

Abstract
Drug interactions due to efflux transporters may result in one drug increasing or decreasing the systemic exposure of a second drug. The potential for in vivo drug interactions is estimated through in vitro cell assays. Variability in in vitro parameter determination (e.g., IC50 values) among laboratories may lead to different conclusions in in vivo interaction predictions. The objective of this study was to investigate variability in in vitro inhibition potency determination that may be due to calculation methods. In a Caco-2 cell assay, the absorptive and secretive permeability of digoxin was measured in the presence of spironolactone, itraconazole and vardenafil. From the permeability data, the efflux ratio and net secretory flux where calculated for each inhibitor. IC50 values were then calculated using a variety of equations and software programs. All three drugs decreased the secretory transport of digoxin in a concentration-dependent manner while increasing digoxin’s absorption to a lesser extent. The resulting IC50 values varied according to the parameter evaluated, whether percent inhibition or percent control was applied, and the computational IC50 equation. This study has shown that multiple methods used to quantitate the inhibition of drug efflux in a cell assay can result in different IC50 values. The variability in the results in this study points to a need to standardize any transporter assay and calculation methods within a laboratory and to validate the assay with a set of known inhibitors and non-inhibitors against a clinically relevant substrate.
Electronic supplementary material
The online version of this article (doi:10.1208/s12248-013-9554-7) contains supplementary material, which is available to authorized users.
KEY WORDS: Caco-2, drug interaction, IC50, inhibition, P-glycoprotein
INTRODUCTION
Recognizing the potential for drug–drug interactions (DDI) is an important factor in the development and regulatory review of a new drug (1–3). Transporter-based DDI may be due to competition for a transporter-binding site (by a competitive substrate or an inhibitor) or a change in level of transporter expression (from an inducer). Competition for the same transport pathways among coadministered drugs can result in significant changes in a drug’s absorption, tissue distribution, metabolism, and excretion profiles (4). The objective of DDI studies is to determine the potential for clinical interactions between an investigational drug and other drugs that may be co-administered.
Early identification of compounds that are transporter substrates or inhibitors has become a routine task during the optimization and selection of drug candidate (5). An integrated in vitro to in vivo approach can aid in determining the need for in vivo drug interaction studies. Variability in in vitro parameter determination, such as IC50 or Ki values, among laboratories may lead to different conclusions for in vivo interaction projections using universal criteria such as those proposed in the FDA draft and EMA drug interaction guidances (2,3). Therefore, predictability of in vitro assays is important, as costly clinical studies might be initiated based solely on the in vitro results (1). Therefore, it is important to understand the sources of variability and to use standardized methods within a laboratory to minimize variability.
Bidirectional assays are the most direct and accepted models for evaluating the potential of new drugs as substrates or inhibitors of efflux transporters (2,5–7). In vitro transport assays utilizing the Caco-2 cell line with digoxin as the probe substrate is a well-established method to determine P-glycoprotein (P-gp) inhibition and mimics intestinal interactions (8,9). However, specific assay methodologies vary between laboratories (10,11) along with how the transport kinetics are calculated (1,12). There is no consensus on how to best calculate IC50 values (50% inhibition of substrate transport) from in vitro efflux assays (1,12–14). The calculation methods vary among laboratories which may lead to misinterpretation of the rank order of P-gp inhibitory potency (13). This lack of uniformity allows flexibility by investigators which may lead to potentially erroneous calculations and possibly erroneous interpretation of results (1).
In this study with a Caco-2 cell assay, digoxin served as the probe P-gp substrate with spironolactone, itraconazole, and vardenafil as inhibitor compounds. Digoxin, a narrow therapeutic index drug, is a recommended probe substrate for in vitro and in vivo assays based on known clinical interactions (2,3,8,11,15–20). Digoxin is negligibly metabolized and renally eliminated unchanged, predominately through P-gp-mediated renal tubular secretion (21).
Spironolactone is a potassium-sparing diuretic that that has been shown to inhibit digoxin efflux in transfected cell lines (6,13,22). Spironolactone, given orally before and after digoxin, reduced digoxin renal and nonrenal clearances and prolonged digoxin’s elimination half-life (23–25). Itraconazole is a triazole antifungal agent that is an inhibitor of several P-gp substrates in wild-type and transfected cell assays (6,13,19,26–28) and is considered to be a P-gp substrate (29,30). Clinically, itraconazole increases digoxin plasma levels and AUC (concentration-time curve) while decreasing digoxin’s renal clearance (31,32). Vardenafil is a phosphodiesterase 5 inhibitor that is a potent inhibitor of P-gp in vitro as determined in cytotoxicity, accumulation and ATPase assays (33,34). Vardenafil is a substrate of the efflux transporters P-gp, breast cancer resistance protein, and multidrug resistant protein-2 in Caco-2 and transfected MDCK cells (35,36). However, vardenafil did not significantly alter the steady-state AUC or plasma concentration of digoxin in vivo (37).
In developing and utilizing efflux transporter models, there are a number of variabilities in the assays that can affect the experimental outcomes. Sources of variabilities include the choice of cell line (11,38,39), culture conditions (e.g., cell passage number and monolayer age) (40,41), control substrate or inhibitor specificity (17,42,43), level of transporter expression (44,45), and data analysis (13,19). The objective of this study is to focus on the variability in in vitro inhibition potency determination that may be caused by different calculation methods (e.g., efflux parameters and software programs) from a bidirectional Caco-2 cell assay. Digoxin was utilized as a probe P-gp substrate in the efflux assay with spironolactone, itraconazole and vardenafil as selected inhibitor compounds. Utilizing several analysis methods for the data generated in the transporter assays, IC50 values were then calculated.
MATERIALS AND METHODS
Materials
Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), nonessential amino acids, sodium pyruvate, penicillin, streptomycin, Hank’s balanced salt solution (HBSS), and hydroxyethyl piperazineethanesulfonic acid (HEPES) were from Invitrogen/Life Technologies (Carlsbad, CA). 2-(N-Morpholino)ethanesulfonic acid (MES), spironolactone, itraconazole, digoxin, dimethyl sulfoxide (DMSO), and ethanol were purchased from Sigma-Aldrich (St. Louis, MO). Labeled [3H]-digoxin was from Perkin-Elmer (Waltham, MA) and vardenafil was from Toronto Research Chemicals (Toronto, Canada).
The DMEM culture media contained 4.5 g/L glucose, 10% heat-inactivated FBS, 1% nonessential amino acids, sodium pyruvate, 100 U/mL penicillin, and 100 μg/mL streptomycin. The transport buffers were comprised of HBSS with either 25 mM HEPES (HBSS/HEPES, pH 7.4) or 10 mM 2-(N-morpholino)ethanesulfonic acid (HBSS/MES, pH 6.8). [3H]-Digoxin (40 Ci/mmol) was diluted in ethanol to a concentration of 1,000 μCi/mL and stored at −80°C. It was then diluted to a working stock of 10 μCi/mL in ethanol for use in the transport assays. The stock solutions of unlabeled digoxin (1.0 mM), spironolactone (100 mM), vardenafil (100 mM), and itraconazole (10 mM) were prepared in DMSO, stored at 4°C, and diluted in distilled water to 10 times their final concentrations for use in the transport assays.
Caco-2 Monolayers
The Caco-2 cell monolayers were obtained from Absorption Systems L.P. (Exton, PA). The cells (CRL-2102, American Type Culture Collection, Manassas, VA) were cultured at 37°C with 5% CO2 and plated for monolayer formation according to previously published reports (46,47). Caco-2 cells were seeded at 60,000 cells/cm2 onto collagen-coated, polycarbonate membranes in 12-well Costar® Transwell® plates (1.13 cm2 area, 0.4 μm pore size; Corning Life Sciences, Lowell, MA). The culture medium was changed 24 h after seeding and then changed every other day. The cells were incubated for up to 3 weeks to form confluent monolayers. The passage number of the Caco-2 cells ranged from 61 to 66 and the monolayer age at the time of the transport study was 23 or 24 days.
Caco-2 Transport Assay
The monolayers were removed from the incubator and the medium aspirated from the apical (AP) and basolateral (BL) chambers. Approximately 0.5 mL of HBSS/HEPES buffer was used to wash the cell monolayers. To the AP and BL chambers, 0.5 mL HBSS/MES and 1.5 mL HBSS/HEPES were added, respectively. The monolayers were incubated at 37°C, 5% CO2 for 10–30 min. The TEER was measured in the cell wells and blanks with an epithelial voltohmmeter (EVOM2; World Precision Instruments, Inc., Sarasota, FL). The average blank resistance (Rblank) measurement was subtracted from the cell monolayer insert (Rsample). TEER was calculated according to the following equation.
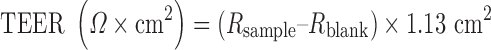 |
1 |
For the Caco-2 cell monolayers, the TEER values were at least 250 Ω × cm2.
The buffer was then removed from the chambers and replaced with HBSSS/MES (AP) or HBSSS/HEPES (BL) containing the inhibitor with control wells containing buffer only. The monolayers were pre-incubated for 30 min at 37°C with the inhibitor solution. The inhibitor solution from the donor chamber was then replaced with a digoxin solution containing the inhibitor. The amount of cold digoxin in each donor chamber was 5 μM with 0.1 μCi/well [3H]-digoxin. During the transport experiment, the receiver chamber contained buffer with or without inhibitor solution and was replaced with the same buffer after a sample was removed.
For absorptive (AP-BL) permeability studies, the buffer solution was aspirated from the AP chamber of the monolayers and replaced with digoxin solution in HBSS/MES, with or without the inhibitor, using four wells for each group. The plates were returned to a 37°C, 5% CO2 incubator on a plate shaker for agitation during the transport experiment. At 30, 60, 90, and 120 min, the insert was transferred to a new well containing HBSS/HEPES buffer, with or without inhibitor. A sample was taken from the donor chamber at 120 min and all receiver and donor samples were stored at −20°C in labeled vials until analysis by liquid scintillation counting (LSC).
For secretive (BL-AP) permeability studies, the buffer was aspirated from the BL chamber of the monolayers and replaced with digoxin solution in HBSS/HEPES, with or without the inhibitor, using four wells for each group. The plates were returned to the incubator on a plate shaker for agitation during the transport experiment. At the time points of 30, 60, 90, and 120 min, 0.5 mL samples were collected from the AP chamber and replenished with an equal volume of HBSS/MES buffer, with or without inhibitor. A sample was taken from the donor chamber at 120 min, and all receiver and donor samples are stored at −20°C in labeled vials until analysis by LSC.
To determine the amount of digoxin in the assay samples, 100 μL was added to 5 mL of scintillation fluid (3a70B™, Research Products International, Corp., Mount Prospect, IL). The samples were analysed in a LS 650 scintillation counter (Beckman Coulter™ Inc., Fullerton, CA) following an internal instrument calibration. The disintegration counts per minute (dpm) were converted to digoxin concentrations (micromolars) taking into account the sample size (100 μL) and volume of the AP (0.5 mL) or BL (1.5 mL) chamber.
Calculations
The apparent permeability (Papp) values were calculated for digoxin in both AP-BL and BL-AP directions in the absence or presence of the inhibitors. Papp (×10−6 cm/s) was calculated from the following equation:
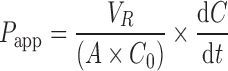 |
2 |
where VR was the volume in the receiver chamber, A the filter surface area (1.13 cm2), C0 the initial digoxin concentration in the donor chamber, and dC/dt was the slope of the linear portion of the concentration vs. time curve. In comparing the Papp results, a two-sample Student’s t test assuming unequal variances was calculated for the rate values with an Excel spreadsheet (Office 2003, Microsoft, Redmond, WA).
The efflux ratio (ER) of digoxin was calculated from the bidirectional Papp values in the BL-AP and AP-BL directions.
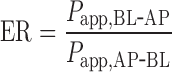 |
3 |
The net secretory flux (NSF; centimeters per second), calculated by subtracting the absorptive from the secretory permeability, measured the net amount of digoxin transported across the monolayers in the BL-AP direction (17,19):
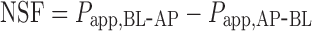 |
4 |
The parameters Papp,BL-AP, ER, and NSF for digoxin were calculated for the experiments in the presence and absence of each inhibitor and designated as original, or non-normalized, data. From these data, percent control and percent inhibition values were calculated for digoxin in the presence of the inhibitors. The percent control was calculated for each inhibitor concentration according to the following equation:
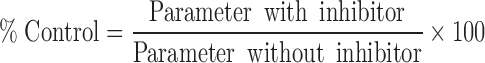 |
5 |
The inhibition of digoxin transport in the presence of the inhibitors was calculated based on the parameter in the absence (negative control) or presence of the inhibitor (1,7,17).
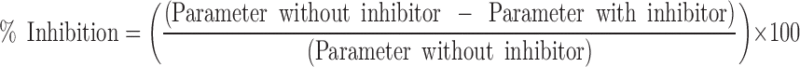 |
6 |
IC50 calculations for Papp, BL-AP, ER, and NSF data with modified Hill equations were completed with Phoenix® WinNonlin® software (version 6.1, Pharsight®, St. Louis, MO), GraphPad Prism® (version 5.0, La Jolla, CA), and SigmaPlot® (version 12.0, Systat Software, Inc., San Jose, CA) software programs. With the WinNonlin® analysis, Eq. 7 was used for the original and %control data and Eq. 8 for the %inhibition data.
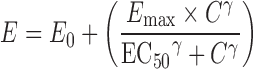 |
7 |
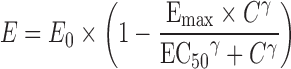 |
8 |
In the equations, E is the effect (original data, %control, or %inhibition), E0 is the baseline value, Emax is the maximal effect, C is the inhibitor concentration, EC50 is the concentration at 50% maximal value, and γ is the sigmoidicity factor. The assumptions are that baseline value (E0) is 0% in the case of %inhibition, and E0 is 100% for the %control data.
In the GraphPad Prism® equation, log-transformed concentration values and the effect data were fitted to a four-parameter logistic equation. The original, %control, or %inhibition data are represented by Y along with their minimal (min) and maximal (max) values. The inhibitor concentration is represented by X, IC50 is the concentration at 50% maximal value, and HillSlope is the slope factor.
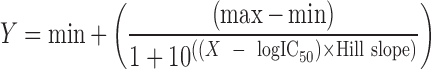 |
9 |
For the SigmaPlot® plot analysis, the same four-parameter logistic nonlinear regression equation, without baseline, was utilized. The original, %control, or %inhibition data are represented by Y along with their minimal (min) and maximal (max) values. The inhibitor concentration is represented by X, IC50 is the concentration at 50% maximal value, and HillSlope is the slope factor.
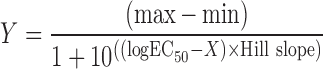 |
10 |
Data fitting for the IC50 values included %coefficient of variance (%CV) and R2 values (see Electronic Supplementary Material).
RESULTS
The average TEER value for the monolayers in the three experiments was 577 ± 56 Ω × cm2. In the experiments, Papp, AP-BL and Papp, BL-AP for digoxin in the absence of an inhibitor was 0.850 ± 0.214 and 8.972 ± 0.404 × 10−6 cm/s, respectively, demonstrating significant efflux through the Caco-2 cell monolayers (p = 0.00004). The ER and NSF for digoxin ranged from 8.60 to 12.67 (10.89 ± 2.06) and 8.00 to 8.34 cm/s (8.12 ± 0.19), respectively, in the absence of the inhibitors (Figs. 1, 2, and 3). All three drugs decreased the Papp, BL-AP, ER, and NSF values for digoxin in a concentration-dependent manner. The drugs increased digoxin’s absorptive permeability with increasing concentrations, although to a lesser extent than their reduction in digoxin’s secretive permeability.
Fig. 1.
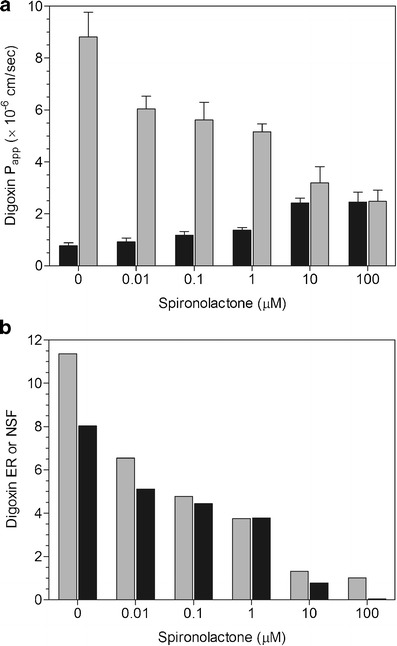
a Inhibition of digoxin P app, AP-BL (black bar) and P app, BL-AP (gray bar) by spironolactone. Mean ± SD of four wells. b Inhibition of digoxin ER (gray bar) and NSF (black bar) by spironolactone
Fig. 2.
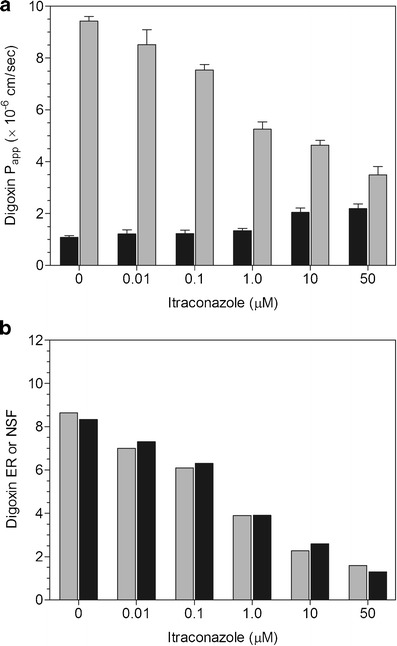
a Inhibition of digoxin P app, AP-BL (black bar) and P app, BL-AP (gray bar) by itraconazole. Mean ± SD of 4 wells. b Inhibition of digoxin ER (gray bar) and NSF (black bar) by itraconazole
Fig. 3.
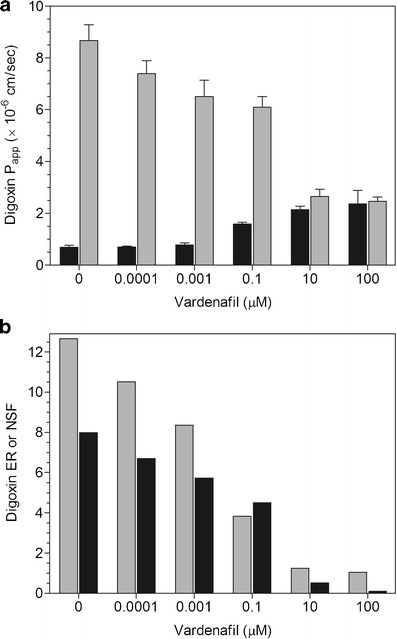
a Inhibition of digoxin P app, AP-BL (black bar) and P app, BL-AP (gray bar) by vardenafil. Mean ± SD of four wells. b Inhibition of digoxin ER (gray bar) and NSF (black bar) by vardenafil
The IC50 values for the inhibitors was highly dependent upon the parameter (Papp, BL-AP, ER, NSF) used in the data analysis along with inclusion or exclusion of the control (absence of inhibitor). When using the four-parameter logistic equation to estimate the IC50 parameter, the inhibitor concentrations are log transformed, and as a result, the zero inhibitor concentration (control) is omitted from the analysis. By contrast, the Hill equation fits untransformed concentration data, inclusive of the baseline effect. Comparing the Hill equation vs. the four-parameter logistic equation, there is significant difference in the estimate of IC50 when the zero concentration of the inhibitor is either included or excluded in the analysis (data not shown). When the zero concentration is included, there was a difference in the IC50 value estimate and goodness of fit measures between the Hill and four-parameter logistic equations as the fit is for a truncated dataset for the four-parameter logistic equation. When the zero concentration was excluded, the Hill and four-parameter logistic equations yielded very similar IC50 estimates and goodness of fit measures.
Spironolactone (0.01–100 μM) caused a concentration-dependent decrease in digoxin Papp, BL-AP while increasing Papp, AP-BL at 10 and 100 μM (Fig. 1a). At 100 μM, there were equivalent AP-BL and BL-AP permeabilities, resulting in ER and NSF values of 1.01 and 0.04, respectively (Fig. 1b). Itraconazole (0.01–50 μM) produced a concentration-dependent decrease in digoxin efflux while increasing Papp, AP-BL at 10 and 50 μM (Fig. 2a). The ER and NSF values for itraconazole were 1.59 and 1.30, respectively at 50 μM (Fig. 2b). Vardenafil (0.0001–100 μM) caused a concentration-dependent decrease in digoxin Papp, BL-AP while increasing Papp, AP-BL at 0.1–100 μM (Fig. 3a). There was unity at 100 μM with equivalent AP-BL and BL-AP permeabilities, generating ER and NSF values of 1.04 and 0.10, respectively (Fig. 3b).
The IC50 values for the drugs varied depending upon which parameter was considered (Papp, BL-AP, ER, and NSF), whether the data was original (non-normalized), %control or %inhibition, and which IC50 software program was utilized. The IC50 values for spironolactone and vardenafil were similar with GraphPad® and SigmaPlot® with values generally lower when estimated with WinNonlin®. For itraconazole, the IC50 values were similar with all three programs.
For spironolactone, more variable IC50 values were noted with WinNonlin® than GraphPad® and SigmaPlot® software (Table I). Overall, IC50 values from the ER data were lower than those from Papp, BL-AP or NSF data. Utilizing the GraphPad® and SigmaPlot® programs, the IC50 values were the same whether derived from the original, %control or %inhibition data with a rank order for spironolactone IC50 values of ER < Papp, BL-AP < NSF.
Table I.
Spironolactone IC50 (micromolars) Values
| Program | Parameter | Original | %Control | %Inhibition |
|---|---|---|---|---|
| WinNonlin® | P app, BL-AP | 9.277 | 3.280 | 1.371 |
| ER | 0.077 | 0.448 | 0.705 | |
| NSF | 9.817 | 2.759 | 0.024 | |
| GraphPad® | P app, BL-AP | 3.279 | 3.308 | 3.308 |
| ER | 0.455 | 0.377 | 0.377 | |
| NSF | 2.760 | 2.840 | 2.840 | |
| SigmaPlot® | P app, BL-AP | 3.280 | 3.280 | 3.280 |
| ER | 0.445 | 0.445 | 0.445 | |
| NSF | 2.761 | 2.761 | 2.761 |
For itraconazole, all three programs had similar IC50 values with a rank order of Papp, BL-AP < ER < NSF (Table II). In the WinNonlin® analyses, IC50 values based on the NSF and Papp, BL-AP data were the same for all calculation sets. There was some variability with the ER IC50 values based on original, %control, or %inhibition data. Utilizing the GraphPad® program, IC50 values for itraconazole using the different datasets were comparable with each parameter. With the SigmaPlot® analysis, the IC50 values were the same whether derived from the original, %control or %inhibition data.
Table II.
Itraconazole IC50 (micromolars) Values
| Program | Parameter | Original | %Control | %Inhibition |
|---|---|---|---|---|
| WinNonlin® | P app, BL-AP | 0.376 | 0.378 | 0.378 |
| ER | 0.572 | 0.686 | 0.629 | |
| NSF | 0.859 | 0.861 | 0.860 | |
| GraphPad® | P app, BL-AP | 0.376 | 0.408 | 0.408 |
| ER | 0.687 | 0.701 | 0.701 | |
| NSF | 0.862 | 0.797 | 0.797 | |
| SigmaPlot® | P app, BL-AP | 0.376 | 0.376 | 0.376 |
| ER | 0.686 | 0.686 | 0.686 | |
| NSF | 0.862 | 0.862 | 0.862 |
For vardenafil, GraphPad® and SigmaPlot® had similar IC50 values while those calculated with WinNonlin® had more variability (Table III). In the WinNonlin® analyses, the IC50 values for the NSF and Papp, BL-AP parameters was higher than the other calculation method. In the SigmaPlot® and GraphPad® analyses, the IC50 values were the same whether derived from the original, %control, or %inhibition data for each parameter. Overall, the rank of vardenafil IC50 values with the three programs was ER < NSF < Papp, BL-AP.
Table III.
Vardenafil IC50 (micromolars) Values
| Program | Parameter | Original | %Control | %Inhibition |
|---|---|---|---|---|
| WinNonlin® | P app, BL-AP | 9.864 | 0.485 | 0.540 |
| ER | 0.005 | 0.004 | 0.005 | |
| NSF | 1.619 | 0.415 | 0.396 | |
| GraphPad® | P app, BL-AP | 0.485 | 0.539 | 0.539 |
| ER | 0.004 | 0.004 | 0.004 | |
| NSF | 0.416 | 0.396 | 0.396 | |
| SigmaPlot® | P app, BL-AP | 0.485 | 0.485 | 0.485 |
| ER | 0.004 | 0.004 | 0.004 | |
| NSF | 0.416 | 0.416 | 0.416 |
DISCUSSION
There is interest in predicting potential clinical interactions between an investigational new drug and other coadministered drugs during drug development and regulatory review (2,3,20). A bidirectional assay in Caco-2 or transporter overexpressed cell (e.g., MDCK or LLC-PK1) lines is a preferred method for in vitro evaluation of a new drug as an efflux substrate or inhibitor. Besides differences in test systems with P-gp-expressing cell lines, differing methodology and data processing approaches play a role in the interlaboratory variability of P-gp inhibition data (48,49).
In a Caco-2 cell assay with digoxin as the substrate, secretory permeability (Papp, BL-AP), ER, and NSF were used as parameters to calculate P-gp inhibitor IC50 values with a number of equations in commercial software packages. Overall, the drugs’ IC50 values for each parameter were the same no matter if original, %control, or %inhibition data were used in the SigmaPlot® analyses. More variability was noted with the WinNonlin® calculation within data sets, especially with spironolactone. For vardenafil and spironolactone, the IC50 values from SigmaPlot® and GraphPad® were similar and usually higher than those with WinNonlin®. With itraconazole, the IC50 values with each parameter were essentially the same for the three programs. In looking at the different parameters, the ER data yielded the lowest IC50 values for spironolactone and vardenafil whereas Papp, BL-AP had the lowest values for itraconazole.
Literature reports on the inhibition of P-gp by compounds have utilized a variety of equations and software packages to calculate IC50 values. These include programs for Excel™ (1,48,50,51), GraphPad® (12,13,39,52–54), WinNonlin® (12,49,55), GraFit (6,12,15–19,56), and SigmaPlot® (12,14). The calculations in these publications were some modification of the Hill equation that included an effect (%inhibition or %control), maximal effect or range of effect, inhibitor concentration, EC50 or IC50, and a Hill coefficient or factor. The effect of an inhibitor on a P-gp substrate was measured based upon the Papp, BL-AP (1,6,12,13,18,57), ER (1,12–14,19,48–51,57), or NSF (1,12,14,17,19,52,53).
Variability in IC50 determinations results in challenges to use an “universal” cutoff criteria as prosposed in the FDA’s draft DDI guidance (2) to project in vivo interaction potential based on in vitro inhibition data. Comparisons have been made in publications as to how the different paramaeters affect the IC50 calculations. Different conclusions concerning the inhibitory potential of drug could be derived from the same experimental dataset if different calculation methods are utilized (1,12). For example, a multi-laboratory study with several cell systems with digoxin as the substrate found great variability in IC50 values with equations based on either Papp, NSF or ER (12). IC50 values for 16 inhibitors derived from ER or Papp, AP-BL data were lower than those from Papp,BL-AP or NSF data (12). Perloff et al. used ER and NSF parameters in their calculation of IC50 values for digoxin transport in Caco-2 and MDR1-LLC-PK1 cells with 16 inhibitor compounds (51). The IC50 values based on NSF were on an average 2.5-fold higher than corresponding values based upon ER (51). Lin et al. calculated IC50 values for ketoconazole with two P-gp substrates, with those based on ER less than those from Papp calculations (57).
Balimane et al. examined different %inhibition calculation methods in a Caco-2 cell P-gp inhibition assay using the parameters Papp, AP-BL, Papp, BL-AP, and ER in the presence and absence of inhibitors (1). For the over 50 tested compounds, the %inhibition results notably differed depending on the calculation method used (1). When the IC50 values were compared from the different %inhibition equations, there was a 4-fold variation based on the calculation method used, with significantly lower values observed with the ER calculation method (1). The commonly used calculation method of ER requires studies in bidirectional mode and may lead to results that are oversensitive in the “low” inhibition range which can potentially lead to false positives (1).
Sugimoto et al. found that the IC50 values generated for over 20 drugs from Papp, BL-AP were larger than those from ER for compounds tested, and there was a positive correlation between these IC50 values (13). Cook et al. found that for the majority of the 30 compounds they tested in a Caco-2 cell assay, IC50 values were 2-fold more potent by the ER method (ER < NSF) (19). For example, the IC50 value for itraconazole was 6 and 2 μM based upon NSF and ER, respectively (19).
The calculation of IC50 values based on Papp, BL-AP provides a simple experimental design with confluent cell monolayers in the presence of increasing inhibitor concentrations (12). It is recommended that multiple inhibitor concentrations are assayed to define both the upper and lower plateaus of the response curve (12). However, it should be noted that a drug’s aqueous solubility may limit the lower plateau (maximal effect) and a positive control inhibitor can be used to define complete inhibition of P-gp activity in the cells (12,56).
Bentz et al. suggest that the differences in IC50 values for ER vs. Papp, BL-AP for digoxin is likely due to the decline of Papp, BL-AP is greater than the increase in Papp, AP-BL for any given inhibitor concentration (12). This minimizes the effect of the increase in Papp, AP-BL on the decline in Papp, BL-AP and thus on the IC50 value obtained for the net flux equation (12). We also found that the change in Papp, BL-AP was greater that for Papp, AP-BL with the three inhibitors (Figs. 1, 2, and 3). IC50 values determined from ER data often appear more potent, which may be due to a mathematical artifact as ER can never numerically achieve zero (19).
Additionally, the difference between the IC50 values generated from ER and Papp, BL-AP may be attributed to the fact that the inhibitory rate based on Papp, BL-AP does not disregard the influence of passive permeability of the substrate (13). Using loperamide as the P-gp substrate, Taur et al. speculated that the ER approach may overestimate the P-gp inhibition potency since ER was calculated from the ratio of Papp, BL-AP normalized by Papp, AP-BL, thus the inhibition effect of inhibitors may have been accounted twice under the calculation (14).
Besides the sources of variability as discussed above, from our study we found that there is some degree of variability in the parameter estimates among different software programs, such as those obtained via WinNonlin® in comparison to SigmaPlot® and GraphPad® (Tables I, II, and III) because of the nature of the software’s fitting equation. SigmaPlot® and GraphPad® utilize a four-parameter logistic equation, while the data was fitted to a Hill equation with cooperativity coefficient in WinNonlin®. If there is a perfectly sigmoidal curve, the software will not matter since the estimate of the IC50 is highly dependent on the plateau regions (both at low response levels and saturation of response). While the fits are expected to be essentially identical for a well characterized sigmoidal dose response, high variability in estimates are often observed with a dose response curve that is not perfectly sigmoidal. In cases where the baseline effect is quite distinct from the effect of the lowest concentration, IC50 estimates are greatly influenced by the omission of the baseline effect. Conversely, this influence is minimal when the data assumes a complete sigmoid shape (i.e., low concentrations of inhibitor have an effect that is similar to baseline). In addition, the log transformation also changes the assumption of normal distribution of error and may result in smaller erroneous estimates. Therefore, it may be appropriate to use a Hill equation rather than a four-parameter logistic equation, especially when the shape of response curve is not perfectly sigmoidal.
Based upon this limited data set, suggestions for the calculation of IC50 values include the use of original (non-normalized) data and the fitting of nontransformed data. The use of original data results in fewer assumptions, i.e., 100% (maximum) or 0% (minimum) activity, which could affect the E0 or Emax estimate, and IC50 by extension. Fitting of untransformed data does not violate the assumption that errors are normally distributed as transformation alters this distribution.
CONCLUSIONS
This study highlights the factors that investigators need to consider when using software programs to calculate IC50 values based on the shape of their inhibition curve. The variability in the IC50 results in this study reinforces the need to standardize any transporter assay and calculation methods within a laboratory (1,12). The assay should be validated with acceptance criteria for a set of known inhibitors and non-inhibitors against a clinically relevant substrate. From such a study, a single parameter (e.g., Papp, BL-AP or ER) and a Hill-type IC50 equation ought to be utilized to determine whether a new drug is a transporter inhibitor and confidently predict if a clinical DDI study is necessary for development and regulatory purposes.
ELECTRONIC SUPPLEMENTARY MATERIAL
(DOCX 70 kb)
ACKNOWLEDGMENTS
The authors wish to thank Drs. Aspandiar G. Katki, Neil R. Hartman, and Robert J. Parker at the FDA and Ms. Lisa Murray at Absorption Systems for their assistance during the conduct of the study.
Disclaimer
The findings and conclusions in this article have not been formally disseminated by the Food and Drug Administration and should not be construed to represent any Agency determination or policy.
REFERENCES
- 1.Balimane PV, Marino A, Chong S. P-gp inhibition potential in cell-based models: which “calculation” method is the most accurate? AAPS J. 2008;10:577–86. doi: 10.1208/s12248-008-9068-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Food and Drug Administration (2012). Draft guidance. Guidance for industry: drug interaction studies—study design, data analysis, and implications for dosing and labeling. Available from http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm064982.htm. Accessed 28 October 2013.
- 3.European Medicine Agency. Guideline on the investigation of drug interactions. 2012. http://www.ema.europa.eu/ema/index.jsp?curl=pages/includes/document/document_detail.jsp?webContentId=WC500129606&mid=WC0b01ac058009a3dc. Accessed 28 October 2013.
- 4.Han HK. Role of transporters in drug interactions. Arch Pharmacol Res. 2011;34:1865–77. doi: 10.1007/s12272-011-1107-y. [DOI] [PubMed] [Google Scholar]
- 5.Balimane PV, Han YH, Chong S. Current industrial practices of assessing permeability and P-glycoprotein interaction. AAPS J. 2006; 8(1):article 1. [DOI] [PMC free article] [PubMed]
- 6.Keogh JP, Kunta JR. Development, validation and utility of an in vitro technique for assessment of potential clinical drug–drug interactions involving P-glycoprotein. Eur J Pharm Sci. 2006;27:543–54. doi: 10.1016/j.ejps.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 7.Kim RB, Wandel C, Leake B, Cvetkovic M, Fromm MF, Dempsey PJ, et al. Interrelationships between substrates and inhibitors of human CYP3A and P-glycoprotein. Pharm Res. 1999;16:408–14. doi: 10.1023/a:1018877803319. [DOI] [PubMed] [Google Scholar]
- 8.Polli JW, Wring SA, Humphreys JE, Huang L, Morgan JB, Webster LO, et al. Rational use of in vitro P-glycoprotein assays in drug discovery. J Pharmacol Exp Ther. 2001;299:620–8. [PubMed] [Google Scholar]
- 9.Collett A, Tanianis-Hughes J, Carlson GL, Harwood MD, Warhurst G. Comparison of P-glycoprotein-mediated drug-digoxin interactions in Caco-2 with human and rodent intestine: relevance to in vivo prediction. Eur J Pharm Sci. 2005;26:386–93. doi: 10.1016/j.ejps.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 10.Volpe DA. Variability in Caco-2 and MDCK cell-based intestinal permeability assays. J Pharm Sci. 2008;97:712–25. doi: 10.1002/jps.21010. [DOI] [PubMed] [Google Scholar]
- 11.Taub ME, Podila L, Ely D, Almeida I. Functional assessment of multiple P-glycoprotein (P-gp) probe substrates: influence of cell line and modulator concentration on P-gp activity. Drug Metab Dispos. 2005;33:1679–87. doi: 10.1124/dmd.105.005421. [DOI] [PubMed] [Google Scholar]
- 12.Bentz J, O’Connor MP, Bednarczyk D, Coleman J, Lee C, Palm J, et al. Variability in P-glycoprotein inhibitory potency (IC50) using various in vitro experimental systems: implications for universal digoxin DDI risk assessment decision criteria. Drug Metab Dispos. 2013;41:1347–66. doi: 10.1124/dmd.112.050500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sugimoto H, Matsumoto S, Tachibana M, Niwa S, Hirabayashi H, Amano N, et al. Establishment of in vitro P-glycoprotein inhibition assay and its exclusion criteria to assess the risk of drug-drug interaction at the drug discovery stage. J Pharm Sci. 2011;100:4013–23. doi: 10.1002/jps.22652. [DOI] [PubMed] [Google Scholar]
- 14.Taur JS, Rodriguez-Proteau R. Effects of dietary flavonoids on the transport of cimetidine via P-glycoprotein and cationic transporters in Caco-2 and LLC-PK1 cell models. Xenobiotica. 2008;38:1536–50. doi: 10.1080/00498250802499467. [DOI] [PubMed] [Google Scholar]
- 15.Zhang L, Zhang YD, Strong JM, Reynolds KS, Huang SM. A regulatory viewpoint on transporter-based drug interactions. Xenobiotica. 2008;38:709–24. doi: 10.1080/00498250802017715. [DOI] [PubMed] [Google Scholar]
- 16.Zhang L, Zhang Y, Zhao P, Huang SM. Predicting drug–drug interactions: an FDA perspective. AAPS J. 2009;11:300–6. doi: 10.1208/s12248-009-9106-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fenner KS, Troutman MD, Kempshall S, Cook JA, Ware JA, Smith DA, et al. Drug-drug interactions mediated through P-glycoprotein: clinical relevance and in vitro–in vivo correlation using digoxin as a probe drug. Clin Pharmacol Ther. 2009;85:173–81. doi: 10.1038/clpt.2008.195. [DOI] [PubMed] [Google Scholar]
- 18.Rautio J, Humphreys JE, Webster LO, Balakrishnan A, Keogh JP, Kunta JR, et al. In vitro P-glycoprotein inhibition assays for assessment of clinical drug interaction potential of new drug candidates: a recommendation for probe substrates. Drug Metab Dispos. 2006;34:786–92. doi: 10.1124/dmd.105.008615. [DOI] [PubMed] [Google Scholar]
- 19.Cook JA, Feng B, Fenner KS, Kempshall S, Liu R, Rotter C, et al. Refining the in vitro and in vivo critical parameters for P-glycoprotein, [I]/IC50 and [I]2/IC50, that allow for the exclusion of drug candidates from clinical digoxin interaction studies. Mol Pharm. 2010;7:398–411. doi: 10.1021/mp900174z. [DOI] [PubMed] [Google Scholar]
- 20.Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KLR, Chu X, et al. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9:215–36. doi: 10.1038/nrd3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gheorghiade M, Adams KF, Colucci WS. Digoxin in the management of cardiovascular disorders. Circulation. 2004;109:2959–64. doi: 10.1161/01.CIR.0000132482.95686.87. [DOI] [PubMed] [Google Scholar]
- 22.Nakamura T, Kakumoto M, Yamashita K, Takara K, Tanigawara Y, Sakaeda T, et al. Factors influencing the prediction of steady state concentrations of digoxin. Biol Pharm Bull. 2001;24:403–8. doi: 10.1248/bpb.24.403. [DOI] [PubMed] [Google Scholar]
- 23.Fenster PE, Hager WD, Goodman MM. Digoxin–quinidine–spironolactone interaction. Clin Pharmacol Ther. 1984;36:70–3. doi: 10.1038/clpt.1984.141. [DOI] [PubMed] [Google Scholar]
- 24.Hedman A, Angelin B, Arvidsson A, Dahlqvist R. Digoxin-interactions in man: spironolactone reduces renal but not biliary digoxin clearance. Eur J Clin Pharmacol. 1992;42:481–5. doi: 10.1007/BF00314854. [DOI] [PubMed] [Google Scholar]
- 25.Waldorff S, Andersen JD, Heebøll-Nielsen N, Nielsen OG, Moltke E, Sørensen U, et al. Spironolactone-induced changes in digoxin kinetics. Clin Pharmacol Ther. 1978;24:162–7. doi: 10.1002/cpt1978242162. [DOI] [PubMed] [Google Scholar]
- 26.Ito S, Woodland C, Sarkadi B, Hockmann G, Walker SE, Koren G. Modeling of P-glycoprotein-involved epithelial drug transport in MDCK cells. Am J Physiol Renal Physiol. 1999;277:F84–96. doi: 10.1152/ajprenal.1999.277.1.F84. [DOI] [PubMed] [Google Scholar]
- 27.Karyekar CS, Eddington ND, Garimella TS, Gubbins PO, Dowling TC. Evaluation of P-glycoprotein-mediated renal drug interactions in an MDR1-MDCK model. Pharmacother. 2003;23:436–42. doi: 10.1592/phco.23.4.436.32125. [DOI] [PubMed] [Google Scholar]
- 28.Takara K, Tanigawara Y, Komada F, Nishiguchi K, Sakaeda T, Okumura K. Cellular pharmacokinetic aspects of reversal effect of itraconazole on P-glycoprotein-mediated resistance of anticancer drugs. Biol Pharm Bull. 1999;22:1355–9. doi: 10.1248/bpb.22.1355. [DOI] [PubMed] [Google Scholar]
- 29.Saad AH, DePestel DD, Carver PL. Factors influencing the magnitude and clinical significance of drug interactions between azole antifungals and select immunosuppressants. Pharmacotherapy. 2006;26:1730–44. doi: 10.1592/phco.26.12.1730. [DOI] [PubMed] [Google Scholar]
- 30.Dodds-Ashley E. Management of drug and food interactions with azole antifungal agents in transplant recipients. Pharmacotherapy. 2010;30:842–54. doi: 10.1592/phco.30.8.842. [DOI] [PubMed] [Google Scholar]
- 31.Jalava KM, Partanen J, Neuvonen PJ. Itraconazole decreases renal clearance of digoxin. Ther Drug Monit. 1997;19:609–13. doi: 10.1097/00007691-199712000-00001. [DOI] [PubMed] [Google Scholar]
- 32.Partanen J, Jalava KM, Neuvonen PJ. Itraconazole increases serum digoxin concentration. Pharmacol Toxicol. 1996;79:274–6. doi: 10.1111/j.1600-0773.1996.tb00273.x. [DOI] [PubMed] [Google Scholar]
- 33.Ding PR, Tiwari AK, Ohnuma S, Lee JW, An X, Dai CL, et al. The phosphodiesterase-5 inhibitor vardenafil is a potent inhibitor of ABCB1/P-glycoprotein transporter. PLoS One. 2011;6:e19329. doi: 10.1371/journal.pone.0019329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi Z, Tiwari AK, Patel AS, Fu LW, Chen ZS. Roles of sildenafil in enhancing drug sensitivity in cancer. Cancer Res. 2011;71:3735–8. doi: 10.1158/0008-5472.CAN-11-0375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jian-Wei D, Song I, Shon J, Shin J. Characterization of efflux transport of PDE5 inhibitors, sildenafil, vardenafil, and udenafil. Clin Pharmacol Ther. 2009;85(Suppl. 1):S49. [Google Scholar]
- 36.Choi MK, Song IS. Characterization of efflux transport of the PDE5 inhibitors, vardenafil and sildenafil. J Pharm Pharmacol. 2012;64:1074–83. doi: 10.1111/j.2042-7158.2012.01498.x. [DOI] [PubMed] [Google Scholar]
- 37.Rohde G, Bauer R-J, Unger S, Ahr G, Wensing G. Vardenafil, a new selective PDE5 inhibitor, produces no interaction with digoxin. Pharmacotherapy. 2001;21:1254. [Google Scholar]
- 38.Tang F, Horie K, Borchardt RT. Are MDCK cells transfected with the human MDR1 gene a good model of the human intestinal mucosa? Pharm Res. 2002;19:765–72. doi: 10.1023/a:1016140429238. [DOI] [PubMed] [Google Scholar]
- 39.Glavinas H, von Richter O, Vojnits K, Mehn D, Nagy T, Janossy J, et al. Calcein assay: a high-throughput method to assess P-gp inhibition. Xenobiotica. 2011;41:712–9. doi: 10.3109/00498254.2011.587033. [DOI] [PubMed] [Google Scholar]
- 40.Siissalo S, Laitinen L, Koljonen M, Vellonen KS, Kortejärvi H, Urtti A, et al. Effect of cell differentiation and passage number on the expression of efflux proteins in wild type and vinblastine-induced Caco-2 cell lines. Eur J Pharm Biopharm. 2007;67:548–54. doi: 10.1016/j.ejpb.2007.03.017. [DOI] [PubMed] [Google Scholar]
- 41.Kamiyama E, Sugiyama D, Nakai D, Miura S, Okazaki O. Culture period-dependent change of function and expression of ATP-binding cassette transporters in Caco-2 cells. Drug Metab Dipos. 2009;37:1956–62. doi: 10.1124/dmd.109.027490. [DOI] [PubMed] [Google Scholar]
- 42.Mease K, Sane R, Podila L, Taub ME. Differential selectivity of efflux transporter inhibitors in Caco-2 and MDCK-MDR1 monolayers: a strategy to assess the interaction of a new chemical entity with P-gp, BCRP, and MRP2. J Pharm Sci. 2012;101:1888–97. doi: 10.1002/jps.23069. [DOI] [PubMed] [Google Scholar]
- 43.Patil AG, D’Souza R, Dixit N, Damre A. Validation of quinidine as a probe substrate for the in vitro P-gp inhibition assay in Caco-2 cell monolayer. Eur J Drug Metab Pharmacokinet. 2011;36:115–9. doi: 10.1007/s13318-011-0046-9. [DOI] [PubMed] [Google Scholar]
- 44.Shirasaka Y, Sakane T, Yamashita S. Effect of P-glycoprotein expression levels on the concentration-dependent permeability of drugs to the cell membrane. J Pharm Sci. 2008;97:553–65. doi: 10.1002/jps.21114. [DOI] [PubMed] [Google Scholar]
- 45.Hayeshi R, Hilgendorf C, Artursson P, Augustijns P, Brodin B, Dehertogh P, et al. Comparison of drug transporter gene expression and functionality in Caco-2 cells from 10 different laboratories. Eur J Pharm Sci. 2008;35:383–96. doi: 10.1016/j.ejps.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 46.Li J, Volpe DA, Wang Y, Zhang W, Bode C, Owen A, et al. Use of transporter knockdown Caco-2 cells to investigate the in vitro efflux of statin drugs. Drug Metab Dispos. 2011;39:1196–202. doi: 10.1124/dmd.111.038075. [DOI] [PubMed] [Google Scholar]
- 47.Darnell M, Karlsson JE, Owen A, Hidalgo IJ, Li J, Zhang W, et al. Investigation of the involvement of P-glycoprotein and multidrug resistance-associated protein 2 in the efflux of ximelagatran and its metabolites by using short hairpin RNA knockdown in Caco-2 cells. Drug Metab Dispos. 2010;38:491–7. doi: 10.1124/dmd.109.029967. [DOI] [PubMed] [Google Scholar]
- 48.Elsby R, Surry DD, Smith VN, Gray AJ. Validation and application of Caco-2 assays for the in vitro evaluation of development candidate drugs as substrates or inhibitors of P-glycoprotein to support regulatory submissions. Xenobiotica. 2008;38:1140–64. doi: 10.1080/00498250802050880. [DOI] [PubMed] [Google Scholar]
- 49.Kamiyama E, Nakai D, Mikkaichi T, Okudaira N, Okazaki O. Interaction of angiotensin II type 1 receptor blockers with P-gp substrates in Caco-2 cells and hMDR1-expressing membranes. Life Sci. 2010;86:52–8. doi: 10.1016/j.lfs.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 50.Elsby R, Gillen M, Butters C, Imisson G, Sharma P, Smith V, et al. The utility of in vitro data in making accurate predictions of human P-glycoprotein-mediated drug-drug interactions: a case study for AZD5672. Drug Metab Dispos. 2011;39:275–82. doi: 10.1124/dmd.110.035881. [DOI] [PubMed] [Google Scholar]
- 51.Perloff ES, Mason AK, Kapadnis S, Khun W, Fox LG, Stresser DM. Comparison of digoxin efflux inhibition in Caco-2 and MDR1-LLC-PK1 cell monolayers. Drug Metab Rev. 2011;43(Suppl 2):193. [Google Scholar]
- 52.Eberl S, Renner B, Neubert A, Reisig M, Bachmakov I, König J, et al. Role of P-glycoprotein inhibition for drug interactions evidence from in vitro and pharmacoepidemiological studies. Clin Pharmacokinet. 2007;46:1039–49. doi: 10.2165/00003088-200746120-00004. [DOI] [PubMed] [Google Scholar]
- 53.Choo EF, Leake B, Wandel C, Imamura H, Wood AJJ, Wilkinson GR, et al. Pharmacological inhibition of P-glycoprotein transport enhances the distribution of HIV-1 protease inhibitors into brain and testes. Drug Metab Dispos. 2000;28:655–60. [PubMed] [Google Scholar]
- 54.von Richter O, Glavinas H, Krajcsi P, Liehner S, Siewert B, Zech K. A novel screening strategy to identify ABCB1 substrates and inhibitors. Naunyn-Schmied Arch Pharmacol. 2009;379:11–26. doi: 10.1007/s00210-008-0345-0. [DOI] [PubMed] [Google Scholar]
- 55.Hsiao P, Bui T, Ho RJY, Unadkat JD. In vitro-to-in vivo prediction of P-glycoprotein-based drug interactions at the human and rodent blood–brain barrier. Drug Metab Dispos. 2008;36:481–4. doi: 10.1124/dmd.107.018176. [DOI] [PubMed] [Google Scholar]
- 56.Weiss J, Haefeli WE. Evaluation of inhibitory potencies for compounds inhibiting P-glycoprotein but without maximum effects: F2 values. Drug Metab Dispos. 2006;34:203–7. doi: 10.1124/dmd.105.007377. [DOI] [PubMed] [Google Scholar]
- 57.Lin J, Grimm S. IC50 determinations in an in vitro P-glycoprotein inhibition assay: Papp values vs. efflux ratios. Drug Metab Rev. 2009;41(Suppl 3):76. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 70 kb)