

Abstract
The plants used in Ayurvedic medicine, which has been practiced in India for thousands of years for the treatment of a variety of disorders, are rich in chemicals potentially useful for prevention and treatment of cancer. Withania somnifera (commonly known as Ashwagandha in Ayurvedic medicine) is one such medicinal plant whose anticancer value was realized over four decades ago after isolation of a crystalline steroidal compound (withaferin A) from the leaves of this shrub. The root and leaf extracts of W. somnifera are shown to confer protection against chemically-induced cancers in experimental rodents, and retard tumor xenograft growth in athymic mice. Anticancer effect of W. somnifera is generally attributable to steroidal lactones collectively referred to as withanolides. Withaferin A (WA) appears most active against cancer among structurally divergent withanolides isolated from the root or leaf of W. somnifera. Cancer-protective role for WA has now been established using chemically-induced and oncogene-driven rodent cancer models. This review summarizes the key in vivo preclinical studies demonstrating anticancer effects of WA. Molecular targets and mechanisms likely contributing to the anticancer effects of WA are also discussed. Finally, challenges in clinical development of WA for the prevention and treatment of cancer are highlighted.
KEY WORDS: apoptosis, autophagy, cell cycle, chemoprevention, signal transduction, withaferin A
INTRODUCTION
Plants are a rich source of chemicals potentially useful for prevention and treatment of cancer as exemplified by clinical success of many natural products including taxol and vinca-alkaloids (1,2). Plants used in Ayurvedic medicine, a system of traditional medicine native to the Indian subcontinent, are particularly attractive for identification of novel cancer preventive and therapeutic agents (3). Withania somnifera (also known as Ashwagandha, Indian ginseng, or Indian winter cherry) is one such shrub of the Solanaceae family with multiple medicinal properties (4–6). Ashwagandha is an essential ingredient in hundreds of Ayurvedic medicine formulations (5). Anticancer value of W. somnifera was realized over four decades ago after isolation of a crystalline steroidal compound (withaferin A; hereafter, abbreviated as WA) from the leaves of this plant (7). Extracts from the leaf, root, and other parts of W. somnifera have been shown to exhibit a variety of pharmacological effects including anticancer property (4–6). Root and leaf extracts of W. somnifera are shown to confer protection against chemically-induced cancers in experimental rodents (8–13), and retard growth of transplanted tumors in mice (14–18). For example, daily administration of W. somnifera root extract (150 mg/kg body weight) for 155 days resulted in 21–23% decrease in mammary tumor multiplicity and burden in rats induced by the chemical carcinogen methylnitrosourea (13). Alleviation of cancer chemotherapy-induced toxicity and fatigue by administration of Withania extract has also been reported (19,20). Anticancer effect of W. somnifera is credited to steroidal lactones collectively known as withanolides (21,22). The chemical constituents of W. somnifera and biosynthesis of withanolides have been elegantly reviewed by Mirjalili et al. (4). WA [(4β,5β,6β,22R)-4,27-dihydroxy-5,6:22,26-diepoxyergosta-2,24-diene-1,26-dione; structure of WA is shown in Fig. 1] appears relatively more active against cancer than other withanolides isolated from this plant. This review summarizes the key in vivo preclinical studies to merit further development of WA for prevention and treatment of cancer. Molecular mechanisms underlying anticancer effects of WA are also discussed.
Fig. 1.
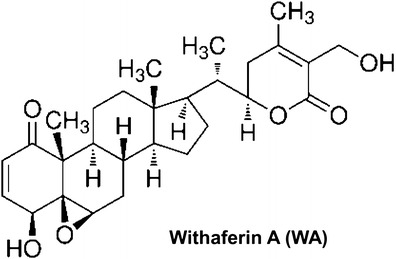
Chemical structure of withaferin A (WA)
IN VIVO EFFICACY AND PHARMACOKINETIC STUDIES
Inhibition of Transplanted Tumor Cell Growth by WA in Mice
Table I summarizes the in vivo studies demonstrating anticancer effects of WA. The first published report on in vivo anticancer effect of WA isolated from the leaves of W. somnifera Dunal dates back to 1967 (7). WA administration was shown to inhibit growth of Ehrlich ascites carcinoma in more than 50% of the mice (7). The LD50 of WA after i.p. injection in Swiss mice weighing 18–22 g was shown to be about 400 mg/kg (7). The same group of investigators demonstrated later that a single i.p. injection of 30 mg WA/kg body weight prolonged survival of S-180 ascites-bearing mice (23). A single i.p. treatment with 10–60 mg WA/kg 24 h after tumor cell injection dose-dependently inhibited growth of mouse Ehrlich ascites carcinoma cells and increased tumor-free survival in Swiss albino mice (24). Radiation sensitization by WA administration in Ehrlich ascites carcinoma bearing mice was also reported in this study (24). Similar growth inhibitory and/or radiosensitizing effects of WA were observed against B16F1 mouse melanoma and fibrosarcoma (25,26). The in vivo efficacy studies with some mechanistic insights began to appear in the literature after 2006. For example, daily i.p. injection of 4 and 8 mg WA/kg to PC-3 human prostate cancer cell-bearing male nude mice resulted in 54–70% growth inhibition (27). Interestingly, complete tumor regression in one WA-treated mouse was observed after 7 days of treatment (27). The WA-mediated growth retardation of PC-3 xenografts in vivo was associated with inhibition of proteasomal chymotrypsin-like activity and CD31 protein expression (a marker for neoangiogenesis), and induction of multidomain proapoptotic protein Bax, nuclear factor-κB (NF-κB) regulator IκB-α, and cyclin-dependent kinase inhibitor p27 (27). Consistent with Bax induction results, tumors from the WA-treated mice exhibited increased apoptosis and caspase-3 activation compared with those from the vehicle-treated control mice (27). In another study, intratumoral injections of WA significantly inhibited growth of PC-3 xenografts in association with prostate apoptosis response-4 (Par-4) upregulation and increased apoptosis (28). Our own laboratory was the first to demonstrate in vivo anticancer activity of WA against a human breast cancer (MDA-MB-231) xenograft (29). The MDA-MB-231 xenograft growth inhibition by WA was accompanied by reduced proliferating cell nuclear antigen expression (indicative of reduced cellular proliferation) and increased apoptosis (29). Daily i.p. administration of 8 mg WA/kg body weight to female nude mice with subcutaneously implanted DRO81-1 medullary thyroid cancer cells caused inhibition of tumor growth and metastasis leading to increased survival (30). The tumors from WA-treated mice exhibited a decrease in levels of total and phosphorylated RET (rearranged during transfection) (30). A decrease in levels of full-length caspase-3 protein in the tumor (suggesting cleavage) and serum calcitonin were also observed in WA-treated group compared with control (30). In vivo growth inhibition of soft tissue sarcoma cells implanted in female SCID mice by WA administration (2 mg/kg, i.p., daily) was accompanied by reduced cell proliferation and neoangiogenesis, increased apoptosis, and degradation of vimentin (31). WA administration (2 and 4 mg/kg, i.p., every other day) to female Balb/c mice orthotopically implanted with 4T1 mouse mammary tumor cells resulted in suppression of primary tumor growth in association with increased ser56 phosphorylation of vimentin (32). Molecular changes indicative of inactivation of human papilloma virus oncoproteins E6 and E7, activation of tumor suppressor p53 and pRb, growth arrest (cyclin B1, p34 cdc2, and p21), reduced cell proliferation (PCNA), and increased apoptosis were observed in WA-mediated suppression of CaSki cervical carcinoma xenograft growth (33). Using a 92.1 uveal melanoma xenograft model, Samadi et al. (34) showed tumor growth inhibition after 8 mg WA/kg/day i.p. with 29% of the mice exhibiting a complete clinical response. In the 12 mg/kg/day group, three mice (43%) exhibited progressive disease after discontinuation of WA treatment, but complete response was noted in one mouse (34). However, the higher dose group demonstrated increased toxicity and mortality (34). Inhibition of mouse mesothelioma xenograft growth in Balb/c mice after daily i.p. injection with 5 mg WA/kg correlated with suppression of the proteasomal chymotrypsin-like activity and c-Myc protein expression but elevation of Bax, p27, and CARP-1 (Cell cycle and Apoptosis Regulatory Protein-1) protein levels (35). Increased apoptosis in the tumors from WA-treated mice relative to vehicle-treated controls was also noted in this study (35). The WA-mediated inhibition of Panc-1 pancreatic tumor xenograft growth was demonstrated in another study (36). Contrary to above mentioned studies, growth of HT1080 fibrosarcoma was not inhibited after oral administration of 10 mg WA/kg body weight (18).
Table I.
Key in vivo Studies Demonstrating Anticancer Effects of WA
| Cell/tumor type | Dose/route | Antitumor effect | Notable in vivo mechanistic correlates | Ref. |
|---|---|---|---|---|
| Inhibition of transplanted tumors | ||||
| S-180 ascites | 30 mg/kg, i.p. | ↑Survival | Vacuolization of cytoplasm, distention or dissolution of mitochondrial cristae, disruption of microtubules of mitotic spindles | (23) |
| ↓Tumor cells | ||||
| PC-3 (prostate) | 4 or 8 mg/kg, i.p., daily | ↓Tumor growth | ↓Proteasomal activity, ↑Bax, ↑IκB-α, ↑p27 ↑caspase-3 activity, ↑apoptosis, ↓CD31 | (27) |
| PC-3 (prostate) | 5 mg/kg, intratumor, 5 days per week | ↓Tumor growth | ↑Par-4, ↑apoptosis | (28) |
| MDA-MB-231 (breast) | 4 mg/kg, i.p., 5 days per week | ↓Tumor growth | ↓PCNA, ↑apoptosis | (29) |
| DRO81-1 (medullary thyroid) | 8 mg/kg, i.p., daily | ↓Tumor growth | ↓Total and phospho-RET | (30) |
| ↑Survival | ↓Serum calcitonin | |||
| HT-1080, SKLMS-1 (soft tissue sarcoma) | 2 mg/kg, i.p., daily | ↓Tumor growth | ↓PCNA, ↓CD31, ↑apoptosis | (31) |
| ↓Tumor recurrence | ↑Caspase-3 activation, vimentin degradation | |||
| 4T1 (mouse breast) | 2 and 4 mg/kg, i.p., every other day | ↓Tumor growth | ↑Ser56 phosphorylation of vimentin | (32) |
| CaSki (cervical) | 8 mg/kg, i.p. every other day | ↓Tumor growth | ↓E6 and E7, ↑p53, ↑pRb, ↑cyclin B1, ↓p34cdc2, | (33) |
| ↑p21, ↓PCNA, ↓phospho-Stat3, ↑Bax, ↓Bcl-2 | ||||
| 92.1 (uveal melanoma) | 8 or 12 mg/kg, i.p., daily | ↓Tumor growth | (34) | |
| AB12 (mesothelioma) | 5 mg/kg, i.p., daily | ↓Tumor growth | ↓Proteasomal chymotrypsin-like activity | (35) |
| ↑Bax, ↑p27, ↑apoptosis, ↓c-Myc, ↑CARP-1 | ||||
| Panc-1 (pancreatic) | 3 and 6 mg/kg, i.p., 2 times per week | ↓Tumor growth | (36) | |
| Cancer prevention | ||||
| DMBA (oral) | 20 mg/kg, oral 3 times/week | ↓Oral cancer (100%) | ↓Lipid peroxidation, ↑antioxidant defense | (37) |
| MMTV-neu (breast) | 100 μg/mouse, i.p., 3 times/week | ↓Macroscopic and ↓microscopic tumor burden | ↑Apoptosis, ↓complex III, ↓glycolysis, | (40) |
| ↓Tricarboxylic acid cycle | ||||
PAR-4 prostate apoptosis response-4; PCNA proliferating cell nuclear antigen; pRb retinoblastoma; RET rearranged during transfection; Cdc2 cell-division cycle 2; Stat3 signal transducer and activator of transcription 3; CARP-1 Cell cycle and Apoptosis Regulatory Protein-1; DMBA 7,12-dimethylbenz[a]anthracene; MMTV-neu mouse mammary tumor virus-neu
The take home message from these in vivo preclinical studies is that: (a) WA administration inhibits in vivo growth of a variety of tumor xenografts, (b) i.p. doses up to 8 mg WA/kg body weight are well-tolerated by the mice, and (c) WA-mediated inhibition of tumor xenograft growth is associated with reduced tumor cell proliferation and increased apoptosis.
Prevention of Chemically-Induced and Oncogene-Driven Cancer by WA in Rodents
A handful of studies have now demonstrated in vivo cancer chemopreventive effects WA (37–40). Oral administration of 20 mg WA/kg body weight amazingly conferred complete protection against 7,12-dimethylbenz[a]anthracene-induced oral cancer in hamsters in a circadian time-dependent manner (37–39). Oral cancer prevention by WA in this model correlated with (a) attenuation of carcinogen-induced lipid peroxidation, (b) restoration of plasma and erythrocyte levels of antioxidant defense molecules (reduced glutathione, vitamin C, and vitamin E) and antioxidative enzymes (superoxide dismutase, catalase and glutathione peroxidase), and (c) alterations in p53 and Bcl-2 (37,38). Our laboratory is the first to document in vivo cancer prevention by WA in a clinically relevant transgenic mouse model of breast cancer (40). In these studies, the incidence and burden of mammary cancer were scored in female mouse mammary tumor virus-neu (MMTV-neu) transgenic mice after 28 weeks of treatment with 100 μg WA/mouse, i.p., three times per week (40). Tumor incidence was not decreased after WA administration, but the mean palpable (macroscopic) tumor weight in the WA treatment group was lower by 50% compared with control (P = 0.03 by two-sided Student’s t test) (40). Furthermore, the mean area of microscopic invasive carcinoma was lower by 95.14% in WA group compared with control group (40). Mechanistically, the breast cancer prevention by WA in MMTV-neu mice was associated with increased apoptosis, inhibition of complex III activity of the mitochondrial respiration, and reduced levels of glycolysis and tricarboxylic acid cycle intermediates (40). Proteomic profiling using tumor tissues confirmed downregulation of many glycolysis-related proteins in the tumor of WA-treated mice compared with control, including M2-type pyruvate kinase, phosphoglycerate kinase, and fructose-bisphosphate aldolase A isoform 2 (40). Cluster analysis of the tumor protein changes indicated statistically significant enrichment of gluconeogenesis as well as annexin repeat proteins (40). On the other hand, cellular proliferation and neoangiogenesis was not affected by WA treatment (40). It is important to determine the efficacy of WA in other rodent models of chemically-induced as well as oncogene-driven cancers for a full appreciation of its preventive role.
Antimetastatic Effect of WA
In a mouse model involving tail veil injection of STS26T soft tissue sarcoma cells, Lahat et al. (31) observed marked inhibition of pulmonary metastasis multiplicity and area after WA administration (2 mg WA/kg/day, i.p.) beginning 10 days after tumor cell injection. Antimetastatic effect of WA (4, 2, 1, 0.5, and 0.1 mg/kg body weight every other day for 1 month) was also evaluated after implantation of 4T1 mouse breast cancer cells into the mammary fat pad of Balb/c mice (32). The number of metastatic lung nodules in WA-treated mice was reduced significantly in a dose-dependent manner starting at the 0.1 mg/kg dose (32). In vivo antimetastatic effect of WA has also been observed in a mouse model of medullary thyroid carcinoma cells (30). Our own breast cancer prevention study in the MMTV-neu mice revealed a significant decrease in incidence of pulmonary metastasis after WA administration (40).
Pharmacokinetics of WA
Thaiparambil et al. (32) were the first to study pharmacokinetic behavior of WA in mice. Following a single i.p. injection of 4 mg WA/kg body weight to female Balb/c mice, the maximum plasma concentration (Cmax) of WA was 1.8 μM (32). The plasma half life of WA was about 1.3 h with an exposure AUC0−t of 1.09 μM · h (32). Pharmacokinetics of WA was also studied after oral administration of W. somnifera extract at a dose of 1,000 mg/kg (equivalent to 0.4585 mg/kg of WA) in mice (41). The Cmax of WA was 16.69 ± 4.02 ng/ml with observed Tmax (time to reach Cmax) of 20 min (41). However, the pharmacokinetic behavior of WA in humans is yet to be determined. This knowledge is essential for further clinical development of WA for prevention and treatment of cancers.
ANTICANCER TARGETS/MECHANISMS OF WA
Cellular proliferation, evasion of apoptosis, and neoangiogenesis are some of the hallmarks of cancer (42), and WA has the ability to alter these processes in vitro as well as in vivo. Known molecular targets of WA and mechanisms potentially contributing to its anticancer effect are briefly summarized below. The purpose of this review is not to catalog every molecular alteration reported in response to WA treatment in cancer cells, but to highlight its suppressive effects on key oncogenic processes (cell growth, apoptosis, migration, and invasion) and pathways likely contributing to prevention and treatment of cancer. We also highlight studies that have incorporated functional experiments (gene knockdown or overexpression and pharmacologic inhibitors) to gain insights into the mechanisms by which WA treatment affects growth, apoptosis, and cell migration/invasion in cancer cells.
Cell Cycle Arrest by WA Treatment
Initial clue for cell cycle arrest by WA emerged from ultrastructural studies of S-180 tumors (23). These investigators observed disruption of microtubules of mitotic spindles (23). Subsequent studies have provided insights into the mechanism by which WA causes cell cycle arrest. For example, exposure of MDA-MB-231 and MCF-7 human breast cancer cells to WA resulted in a concentration- and time-dependent increase in G2-M fraction, that was accompanied by a marked decrease in levels of cyclin-dependent kinase 1 (Cdk1), cell division cycle 25C (Cdc25C), and/or Cdc25B proteins (43). Overexpression of Cdc25C conferred partial but significant protection against WA-mediated G2-M phase arrest (43). The breast cancer cells exposed to WA were also arrested in mitosis as evidenced by increased Ser10 phosphorylated histone H3 and accumulation of securin (43). Our observations of G2-M arrest by WA treatment in MCF-7 cells were since then confirmed by other investigators (44). The S and/or G2-M phase arrest upon treatment with WA has also been reported in CaSki cervical cancer cell line (33), 92.1 and MEL290 uveal melanoma cells (34), human head and neck cancer cells (JMAR, MDA1986, UMSCC-2, and JHU011) (45), glioblastoma cells (46,47), and CaOV3 and SKOV3 ovarian carcinoma cells (48). The mechanism(s) underlying cell cycle arrest by WA was not investigated in most of these studies (44–47), but consistent with our data in breast cancer cells (43) the CaSki cells exhibited dose-dependent accumulation of cyclin B1 (indicative of M phase arrest), downregulation of Cdk1, and a decrease in complex formation between cyclin B1 and Cdk1 upon treatment with WA (33). Induction of the Cdk inhibitor p21 was also observed in WA-treated CaSki cells (33). Likewise, in agreement with the results in MDA-MB-231 and MCF-7 breast cancer cells (43), WA treatment caused downregulation of Cdc25C in CaOV3 and SKOV3 cells (48), and induction of cyclin B1 in glioblastoma and ovarian cancer cells (47,48). Furthermore, WA treatment synergized with sorafenib (a multikinase-targeted inhibitor) in causing G2-M arrest in papillary and anaplastic thyroid cancer cells (49). Together, these results indicate that G2-M arrest by WA treatment is not a cell line-specific phenomenon.
Apoptosis Induction by WA
Apoptosis induction by WA was initially observed in leishmania, a unicellular kinetoplastid protozoan parasite (50). Since then, in vitro and/or in vivo apoptosis induction upon treatment with WA has been reported for a variety of cancer types including prostate cancer (27,28), breast cancer (29,44,51,52), soft tissue sarcoma (31), cervical carcinoma (33), uveal melanoma (34), mesothelioma (35), pancreatic cancer (36), head and neck carcinoma (45), glioblastoma (47), leukemia (53–56), renal carcinoma (57), and melanoma (58). Several different mechanisms have been proposed to explain proapoptotic response to WA in cancer cells (27–29,31,33–35,53–58). Instead of indexing each of the molecular alterations associated with WA-induced apoptosis, this article summarizes mechanisms that have been confirmed in more than one cancer cell type and/or substantiated by functional experiments using genetic and/or pharmacological approaches. Some of the existing discrepancies in results from different laboratories are also highlighted.
Role of Reactive Oxygen Species (ROS) in Apoptosis Induction by WA
Pro-oxidant effect of WA was initially shown in leishmanial cells where loss of membrane potential after treatment with this agent resulted in reactive oxygen species (ROS) production leading to depletion of reduced glutathione and oxidative DNA lesions (50). A role for ROS in pro-apoptotic response to WA has also been established in several different types of cancer cells, including leukemia, breast, cervical, and renal cancer cells (51,53,58–61). For example, WA treatment resulted in ROS generation in myeloid leukemia (HL-60), acute lymphoblastic leukemia (Molt-4), prostate carcinoma (PC-3 and DU145), and cervical carcinoma (HeLa) cells (53). A role for ROS in WA-mediated sensitization of cancer cells to apoptosis induction by radiation and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) has also been suggested (60,61). Sensitization of cancer cells to TRAIL-induced apoptosis by WA treatment was associated with ROS-mediated upregulation of death receptor 5, but not death receptor 4, and these effects were attenuated by exogenous catalase and superoxide dismutase (60). Endoplasmic reticulum stress induced by WA in renal cells was associated with ROS production (57). However, three critical issues related to the role of ROS in WA-induced apoptosis deserve attention:
Several studies have utilized N-acetylcysteine (NAC) to establish the role of ROS in proapoptotic effect of WA (53,58,60,61). Because WA is electrophilic (62), NAC is not ideal for this purpose. The protection conferred by NAC may simply be a consequence of the non-availability of free WA. We have employed genetic approaches involving overexpression of Cu,Zn-superoxide dismutase (SOD) to study the role of ROS in apoptosis induction by WA using breast cancer cells (51). Overexpression of Cu,Zn-SOD conferred significant protection against WA-mediated ROS generation as well as apoptosis in MDA-MB-231 and MCF-7 cells (51).
It is still unclear if ROS generation by WA is selective for cancer cells. Widodo et al. (59) showed ROS generation upon WA treatment in both MCF-7 cells and in a normal human fibroblast line (TIG-3). On the other hand, studies from our own laboratory have indicated that a normal human mammary epithelial cell line (HMEC) is resistant to ROS production by WA in comparison with MCF-7 cells (51). We also found different levels of oxidative stress markers in normal mammary ductal epithelium and breast tumor cells in the WA-treated MMTV-neu mice (40). A normal human prostate epithelial cell line (Pz-HPV-7) immortalized by human papilloma virus 18 also exhibited resistance to WA-induced apoptosis compared with prostate cancer cells (28). It is possible that normal fibroblasts are more sensitive to ROS production by WA compared with normal epithelial cells (51,59). Of note, suicidal death of erythrocytes upon treatment with WA has been observed at a concentration (≥10 μM) (63) that is pharmacologically not attainable (32). Nevertheless, further studies are needed to ascertain whether resistance to ROS generation upon treatment with WA is unique to epithelial cells such as HMEC (51).
The mechanism underlying ROS generation by WA has been studied thoroughly only in breast cancer cells (51). A model emerging from our own studies using wild-type and mitochondrial DNA deficient (Rho0) variants of MDA-MB-231 and MCF-7 cells indicates that WA treatment inhibits complex III activity and oxidative phosphorylation leading to ROS-dependent activation of Bak and ensuing cell death (51). The mammary cancer prevention by WA in MMTV-neu transgenic mice was also associated with in vivo inhibition of complex III activity in the tumor (40). However, the mechanism by which WA inhibits complex III activity is still elusive.
Role of Par-4 in WA-Induced Apoptosis
Several studies have suggested involvement of Par-4 in apoptosis regulation by WA (28,64,65). A role for Par-4 protein in apoptosis induction by WA was first proposed by Srinivasan et al. (28) in androgen receptor (AR) negative prostate cancer cells. These investigators also proposed a model where WA-mediated induction of Par-4 protein led to inhibition of NF-κB and subsequently downregulation of the anti-apoptotic protein Bcl-2 (28). Interestingly, the Par-4 induction and apoptotic cell death resulting from WA exposure was attenuated by overexpression of wild-type AR (28). WA-mediated induction of PAR-4 has also been demonstrated in neuroblastoma and cholangiocarcinoma cells (64,65). However, the precise role of Par-4 in apoptosis regulation by WA was not thoroughly investigated in neuroblastoma and cholangiocarcinoma cells (64,65). It is also unclear if Par-4 protein is involved in apoptosis regulation by WA in other cancer types.
Inhibition of Proteasome in WA-Induced Apoptosis
Mohan et al. (66) were the first to show inhibition of ubiquitin-mediated proteasome pathway by WA treatment in human umbilical vein endothelial cells. Yang et al. showed convincingly that tumor proteasome is a target of WA in prostate cancer cells (27), and these observations were subsequently extended to mesothelioma cells (35). WA was shown to be a potent inhibitor of chymotrypsin-like activity in human prostate cancer cultures and xenografts (27). Inhibition of prostate tumor proteasome activity by WA resulted in accumulation of proteasome target protein Bax (35). Thus, inhibition of proteasome and proapoptotic effect of WA may be linked through Bax stabilization. In this context, we have shown previously that knockdown of Bax and Bak protein confers significant protection against WA-induced apoptosis in MCF-7 cells (51). Carefully designed kinetic studies are needed to test whether inhibition of proteasome by WA treatment is linked to its pro-oxidant effect. Molecular docking studies have suggested that WA likely inhibits proteasome irreversibly and with a high rate due to acylation of the N-terminal Thr1 of the β-5 subunit (67).
Role of Bcl-2 Family Proteins in WA-Induced Apoptosis
The Bcl-2 family proteins play an important role in regulation of apoptosis by different stimuli (68). Induction of pro-apoptotic Bcl-2 family members (Bax, Bak, and Bim), translocation of Bax to mitochondria and/or downregulation of anti-apoptotic Bcl-2 proteins (Bcl-2, Bcl-xL) upon WA treatment has been observed in a variety of cultured cancer cells (27–29,33,35,48,53–55,58). However, the in vivo evidence for WA-mediated induction of Bax and downregulation of Bcl-2 is available only in cervical cancer xenografts (33). In leukemia cells, WA-mediated apoptosis was attenuated by Bcl-2 overexpression (54). Similar protective effect of Bcl-2 overexpression on apoptosis induction by WA was observed in Caki renal cancer cells (61). In human breast cancer cells, FOXO3a-dependent induction of Bim was implicated in apoptosis regulation by WA (29). WA-mediated apoptosis in breast cancer cells was significantly attenuated by RNA interference of both Bim and FOXO3a (29). Once again, similar functional studies are needed in other cancer cell types to establish the generality of these mechanisms. In addition, it remains to be seen if WA administration affects protein levels of Bcl-2 family members in vivo in different types of cancer cells.
Role of p53 Tumor Suppressor in Apoptosis Regulation by WA
The tumor suppressor role for p53 and its involvement in apoptosis regulation is well established (69). WA-mediated stabilization of p53 protein level and/or increased Ser15 phosphorylation of this tumor suppressor have been observed in breast, cervical, and renal cancer cells (33,44,61,70). WA-mediated induction of p53 in vivo has been reported in cervical cancer xenografts (33). In cervical cancer cells, apoptosis induction by WA was partially but statistically significantly attenuated by small interfering RNA (siRNA) knockdown of p53 (33). We also observed a modest but significant protection against proapoptotic effect of WA by RNA interference of p53 in MCF-7 human breast cancer cell line (70). Collectively, these results indicate that p53 status can influence cell death induction by WA.
Suppression of Inhibitor of Apoptosis Family Proteins by WA
The inhibitor of apoptosis (IAP) family proteins, including X-linked inhibitor of apoptosis, survivin, and cIAP1/cIAP2 play an important role in apoptosis regulation by inhibiting caspases (71,72). WA-mediated suppression of IAPs has been shown in breast cancer cells in vitro (XIAP, survivin, and cIAP2) and in vivo (survivin) (44,52) and leukemia cells (XIAP) (54). However, the functional evidence for involvement of IAPs in apoptosis regulation by WA is limited to MCF-7 and MDA-MB-231 breast cancer cells (52). WA treatment decreases protein and mRNA levels of XIAP, survivin, and cIAP2 in a dose-dependent manner (52). Overexpression of these proteins was protective to varying extent against apoptosis induction by WA (52). Docking studies suggested strong binding affinity for WA with cIAP1 (73). Further work is needed to determine the contribution of IAP family proteins in apoptotic cell death induction by WA in other types of cancer cells.
Role of Mitogen-Activated Protein Kinases in Apoptosis Regulation by WA
Mitogen-activated protein kinases (MAPK) are known to play an important role in apoptosis regulation by different agents (74,75). Mandal et al (55) were the first to study role of MAPK in apoptotic response to WA using leukemic cells of lymphoid and myeloid origin. This study showed activation of p38 MAPK upon treatment with WA (55). Moreover, inhibition of p38 activation by an inhibitor as well as p38 protein knockdown resulted in significant protection against apoptosis by WA (55). Contribution of extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinases (JNK), and/or p38 MAPK in pro-apoptotic response to WA has also been suggested in breast cancer cells, head and neck cancer cells, and glioblastoma cells (44,45,47,76). One study showed transient activation (increased phosphorylation) of p38 MAPK after 1–3 h treatment with 2.5 μM WA in MCF-7 breast cancer cells that was not due to an increase in level of p38 MAPK protein (44). In fact, the protein level of p38 MAPK was decreased after 6-h treatment with WA (44). The effect of WA on ERK or JNK was not studied by these investigators (44). The same group of investigators noted activation of ERK1/2 without any effect on protein level upon treatment of some head and neck cancer cells and glioblastoma cells with WA (45,47). Once again, the effect of WA on other MAPK was not determined in head and neck cancer cells or in glioblastoma cells (45). In addition, these studies lacked functional experiments (pharmacological inhibitors or overexpression of dominant negative mutants) to allow meaningful conclusions concerning the role of MAPK in apoptosis by WA (44,45). WA treatment resulted in increased phosphorylation of ERK1/2, JNK, and p38 MAPK in U937 human leukemia cells (54). Pharmacological inhibition of JNK resulted in potentiation of WA-induced apoptosis as evidenced by enrichment of sub-G0-G1 population whereas p38 MAPK inhibition had no effect of cell death resulting from WA treatment (54). On the other hand, partial but significant attenuation of WA-mediated apoptosis was discernible in the presence of an inhibitor of ERK (54). These observations indicated opposing roles of MAPK activation in apoptosis induction by WA in U937 cells (54).
More recently, we have used MCF-7 (estrogen responsive) and SUM159 (triple negative) human breast cancer cells to systematically study the role of MAPK in apoptosis induction by WA (76). Exposure of both cells to WA resulted in increased phosphorylation of ERK, JNK, and p38 MAPK, but these effects were relatively more pronounced in the former cell line than in SUM159 (76). Activation of ERK, but not JNK or p38 MAPK, resulting from WA treatment was partially attenuated by overexpression of Mn-SOD (76). Cell death resulting from WA treatment in MCF-7 cells was significantly augmented by pharmacological inhibition of ERK and p38 MAPK (76). Interestingly, the WA-induced apoptosis in MCF-7 cells was partially but significantly blocked in the presence of a JNK-specific inhibitor (76). Inhibition of ERK or JNK had no effect on WA-induced apoptosis in SUM159 cells (76). Collectively, it is reasonable to propose that role of MAPK in apoptosis induction by WA is cell line dependent.
Suppression of Oncogenic Signaling Pathways by WA Treatment
WA has the ability to target multiple oncogenic signaling pathways often hyperactive in human cancers, including NF-κB, Akt, signal transducer and activator of transcription 3 (Stat3), and estrogen receptor-α (ER-α) to name a few. Most of these studies have relied on protein expression changes (total and phosphorylated proteins) to draw major conclusions. Furthermore, data on in vivo relevance of the WA-mediated changes in oncogenic pathways is still limited. A brief review of the effects of WA on various oncogenic pathways follows:
WA-Mediated Inhibition of NF-κB and Akt in Cancer Cells
The role for NF-κB and Akt in cancer is well-established and reviewed elsewhere (77,78). WA-mediated inhibition of NF-κB, suppression of nuclear translocation of p65 subunit of NF-κB, and/or downregulation of p65 has been reported in prostate cancer cells and soft tissue sarcoma cells (27,28,31). Likewise, inhibition of total and/or Ser473 phosphorylated Akt upon treatment with WA has been shown in medullary thyroid cancer cells (30), soft tissue sarcoma cells (31), uveal melanoma cells (34), pancreatic cancer cells (36), head and neck cancer cells (45), glioblastoma cells (47), ovarian cancer cells (48), an leukemia cells (54). In some models, the WA-mediated suppression of phospho-Akt levels seems cell line specific phenomenon. For example, WA treatment caused a decrease in level of phospho-Akt in U87 but not in U251 glioblastoma cells (47). The mechanism by which WA treatment inhibits phosphorylation of Akt is not fully understood, but in soft tissue sarcoma cells this effect was dependent on degradation of vimentin (31). It is unclear if the vimentin-mediated suppression of phospho-Akt levels by WA treatment is unique to soft tissue sarcoma. WA-mediated increase in Ser473 phosphorylation of Akt was visually obvious in some cancer cells (34) but lack of quantitation hindered clear data interpretation. Nevertheless, overexpression of dominant active Akt conferred protection against apoptosis induction by WA in leukemia cells (54). Similar functional studies are needed in other types of cancer cells to establish if inhibition of Akt and/or NF-κB are key determinants of apoptosis induction by WA.
Suppression of Stat3 by WA Treatment
Stat3 activation plays an important role in tumorigenesis (79). We were the first to systematically study the role of Stat3 in anticancer effects of WA in human breast cancer cells (80). We found that WA treatment inhibits constitutive as well as interkeukin-6 (IL-6) inducible activation of Stat3 and its upstream kinase Jak2 (80). The IL-6 stimulation, either before or after treatment with WA, did not have any appreciable effect on WA-induced apoptosis (80). On the other hand, IL-6 stimulated activation of Stat3 conferred modest protection against inhibition of cell invasion by WA (80). WA-mediated suppression of constitutive or IL-6 inducible activation of Stat3 has also been reported in cervical and renal cancer cells (33,81). However, in vivo suppression of Stat3 phosphorylation by WA treatment has only been shown in cervical cancer xenografts (33). Unlike our conclusions in breast cancer cells (80), WA-induced apoptosis was significantly suppressed by overexpression of Stat3 in Caki renal cancer cells (81).
Downregulation of ER-α by WA Treatment in Breast Cancer Cells
The ER-α is a well-accepted therapeutic target for breast cancer (82). Our laboratory was the first to show suppression of ER-α by WA in human breast cancer cells (70), and the major conclusions from this study were: (a) growth inhibition as well as apoptosis induction by WA in MCF-7 cells was significantly attenuated in the presence of 17β-estradiol (E2), (b) WA treatment downregulated ER-α protein (but not ER-β) and this effect was markedly abolished in the presence of E2, and (c) ectopic expression of ER-α in the MDA-MB-231 cell line conferred partial but significant protection against WA-mediated apoptosis (70). Our observation of WA-mediated suppression of ER-α protein has since been confirmed by another laboratory (44). In our study, we were unable to rescue loss of ER-α protein in the presence of a proteasomal inhibitor (MG132) indicating transcriptional repression of this hormone receptor (70). Indeed, we found transcriptional repression of ER-α in WA-treated breast cancer cells. On the other hand, Zhang et al (44) have concluded that WA treatment causes aggregation followed by proteasomal degradation of ER-α (44). Zhang et al (44) did not determine the effect of WA on ER-α mRNA, but further investigation is needed to resolve this discrepancy.
Effect of WA on Notch Signaling
The Notch pathway is implicated in cell fate determination (proliferation and differentiation) and tumorigenesis (83,84). The Notch signaling is mediated by four receptors (Notch1-4) and five ligands (83,84). Koduru et al (85) observed suppression of full-length Notch-1 after treatment with WA in human colon cancer cells. We also noticed a decrease in levels of both full-length and cleaved (active form) Notch-1 in WA-treated breast cancer cells (86). At the same time, exposure of MDA-MB-231 and MCF-7 human breast cancer cells to pharmacological concentrations of WA resulted in cleavage of Notch2 as well as Notch4, which was accompanied by transcriptional activation of Notch (86). The WA-mediated activation of Notch in breast cancer cells was associated with induction of γ-secretase complex components Presenilin1 and/or Nicastrin (86). Inhibition of MDA-MB-231 and MDA-MB-468 cell migration resulting from WA exposure was significantly augmented by RNA interference of Notch2 and Notch4 (86). Thus, activation of Notch2 and Notch4 by WA impeded its inhibitory effect on breast cancer cell migration. Zhang et al (48) reported either increase or decrease in levels of cleaved Notch1 and suppression of cleaved Notch3 in WA-treated ovarian cancer cells. However, activation of Notch 2 or Notch4 by WA has not been demonstrated in other types of cancer cells.
Role of Autophagy in Anticancer Effects of WA
Autophagy is an evolutionarily conserved physiological process for bulk degradation of cellular macromolecules and various organelles and a cancer therapeutic target (87). For some agents, autophagy induction is cytoprotective against apoptotic cell death (88). On the other hand, autophagy has been shown to contribute to overall cell death by some agents (87,89). We have shown recently that exposure of MDA-MB-231 and MCF-7 human breast cancer cells as well as a spontaneously immortalized and non-tumorigenic normal human mammary epithelial cell line (MCF-10A) to WA results in autophagy (90). The MDA-MB-231 xenografts from WA-treated mice also exhibit markers of autophagy (90). Interestingly, the WA-mediated inhibition of MDA-MB-231 and MCF-7 cell viability was not affected either by pharmacological suppression of autophagy or genetic repression of autophagy by transfection with Atg5 siRNA (90). In another study, WA treatment sensitized ovarian cancer cells to doxorubicin by causing ROS-dependent autophagy in vitro and in vivo (91). However, the role of autophagy, if any, in anticancer effects of WA in other types of cancer cells remain to be determined.
WA Causes Endoplasmic Reticulum Stress
Treatment of renal carcinoma Caki cells with WA resulted in endoplasmic reticulum (ER) stress characterized by phosphorylation of eukaryotic initiation factor-2α (eIF-2α), ER stress-specific XBP1 splicing, and upregulation of glucose-regulated protein-78 (57). In addition, WA treatment caused upregulation of CAAT/enhancer-binding protein-homologous protein (CHOP) (57). Transfection with CHOP siRNA or inhibition of caspase-4 activity attenuated WA-induced apoptosis (57). However, WA-mediated induction of ER stress has not yet been reported in other types of cancer cells.
Combination Chemotherapies with WA
Several studies have shown sensitization of cultured cancer cells to chemotherapy drugs and radiation in the presence of WA (49,60,92–94). For example, WA and sorafenib exhibited synergistic efficacy in thyroid cancer cells (49). Likewise, WA sensitized Caki renal cancer cells to TRAIL-induced apoptosis due to ROS-dependent upregulation of DR5 and downregulation of c-FLIP (60). WA-mediated sensitization of ovarian cancer cells to cisplatin-induced apoptosis in vitro has also been shown (92). However, none of these studies provided any in vivo evidence for synergy between WA and chemotherapy drugs (49,60,92), without which the therapeutic value of these observations is still unknown.
CONCLUDING REMARKS AND FUTURE DIRECTIONS
Despite promising preclinical cellular and in vivo data summarized in this article, several steps are necessary for clinical development of WA for prevention and/or therapy of cancers. First, long-term toxicological evaluation of WA is necessary to establish its safety profile. Second, pharmacodynamics biomarkers predictive of WA tissue exposure and possibly response are necessary. Many studies have shown chemotherapy sensitization by WA in cultured cancer cells, but the in vivo significance of these findings is still unclear. Finally, it is clear that WA targets multiple molecules/pathways that may be cell line-specific. Further work is also needed to systematically explore this possibility. For example, a role for ROS in WA-induced apoptosis has been suggested for many types of cancer cells, but the knowledge on mechanisms leading to ROS production or mechanisms downstream of ROS in execution of apoptosis is limited.
ACKNOWLEDGMENTS
The work cited in this article from the authors’ laboratory was supported by the United States Public Health Service Grant CA142604-04 (to SVS).
Conflict of Interest
None
REFERENCES
- 1.Newman DJ, Cragg GM, Snader KM. Natural products as sources of new drugs over the period 1981–2002. J Nat Prod. 2003;66(7):1022–1037. doi: 10.1021/np030096l. [DOI] [PubMed] [Google Scholar]
- 2.Pezzuto JM, Kosmeder JW, II, Park EJ, Lee SK, Cuendet M, Gills J, et al. Characterization of natural product cancer chemopreventive agents. In: Kelloff GJ, Hawk ET, Sigman CC, et al., editors. Cancer chemoprevention, vol. 2. Strategies for cancer chemoprevention. Totowa, NJ: Humana Press; 2005. pp. 3–37. [Google Scholar]
- 3.Garodia P, Ichikawa H, Malani N, Sethi G, Aggarwal BB. From ancient medicine to modern medicine: ayurvedic concepts of health and their role in inflammation and cancer. J Soc Integr Oncol. 2007;5(1):25–37. doi: 10.2310/7200.2006.029. [DOI] [PubMed] [Google Scholar]
- 4.Mirjalili MH, Moyano E, Bonfill M, Cusido RM, Palazón J. Steroidal lactones from Withania somnifera, an ancient plant for novel medicine. Molecules. 2009;14(7):2373–2393. doi: 10.3390/molecules14072373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mishra LC, Singh BB, Dagenais S. Scientific basis for the therapeutic use of Withania somnifera (Ashwagandha): a review. Altern Med Rev. 2000;5(4):334–346. [PubMed] [Google Scholar]
- 6.Winters M. Ancient medicine, modern use: Withania somnifera and its potential role in integrative oncology. Altern Med Rev. 2006;11(4):269–277. [PubMed] [Google Scholar]
- 7.Shohat B, Gitter S, Abraham A, Lavie D. Antitumor activity of withaferin A (NSC-101088) Cancer Chemother Rep. 1967;51(5):271–276. [PubMed] [Google Scholar]
- 8.Davis L, Kuttan G. Effect of Withania somnifera on 20-methylcholanthrene induced fibrosarcoma. J Exp Clin Cancer Res. 2000;19(2):165–167. [PubMed] [Google Scholar]
- 9.Davis L, Kuttan G. Effect of Withania somnifera on DMBA induced carcinogenesis. J Ethnopharmacol. 2001;75(2–3):165–168. doi: 10.1016/s0378-8741(00)00404-9. [DOI] [PubMed] [Google Scholar]
- 10.Prakash J, Gupta SK, Kochupillai V, Singh N, Gupta YK, Joshi S. Chemopreventive activity of Withania somnifera in experimentally induced fibrosarcoma tumours in Swiss albino mice. Phytother Res. 2001;15(3):240–244. doi: 10.1002/ptr.779. [DOI] [PubMed] [Google Scholar]
- 11.Prakash J, Gupta SK, Dinda AK. Withania somnifera root extract prevents DMBA-induced squamous cell carcinoma of skin in Swiss albino mice. Nutr Cancer. 2002;42(1):91–97. doi: 10.1207/S15327914NC421_12. [DOI] [PubMed] [Google Scholar]
- 12.Padmavathi B, Rath PC, Rao AR, Singh RP. Roots of Withania somnifera inhibit forestomach and skin carcinogenesis in mice. Evid Based Complement Alternat Med. 2005;2(1):99–105. doi: 10.1093/ecam/neh064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khazal KF, Samuel T, Hill DL, Grubbs CJ. Effect of an extract of Withania somnifera root on estrogen receptor-positive mammary carcinomas. Anticancer Res. 2013;33(4):1519–1523. [PMC free article] [PubMed] [Google Scholar]
- 14.Devi PU, Sharada AC, Solomon FE, Kamath MS. In vivo growth inhibitory effect of Withania somnifera (Ashwagandha) on a transplantable mouse tumor, Sarcoma 180. Indian J Exp Biol. 1992;30(3):169–172. [PubMed] [Google Scholar]
- 15.Devi PU, Sharada AC, Solomon FE. Antitumor and radiosensitizing effects of Withania somnifera (Ashwagandha) on a transplantable mouse tumor, Sarcoma-180. Indian J Exp Biol. 1993;31(7):607–611. [PubMed] [Google Scholar]
- 16.Christina AJM, Joseph DG, Packialakshmi M, Kothai R, Robert SJH, Chidambaranathan N, et al. Anticarcinogenic activity of Withania somnifera Dunal against Dalton’s ascitic lymphoma. J Ethnopharmacol. 2004;93(2–3):359–361. doi: 10.1016/j.jep.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 17.Leyon PV, Kuttan G. Effect of Withania somnifera on B16F-10 melanoma induced metastasis in mice. Phytother Res. 2004;18(2):118–122. doi: 10.1002/ptr.1378. [DOI] [PubMed] [Google Scholar]
- 18.Widodo N, Kaur K, Shrestha BG, Takagi Y, Ishii T, Wadhwa R, et al. Selective killing of cancer cells by leaf extract of Ashwagandha: identification of a tumor-inhibitory factor and the first molecular insights to its effect. Clin Cancer Res. 2007;13(7):2298–2306. doi: 10.1158/1078-0432.CCR-06-0948. [DOI] [PubMed] [Google Scholar]
- 19.Hamza A, Amin A, Daoud S. The protective effect of a purified extract of Withania somnifera against doxorubicin-induced cardiac toxicity in rats. Cell Biol Toxicol. 2008;24(1):63–73. doi: 10.1007/s10565-007-9016-z. [DOI] [PubMed] [Google Scholar]
- 20.Biswal BM, Sulaiman SA, Ismail HC, Zakaria H, Musa KI. Effect of Withania somnifera (Ashwagandha) on the development of chemotherapy-induced fatigue and quality of life in breast cancer patients. Integr Cancer Ther. 2013;12(4):312–322. doi: 10.1177/1534735412464551. [DOI] [PubMed] [Google Scholar]
- 21.Jayaprakasam B, Zhang Y, Seeram NP, Nair MG. Growth inhibition of human tumor cell lines by withanolides from Withania somnifera leaves. Life Sci. 2003;74(1):125–132. doi: 10.1016/j.lfs.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 22.Ichikawa H, Takada Y, Shishodia S, Jayaprakasam B, Nair MG, Aggarwal BB. Withanolides potentiate apoptosis, inhibit invasion, and abolish osteoclastogenesis through suppression of nuclear factor-κB (NF-κB) activation and NF-κB-regulated gene expression. Mol Cancer Ther. 2006;5(6):1434–1445. doi: 10.1158/1535-7163.MCT-06-0096. [DOI] [PubMed] [Google Scholar]
- 23.Shohat B, Shaltiel A, Ben-Bassat M, Joshua H. The effect of withaferin A, a natural steroidal lactone, on the fine structure of S-180 tumor cells. Cancer Lett. 1976;2(2):71–78. doi: 10.1016/s0304-3835(76)80014-6. [DOI] [PubMed] [Google Scholar]
- 24.Devi PU, Sharada AC, Solomon FE. In vivo growth inhibitory and radiosensitizing effects of withaferin A on mouse Ehrlich ascites carcinoma. Cancer Lett. 1995;95(1–2):189–193. doi: 10.1016/0304-3835(95)03892-z. [DOI] [PubMed] [Google Scholar]
- 25.Devi PU, Kamath R, Rao BS. Radiosensitization of a mouse melanoma by withaferin A: in vivo studies. Indian J Exp Biol. 2000;38(5):432–437. [PubMed] [Google Scholar]
- 26.Devi PU, Kamath R. Radiosensitizing effect of withaferin A combined with hyperthermia on mouse fibrosarcoma and melanoma. J Radiat Res. 2003;44(1):1–6. doi: 10.1269/jrr.44.1. [DOI] [PubMed] [Google Scholar]
- 27.Yang H, Shi G, Dou QP. The tumor proteasome is a primary target for the natural anticancer compound Withaferin A isolated from "Indian winter cherry". Mol Pharmacol. 2007;71(2):426–437. doi: 10.1124/mol.106.030015. [DOI] [PubMed] [Google Scholar]
- 28.Srinivasan S, Ranga RS, Burikhanov R, Han SS, Chendil D. Par-4-dependent apoptosis by the dietary compound withaferin A in prostate cancer cells. Cancer Res. 2007;67(1):246–253. doi: 10.1158/0008-5472.CAN-06-2430. [DOI] [PubMed] [Google Scholar]
- 29.Stan SD, Hahm ER, Warin R, Singh SV. Withaferin A causes FOXO3a- and Bim-dependent apoptosis and inhibits growth of human breast cancer cells in vivo. Cancer Res. 2008;68(18):7661–7669. doi: 10.1158/0008-5472.CAN-08-1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Samadi AK, Mukerji R, Shah A, Timmermann BN, Cohen MS. A novel RET inhibitor with potent efficacy against medullary thyroid cancer in vivo. Surgery. 2010;148(6):1228–1236. doi: 10.1016/j.surg.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lahat G, Zhu QS, Huang KL, Wang S, Bolshakov S, Liu J, et al. Vimentin is a novel anti-cancer therapeutic target; insights from in vitro and in vivo mice xenograft studies. PLoS One. 2010;5(4):e10105. doi: 10.1371/journal.pone.0010105. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Thaiparambil JT, Bender L, Ganesh T, Kline E, Patel P, Liu Y, et al. Withaferin A inhibits breast cancer invasion and metastasis at sub-cytotoxic doses by inducing vimentin disassembly and serine 56 phosphorylation. Int J Cancer. 2011;129(11):2744–2755. doi: 10.1002/ijc.25938. [DOI] [PubMed] [Google Scholar]
- 33.Munagala R, Kausar H, Munjal C, Gupta RC. Withaferin A induces p53-dependent apoptosis by repression of HPV oncogenes and upregulation of tumor suppressor proteins in human cervical cancer cells. Carcinogenesis. 2011;32(11):1697–1705. doi: 10.1093/carcin/bgr192. [DOI] [PubMed] [Google Scholar]
- 34.Samadi AK, Cohen SM, Mukerji R, Chaguturu V, Zhang X, Timmermann BN, et al. Natural withanolide withaferin A induces apoptosis in uveal melanoma cells by suppression of Akt and c-MET activation. Tumor Biol. 2012;33(4):1179–1189. doi: 10.1007/s13277-012-0363-x. [DOI] [PubMed] [Google Scholar]
- 35.Yang H, Wang Y, Cheryan VT, Wu W, Cui CQ, Polin LA, et al. Withaferin A inhibits the proteasome activity in mesothelioma in vitro and in vivo. PLoS One. 2012;7(8):e41214. doi: 10.1371/journal.pone.0041214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu Y, Hamza A, Zhang T, Gu M, Zou P, Newman B, et al. Withaferin A targets heat shock protein 90 in pancreatic cancer cells. Biochem Pharmacol. 2010;79(4):542–551. doi: 10.1016/j.bcp.2009.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Manoharan S, Panjamurthy K, Menon VP, Balakrishnan S, Alias LM. Protective effect of Withaferin-A on tumour formation in 7,12-dimethylbenz[a]anthracene induced oral carcinogenesis in hamsters. Indian J Exp Biol. 2009;47(1):16–23. [PubMed] [Google Scholar]
- 38.Panjamurthy K, Manoharan S, Nirmal MR, Vellaichamy L. Protective role of Withaferin-A on immunoexpression of p53 and bcl-2 in 7,12-dimethylbenz(a)anthracene-induced experimental oral carcinogenesis. Invest New Drugs. 2009;27(5):447–452. doi: 10.1007/s10637-008-9199-z. [DOI] [PubMed] [Google Scholar]
- 39.Manoharan S, Panjamurthy K, Balakrishnan S, Vasudevan K, Vellaichamy L. Circadian time-dependent chemopreventive potential of withaferin-A in 7,12-dimethylbenz[a]anthracene-induced oral carcinogenesis. Pharmacol Rep. 2009;61(4):719–726. doi: 10.1016/s1734-1140(09)70125-2. [DOI] [PubMed] [Google Scholar]
- 40.Hahm ER, Lee J, Kim SH, Sehrawat A, Arlotti JA, Shiva SS, et al. Metabolic alterations in mammary cancer prevention by withaferin A in a clinically relevant mouse model. J Natl Cancer Inst. 2013;105(15):1111–1122. doi: 10.1093/jnci/djt153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patil D, Gautam M, Mishra S, Karupothula S, Gairola S, Jadhav S, et al. Determination of withaferin A and withanolide A in mice plasma using high-performance liquid chromatography-tandem mass spectrometry: application to pharmacokinetics after oral administration of Withania somnifera aqueous extract. J Pharm Biomed Anal. 2013;80:203–212. doi: 10.1016/j.jpba.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 42.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 43.Stan SD, Zeng Y, Singh SV. Ayurvedic medicine constituent withaferin a causes G2 and M phase cell cycle arrest in human breast cancer cells. Nutr Cancer. 2008;60(Suppl 1):51–60. doi: 10.1080/01635580802381477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang X, Mukerji R, Samadi AK, Cohen MS. Down-regulation of estrogen receptor-alpha and rearranged during transfection tyrosine kinase is associated with withaferin A-induced apoptosis in MCF-7 breast cancer cells. BMC Complement Altern Med. 2011;11:84. doi: 10.1186/1472-6882-11-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Samadi AK, Tong X, Mukerji R, Zhang H, Timmermann BN, Cohen MS. Withaferin A, a cytotoxic steroid from Vassobia breviflora, induces apoptosis in human head and neck squamous cell carcinoma. J Nat Prod. 2010;73(9):1476–1481. doi: 10.1021/np100112p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shah N, Kataria H, Kaul SC, Ishii T, Kaur G, Wadhwa R. Effect of the alcoholic extract of Ashwagandha leaves and its components on proliferation, migration, and differentiation of glioblastoma cells: combinational approach for enhanced differentiation. Cancer Sci. 2009;100(9):1740–1747. doi: 10.1111/j.1349-7006.2009.01236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grogan PT, Sleder KD, Samadi AK, Zhang H, Timmermann BN, Cohen MS. Cytotoxicity of withaferin A in glioblastomas involves induction of an oxidative stress-mediated heat shock response while altering Akt/mTOR and MAPK signaling pathways. Invest New Drugs. 2013;31(3):545–557. doi: 10.1007/s10637-012-9888-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang X, Samadi AK, Roby KF, Timmermann B, Cohen MS. Inhibition of cell growth and induction of apoptosis in ovarian carcinoma cell lines CaOV3 and SKOV3 by natural withanolide Withaferin A. Gynecol Oncol. 2012;124(3):606–612. doi: 10.1016/j.ygyno.2011.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cohen SM, Mukerji R, Timmermann BN, Samadi AK, Cohen MS. A novel combination of withaferin A and sorafenib shows synergistic efficacy against both papillary and anaplastic thyroid cancers. Am J Surg. 2012;204(6):895–900. doi: 10.1016/j.amjsurg.2012.07.027. [DOI] [PubMed] [Google Scholar]
- 50.Sen N, Banerjee B, Das BB, Ganguly A, Sen T, Pramanik S, et al. Apoptosis is induced in leishmanial cells by a novel protein kinase inhibitor withaferin A and is facilitated by apoptotic topoisomerase I-DNA complex. Cell Death Differ. 2007;14(2):358–367. doi: 10.1038/sj.cdd.4402002. [DOI] [PubMed] [Google Scholar]
- 51.Hahm ER, Moura MB, Kelley EE, Van Houten B, Shiva S, Singh SV. Withaferin A-induced apoptosis in human breast cancer cells is mediated by reactive oxygen species. PLoS One. 2011;6(8):e23354. doi: 10.1371/journal.pone.0023354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hahm ER, Singh SV. Withaferin A-induced apoptosis in human breast cancer cells is associated with suppression of inhibitor of apoptosis family protein expression. Cancer Lett. 2013;334(1):101–108. doi: 10.1016/j.canlet.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Malik F, Kumar A, Bhushan S, Khan S, Bhatia A, Suri KA, et al. Reactive oxygen species generation and mitochondrial dysfunction in the apoptotic cell death of human myeloid leukemia HL-60 cells by a dietary compound withaferin A with concomitant protection by N-acetyl cysteine. Apoptosis. 2007;12(11):2115–2133. doi: 10.1007/s10495-007-0129-x. [DOI] [PubMed] [Google Scholar]
- 54.Oh JH, Lee TJ, Kim SH, Choi YH, Lee SH, Lee JM, et al. Induction of apoptosis by withaferin A in human leukemia U937 cells through down-regulation of Akt phosphorylation. Apoptosis. 2008;13(12):1494–1504. doi: 10.1007/s10495-008-0273-y. [DOI] [PubMed] [Google Scholar]
- 55.Mandal C, Dutta A, Mallick A, Chandra S, Misra L, Sangwan RS, et al. Withaferin A induces apoptosis by activating p38 mitogen-activated protein kinase signaling cascade in leukemic cells of lymphoid and myeloid origin through mitochondrial death cascade. Apoptosis. 2008;13(12):1450–1464. doi: 10.1007/s10495-008-0271-0. [DOI] [PubMed] [Google Scholar]
- 56.Mehrotra A, Kaul D, Joshi K. LXR-α selectively reprogrammes cancer cells to enter into apoptosis. Mol Cell Biochem. 2011;349(1–2):41–55. doi: 10.1007/s11010-010-0659-3. [DOI] [PubMed] [Google Scholar]
- 57.Choi MJ, Park EJ, Min KJ, Park JW, Kwon TK. Endoplasmic reticulum stress mediates withaferin A-induced apoptosis in human renal carcinoma cells. Toxicol In Vitro. 2011;25(3):692–698. doi: 10.1016/j.tiv.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 58.Mayola E, Gallerne C, Esposti DD, Martel C, Pervaiz S, Larue L, et al. Withaferin A induces apoptosis in human melanoma cells through generation of reactive oxygen species and down-regulation of Bcl-2. Apoptosis. 2011;16(10):1014–1027. doi: 10.1007/s10495-011-0625-x. [DOI] [PubMed] [Google Scholar]
- 59.Widodo N, Priyandoko D, Shah N, Wadhwa R, Kaul SC. Selective killing of cancer cells by Ashwagandha leaf extract and its component withanone involves ROS signaling. PLoS One. 2010;5(10):e13536. doi: 10.1371/journal.pone.0013536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee TJ, Um HJ, Do Min S, Park JW, Choi KS, Kwon TK. Withaferin A sensitizes TRAIL-induced apoptosis through reactive oxygen species-mediated up-regulation of death receptor 5 and down-regulation of c-FLIP. Free Radic Biol Med. 2009;46(12):1639–1649. doi: 10.1016/j.freeradbiomed.2009.03.022. [DOI] [PubMed] [Google Scholar]
- 61.Yang ES, Choi MJ, Kim JH, Choi KS, Kwon TK. Withaferin A enhances radiation-induced apoptosis in Caki cells through induction of reactive oxygen species, Bcl-2 downregulation and Akt inhibition. Chem Biol Interact. 2011;190(1):9–15. doi: 10.1016/j.cbi.2011.01.015. [DOI] [PubMed] [Google Scholar]
- 62.Fuska J, Fusková A, Rosazza JP, Nicholas AW. Novel cytotoxic and antitumor agents. IV. Withaferin A: relation of its structure to the in vitro cytotoxic effects on P388 cells. Neoplasma. 1984;31(1):31–36. [PubMed] [Google Scholar]
- 63.Jilani K, Lupescu A, Zbidah M, Shaik N, Lang F. Withaferin A-stimulated Ca2+ entry, ceramide formation and suicidal death of erythrocytes. Toxicol In Vitro. 2013;27(1):52–58. doi: 10.1016/j.tiv.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 64.Raina A, Kaul D. LXR-α genomics programmes neuronal death observed in Alzheimer’s disease. Apoptosis. 2010;15(12):1461–1469. doi: 10.1007/s10495-010-0541-5. [DOI] [PubMed] [Google Scholar]
- 65.Franchitto A, Torrice A, Semeraro R, Napoli C, Nuzzo G, Giuliante F, et al. Prostate apoptosis response-4 is expressed in normal cholangiocytes, is down-regulated in human cholangiocarcinoma, and promotes apoptosis of neoplastic cholangiocytes when induced pharmacologically. Am J Pathol. 2010;177(4):1779–1790. doi: 10.2353/ajpath.2010.091171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mohan R, Hammers HJ, Bargagna-Mohan P, Zhan XH, Herbstritt CJ, Ruiz A, et al. Withaferin A is a potent inhibitor of angiogenesis. Angiogenesis. 2004;7(2):115–122. doi: 10.1007/s10456-004-1026-3. [DOI] [PubMed] [Google Scholar]
- 67.Grover A, Shandilya A, Bisaria VS, Sundar D. Probing the anticancer mechanism of prospective herbal drug Withaferin A on mammals: a case study on human and bovine proteasomes. BMC Genomics. 2010;11(Suppl 4):S15. doi: 10.1186/1471-2164-11-S4-S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. 2007;26(9):1324–1337. doi: 10.1038/sj.onc.1210220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat Rev Cancer. 2002;2(8):594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 70.Hahm ER, Lee J, Huang Y, Singh SV. Withaferin A suppresses estrogen receptor-α expression in human breast cancer cells. Mol Carcinog. 2011;50(8):614–624. doi: 10.1002/mc.20760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.LaCasse EC, Baird S, Korneluk RG, MacKenzie AE. The inhibitors of apoptosis (IAPs) and their emerging role in cancer. Oncogene. 1998;17(25):3247–3259. doi: 10.1038/sj.onc.1202569. [DOI] [PubMed] [Google Scholar]
- 72.Deveraux QL, Reed JC. IAP family proteins—suppressors of apoptosis. Genes Dev. 1999;13(3):239–252. doi: 10.1101/gad.13.3.239. [DOI] [PubMed] [Google Scholar]
- 73.Wadegaonkar VP, Wadegaonkar PA. Withaferin A targets apoptosis inhibitor cIAP1: a potential anticancer candidate. J Appl Pharm Sci. 2012;2(5):154–7. [Google Scholar]
- 74.Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26(22):3279–3290. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- 75.Dhanasekaran DN, Johnson GL. MAPKs: function, regulation, role in cancer and therapeutic targeting. Oncogene. 2007;26(22):3097–3099. doi: 10.1038/sj.onc.1210395. [DOI] [PubMed] [Google Scholar]
- 76.Hahm E, Lee J, Singh SV. Role of mitogen-activated protein kinases and Mcl-1 in apoptosis induction by withaferin A in human breast cancer cells. Mol Carcinog. 2013 (in press). [DOI] [PMC free article] [PubMed]
- 77.Karin M, Cao Y, Greten FR, Li ZW. NF-κB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2(4):301–310. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- 78.Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase–AKT pathway in human cancer. Nat Rev Cancer. 2002;2(7):489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 79.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9(11):798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lee J, Hahm ER, Singh SV. Withaferin A inhibits activation of signal transducer and activator of transcription 3 in human breast cancer cells. Carcinogenesis. 2010;31(11):1991–1998. doi: 10.1093/carcin/bgq175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Um HJ, Min KJ, Kim DE, Kwon TK. Withaferin A inhibits JAK/STAT3 signaling and induces apoptosis of human renal carcinoma Caki cells. Biochem Biophys Res Commun. 2012;427(1):24–29. doi: 10.1016/j.bbrc.2012.08.133. [DOI] [PubMed] [Google Scholar]
- 82.Ali S, Coombes RC. Estrogen receptor alpha in human breast cancer: occurrence and significance. J Mammary Gland Biol Neoplasia. 2000;5(3):271–281. doi: 10.1023/a:1009594727358. [DOI] [PubMed] [Google Scholar]
- 83.Leong KG, Karsan A. Recent insights into the role of Notch signaling in tumorigenesis. Blood. 2006;107(6):2223–2233. doi: 10.1182/blood-2005-08-3329. [DOI] [PubMed] [Google Scholar]
- 84.Mumm JS, Kopan R. Notch signaling: from the outside in. Dev Biol. 2000;228(2):151–165. doi: 10.1006/dbio.2000.9960. [DOI] [PubMed] [Google Scholar]
- 85.Koduru S, Kumar R, Srinivasan S, Evers MB, Damodaran C. Notch-1 inhibition by Withaferin-A: a therapeutic target against colon carcinogenesis. Mol Cancer Ther. 2010;9(1):202–210. doi: 10.1158/1535-7163.MCT-09-0771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lee J, Sehrawat A, Singh SV. Withaferin A causes activation of Notch2 and Notch4 in human breast cancer cells. Breast Cancer Res Treat. 2012;136(1):45–56. doi: 10.1007/s10549-012-2239-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Su M, Mei Y, Sinha S. Role of the crosstalk between autophagy and apoptosis in cancer. J Oncol. 2013;2013:102735. doi: 10.1155/2013/102735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Herman-Antosiewicz A, Johnson DE, Singh SV. Sulforaphane causes autophagy to inhibit release of cytochrome C and apoptosis in human prostate cancer cells. Cancer Res. 2006;66(11):5828–5835. doi: 10.1158/0008-5472.CAN-06-0139. [DOI] [PubMed] [Google Scholar]
- 89.Bommareddy A, Hahm ER, Xiao D, Powolny AA, Fisher AL, Jiang Y, et al. Atg5 regulates phenethyl isothiocyanate-induced autophagic and apoptotic cell death in human prostate cancer cells. Cancer Res. 2009;69(8):3704–3712. doi: 10.1158/0008-5472.CAN-08-4344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hahm ER, Singh SV. Autophagy fails to alter withaferin A-mediated lethality in human breast cancer cells. Curr Cancer Drug Targets. 2013;13(6):640–650. doi: 10.2174/15680096113139990039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fong MY, Jin S, Rane M, Singh RK, Gupta R, Kakar SS. Withaferin A synergizes the therapeutic effect of doxorubicin through ROS-mediated autophagy in ovarian cancer. PLoS One. 2012;7(7):e42265. doi: 10.1371/journal.pone.0042265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kakar SS, Jala VR, Fong MY. Synergistic cytotoxic action of cisplatin and withaferin A on ovarian cancer cell lines. Biochem Biophys Res Commun. 2012;423(4):819–825. doi: 10.1016/j.bbrc.2012.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Suttana W, Mankhetkorn S, Poompimon W, Palagani A, Zhokhov S, Gerlo S, et al. Differential chemosensitization of P-glycoprotein overexpressing K562/Adr cells by withaferin A and Siamois polyphenols. Mol Cancer. 2010;9:99. doi: 10.1186/1476-4598-9-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yang ES, Choi MJ, Kim JH, Choi KS, Kwon TK. Combination of withaferin A and X-ray irradiation enhances apoptosis in U937 cells. Toxicol in Vitro. 2011;25(8):1803–1810. doi: 10.1016/j.tiv.2011.09.016. [DOI] [PubMed] [Google Scholar]