10.1172/JCI32103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Berger C, Berger M, Anderson D, Riddell SR. A non-human primate model for analysis of safety, persistence, and function of adoptively transferred T cells. J Med Primatol. doi: 10.1111/j.1600-0684.2010.00451.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, Robbins PF, Huang J, Citrin DE, Leitman SF, Wunderlich J, Restifo NP, Thomasian A, Downey SG, Smith FO, Klapper J, Morton K, Laurencot C, White DE, Rosenberg SA. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. 2008;26(32):5233–9. doi: 10.1200/JCO.2008.16.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, Debets R, Gratama JW, Stoter G, Oosterwijk E. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. 2006;24(13):e20–2. doi: 10.1200/JCO.2006.05.9964. [DOI] [PubMed] [Google Scholar]
- 64.Lamers CH, Willemsen R, van Elzakker P, van Steenbergen-Langeveld S, Broertjes M, Oosterwijk-Wakka J, Oosterwijk E, Sleijfer S, Debets R, Gratama JW. Immune responses to transgene and retroviral vector in patients treated with ex vivo engineered T cells. Blood. 2010 doi: 10.1182/blood-2010-07-294520. [DOI] [PubMed] [Google Scholar]
- 65.Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, White DE, Wunderlich JR, Canevari S, Rogers-Freezer L, Chen CC, Yang JC, Rosenberg SA, Hwu P. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006;12(20 Pt 1):6106–15. doi: 10.1158/1078-0432.CCR-06-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Manuri SOPR, Dara N, Dawson MJ, Huls H, Lee DA, Shpall EJ, Champlin RE, Cooper LJN. A Fully-Human Chimeric Antigen Receptor for Redirecting Specificity of T Cells to B-Lineage Tumors. American Society for Blood and Marrow Transplantation, Tandem Meetings; San Diego, California. 2008. [Google Scholar]
- 67.Carrio R, Rolle CE, Malek TR. Non-redundant role for IL-7R signaling for the survival of CD8+ memory T cells. Eur J Immunol. 2007;37(11):3078–88. doi: 10.1002/eji.200737585. [DOI] [PubMed] [Google Scholar]
- 68.Powell DJ, Jr, Dudley ME, Robbins PF, Rosenberg SA. Transition of late-stage effector T cells to CD27+ CD28+ tumor-reactive effector memory T cells in humans after adoptive cell transfer therapy. Blood. 2005;105(1):241–50. doi: 10.1182/blood-2004-06-2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vera JF, Hoyos V, Savoldo B, Quintarelli C, Giordano Attianese GM, Leen AM, Liu H, Foster AE, Heslop HE, Rooney CM, Brenner MK, Dotti G. Genetic manipulation of tumor-specific cytotoxic T lymphocytes to restore responsiveness to IL-7. Mol Ther. 2009;17(5):880–8. doi: 10.1038/mt.2009.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sogo T, Kawahara M, Tsumoto K, Kumagai I, Ueda H, Nagamune T. Selective expansion of genetically modified T cells using an antibody/interleukin-2 receptor chimera. J Immunol Methods. 2008;337(1):16–23. doi: 10.1016/j.jim.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 71.Markley JC, Sadelain M. IL-7 and IL-21 are superior to IL-2 and IL-15 in promoting human T cell-mediated rejection of systemic lymphoma in immunodeficient mice. Blood. 115(17):3508–19. doi: 10.1182/blood-2009-09-241398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu K, Rosenberg SA. Transduction of an IL-2 gene into human melanoma-reactive lymphocytes results in their continued growth in the absence of exogenous IL-2 and maintenance of specific antitumor activity. J Immunol. 2001;167(11):6356–65. doi: 10.4049/jimmunol.167.11.6356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hsu C, Jones SA, Cohen CJ, Zheng Z, Kerstann K, Zhou J, Robbins PF, Peng PD, Shen X, Gomes TJ, Dunbar CE, Munroe DJ, Stewart C, Cornetta K, Wangsa D, Ried T, Rosenberg SA, Morgan RA. Cytokine-independent growth and clonal expansion of a primary human CD8+ T-cell clone following retroviral transduction with the IL-15 gene. Blood. 2007;109(12):5168–77. doi: 10.1182/blood-2006-06-029173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, Heslop HE, Rooney CM, Brenner MK, Dotti G. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. 24(6):1160–70. doi: 10.1038/leu.2010.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rowley J, Monie A, Hung CF, Wu TC. Expression of IL-15RA or an IL-15/IL-15RA fusion on CD8+ T cells modifies adoptively transferred T-cell function in cis. Eur J Immunol. 2009;39(2):491–506. doi: 10.1002/eji.200838594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Heemskerk B, Liu K, Dudley ME, Johnson LA, Kaiser A, Downey S, Zheng Z, Shelton TE, Matsuda K, Robbins PF, Morgan RA, Rosenberg SA. Adoptive cell therapy for patients with melanoma, using tumor-infiltrating lymphocytes genetically engineered to secrete interleukin-2. Hum Gene Ther. 2008;19(5):496–510. doi: 10.1089/hum.2007.0171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bollard CM, Rossig C, Calonge MJ, Huls MH, Wagner HJ, Massague J, Brenner MK, Heslop HE, Rooney CM. Adapting a transforming growth factor beta-related tumor protection strategy to enhance antitumor immunity. Blood. 2002;99(9):3179–87. doi: 10.1182/blood.v99.9.3179. [DOI] [PubMed] [Google Scholar]
- 78.Loskog A, Giandomenico V, Rossig C, Pule M, Dotti G, Brenner MK. Addition of the CD28 signaling domain to chimeric T-cell receptors enhances chimeric T-cell resistance to T regulatory cells. Leukemia. 2006;20(10):1819–28. doi: 10.1038/sj.leu.2404366. [DOI] [PubMed] [Google Scholar]
- 79.Palendira U, Chinn R, Raza W, Piper K, Pratt G, Machado L, Bell A, Khan N, Hislop AD, Steyn R, Rickinson AB, Buckley CD, Moss P. Selective accumulation of virus-specific CD8+ T cells with unique homing phenotype within the human bone marrow. Blood. 2008;112(8):3293–302. doi: 10.1182/blood-2008-02-138040. [DOI] [PubMed] [Google Scholar]
- 80.Brown CE, Vishwanath RP, Aguilar B, Starr R, Najbauer J, Aboody KS, Jensen MC. Tumor-derived chemokine MCP-1/CCL2 is sufficient for mediating tumor tropism of adoptively transferred T cells. J Immunol. 2007;179(5):3332–41. doi: 10.4049/jimmunol.179.5.3332. [DOI] [PubMed] [Google Scholar]
- 81.Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, Foster AE, Heslop HE, Brenner MK, Dotti G, Savoldo B. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood. 2009;113(25):6392–402. doi: 10.1182/blood-2009-03-209650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Heyman RA, Borrelli E, Lesley J, Anderson D, Richman DD, Baird SM, Hyman R, Evans RM. Thymidine kinase obliteration: creation of transgenic mice with controlled immune deficiency. Proc Natl Acad Sci U S A. 1989;86(8):2698–702. doi: 10.1073/pnas.86.8.2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yaghoubi SS, Jensen MC, Satyamurthy N, Budhiraja S, Paik D, Czernin J, Gambhir SS. Noninvasive detection of therapeutic cytolytic T cells with 18F-FHBG PET in a patient with glioma. Nat Clin Pract Oncol. 2009;6(1):53–8. doi: 10.1038/ncponc1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Singh H, Najjar AM, Olivares S, Nishii R, Mukhopadhyay U, Alauddin M, Manuri PR, Huls H, Lee DA, Dotti G, Bollard C, Simmons PJ, Shpall EJ, Champlin RE, Gelovani JG, Cooper LJ. PET imaging of T cells derived from umbilical cord blood. Leukemia. 2009;23(3):620–2. doi: 10.1038/leu.2008.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Munshi NC, Govindarajan R, Drake R, Ding LM, Iyer R, Saylors R, Kornbluth J, Marcus S, Chiang Y, Ennist D, Kwak L, Reynolds C, Tricot G, Barlogie B. Thymidine kinase (TK) gene-transduced human lymphocytes can be highly purified, remain fully functional, and are killed efficiently with ganciclovir. Blood. 1997;89(4):1334–40. [PubMed] [Google Scholar]
- 86.Serrano LM, Pfeiffer T, Olivares S, Numbenjapon T, Bennitt J, Kim D, Smith D, McNamara G, AlKadhimi Z, Rosenthal J, Forman SJ, Jensen MC, Cooper LJ. Differentiation of naive cord-blood T cells into CD19-specific cytolytic effectors for posttransplantation adoptive immunotherapy. Blood. 2006;107(7):2643–52. doi: 10.1182/blood-2005-09-3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ponomarev V, Doubrovin M, Shavrin A, Serganova I, Beresten T, Ageyeva L, Cai C, Balatoni J, Alauddin M, Gelovani J. A human-derived reporter gene for noninvasive imaging in humans: mitochondrial thymidine kinase type 2. J Nucl Med. 2007;48(5):819–26. doi: 10.2967/jnumed.106.036962. [DOI] [PubMed] [Google Scholar]
- 88.Shu CJ, Campbell DO, Lee JT, Tran AQ, Wengrod JC, Witte ON, Phelps ME, Satyamurthy N, Czernin J, Radu CG. Novel PET probes specific for deoxycytidine kinase. J Nucl Med. 51(7):1092–8. doi: 10.2967/jnumed.109.073361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Likar Y, Zurita J, Dobrenkov K, Shenker L, Cai S, Neschadim A, Medin JA, Sadelain M, Hricak H, Ponomarev V. A new pyrimidine-specific reporter gene: a mutated human deoxycytidine kinase suitable for PET during treatment with acycloguanosine-based cytotoxic drugs. J Nucl Med. 51(9):1395–403. doi: 10.2967/jnumed.109.074344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.June CH. Adoptive T cell therapy for cancer in the clinic. J Clin Invest. 2007;117(6):1466–76. doi: 10.1172/JCI32446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, Zheng Z, Nahvi A, de Vries CR, Rogers-Freezer LJ, Mavroukakis SA, Rosenberg SA. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314(5796):126–9. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Brentjens R, Yeh R, Bernal Y, Riviere I, Sadelain M. Treatment of chronic lymphocytic leukemia with genetically targeted autologous T cells: case report of an unforeseen adverse event in a phase I clinical trial. Mol Ther. 18(4):666–8. doi: 10.1038/mt.2010.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E, Clappier E, Caccavelli L, Delabesse E, Beldjord K, Asnafi V, MacIntyre E, Dal Cortivo L, Radford I, Brousse N, Sigaux F, Moshous D, Hauer J, Borkhardt A, Belohradsky BH, Wintergerst U, Velez MC, Leiva L, Sorensen R, Wulffraat N, Blanche S, Bushman FD, Fischer A, Cavazzana-Calvo M. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118(9):3132–42. doi: 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Baum C, Kustikova O, Modlich U, Li Z, Fehse B. Mutagenesis and oncogenesis by chromosomal insertion of gene transfer vectors. Hum Gene Ther. 2006;17(3):253–63. doi: 10.1089/hum.2006.17.253. [DOI] [PubMed] [Google Scholar]
- 95.Bonini C, Grez M, Traversari C, Ciceri F, Marktel S, Ferrari G, Dinauer M, Sadat M, Aiuti A, Deola S, Radrizzani M, Hagenbeek A, Apperley J, Ebeling S, Martens A, Kolb HJ, Weber M, Lotti F, Grande A, Weissinger E, Bueren JA, Lamana M, Falkenburg JH, Heemskerk MH, Austin T, Kornblau S, Marini F, Benati C, Magnani Z, Cazzaniga S, Toma S, Gallo-Stampino C, Introna M, Slavin S, Greenberg PD, Bregni M, Mavilio F, Bordignon C. Safety of retroviral gene marking with a truncated NGF receptor. Nat Med. 2003;9(4):367–9. doi: 10.1038/nm0403-367. [DOI] [PubMed] [Google Scholar]
- 96.Zhao Y, Zheng Z, Cohen CJ, Gattinoni L, Palmer DC, Restifo NP, Rosenberg SA, Morgan RA. High-efficiency transfection of primary human and mouse T lymphocytes using RNA electroporation. Mol Ther. 2006;13(1):151–9. doi: 10.1016/j.ymthe.2005.07.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Choi Y, Yuen C, Maiti SN, Olivares S, Gibbons H, Huls H, Raphael R, Killian TC, Stark DJ, Lee DA, Torikai H, Monticello D, Kelly SS, Kebriaei P, Champlin RE, Biswal SL, Cooper LJ. A high throughput microelectroporation device to introduce a chimeric antigen receptor to redirect the specificity of human T cells. Biomed Microdevices. 12(5):855–63. doi: 10.1007/s10544-010-9440-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.James SE, Orgun NN, Tedder TF, Shlomchik MJ, Jensen MC, Lin Y, Greenberg PD, Press OW. Antibody-mediated B-cell depletion before adoptive immunotherapy with T cells expressing CD20-specific chimeric T-cell receptors facilitates eradication of leukemia in immunocompetent mice. Blood. 2009;114(27):5454–63. doi: 10.1182/blood-2009-08-232967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Singh H, Serrano LM, Pfeiffer T, Olivares S, McNamara G, Smith DD, Al-Kadhimi Z, Forman SJ, Gillies SD, Jensen MC, Colcher D, Raubitschek A, Cooper LJ. Combining adoptive cellular and immunocytokine therapies to improve treatment of B-lineage malignancy. Cancer Res. 2007;67(6):2872–80. doi: 10.1158/0008-5472.CAN-06-2283. [DOI] [PubMed] [Google Scholar]