

Abstract
Efficient energy conversion often requires stabilization of one-electron intermediates within catalytic sites of redox enzymes. While quinol oxidoreductases are known to stabilize semiquinones, one of the famous exceptions includes the quinol oxidation site of cytochrome bc1 (Qo), for which detection of any intermediate states is extremely difficult. Here we discover a semiquinone at the Qo site (SQo) that is coupled to the reduced Rieske cluster (FeS) via spin–spin exchange interaction. This interaction creates a new electron paramagnetic resonance (EPR) transitions with the most prominent g = 1.94 signal shifting to 1.96 with an increase in the EPR frequency from X- to Q-band. The estimated value of isotropic spin–spin exchange interaction (|J0| = 3500 MHz) indicates that at a lower magnetic field (typical of X-band) the SQo–FeS coupled centers can be described as a triplet state. Concomitantly with the appearance of the SQo–FeS triplet state, we detected a g = 2.0045 radical signal that corresponded to the population of unusually fast-relaxing SQo for which spin–spin exchange does not exist or is too small to be resolved. The g = 1.94 and g = 2.0045 signals reached up to 20% of cytochrome bc1 monomers under aerobic conditions, challenging the paradigm of the high reactivity of SQo toward molecular oxygen. Recognition of stable SQo reflected in g = 1.94 and g = 2.0045 signals offers a new perspective on understanding the mechanism of Qo site catalysis. The frequency-dependent EPR transitions of the SQo–FeS coupled system establish a new spectroscopic approach for the detection of SQo in mitochondria and other bioenergetic systems.
Biological energy conversion faces an engineering problem of joining the one- and two-electron stoichiometry of redox reactions between substrates and cofactors. Most catalytic sites accomplish this by supporting two sequential one-electron transfers toward a single cofactor chain involving a stable intermediate radical.1,2 The catalytic Qo site of cytochrome bc1 (respiratory complex III) is different and unique in that it changes the electronic stoichiometry by steering two electrons from ubiquinol (QH2) to two separate chains of cofactors: it delivers one electron to the Rieske cluster (FeS) in the high-potential chain and the second electron to heme bL in the low-potential chain (Figure S1 of the Supporting Information).3−6 The common view of this bifurcation process is that the intermediate semiquinone radical (SQo), formed by one-electron oxidation of QH2 by FeS, is highly unstable5,7 and reduces heme bL very rapidly before it can react with dioxygen to generate superoxide.8−11 This concept has been supported by a general difficulty to detect SQo under aerobic conditions. In fact, the only report of detection of SQo under those conditions comes from early studies with submitochondrial particles (SOM).12 The origin of this signal was, however, questioned by later studies showing the insensitivity of the SQ signals in SOM to specific inhibitors of the Qo site.13 More recent studies reported either detection of small amounts of SQo under anaerobic conditions14,15 or a lack of detection of SQo under aerobic conditions,16 which further supported the concept of the high instability of SQo and its high reactivity with oxygen. Apart from those examples, there have been no other studies reporting detection of intermediate states for Qo site catalysis, which leaves the mechanism of electronic bifurcation largely unknown.
Here, we explore a possibility that the intriguing lack of SQo detection is a result of its magnetic interactions with metal centers of the Qo site rather than an effect of its high instability. In principle, a strong antiferromagnetic coupling of SQo with a metal center could result in the elimination of the SQo electron paramagnetic resonance (EPR) signal, as proposed by Link.17 However, if the coupling is ferromagnetic and/or weak (in comparison to the thermal energy of the lattice), it may be expected that it will manifest itself as a new spectroscopic identity.18,19 Indeed, by exposing the purified enzyme to its substrates (oxidized cytochrome c and QH2), we have detected new transitions in EPR spectra assigned to a SQo magnetically coupled to reduced FeS via spin–spin exchange interaction. We also detected a separate radical signal of SQo with relaxation properties consistent with its location between the metal centers of the Qo site. This discovery offers a new perspective on understanding the mechanism of quinol oxidation at the Qo site. It also provides new insight into side reactions of the catalytic cycle involved in the production of superoxide by cytochrome bc1.
Materials and Methods
Biochemical Procedures
The cytochrome bc1 complex was isolated from the purple bacterium Rhodobacter capsulatus strain grown semiaerobically as described previously.20 Bovine cytochrome c, 2,3-dimethoxy-5-decyl-6-methyl-1,4-benzoquinone (DB), and inhibitors (antimycin, myxothiazol, atovaquone, azoxystrobin, kresoxim-methyl, and famoxadone) were purchased from Sigma-Aldrich and used without further purifications. Tridecyl-stigmatellin was a generous gift from N. Fisher. DB was dissolved in an HCl/DMSO solution and then reduced to its hydroquinone form (DBH2) with sodium borohydride. Inhibitors were used in 5-fold molar excess over the concentration of cytochrome bc1 monomers. Cytochrome bc1 and cytochrome c solutions were dialyzed against the reaction buffer composed of 50 mM Tris (pH 8.0), 100 mM NaCl, 20% glycerol (v/v), 0.01% (m/m) dodecyl maltoside, and 1 mM EDTA. All buffers were in equilibrium with air. Glycerol, added as a cryoprotective agent, increased the viscosity of the reaction buffer, which resulted in a deceleration of the overall catalytic turnover rate of the enzyme by decreasing diffusion rates of the substrates.
Freeze-quench experiments were performed using a Biologic SFM-300 stopped-flow mixer equipped with an MPS-70 programmable syringe control. The system was equipped with EPR FQ accessories. One syringe contained a cytochrome bc1/cytochrome c solution, and the second syringe contained DBH2 in reaction buffer. Steady-state reduction of cytochrome c by cytochrome bc1 was initiated by mixing the cytochrome bc1/cytochrome c solution with DBH2 in a 1:1 volume ratio to obtain final concentrations of cytochrome bc1, cytochrome c, and DBH2 of 50, 393, and 665 μM, respectively. The reaction mixture was incubated at room temperature in a delay line for a programmed number of milliseconds and then injected into an isopentane bath cooled to 100 K. Samples with higher cytochrome bc1 concentrations required for hemes b measurements were prepared by manual injection of DBH2 into the cytochrome bc1/cytochrome c solution inside EPR tube. The reaction was stopped by immersing the tube into cold ethanol glue.
EPR Spectroscopy and Data Analysis
All measurements were performed using a Bruker Elexsys E580 spectrometer. X-Band continuous wave electron paramagnetic resonance (CW EPR) spectra of hemes and FeS were measured at 10 and 20 K, respectively, using a SHQE0511 resonator and ESR900 cryostat (Oxford Instruments). X-Band spectra of semiquinones were recorded using a TM9103 resonator equipped with a temperature controller system (Bruker). Q-Band spectra of semiquinones were measured at 200 K by CW EPR using an ER507D2 resonator (Bruker) equipped with homemade modulation coils using a 0.6 mT modulation amplitude, a 90 kHz frequency, and a 1.92 mW microwave power. Q-Band echo-detected EPR (ED EPR) spectra of FeS were measured at 10 K using a π/2–148 ns−π sequence with a π pulse of 48 ns and a shot repetition time of 300 μs. First-derivative spectra of FeS were generated by applying the pseudomodulation procedure21 on ED EPR spectra using Eleana (http://www.wbbib.uj.edu.pl/web/gbm/eleana). The magnitude of the external magnetic field was controlled using a Bruker NMR teslameter.
The microwave power saturation profiles of semiquinones were fit using formulas described in ref (22). The data for chemically induced semiquinone (SQCH) were fit assuming a contribution from one saturable component, while data for SQo were fit assuming the presence of two species: major, nonsaturable component and minor, saturable component. The temperature dependencies of the amplitude of SQCH were fit with the well-known Curie law. The data for SQo were fit assuming the presence of the Leigh effect23 in which the correlation time of the fluctuating dipolar field increases with a decrease in temperature. Q-Band spectra of semiquinones were simulated with Easy-spin24 using the anisotropic g tensor, assuming homogeneous and inhomogeneous line broadening.
Spectral simulations based on a spin Hamiltonian including Zeeman interaction of spins of FeS and SQo centers with the external static magnetic field and a general bilinear spin–spin interaction term were performed as described in the Supporting Information.
Results
Detection of New EPR Transitions Associated with the Qo Site of Cytochrome bc1
In searching for intermediates of the Qo site, we performed series of experiments in which isolated cytochrome bc1 in equilibrium with air catalyzed steady-state electron transfer from the water-soluble QH2 analogue [2,3-dimethoxy-5-decyl-6-methyl-1,4-benzohydroquinone (DBH2)] to oxidized cytochrome c, and the time course of spin states of redox centers was monitored by EPR. The time points of freezing the samples were selected to cover the range from the beginning of the reaction until an equilibrium between the substrates and the products was reached. As a measure of the reaction progress, the amount of oxidized cytochrome c available for reaction was determined from the amplitude of the EPR signal of heme c (not shown). We compared two cases: the reaction catalyzed by the noninhibited enzyme and that catalyzed by the enzyme inhibited with antimycin. These two cases differ by the way in which the heme bL undergoes reoxidation (after its initial reduction by an electron derived from quinol) to support the turnover of the Qo site. In the noninhibited enzyme, heme bH rapidly reoxidizes heme bL and then transfers an electron to the Qi site (see Figure S1 of the Supporting Information). This reaction sequence continues until the equilibrium is reached (the substrates are used up). In the antimycin-inhibited enzyme, the Qi site is blocked by the inhibitor, and after the first QH2 oxidation at the Qo site, heme bH remains reduced, preventing fast reoxidation of heme bL after the oxidation of a second QH2 at the Qo site. Nevertheless, this heme can undergo slow reoxidation by the back electron transfer to SQo that re-forms QH225 or electron transfer to Q that forms SQo.5,26−28 With these reactions, the Qo site can also keep the turnover until the equilibrium is reached, although the overall rate is significantly slower than that of the noninhibited enzyme.
As shown in Figure 1a, in the noninhibited enzyme, the level of reduced FeS increased within the first 7 s, reflecting the expected progress of the reaction, and after an equilibrium had been reached, the amplitude of the FeS signal remained constant. In the antimycin-inhibited enzyme, the rate of reaching the equilibrium level of reduced FeS decreased, as expected, but at the same time, quite unexpectedly we observed an additional EPR transition at g = 1.94 (Figure 1b). Its amplitude reached a maximum at 10 s and then gradually decreased to zero. A comparison of amplitudes of EPR signals of hemes b shown in Figure 1c indicates that in the samples exhibiting a g = 1.94 signal, heme bL remained fully oxidized. The presence of a g = 1.94 signal correlated with the presence of another weak signal of organic radical at g = 2.0 (exact value of 2.0045) detected with the use of a high microwave power (Figure 1b). Both g = 1.94 and g = 2.0 signals arose during the enzymatic turnover to reach their maximal amplitudes at the time where the gy (1.89) transition of reduced FeS reached approximately half of its maximal amplitude. After the maximum had been reached, the amplitude of both g = 1.94 and g = 2.0 signals gradually decreased, and when the system reached equilibrium (gy of FeS remains at its maximum), both signals disappeared completely.
Figure 1.

Detection of new g = 1.94 and g = 2.0 EPR transitions in cytochrome bc1. Monitoring changes in paramagnetic states of redox centers in WT cytochrome bc1 by X-band EPR during steady-state reduction of cytochrome c and oxidation of DBH2. Samples were frozen at different time points after addition of DBH2 to the mixture containing enzyme and cytochrome c. (a) Spectra of FeS in the noninhibited enzyme and time dependence of the amplitude (measured for the gy transition of FeS indicated by the dashed line). (b) Appearance of a new g = 1.94 transition (arrow) and a g = 2.0 radical signal in the antimycin-inhibited enzyme. The plot on the left shows time dependencies of the amplitude of the g = 1.94 signal (red line) and of gy of the FeS cluster (gray line). The plot on the right shows the time dependence of the amplitude of the g = 2.0 signal. (c) Spectra of hemes bL and bH (left) and FeS (right) for mixtures with antimycin-inhibited (red) or myxothiazol- and antimycin-inhibited cytochrome bc1 (green), respectively, frozen 12 s after addition of DBH2. Hemes, FeS, and SQ signals were measured at 10, 20, and 200 K, respectively. In panels a and b, the numbers on the left correspond to the reaction time in seconds (time before freezing).
The experiments described in Figures 1c and 2 asserted that g = 1.94 and g = 2.0 signals originate specifically from the Qo site. Both signals were sensitive to inhibitors of the Qo site and to point mutations that abolish the activity of the site7,29 and were not present in the b-c1 subcomplex lacking the FeS subunit.20 On the other hand, the amplitude of the g = 1.94 and g = 2.0 signals was larger in the mutants with affected motion of the FeS head domain (+2Ala)30 (see Figure S2 of the Supporting Information). As +2Ala arrests this domain at the Qo site for seconds with FeS in the reduced state30 (this way it abolishes the natural submillisecond electronic connection between the Qo site and heme c1), the observed enhancement of the signals immediately suggests that they must be associated with paramagnetism of FeS occupying the Qo site. Furthermore, in light of all of the results described above, the g = 2.0 signal must report SQo. We note that g = 1.94 and g = 2.0 signals were not present in samples reduced with dithionite (not shown), precluding the possibility that they originate from a contamination of the sample with low-potential iron–sulfur centers.
Figure 2.
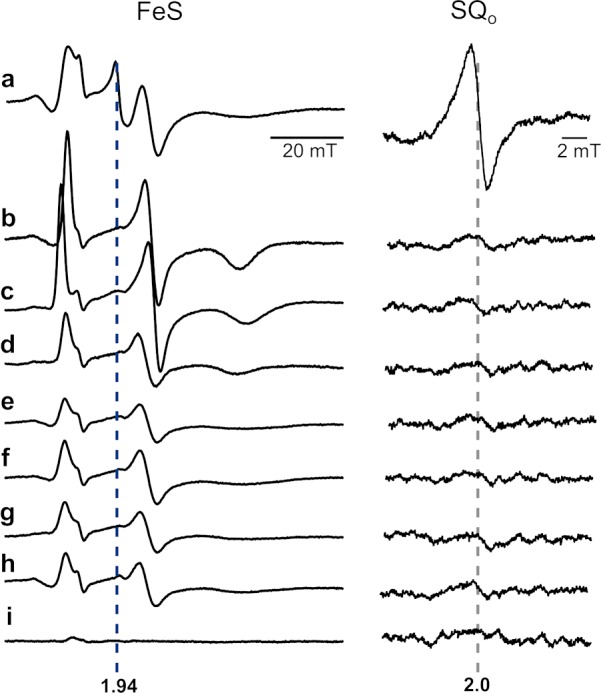
Testing the sensitivity of g = 1.94 and g = 2.0 signals to inhibitors and mutations that abolish the activity of the Qo site. X-Band EPR spectra of isolated WT cytochrome bc1 obtained under the conditions described for Figure 1c in the presence of antimycin alone (a) or antimycin and one of the Qo site inhibitors: tridecyl-stigmatellin (b), atovaquone (c), famoxadone (d), myxothiazol (e), azoxystrobin (f), or kresoxim-methyl (g). Spectra of antimycin-inhibited mutants G158W (h) and the b-c1 subcomplex (i). The left panel (FeS) shows spectra measured at 20 K in a magnetic field range of the FeS signal, and the right panel (SQo) shows spectra measured at 200 K in a magnetic field range typical of organic radicals.
Identification of the Semiquinone–Rieske Cluster Coupled System
Chemicals, such as DMSO or glycerol, and some point mutations have been reported to induce small changes in the EPR spectra of iron–sulfur clusters in proteins (Rieske or ferredoxins) with shifts in the gy values of <0.01.29,31−34 The new g = 1.94 transition does not fall into this category, because the observed difference between the gy of Rieske and the new signal was 1 order of magnitude larger (Δg ∼ 0.05) and the signal disappeared over time. Most importantly, the g = 1.94 signal detected at X-band (9.46 GHz) shifted to a g = 1.96 when the same samples were measured at Q-band (33.5 GHz) (Figure 3, black). This excludes the possibility that this signal originated from a new paramagnetic center. It thus must be a result of magnetic interactions between two closely separated paramagnetic species. An assumption that reduced FeS at the Qo site is one of them leaves SQo as the only possible candidate for the other.
Figure 3.
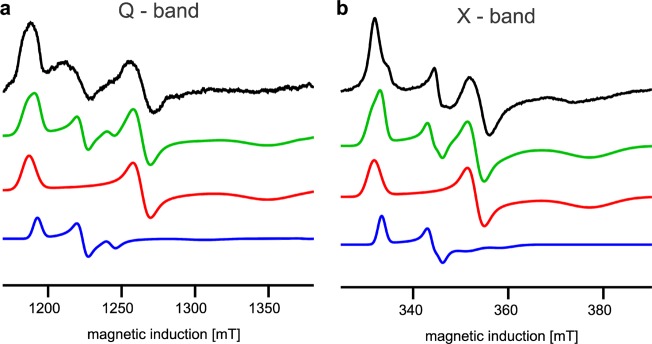
Simulating EPR spectra to define the physical nature of the g = 1.94 transition. Analysis of EPR transitions in a magnetic field range of the FeS signal at Q-band (a) and X-band (b) for the antimycin-inhibited +2Ala mutant. In panels a and b, experimental spectra (black) were simulated (green) as a sum of the FeS spectra (red) and the spectra resulting from exchange coupling between FeS and SQo (blue), assuming |J0| ∼ 3500 MHz. Blue and red represent 17 and 83%, respectively, of the total number of spins in green. The blue spectrum in panel b represents the spectrum of the SQo–FeS triplet state.
To verify that both FeS and SQo do interact with one another and to identify the dominant mechanism responsible for the appearance of a new EPR spectrum, we performed simulations based on a spin Hamiltonian including isotropic (scalar exchange) and anisotropic (exchange and dipolar) terms of spin coupling between SQo and the FeS cluster (Figure 3) (see details in the Supporting Information).18 Dipolar interaction alone appeared to be too weak to produce the g = 1.94 transition. However, when spin–spin exchange interaction was taken into account and its frequency |J0| was on the order of 3500 MHz (∼0.1 cm–1), the simulations neatly reproduced experimental spectra (Figure 3). We thus identified the SQo–FeS coupled system that at lower magnetic fields (those used at X-band) exists as a triplet state (and will be termed as such, in the remaining text). The SQo–FeS triplet emerges as a new intermediate of the reactions at the Qo site that when formed averages the g transitions of SQo and FeS.
Distinct Population of SQo without Spin–Spin Exchange Interaction
The presence of a separate g = 2.0 SQo transition identified a distinct population of SQo centers for which spin–spin exchange with FeS does not exist or is too small to be resolved. Nevertheless, fast-relaxing paramagnetic metals of the Qo site (oxidized heme bL and reduced FeS) still exerted a profound impact on SQo, resulting in its unusually fast relaxation compared to the relaxation of chemically induced semiquinone in buffer (SQCH) or well-known Qi site semiquinone (SQi).13,35 This manifested itself in significant homogeneous line broadening of the SQo signal (Figure 4a,b and Table 1), an inability to saturate it with microwaves (Figure 4c), and the presence of a Leigh effect (Figure 4d).23 We note that the fast relaxation makes this SQo signal different from other reported SQo signals12,14,15 that did not show signs of interactions with the FeS and/or heme bL metal centers of the Qo site.
Figure 4.

Unusual magnetic properties of the SQo center. (a) The Q-band spectrum of SQo (red) shows significant homogeneous broadening in comparison to the spectrum of SQi generated in myxothiazol-inhibited enzyme (green) or SQCH generated chemically in buffer (black). The spectra were simulated using the rhombic g tensor (dashed lines). (b) The same samples as in panel a measured at X-band. (c) X-band microwave power dependence of the amplitude of SQo at 200 K (red) compared with that of SQCH (black). (d) Temperature dependence of the SQo amplitude showing the Leigh effect (red) while SQCH obeys the Curie law (black). Solid lines in panels c and d represent appropriate fits (see Materials and Methods). a–d refer to the SQo signal generated in the +2Ala mutant for which the signal is the strongest (see Figure S2 of the Supporting Information).
Table 1. Parameters of Q-Band Semiquinone Spectra Obtained by Simulation.
| gz | gy | gx | homogeneous line broadeninga (mT) | |
|---|---|---|---|---|
| SQo | 2.0059 | 2.0045 | 2.0010 | 0.718 |
| SQi | 2.0052 | 2.0043 | 2.0013 | 0.306 |
| SQCH | 2.0052 | 2.0043 | 2.0009 | 0.330 |
The contribution from Gaussian broadening was set to 0.03 mT and kept constant in all simulations.
Discussion
Conditions of Formation of SQo and SQo–FeS Coupled Centers Detected by EPR
In our experiments, the SQo–FeS coupled centers (g = 1.94) and SQo (g = 2.0) were detected during the continuous turnover of quinol oxidation and cytochrome c reduction when fast reoxidation of heme bL through heme bH and the Qi site was prevented. Under such conditions, the reoxidation of heme bL required to maintain the progress of oxidant-induced (by oxidized Rieske) heme bL reduction is achieved by the transfer of an electron from heme bL back to the Qo site. Because the formation of the g = 1.94 signal requires the concomitant presence of the reduced FeS and SQo, the back electron transfer may predispose the Qo site to generate the g = 1.94 signal if heme bL reduces Q to form SQo (via semireverse reaction26−28) at the time when reduced FeS is already present in the site.
Indeed, this appeared to be the dominant way through which the SQo–FeS triplet and SQo signals were trapped in our experiments. The first indication of that comes from the observation that the signals were detected along with oxidized heme bL (Figure 1c). Furthermore, the signals reached maximal amplitudes when FeS and cytochrome c (acting as the oxidizing pool) were approximately half-reduced (Figure 1b). This suggests that the probability of trapping the g = 1.94 and g = 2.0 intermediates comes as a result of competition between the rate of oxidant-induced heme bL reduction and the rate of its oxidation by the transfer of an electron from heme bL to Q to form SQo at the time when FeS is reduced. It follows that the conditions of the formation of SQo–FeS coupled centers are not favored at the beginning of the reaction, when the population of Rieske clusters is largely oxidized and capable of “consuming” electrons from SQo (time points before appearance of the g = 1.94 and g = 2.0 signals in Figure 1b). On the other hand, as the system reaches equilibrium, the populations of Rieske clusters and cytochrome c become largely reduced and the average oxidant-induced reduction of the heme bL rate decreases, diminishing the amount of electron donor for Q at the Qo site. This leads to the loss of SQo–FeS and SQo signals (Figure 1b).
Incorporation of the SQo–FeS Triplet State in the Electronic Reactions of the Qo Site
Detection of the SQo–FeS triplet state along with the residual SQo sets a new stage for understanding the mechanism of reactions catalyzed by the Qo site from both kinetic and thermodynamic points of view (Figure 5). It can be envisaged that the SQo–FeS triplet forms as an initial step of oxidation of QH2 when oxidized FeS withdraws an electron from QH2 (state b in Figure 5). Evolution of this state into the state where SQo and reduced FeS exist as separate spectral identities (state c in Figure 5) leads to immediate reduction of heme bL by SQo, which completes the reaction generating Q (state d in Figure 5). In this scheme, a direct transition from state b to state d cannot be ruled out and might be even rapid enough to consider the two-electron oxidation of QH2 at the Qo site as a virtually concerted process. The flow of electrons out from the cofactor chains (state e in Figure 5) allows the enzyme to regain state a to complete the cycle.
Figure 5.
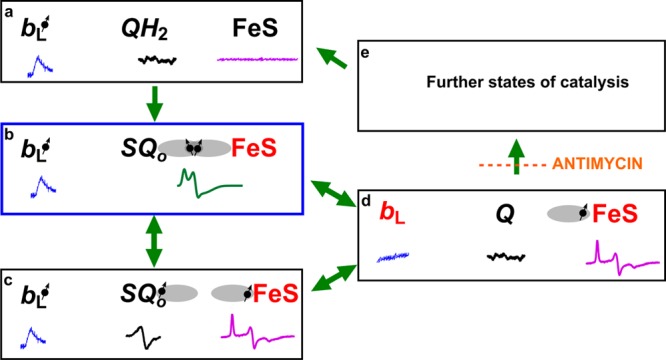
Model of electronic bifurcation of the Qo site accommodating the SQo–FeS coupled system. (a) Bound QH2 is flanked by oxidized heme bL and oxidized FeS. (b) FeS withdraws an electron from QH2, which leads to the formation of the SQo–FeS triplet state. (c) The SQo–FeS distance increases (by movement of the FeS head domain and/or SQo), breaking spin exchange interaction, exposing separate spectra of SQo and reduced FeS. (d) Heme bL is reduced by SQo generating Q. (e) In the noninhibited enzyme, heme bL rapidly transfers an electron across the membrane to heme bH directly or through heme bL in the other monomer44,45 (not shown). The enzyme goes through further states to reach the initial state a. Antimycin prevents oxidation of heme bH, interrupting the transition from state d to state e. Black and red denote the oxidized and reduced cofactor, respectively, while the dot with an arrow indicates the paramagnetic state of the center. Orbitals engaged in spin exchange are shown as gray ovals. Blue, black, magenta, and green spectra are EPR spectra of heme bL, SQo, FeS, and the SQo–FeS triplet state, respectively. Green arrows show transitions between the enzyme states. The blue box denotes the state that was detected as a major fraction of SQ. The scheme does not consider the still unknown proton transfers that may influence transitions between the states.
For this scheme, the measured g = 1.94 (the SQo–FeS triplet) and g = 2.00 (SQo) signals are spectroscopic signatures of states b and c, respectively. These states were detected only when the flow of electrons out from the Qi site was blocked by antimycin (interrupted transition from state d to e) that, in the context of full reversibility of Qo site reactions, indirectly increased the probability of transfer of an electron from reduced heme bL to Q to form SQo (bringing the site back to state b or c).5,26−28
One may ask why a significant amount of SQo cannot be detected in the noninhibited enzyme. At this stage, the precise answer is difficult. Nevertheless, we may propose that if electron transfer among SQo, heme bL, and heme bH is a pure tunneling process, not coupled to any chemical event (like protonation/deprotonation, conformational change, etc.), then freezing the samples will not prevent the transfer of the electron from SQo to heme bH involving a transient step through heme bL. However, in the antimycin-inhibited enzyme, heme bH remains reduced; thus, in frozen samples containing a reduced FeS cluster, an electron may circulate only between SQo and heme bL. Under these conditions, the highest probability of finding unpaired electrons is on SQo–FeS coupled centers, and as long as the electron circulation is significantly slower than the Larmor frequency (∼9.5 GHz), it exerts no effect on the EPR spectra of SQo–FeS coupled centers at the Qo site.
Thermodynamic Properties of the SQo–FeS Couple
While the quantity of residual SQo (from state c) cannot be determined because of the presence of the Leigh effect,23 the estimated maximal abundance of the SQo–FeS triplet state (state b) reaches as much as ∼9 and ∼17% of the total concentration of FeS in WT and +2Ala cytochrome bc1, respectively (Figure 3). This indicates that SQo may not be as highly unstable as the models of the Qo site assume.5,10,11,13 This raises the question of how much the stability constant (Kstab) of SQo detected in this work differs from the Kstab of ≪10–7 typically reported in the literature.7,14,15,25,36 Any temptations to estimate this difference must consider the fact that in our experiments the new intermediates were detected under nonequilibrium conditions of continuous turnover; thus, the use of Kstab for a description of the stability of SQo may be invalid, as this parameter is used to define stability in systems under thermodynamic equilibrium conditions. Nevertheless, the use of this parameter for the description of SQo–FeS triplet stability at the time point (tmax) where the amount of SQo is the highest yields a Kstab on the order of 10–2.6.a This is more than 3 orders of magnitude larger than the previously defined upper limit of Kstab for SQo. Such a value of Kstab makes the stability of SQo comparable to stabilities of other semiquinones in proteins, such as that of the Qi site.35
Until now, the Qo site has been considered exceptional in that, unlike other quinol oxidation–reduction sites, it did not stabilize semiquinones that were naturally volatile outside the protein matrix.2 Our work suggests that the instability of SQo is apparent and is a consequence of the simultaneous accessibility of two redox partners rather than a lack of an influence of the site on the stability of SQo.
Relation of SQo to the Superoxide-Generating Activity of Cytochrome bc1
The observation that large quantities of SQo can be detected under aerobic conditions indicates that SQo is not as highly reactive with oxygen as current mechanisms of superoxide production by cytochrome bc1 assume.10,11,14,37 In fact, high levels of the SQo–FeS triplet state signal observed in the +2Ala mutant, which does not produce any detectable superoxide,27,28 indicate that conditions of triplet formation (when SQo is likely to be hydrogen-bonded to histidine liganding the FeS cluster) do not impose a risk of electron leaks on oxygen. This, however, does not preclude the possibility that the enzyme faces such a risk if SQo is present at the time when FeS is remote from the Qo site27,28 (and the hydrogen bond is not formed). This could be explained in analogy to the reactions of 1,4-semiquinones with oxygen in solution. In such chemical systems, it was found that “hydrogen bonding of the -OH moiety in the semiquinone radical to the HBA (hydrogen-bond-accepting) solvent prevents reaction of the semiquinone with O2”.38
Possible Contribution of bc-Type Complexes to the g = 1.94 Signal in Other Bioenergetic Systems
Signals near g = 1.94 often reported in studies on mitochondrial and bacterial respiration have usually been attributed to iron–sulfur clusters of complex I and II, even though their origin was not always clear.39−42 Our work implies that the Qo site of complex III, so far beyond consideration, should in fact be regarded as one of the possible contributors to the mitochondrial g = 1.94 signal. The diagnostic feature of the Qo site-deriving g = 1.94 signal at X-band is its shift to larger values with an increase in EPR frequency, as observed in cases of weak exchange between two paramagnetic centers.19 We anticipate that knowledge of spectroscopic properties of the SQo–FeS triplet signal will allow us to examine whether it can accumulate in mitochondria to relate SQo levels with other radicals, including ROS, formed during respiration.43
Conclusions
In this work, we identify new EPR transitions (g = 1.94 and g = 2.0) associated with the enzymatic activity of cytochrome bc1. Those two transitions revealed the presence of two distinct populations of semiquinone (SQo) formed at the quinol oxidation site (the Qo site). The g = 1.94 signal was assigned as one of the transitions originating from SQo coupled to the Rieske cluster (FeS) by spin–spin exchange interaction. By analyzing the Q- and X-band EPR spectra of this coupled system, we estimated the 3500 MHz value of the isotropic exchange coupling constant, |J0|, which is strong enough to create the SQo–FeS triplet state at the lower magnetic field typical of X-band. The radical signal centered at g = 2.0 corresponded to the population of fast-relaxing SQo for which spin–spin exchange does not exist or is too weak to be resolved. The paramagnetic properties of this signal were strongly affected by metal centers, consistent with its location between two fast-relaxing metal centers of the Qo site (FeS and heme bL). The detection of SQo together with oxidized heme bL in samples containing antimycin suggests that the dominant way of generating SQo that can be detected under nonequilibrium conditions is the transfer of an electron from heme bL to Q bound at the Qo site. Under these conditions, the amount of SQo is comparable to the amount of stable semiquinones detected in catalytic sites of other bioenergetic enzymes.
Glossary
Abbreviations
- SQo
semiquinone at the Qo site
- SQCH
chemically generated semiquinone
- QH2
ubihydroquinone
- Q
ubiquinone
- DBH2
2,3-dimethoxy-5-decyl-6-methyl-1,4-benzohydroquinone
- EPR
electron paramagnetic resonance
- FeS
two-iron, two-sulfur Rieske cluster
- WT
wild type.
Supporting Information Available
Simulations of EPR spectra, outline of catalytic cycle of cytochrome bc1 (Figure S1), comparison of EPR spectra of wild type and +2Ala mutant (Figure S2) and references. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by The Wellcome Trust International Senior Research Fellowship (to A.O.).
The authors declare no competing financial interest.
Footnotes
Given that ∼20% of the total Rieske clusters is coupled to SQo, the total concentration of SQo is ∼10 μM. This means that the total concentrations of QH2 and Q at tmax are ∼75 and ∼580 μM, respectively. For these values, the Kstab calculated from the formula Kstab = [SQo]2 × [Q]−1 × [QH2]−1 is 10–2.6.
Supplementary Material
References
- Holzenburg A., and Scrutton N. S., Eds. (2000) Enzyme-Catalyzed Electron and Radical Transfer, Kluwer Academic Publishers, New York. [Google Scholar]
- Gunner M. R.; Madeo J.; Zhu Z. (2008) Modification of quinone electrochemistry by the proteins in the biological electron transfer chains: Examples from photosynthetic reaction centers. J. Bioenerg. Biomembr. 40, 509–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell P. (1975) The protonmotive Q cycle: A general formulation. FEBS Lett. 59, 137–139. [DOI] [PubMed] [Google Scholar]
- Berry E. A.; Guergova-Kuras M.; Huang L.; Crofts A. R. (2000) Structure and function of cytochrome bc complexes. Annu. Rev. Biochem. 69, 1005–1075. [DOI] [PubMed] [Google Scholar]
- Osyczka A.; Moser C. C.; Dutton P. L. (2005) Fixing the Q cycle. Trends Biochem. Sci. 30, 176–182. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Chobot S. E.; Osyczka A.; Wraight C. A.; Dutton P. L.; Moser C. C. (2008) Quinone and non-quinone redox couples in Complex III. J. Bioenerg. Biomembr. 40, 493–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H.; Moser C. C.; Robertson D. E.; Tokito M. K.; Daldal F.; Dutton P. L. (1995) Ubiquinone Pair in the Qo Site Central to the Primary Energy Conversion Reactions of Cytochrome bc1 Complex. Biochemistry 34, 15979–15996. [DOI] [PubMed] [Google Scholar]
- Boveris A.; Cadenas E. (1975) Mitochondrial production of superoxide anions and its relationship to the antimycin insensitive respiration. FEBS Lett. 54, 311–314. [DOI] [PubMed] [Google Scholar]
- Muller F.; Crofts A. R.; Kramer D. M. (2002) Multiple Q-cycle bypass reactions at the Qo site of the cytochrome bc1 complex. Biochemistry 41, 7866–7874. [DOI] [PubMed] [Google Scholar]
- Cape J. L.; Bowman M. K.; Kramer D. M. (2006) Understanding the cytochrome bc complexes by what they don’t do. The Q-cycle at 30. Trends Biochem. Sci. 11, 46–54. [DOI] [PubMed] [Google Scholar]
- Rutherford A. W.; Osyczka A.; Rappaport F. (2012) Back-reactions, short-circuits, leaks and other energy wasteful reactions in biological electron transfer: Redox tuning to survive life in O2. FEBS Lett. 586, 603–616. [DOI] [PubMed] [Google Scholar]
- De Vries S.; Albracht S. P. J.; Berden J. A.; Slater E. C. (1981) A New Species of Bound Ubisemiquinone Anion in QH2:Cytochrome c Oxidoreductase. J. Biol. Chem. 256, 11996–11998. [PubMed] [Google Scholar]
- Junemann S.; Heathcote P.; Rich P. R. (1998) On the mechanism of quinol oxidation in the bc1 complex. J. Biol. Chem. 273, 21603–21607. [DOI] [PubMed] [Google Scholar]
- Cape J. L.; Bowman M. K.; Kramer D. M. (2007) A semiquinone intermediate generated at the Qo site of the cytochrome bc1 complex: Importance for the Q-cycle and superoxide production. Proc. Natl. Acad. Sci. U.S.A. 104, 7887–7892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Osyczka A.; Dutton P. L.; Moser C. C. (2007) Exposing the complex III Qo semiquinone radical. Biochim. Biophys. Acta 1767, 883–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J.; Egawa T.; Yeh S.-R.; Yu L.; Yu C.-A. (2007) Simultaneous reduction of iron-sulfur protein and cytochrome bL during ubiquinol oxidation in cytochrome bc1 complex. Proc. Natl. Acad. Sci. U.S.A. 104, 4864–4869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link T. A. (1997) The role of the “Rieske” iron sulfur protein in the hydroquinone oxidation (Qp) site of the cytochrome bc1 complex. The “proton-gated affinity change” mechanism. FEBS Lett. 412, 257–264. [DOI] [PubMed] [Google Scholar]
- Fournel A.; Gambarelli S.; Guigliarelli B.; More C.; Asso M.; Chouteau G.; Hille R.; Bertrand P. (1998) Magnetic interactions between a [4Fe–4S]1+ cluster and a flavin mononucleotide radical in the enzyme trimethylamine dehydrogenase: A high-field electron paramagnetic resonance study. J. Chem. Phys. 109, 10905–10913. [Google Scholar]
- Calvo R. (2007) EPR measurements of weak exchange interactions coupling unpaired spins in model compounds. Appl. Magn. Reson. 299, 271–299. [Google Scholar]
- Valkova-Valchanova M. B.; Saribas A. S.; Gibney B. R.; Dutton P. L.; Daldal F. (1998) Isolation and Characterization of a Two-Subunit Cytochrome b-c1 Subcomplex from Rhodobacter capsulatus and Reconstitution of Its Ubihydroquinone Oxidation (Qo) Site with Purified Fe-S Protein Subunit. Biochemistry 37, 16242–16251. [DOI] [PubMed] [Google Scholar]
- Hyde J. S.; Pasenkiewicz-Gierula M.; Jesmanowicz A.; Antholine W. E. (1990) Pseudo Field Modulation in EPR Spectroscopy. Appl. Magn. Reson. 1, 483–496. [Google Scholar]
- Altenbach C.; Froncisz W.; Hemker R.; McHaourab H.; Hubbell W. L. (2005) Accessibility of nitroxide side chains: Absolute Heisenberg exchange rates from power saturation EPR. Biophys. J. 89, 2103–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leigh J. S. (1970) ESR Rigid-Lattice Line Shape in a System of Two Interacting Spins. J. Chem. Phys. 52, 2608–2612. [Google Scholar]
- Stoll S.; Schweiger A. (2006) EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 178, 42–55. [DOI] [PubMed] [Google Scholar]
- Malnoë A.; Wollman F.-A.; De Vitry C.; Rappaport F. (2011) Photosynthetic growth despite a broken Q-cycle. Nat. Commun. 2, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dröse S.; Brandt U. (2008) The mechanism of mitochondrial superoxide production by the cytochrome bc1 complex. J. Biol. Chem. 283, 21649–21654. [DOI] [PubMed] [Google Scholar]
- Borek A.; Sarewicz M.; Osyczka A. (2008) Movement of the iron-sulfur head domain of cytochrome bc1 transiently opens the catalytic Qo site for reaction with oxygen. Biochemistry 47, 12365–12370. [DOI] [PubMed] [Google Scholar]
- Sarewicz M.; Borek A.; Cieluch E.; Świerczek M.; Osyczka A. (2010) Discrimination between two possible reaction sequences that create potential risk of generation of deleterious radicals by cytochrome bc1. Implications for the mechanism of superoxide production. Biochim. Biophys. Acta 1797, 1820–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czapla M.; Borek A.; Sarewicz M.; Osyczka A. (2012) Enzymatic activities of isolated cytochrome bc1-like complexes containing fused cytochrome b subunits with asymmetrically inactivated segments of electron transfer chains. Biochemistry 51, 829–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darrouzet E.; Valkova-Valchanova M.; Moser C. C.; Dutton P. L.; Daldal F. (2000) Uncovering the [2Fe2S] domain movement in cytochrome bc1 and its implications for energy conversion. Proc. Natl. Acad. Sci. U.S.A. 97, 4567–4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cammack R.; Rao K. K. K.; Hall D. O. O. (1971) Effects of chaotropic agents on the spectroscopic properties of spinach feredoxin. Biochem. Biophys. Res. Commun. 44, 8–14. [DOI] [PubMed] [Google Scholar]
- Sharp R. E.; Moser C. C.; Gibney B. R.; Dutton P. L. (1999) Primary steps in the energy conversion reaction of the cytochrome bc1 complex Qo site. J. Bioenerg. Biomembr. 31, 225–233. [DOI] [PubMed] [Google Scholar]
- Leggate E. J.; Hirst J. (2005) Roles of the Disulfide Bond and Adjacent Residues in Determining the Reduction Potentials and Stabilities of Respiratory-Type Rieske Clusters. Biochemistry 44, 7048–7058. [DOI] [PubMed] [Google Scholar]
- Zu Y.; Fee J. A.; Hirst J. (2002) Breaking and Re-Forming the Disulfide Bond at the High-Potential, Respiratory-Type Rieske [2Fe-2S] Center of Thermus thermophilus: Characterization of the Sulfhydryl State by Protein-Film Voltammetry. Biochemistry 41, 14054–14065. [DOI] [PubMed] [Google Scholar]
- Robertson D. E.; Prince R. C.; Bowyers J. R.; Katsumi M.; Dutton P. L.; Ohnishi T. (1984) Thermodynamic Properties of the Semiquinone and its Binding Site in the Ubiquinol-Cytochrome c (c2) Oxidoreductase of Respiratory and Photosynthetic Systems. J. Biol. Chem. 259, 1758–1763. [PubMed] [Google Scholar]
- Cape J. L.; Bowman M. K.; Kramer D. M. (2006) Understanding the cytochrome bc complexes by what they don’t do. The Q cycle at 30. Trends Plant Sci. 11, 46–55. [DOI] [PubMed] [Google Scholar]
- Quinlan C. L.; Gerencser A. a.; Treberg J. R.; Brand M. D. (2011) The mechanism of superoxide production by the antimycin-inhibited mitochondrial Q-cycle. J. Biol. Chem. 286, 31361–31372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valgimigli L.; Amorati R.; Funo M. G.; DiLabio G. A.; Pedulli G. F.; Ingold K. U.; Pratt D. A. (2008) The unusual reaction of semiquinone radicals with molecular oxygen. J. Org. Chem. 73, 1830–1841. [DOI] [PubMed] [Google Scholar]
- Orme-Johnson N. R.; Hansen R. E.; Beinert H. (1974) Electron paramagnetic Resonance-detectable Electron Acceptors in Beef Heart Mitochondria. J. Biol. Chem. 249, 1928–1939. [PubMed] [Google Scholar]
- Baker J. E.; Felix C.; Olinger G. N.; Kalyanaraman B. (1988) Myocardial ischemia and reperfusion: Direct evidence for free radical generation by electron spin resonance spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 85, 2786–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shergill J. K.; Cammack R.; Chen J.-H.; Fisher M. J.; Madden S.; Rees H. H. (1995) EPR spectroscopic characterization of the iron-sulphur proteins and cytochrome P-450 in mitochondria from the insect Spodoptera littoralis (cotton leafworm). Biochem. J. 307, 719–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki T.; Wakagi T.; Oshima T. (1995) Resolution of the Aerobic Respiratory System of the Thermoacidophilic Archaeon, Sulfolobus sp. Strain 7. J. Biol. Chem. 270, 30902–30908. [DOI] [PubMed] [Google Scholar]
- Murphy M. P. (2009) How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanciano P.; Lee D.-W.; Yang H.; Darrouzet E.; Daldal F. (2011) Intermonomer electron transfer between the low-potential hemes of cytochrome bc1. Biochemistry 50, 1651–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Świerczek M.; Cieluch E.; Sarewicz M.; Borek A.; Moser C. C.; Dutton P. L.; Osyczka A. (2010) An Electronic Bus Bar Lies in the Core of Cytochrome bc1. Science 329, 451–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.