

Abstract
Licorice has been shown to affect the activities of several cytochrome P450 enzymes. This study aims to identify the key constituents in licorice which may affect these activities. Bioactivity assay was combined with metabolic profiling to identify these compounds in several complex licorice extracts. Firstly, the inhibition potencies of 40 pure licorice compounds were tested using an liquid chromatography/tandem mass spectrometry cocktail method. Significant inhibitors of human P450 isozymes 1A2, 2C9, 2C19, 2D6, and 3A4 were then selected for examination of their structural features by molecular docking to determine their molecular interaction with several P450 isozymes. Based on the present in vitro inhibition findings, along with our previous in vivo metabolic studies and the prevalence of individual compounds in licorice extract, we identified several licorice constituents, viz., liquiritigenin, isoliquiritigenin, together with seven isoprenylated flavonoids and arylcoumarins, which could be key components responsible for the herb–drug interaction between cytochrome P450 and licorice. In addition, hydrophilic flavonoid glycosides and saponins may be converted into these P450 inhibitors in vivo. These studies represent a comprehensive examination of the potential effects of licorice components on the metabolic activities of P450 enzymes.
Electronic supplementary material
The online version of this article (doi:10.1208/s12248-013-9544-9) contains supplementary material, which is available to authorized users.
KEY WORDS: CYP450, herb–drug interaction, isoliquiritigenin, licorice, liquiritigenin
INTRODUCTION
Licorice is one of the most frequently used herbal medicines worldwide. Recently, it is used as an adjuvant agent to improve the therapeutic effects of certain drugs (1). For instance, licorice could lower the risk of apthous ulcers caused by anti-inflammatory drugs like aspirin (2); reduce side effects of spironolactone (3); improve the efficacy and lower the adverse effects of glucocorticoids (4); improve excretion rates and decrease side effects of nitrofurantoin in patients with urinary tract infections (5); and treat cough, asthma, and gastric ulcer during chemotherapies (6). While licorice shows synergistic effects with some drugs, it could also decrease the therapeutic effects of other drugs, or even lead to side effects (7,8). One major reason for these herb–drug interactions (HDI) is cytochrome P450 enzymes. CYP450 superfamily is a large and diverse group of metabolic enzymes. Among them, CYP1A2, 2C9, 2C19, 2D6, and 3A4 isoforms are responsible for 80% of known drug metabolism (9). Inactivation of P450 enzymes could slow down drug metabolism, enhance their plasma concentration, and thus increase the risk of side effects (10). For instance, oral intake of 7.5 g/kg licorice could increase the area under plasma concentration–time curve (AUC) of methotrexate by 127% and extend the mean residence time from 240 to 921 min (11). Licorice has also been reported to alter the pharmacokinetics of drugs like warfarin (8), cyclosporine (12), and cortisol (13). Therefore, NIH has indicated that licorice “might have moderate effect on medications changed by the liver cytochrome P450 2C9 and 3A4” (10).
However, little is known about the identity of licorice components that are responsible for P450 modulation. Previous reports have revealed that glycyrrhetinic acid could inactive CYP 2E1 (14), glabridin (form Glycyrrhiza glabra) could inhibit the activities of 3A4, 2B6, and 2C9 (15), and that glycyrrhizic acid could induce (16) or inhibit (17) human CYP 3A4. Unfortunately, results from single compounds could not represent the action of licorice, which is a multicomponent mixture containing more than 400 compounds (18).
The present study attempts to identify the P450 modulators in licorice. To achieve this goal, the P450 inhibition potencies of 40 major licorice compounds were investigated using a liquid chromatography/tandem mass spectrometry (LC/MS/MS) cocktail assay. Their effects on the activities of several human P450 isozymes, viz., 1A2, 2C9, 2C19, 2D6, and 3A4 were evaluated. Molecular docking models were employed to interpret the relationship between chemical structure and bioactivity. Furthermore, chemical–bioactivity fingerprints were established to examine the occurrence of licorice compounds with their bioactivities. Combining the results of in vitro bioactivity, in vivo metabolism (19,20) and natural occurrence, the key licorice compounds which interact with cytochrome P450 were elucidated.
EXPERIMENTAL
Chemicals and Reagents
β-Nicotinamide adenine dinucleotide phosphate hydrate, d-glucose 6-phosphate sodium salt (G-6-P), glucose-6-phosphate dehydrogenase (G-6-P-DE), and tolbutamide (TOL) were purchased from Sigma-Aldrich (MO, USA). Pooled male human hepatic microsomes (20 mg/mL), testosterone (TES), dextrorphan (dMeDEX), 1′-hydroxymidazolam (OHMID), 6β-hydroxytestosterone (OHTES), hydroxytolbutamide (OHTOL), α-naphthoflavone (NAP), ticlopidine (TIC), and quinidine (QUI) were from iPhase Biosciences (Beijing, China). Phenacetin (PHE) was purchased from Aladdin Chemistry (Shanghai, China); 5-hydroxyomeprazole (OHOME) was from Toronto Research Chemicals Inc. (Toronto, Canada). Cimetidine (CIM), ketoconazole (KET), fluconazole (FLU), dextromethorphan (DEX), omeprazole (OME), and acetaminophen (dEtPHE) were obtained from China National Institutes for Food and Drug Control (Beijing, China). Midazolam (MID) was from Nhwa Pharmaceutical Co. (Xuzhou, Jiangsu, China).
Acetonitrile, methanol, and formic acid were of high-performance liquid chromatography (HPLC) grade (Mallinckrodt Baker, Phillipsburg, NJ, USA). Deionized water was obtained from a Milli-Q system (Millipore, MA, USA). High-purity nitrogen (99.9%) and argon (99.99%) were purchased from Haike Yuanchang Co. (Beijing, China). Dimethyl sulfoxide (DMSO, ACS grade) was purchased from Solarbio (Beijing, China).
Licorice Compounds and Extracts
Licorice (dried roots and rhizomes of Glycyrrhiza uralensis Fisch.) was purchased from Elion Resources Group Company (Inner Mongolia, China) and was authenticated by comparing the HPLC fingerprint with a reference sample obtained from China National Institutes for Food and Drug Control (Beijing, China). A total of 42 pure compounds (1–40, C15, C17) were isolated from G. uralensis by the authors. Their structures (shown in Fig. 1 and Electronic Supplementary Material (ESM) Fig. 1S) were characterized by ultraviolet (UV), nuclear magnetic resonance (NMR), and mass spectrometry (MS). The purities were higher than 98% according to HPLC/UV analysis.
Fig. 1.
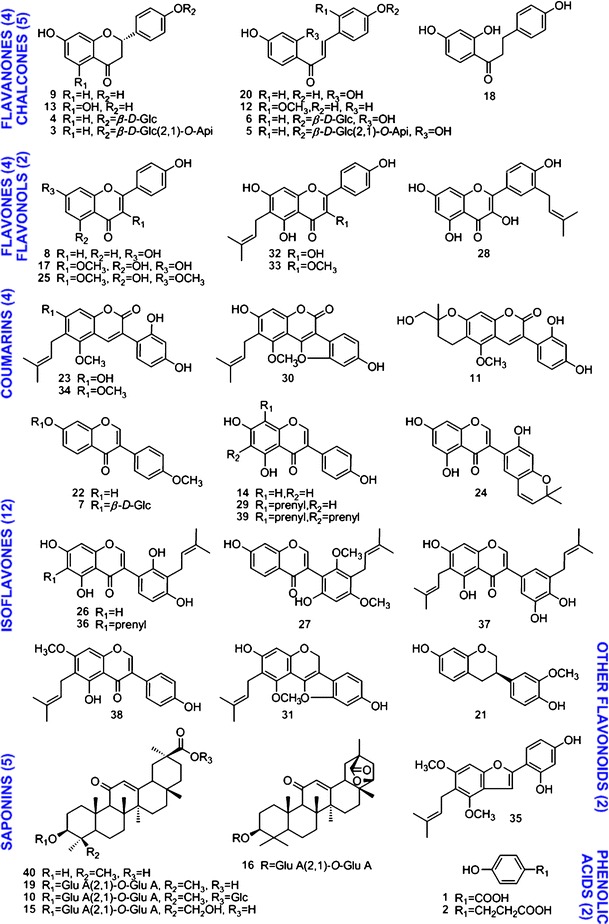
Chemical structures of licorice compounds (isolated from Glycyrrhiza uralensis Fisch.). The compounds were 4-hydroxybenzoic acid (1), phloretic acid (2), liquiritin apioside (3), liquiritin (4), isoliquiritin apioside (5), isoliquiritin (6), ononin (7), 4′,7-dihydroxyflavone (8), liquiritigenin (9), licorice-saponin A3 (10), licopyranocoumarin (11), echinatin (12), naringenin (13), genistein (14), licorice-saponin G2 (15), licorice-saponin E2 (16), 3-methylkaempferol (17), davidigenin (18), glycyrrhizic acid (19), isoliquiritigenin (20), 7,4′-dihydroxy-3′-methoxyisoflavan (21), formononetin (22), glycycoumarin (23), semilicoisoflavone B (24), kumatakenin (25), licoisoflavone A (26), licoricone (27), isolicoflavonol (28), lupiwighteone (29), glycyrol (30), glyurallin A (31), licoflavonol (32), topazolin (33), glycyrin (34), gancaonin I (35), angustone A (36), isoangustone A (37), gancaonin G (38), 6,8-diprenylgenistein (39), and 18β-glycyrrhetinic acid (40). Api apinose, GluA glucuronic acid, Glc glucose, Prenyl isoprenoid. For structures of the substituent groups could be referred to in reference (18)
Licorice water extract (LWE) was prepared by decocting licorice crude drug materials (20 g, fine powder) with water (100 mL × 2, 1 h) at 100°C. Likewise, licorice was extracted with 95% ethanol at 80°C to obtain the licorice ethanol extract (LEE). To prepare liquid–liquid extraction fractions, 20 g of licorice was decocted at 80°C for four times (100 mL × 4, 1 h) using 95%, 95%, 75%, and 75% (v/v) ethanol, successively. The extract was dried, suspended in water (100 mL), and extracted at room temperature with ethyl acetate (100 mL × 4) and n-butanol (100 mL × 4), successively, to obtain the ethyl acetate fraction (LEA) and the n-butanol fraction (LBU). The fractions were concentrated in vacuum at 50°C and then freeze-dried to obtain loose fine powder.
Incubation Procedure and P450 Activity Assay
The effect of licorice compounds on P450 enzyme activities was investigated using a pool of human liver microsomes following the procedure described by Walsky et al. (21). In brief, incubations were conducted at 37 ± 1°C in 300 μL of incubation mixtures containing human hepatic microsome (0.2 mg/mL), potassium phosphate buffer (pH 7.4, 0.1 mM) and MgCl2 (5 mM). The incubation mixture also contained an nicotinamide adenine dinucleotide phosphate-oxidase (NADPH)-generating system (1 mM of NADP, 6 mM of G-6-P, and 2 unit/mL of G-6-P-DE), P450 probe substrates (20/10/40/6/2/40 μM of PHE/OME/TOL/DEX/MID/TES), and licorice compounds or extracts (0.1–200 μM, three to five concentrations for each sample). Compounds 8, 14, 22, 24, 25, 27, 30, and 34 were diluted in a mixture of MeOH and DMSO (1:1, v/v). The other compounds and the four extracts were dissolved in methanol. The stock solutions were then diluted with PBS. All samples were dissolved before adding into the system. The total amount of organic solvent was lower than 1% (v/v). P450 inhibitors (NAP/TIC/FLU/QUI/KET for CYP 1A2/2C19/2C9/2D6/3A4, respectively) were added as positive control, and blank solvents (PBS containing methanol or MeOH/DMSO) were used as negative control. Reactions were initiated by adding the NADPH-generating system and were terminated after 30 min by 200 μL of cold acetonitrile containing 0.5 μM internal standard (CIM). The mixture was kept at 4°C for 30 min, and the precipitated protein was removed by centrifugation (10,000 g for 10 min at 4°C).
Validation by Enzyme Kinetics Analysis
Enzyme kinetics was determined using six concentrations of known P450 substrates (n = 4). The Km values were determined by nonlinear regression analysis of the enzyme activity–substrate concentration data using the Michaelis–Menten model. Half maximal inhibitory concentration (IC50) values of known inhibitors were measured as positive control.
Calibration and Method Validation Samples
The major metabolites (dEtPHE/OHOME/OHTOL/dMeDEX/OHMID/OHTES of six probe substrates) were mixed, diluted, and spiked with the incubation matrix (n = 3) to prepare a series of calibration samples (2.50/2.13/1.88/1.19/1.75/2.50 to 0.0001/0.000085/0.000075/0.000047/0.00007/0.0001 μM). Quality control (QC) samples were similarly prepared at three concentrations (high concentration, HQC; middle concentration, MQC; lower limit of quantitation, LLOQ) as shown in ESM Table 1S. For stability test, pooled metabolites were obtained by following the incubation procedure and were analyzed before and after storage to evaluate their stability.
LC/MS/MS Assay
The LC/MS/MS system consisted of a Finnigan Surveyor HPLC instrument connected to a Finnigan TSQ Quantum triple quadrupole mass spectrometer via ESI interface (ThermoFisher, CA, USA). The mobile phase consisted of acetonitrile (A) and water (B), each containing 0.1% of formic acid. An Atlantis T3 column (3 μm, ID 2.1 × 150 mm) equipped with an XTerra MS C18 guard column (5 μm, ID 3.9 × 20 mm) (Waters, MA, USA) was used. The gradient elution program was 20–50% A in 0–8 min; 50–90% A in the next 0.5 min; and 90% A from 8.5 to 12.5 min. The flow rate was 200 μL/min. The column temperature was 20°C and the sample tray temperature was 25°C. The HPLC effluent was introduced into the mass spectrometer without splitting.
The mass spectrometer was operated in the positive ion mode. High purity nitrogen was used as the sheath (40 arb) and auxiliary (5 arb) gas; high purity argon was used as the collision gas (1.5 mTorr). Other parameters were as follows: spray voltage, 4.0 kV; capillary temperature, 350°C; and capillary offset, 35 V. Quantitative analyses were monitored in selected reaction monitoring (SRM) mode. Scan time for each SRM channel was set at 0.1–0.2 s, and the mass width was set at 1.5 μ. Q1 and Q3 quadrupoles were set at unit resolution. SRM transitions and related parameters are given in ESM Table 2S.
Molecular Modeling
Crystal structures of human CYP 1A2, 2C19, 2C9, 2D6, and 3A4 (PDB# 2HI4, 4GQS, 1R9O, 3TDA, and 2VM0, respectively) were retrieved from the PDB database (http://www.rcsb.org/pdb/home/home.do). The substrate recognition sites were indicated by the interaction of the cocrystallized ligands. All water molecules were removed. Hydrogens were added and then the stripped protein structures were optimized with Discovery Studio v2.5 (Accelrys Software Inc., USA) using CHARMm-based algorithm under a pH condition of 7.4. Licorice compounds were docked using Genetic Optimization for Ligand Docking v5.1 and Hermes v1.5 (CCDC Software Ltd., Cambridge, UK) with a slightly modified genetic algorithm. The docking was performed with the CYP dedicated scoring function (goldscore.p450_pdb.params) and the active site was defined as all residues within 10 Å. Default parameters were used unless otherwise stated.
Data Processing
Calibration was accomplished with Xcalibur 2.0.7 software (ThermoFisher). Data were fit to linear calibration curves using 1/x2 weighting. Substrate inhibition data were analyzed using OriginPro 8 (v8.0951, OriginLab, Northampton, MA, USA) in logistic regression.
RESULTS
Instrument Method Validation
An LC/MS/MS method was used to determine the metabolites of probe substrates. Due to multiple substrates in the cocktail system, the mass analyzer was operated in SRM mode. Analytes were recognized by the mass-to-charge ratio (m/z) of their pseudomolecular ion and a specific fragment ion. By matching these ion pairs, SRM mode ensures the specificity of target analytes and excludes the inference from other substrates/metabolites which could not be separated by HPLC (21,22).
The method validation was conducted following the US Food and Drug Administration (FDA) guidance on bioanalytical method validation and drug interaction studies (23,24). Selectivity, linearity, accuracy, precision, recovery (extraction efficiency), and stability were evaluated for the assay. The calibration curves (ESM Table 3S) covered a wide dynamic range (167-fold for OHTES, 500-fold for dEtPHE and OHTOL, 833-fold for dMeDEX, and 1,000-fold for OHOME and OHMID) with good linearity (r2 > 0.99). Accuracy and precision was assessed by sample analysis at three concentration levels in the same day (n = 5) and in five consecutive days. The measured concentrations were compared to the nominal concentrations, and the relative standard deviations (RSD) of parallel analyses were calculated. Since the calibration curve was established using standard spiked biomatrix, standard addition tests were conducted using the same biomatrix (incubation mixture). Accuracy and precision was assessed by adding known amounts of the analyte to the matrix, and analyze the samples in the same day (n = 5) and in five consecutive days. As shown in ESM Table 1S, the accuracy ranged from 91% to 110% for LLOQ, 96−109% for MQC, and 92−108% for HQC. RSD values were no higher than 10.4% for all three concentrations, indicating acceptable variation of the method.
Matrix effects and extraction efficiency were examined in duplicate by three groups of standard addition experiments. Each group included three concentrations. In group 1, analytes were added to the incubation matrix and then extracted as QC samples; in group 2, analytes were added into pre-extracted incubation matrix; in group 3, analytes were dissolved in methanol/water (1:1, v/v). Extraction efficiency was calculated as: EE (%) = [measured concentration]gp1/[measured concentration]gp2 × 100%. The matrix effect was calculated as: ME (%) = [measured concentration]gp2/[measured concentration]gp3 × 100%. The extraction efficiency of six analytes ranged from 84% to 101% at all three concentrations. The matrix effect led to weak ion suppression, ranging from −2.4% to 15.4% (ESM Table 4S) for all three concentrations. Stability was determined at high and low concentrations using pooled samples after 24 h of ambient temperature storage, three freeze–thaw cycles, and 20 days of −80°C storage, respectively. The analytes were proved stable with variations of −10.6% to 12.0% at low concentration and −6.13% and 13.5% at high concentration (ESM Table 5S).
Optimization and Verification of the Incubation System
Activities of P450 isozymes were tested by adding mixed probe substrates into the incubation system. If the test compound did not inhibit the CYP isozymes, the probe substrates would be metabolized to their corresponding metabolites, which can then be measured by LC/MS/MS (9,22). The probe compounds, viz. phenacetin, omeprazole, tolbutamide, dextromethorphan, midazolam, and testosterone were chosen following the guidance of FDA (24). Their LC/MS/MS chromatograms, metabolic reactions, and corresponding SRM transitions are given in Fig. 2. The incubation system was optimized in the aspects of enzyme concentration, incubation time, and substrate concentration. All incubations were performed in triplicate. According to the results, to assure the linear relationship between enzyme activity and metabolic transformation, the protein concentration should be in the range of 0.05−0.20 mg/mL, and the incubation time should be among 0−35 min. Therefore, the incubation was conducted using 0.20 mg/mL protein for 30 min.
Fig. 2.
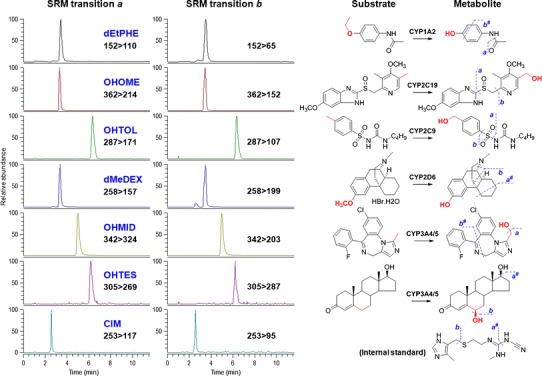
LC/MS/MS chromatograms (left) and substrate reactions (right) of the cocktail assay. Fragment ions and their selected reaction monitoring (SRM) transitions were suggested. dEtPHE acetaminophen (metabolite of phenacetin by 1A2); OHOME 5-hydroxyomeprazole (metabolite of omeprazole by 2C19), OHTOL hydroxytolbutamide (metabolite of tolbutamide by 2C9), dMeDEX dextrorphan (metabolite of dextromethorphan by 2D6), OHMID 1′-hydroxymidazolam (metabolite of midazolam by 3A4/5), OHTES 6β-hydroxytestosterone (metabolite of testosterone by 3A4/5). # indicated cleavages which are subsequent to the other fragmentation
The incubation system was also optimized in terms of substrate concentration (n = 4). The Km and IC50 values for known CYP substrates and inhibitors are shown in Table I. The measured values were in good agreement with recently published literatures (21,24–28), demonstrating applicability of the assay. Therefore, PHE/OME/TOL/DEX/MID/TES at 20/10/40/6/2/40 μM were chosen as probe substrates to evaluate candidate compounds for their inhibition of CYP1A2, 2C9, 2C19, 2D6, and 3A4/5 (MID and TES), respectively.
Table I.
K m Values of Probe Substrates and Inhibition IC50 Values of Positive Inhibitors to CYP Isoforms
| CYP | Substrate/metabolite/inhibitor | K m tested (μM) | K m reported (μM) (24) | IC50 tested (μM) | IC50 reported (μM) |
|---|---|---|---|---|---|
| 1A2 | PHE/dEtPHE/NAP | 13.6 (1.7) | 1.7–152 | 0.054 (0.009) | 0.041 (25), 0.07 (26) |
| 2C19 | OME/OHOME/TIC | 11.4 (0.5) | 17–26 | 2.80 (0.49) | 1.2–10 (24,25) |
| 2C9 | TOL/OHTOL/FLU | 80.0 (11.3) | 67–838 | 6.85 (1.42) | 7–8 (27) |
| 2D6 | DEX/dMeDEX/QUI | 8.0 (3.3) | 0.44–8.5 | 0.16 (0.02) | 0.05 (25), 0.0579 (21), 0.06 (26), 0.12 (28) |
| 3A4 | MID/OHMID/KET | 1.2 (0.3) | 1–14 | 0.075 (0.018) | 0.0187 (22), 0.045 (28), 0.10 (26), 0.0037–0.18 (24) |
| 3A4 | TES/OHTES/KET | 55.1 (2.6) | 52–94 | 0.024 (0.004) | 0.0261 (21), 0.04 (25), 0.05 (28), 0.0037–0.18 (24) |
Data in the brackets suggested standard error of the results (n = 4). For abbreviations of substrates, metabolites, and inhibitors please refer to the “Chemicals and reagents” section
P450 Inhibition Assay for 40 Licorice Compounds
In total, 40 licorice compounds were evaluated by the LC/MS/MS assay. The compounds represented major structural types of licorice compounds including 29 flavonoids (6 flavones, 4 flavanones, 5 chalcones, 12 isoflavones, and 2 other flavonoids), 5 saponins, 4 coumarins, and 2 phenolic acids. The chemical bioactivity fingerprint was established by arraying the inhibition/activation rates of the 40 compounds at 10 μM in the order of their chromatographic retention (Fig. 3). IC50 values were provided in Table II. Different classes of licorice compounds showed distinct activities on P450 isozymes. Flavonoid glycosides (3–7), saponins (10,15,16,19), and phenolic acids (1,2) showed weak regulatory activities, with inhibition or activation rates of less than 35% at 10 μM. Most of their IC50 values were higher than 200 μM. Free flavonoids and arylcoumarins, in contrast, generally exhibited potent inhibitory activities against the five CYP isozymes, especially 1A2 and 2C9. Moreover, their activities could vary due to slight structural difference.
Fig. 3.
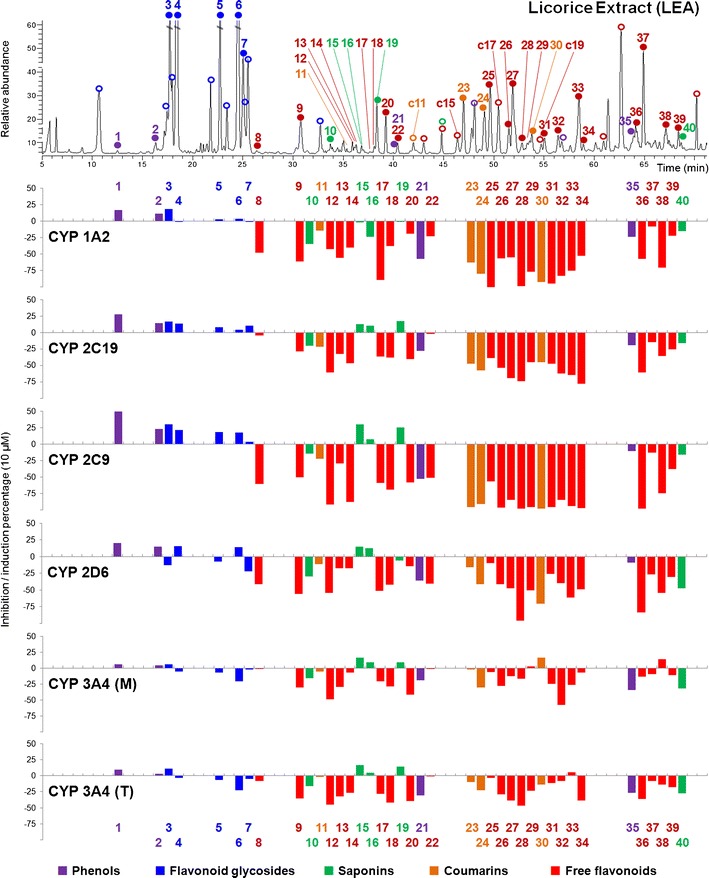
Chemistry–bioactivity fingerprint for licorice (10 μM). Chemistry fingerprint was obtained from licorice ethyl acetate (LEA) at 260 nm. Peaks 3–6 (intensity 65%, 100%, 78%, and 100%) were not fully displayed due to the limited space. Tested compounds and predicted compounds (inhibition percentage not shown) were marked with solid and hollow dots, respectively. 3A4 (M) and 3A4 (T) suggest activities evaluated by using midazolam and testosterone, respectively. Integrate chromatogram of LEA please refer to Fig. 6. The analytical method was described in ESM
Table II.
IC50 (In Micromolar) of 40 Licorice Compounds to Inhibit CYP450 Isozymes
| Compounds | Cytochrome P450 isozyme inhibition (IC 50 in μM) a | ||||||
|---|---|---|---|---|---|---|---|
| 1A2 | 2C19 | 2C9 | 2D6 | 3A4 (M) | 3A4 (T) | ||
| 1 | >200bc | >200bc | >200bc | >200bc | >200bc | >200bc | |
| 2 | >200bc | >200bc | >200bc | >200bc | >200bc | >200bc | |
| 3 | >200bc | >200bc | >200bc | >200b | >200b | >200b | |
| 4 | >200b | >200bc | >200bc | >200bc | >200b | >200b | |
| 5 | >200bc | >200bc | >200bc | >200b | 111.89 (17.8) | 98.82 (11.6) | |
| 6 | >200bc | >200bc | >200bc | >200bc | 48.83 (5.6) | 59.90 (2.8) | |
| 7 | >200b | >200bc | >200bc | >200b | >200b | >200b | |
| 8 | 10.93 (4.1) | >200b | 37.96 (3.3) | 50.84 (21.2) | >200bc | 123.12 (31.8) | |
| 9 | 2.68 (0.5) | 59.15 (11.6) | 22.93 (2.3) | 5.43 (2.0) | 151.15 (88.2) | 43.04 (10.2) | |
| 10 | >200b | >200b | >200b | >200b | >200b | >200b | |
| 11 | 86.59 (9.6) | 50.54 (3.3) | 42.76 (5.2) | 59.34 (7.1) | 154.35 (75.9) | 131.61 (33.1) | |
| 12 | 22.21 (6.6) | 9.76 (3.4) | 2.46 (0.8) | 4.19 (1.7) | 15.95 (6.2) | 34.36 (13.8) | |
| 13 | 27.72 (11.6) | 49.95 (10.6) | 31.56 (7.2) | 191.44 (57.4) | 51.29 (16.6) | 55.54 (8.5) | |
| 14 | 12.92 (4.7) | 24.16 (9.6) | 7.10 (0.7) | 52.00 (32.5) | >200b | >200b | |
| 15 | >200b | >200bc | >200bc | >200bc | >200bc | >200bc | |
| 16 | >200b | >200bc | >200bc | >200bc | >200bc | >200bc | |
| 17 | 1.44 (0.3) | 50.75 (9.5) | 28.84 (3.1) | 33.86 (1.4) | >200b | >200b | |
| 18 | 21.80 (4.1) | 39.15 (9.2) | 8.46 (5.2) | 4.61 (2.6) | 57.88 (27.3) | 22.33 (9.6) | |
| 19 | >200b | >200bc | >200bc | >200b | >200bc | >200bc | |
| 20 | 49.80 (17.0) | 13.55 (1.8) | 17.63 (6.7) | >200b | 10.96 (4.3) | 14.51 (2.5) | |
| 21 | 1.14 (0.1) | 15.02 (1.1) | 6.42 (0.8) | 5.94 (0.7) | 76.13 (10.5) | 22.88 (5.9) | |
| 22 | 18.33 (2.9) | 105.90 (20.1) | 30.76 (4.0) | 18.27 (3.5) | >200b | >200b | |
| 23 | 1.99 (0.5) | 6.77 (1.5) | 0.70 (0.2) | 10.27 (5.0) | 90.66 (8.5) | 41.13 (9.8) | |
| 24 | 2.83 (1.4) | 22.78 (7.0) | 19.22 (7.8) | 3.86 (1.3) | 12.32 (7.6) | 38.44 (3.7) | |
| 25 | 1.45 (0.1) | 59.02 (14.2) | 8.24 (1.4) | >200b | >200b | >200b | |
| 26 | 5.33 (0.7) | 19.46 (3.8) | 2.52 (0.9) | 6.18 (4.1) | 21.31 (11.1) | 31.79 (8.1) | |
| 27 | 8.38 (2.5) | 8.58 (2.4) | 19.41 (1.5) | 20.19 (3.5) | 23.48 (1.3) | 52.58 (12.9) | |
| 28 | 0.72 (0.4) | 12.14 (3.3) | 7.18 (2.8) | 1.75 (0.1) | 12.58 (1.1) | 29.95 (7.3) | |
| 29 | 8.11 (1.8) | 66.48 (11.3) | 16.03 (2.8) | 22.10 (4.2) | >200b | >200b | |
| 30 | 1.34 (0.4) | 31.75 (2.6) | 0.40 (0.2) | 26.46 (1.8) | >200b | 79.25 (4.9) | |
| 31 | 0.48 (0.1) | 5.91 (1.6) | 0.90 (0.1) | 3.15 (0.1) | 7.99 (1.3) | 41.36 (10.2) | |
| 32 | 1.99 (0.6) | 12.50 (0.3) | 18.97 (0.4) | 3.39 (1.0) | 6.00 (1.3) | 49.36 (7.0) | |
| 33 | 2.16 (0.2) | 2.89 (0.3) | 8.83 (2.2) | 3.26 (0.3) | 9.20 (1.0) | >200b | |
| 34 | 4.51 (1.3) | 3.91 (1.1) | 0.65 (0.1) | 15.02 (6.9) | 20.57 (4.3) | 26.14 (1.8) | |
| 35 | 33.56 (6.5) | 18.20 (5.1) | 38.69 (11.3) | 98.79 (24.2) | 26.59 (2.2) | 44.62 (11.9) | |
| 36 | 3.12 (0.9) | 17.68 (10.8) | 4.52 (2.7) | 1.34 (0.4) | 7.97 (1.6) | 30.69 (0.5) | |
| 37 | 57.87 (3.8) | 59.01 (8.7) | 42.12 (17.9) | 19.32 (11.7) | 142.26 (43.4) | 78.24 (36.2) | |
| 38 | 1.19 (0.1) | 29.10 (4.4) | 0.12 (0.08) | 29.61 (10.2) | >200b | >200b | |
| 39 | 9.44 (1.6) | 16.35 (6.3) | 22.03 (2.0) | 6.49 (1.3) | 44.49 (21.7) | 26.72 (7.4) | |
| 40 | 40.96 (5.5) | 71.89 (16.5) | 27.71 (12.3) | 43.53 (25.9) | 25.83 (11.0) | 34.48 (4.9) | |
Potent inhibitors (IC50 > 5 μM for 1A2, 2C9, 2C19, 2D6, and IC50 > 15 μM for 3A4) were italicized. Data in the brackets suggested standard error
3A4 (M) midazolam, 3A4 (T) testosterone
aStandard deviation of IC50 were presented in brackets. Values were obtained from triplicate tests
bIC50 >200 μM was agreed by triplicate tests
cWeak activation was observed
To better elucidate the structure–activity relationship of licorice compounds binding with P450 isozymes, molecular docking was employed to investigate the fitness of ligand structure to the active binding site. Docking models were established using the reported crystal structures of human P450 enzymes (PDB# 2HI4, 4GQS, 1R9O, 3TDA, and 2VM0). Reliability of the models were evaluated by the fitness of reported ligands, the root mean square deviation (RMSD) between docked and reported ligand positions, and the correlation coefficient (R) of scored and measured values as shown in Table III. All RMSD values were lower than 1.5 Å. Licorice compounds which received positive ranks in ligand docking were included in the model. The number of compounds considered in each model is shown in Table III and the R were no less than 0.65 for 1A2, 2C9, 2C19, and 2D6. The R value for 3A4 might be compromised by its substrate-dependent property (29). The linear regressions established for CYP 2C19 ligands are shown in ESM Fig. 2S. For most licorice compounds, particularly free flavonoids and arylcoumarins, the docking results were in good agreement with the experiments.
Table III.
Reliability of CYP Molecular Docking Models. Fitness of Cocrystallized Ligands, RMSD Values, and Correlation Coefficient (R) of Predicted Potency and Measured Values
| PDB ID | Ligand (positive) | PLP fitness | Goldscore fitness | RMSD | Ligand n | R |
|---|---|---|---|---|---|---|
| 2HI4 | α-Naphthoflavone | 93.9 | 67.6 | 0.51 | 21 | 0.764 |
| 1R9O | Flurbiprofen | 73.3 | 52.6 | 0.39 | 25 | 0.673 |
| 3TDA | Prinomastat | 87.4 | 81.4 | 0.59 | 24 | 0.687 |
| 2V0M | Ketoconazole | 96.9 | 70.3 | 1.03 | 24 | 0.473 |
| 4GQS | (4-hydroxy-3,5-dimethylphenyl)(2-methyl-1-benzofuran-3-yl)methanone | 60.2 | 49.1 | 0.60 | 25 | 0.778 |
PLP fitness and Goldscore fitness refers to scoring functions in Genetic Optimization for Ligand Docking v5.1; RMSD root mean square deviation between the docked and reported ligand positions; Ligand n number of ligands used to establish the linear regression; R coefficient of correlation
For CYP 1A2, 21 of the 40 licorice compounds exhibited IC50 values of <20 μM. These active compounds included free flavones, flavanones, isoflavones, and arylcoumarins. The most potent inhibitors were compounds 17, 21, 23, 25, 28, 30, 31, 32, and 38 (their IC50 values were listed in Table II). With the exception of compound 21, all these compounds had the similar planar structure. This result was consistent with previous report that CYP1A2 inhibitors were generally planar molecules as the active site cavity of CYP1A2 is narrow and flat (30). In molecular docking analysis, compounds 28, 30, 31, and α-naphthoflavone (positive control) showed strong interactions with the phenylalanine residues (Phe125, Phe226, Phe256, Phe260, and Phe319) as shown in ESM Fig. 3S. Most of these potent inhibitors (23,28,30–32,38) contained an isoprenyl group, which was very close to the heme region of CYP1A2, and could contribute to their inhibitory activities (ESM Fig. 3S). Compound 21 was the only exception to the above rules. According to the docking analysis, its structure could also bind with the enzyme pocket. Interestingly, liquiritigenin (9; IC50 2.68 ± 0.5 μM) showed remarkably stronger inhibitory activities than its isomer isoliquiritigenin (20; IC50 49.8 ± 17 μM). Liquiritigenin could more tightly fit into the binding pocket than isoliquiritigenin (ESM Fig. 4S).
Potent inhibitors of CYP2C9 mainly involved flavonoids and arylcoumarins bearing prenyl groups (−PR) adjacent to hydroxyl (−OH) and/or methoxyl (−OMe) substitution. In addition, C=O or –C–O–C– substructures were generally observed in potent inhibitors, such as 23, 34, 26, 30, 31, and 38 (IC50 = 0.70 ± 0.2, 0.65 ± 0.1, 2.52 ± 0.9, 0.40 ± 0.2, 0.90 ± 0.1, and 0.12 ± 0.08 μM; Table II). Exceptional cases were found for prenylated flavonoids such as 37 and 39 (IC50 = 42.1 ± 18 and 22.0 ± 2 μM). These compounds also received lower scores (<30) in docking experiments. Though the binding site has sufficient space to contain small molecules, dual prenyl groups might restrict their interaction with the enzyme. Our results were consisted with previous QSAR studies for CYP 2C9 inhibitors, where hydrophobes (–PR), hydrogen bond donors (–OH), and hydrogen bond acceptors were functional groups for inhibition (31,32). Weak and noninhibitors in licorice included glycosides, saponins, and phenolic compounds without hydrophobic features (1 and 2) and/or powerful hydrogen bond donors (35).
Inhibitors of 2C19 were structurally similar to those of 2C9, the homogenous P450 isoenzyme (33). Potent 2C19 inhibitors mainly included prenylated flavonoids and coumarins substituted with –OH or –OMe (27, 31, 32, 33, 36, 38, 39), or compounds containing m-dihydroxybenzene substructure (12,14,20,24). Compounds 23, 26, and 34 contain both structural features, and showed IC50 of 0.70 ± 0.2, 2.52 ± 0.9, and 0.65 ± 0.1 μM. It is worth noting that 15 of 17 inhibitors (IC50 < 30 μM) possessed C=O groups, including 3-carbonyl (13 flavonoids) and 1-carbonyl (2 coumarins) aglycones. In agreement with the experimental data, molecular docking resulted a correlation of R = 0.778 between predictive and experimental values (ESM Fig. 2S). The docking model showed a hydrogen bond with 7-OH (Asp293), several amino acid residues closed to 6-PR (e.g., Leu237 and Ala103), and similar chemical environments for rings B and C (Fig. 4c–f; 33). Based on this model, we predicted the binding activity of licoisoflavone B (C15) and licocoumarone (C17). Their structures were very similar to 24 and 23, respectively (Fig. 1; ESM Fig. 1S). IC50 values of C15, C17, 24, and 23 were 4.2 μM (pred.), 17.9 μM (pred.), 22.8 ± 7.0 μM, and 6.77 ± 1.5 μM, respectively. The results also suggested that cyclization of isoprenyl group could compromise the inhibition activity.
Fig. 4.
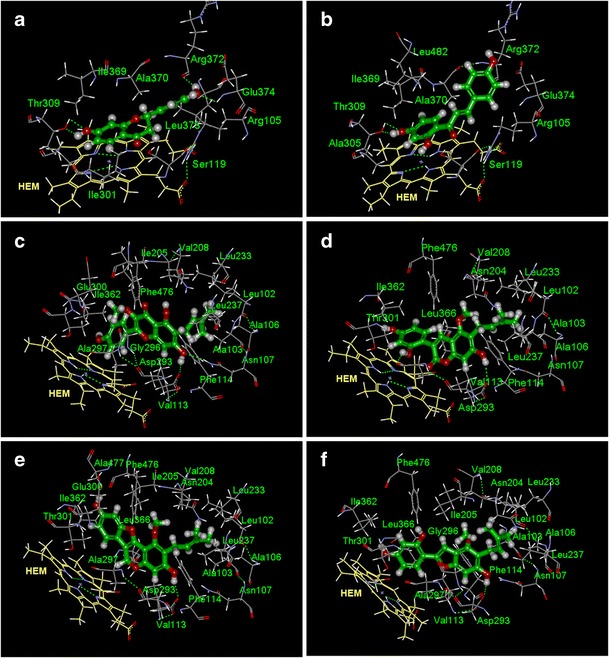
Docking simulation of 9 (a) and 20 (b) into CYP 3A4 protein and 33 (c), 23 (d), 31 (e), and C17 (f) into CYP 2C19 protein. Residues close to the pose (cutoff distance, 4 Å) were assigned by DS 2.5 software; HEM heme group
In total, 15 out of 20 inhibitors (IC50 > 30 μM) of CYP 2D6 were isoprenylated flavones or isoflavones, with adjacent hydroxyl substitution (26, 28, 31, 32, 33, 36, 39, and so on). The other five inhibitors (9, 12, 18, 21, and 22) belonged to flavanone, chalcone, dihydrochlcone, isoflavanone, and isoflavone, respectively. Molecular docking suggested that one phenyl ring could interact with close phenylalanine residues (e.g., Phe120; ESM Fig. 5S). In addition, isoprenylated flavonoids could establish π–π interactions with other aromatic residues (e.g., Phe 247; ESM Fig. 5SA), while unprenylated ones could only interact with closer, non-aromatic residues around the binding pocket (ESM Fig. 5SB). As previously reported, ligand-bound structure of 2D6 varied significantly from its ligand-free structure. Therefore, known 2D6 substrates included a variety of chemical structures (34,35). Our results showed that various types of flavonoids could inactive CYP 2D6, which was consistent with literature.
Following FDA recommendation (24), two structurally unrelated substrates, viz. midazolam and testosterone, were used to evaluate CYP3A4/5 inhibition. The P450 inhibition activities determined using MID and TES showed good correlations except for a few compounds, and the inhibition percentage determined with MID was generally higher than TES for the same compound. We observed that chalcones showed stronger inhibitory activities than flavanones, which could be evidenced by compounds 20/12/6/5 and their isomers 9/13/4/3 (Fig. 5). As suggested by docking simulation (Fig. 4a, b), hydrogen bonds could be observed between A-ring (7-OH of 9, or 4′-OH of 20) and Thr 309. The 2′-OH of 20 was close to the center of heme group, which might affect enzyme activity (36). Besides, chemical environments of their B-ring were also different. CYP 3A4 molecule has multiple active sites (37). These sites could change shape to fit specific molecules (29), and the substrate-dependent property led to poor correlation between predicted and determined values. Therefore, the structure–activity relationships for 3A4 inhibitors were not further discussed.
Fig. 5.
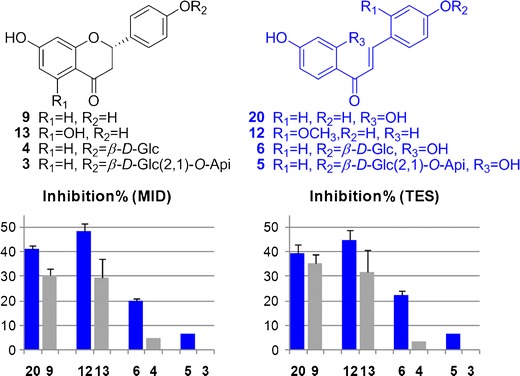
Chemical structures and CYP 3A4 inhibition activities of representative flavanones (9, 13, 4, 3) and chalcones (20, 12, 6, 5) at 10 μM
Based on the above experimental data and molecular docking analysis, free flavonoids and coumarins were identified as significant CYP inhibitors, especially the ones with rigid planar structure (for CYP 1A2), isoprenyl group and adjacent hydroxyl/methoxyl substitution (for CYP 2C9, 2C19, and 2D6), carbonyl group (for CYP 2C9 and 2C19), aromatic system on both sides (for CYP 2D6), and m-dihydroxybenzene substructure (for CYP 2C19 and 3A4).
P450 Inhibition Assay for Licorice Extracts
By using the same LC/MS/MS cocktail assay, the effects of licorice extracts on P450 enzyme activities were also evaluated (Fig. 6). To facilitate the comparison with pure compounds, mean molecular weights for these extracts were estimated according to the Licorice Compounds Database we had established (19). A 500-Da mean molecular weight was used for LWE, LEE, and LEA; 700 Da was used for LBU as described in ESM Fig. 6S. LWE and the LBU mainly contain flavonoid glycosides and saponins, and they did not affect P450s remarkably. In contrast, the LEA and LEE contained abundant free flavonoids and coumarins, and exhibited potent or moderate inhibition to the P450 isozymes except for 3A4. It was noteworthy that LBU exhibited 74.1 ± 14% activation of 2C9 at 10 μM. Given that none of the 40 tested licorice compounds showed strong activation activities on 2C9, LBU may contain unknown potent 2C9 activators.
Fig. 6.
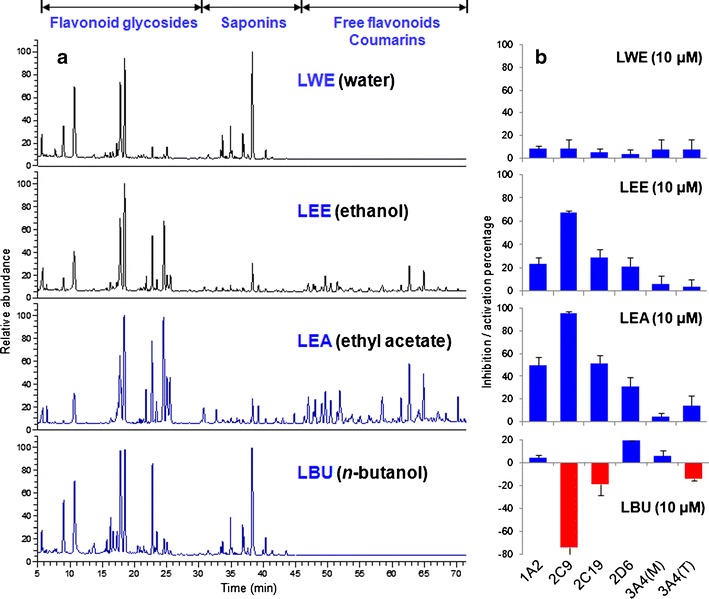
HPLC chromatograms of different licorice extracts a and their inhibition or activation rates on P450 isozymes at 10 μM b. LWE licorice water extract, LEE licorice ethanol extract, LBU licorice n-butanol fraction, LEA licorice ethyl acetate fraction, 3A4 (M) midazolam, and 3A4 (T) testosterone. Real concentration for 10 μM LWE, LEE, LEA, and LBU were 5, 5, 5, and 7 μg per mL of the extracted powder. The analytical method was described in ESM
The above results on licorice extracts were consistent with the assay of 40 single licorice compounds. Given the complex chemical composition of licorice, other components could also contribute to the P450 inhibition activities of licorice extracts. In total, 23 minor compounds were tentatively identified in LEA by LC/ion trap-time-of-flight mass-MS analysis (ESM Table 6S) and their presence in the HPLC fingerprint of licorice was labeled as circles in Fig. 3. Activities of these compounds including ten flavonoid glycosides; seven isoflavones; three coumarins; and a saponin, an isoflavanone, a phenol compound were predicted by molecular docking analysis. The result showed that saponin and most glycosides could not affect P450 isozyme, while isoflavones and coumarins were moderate and strong inhibitors (ESM Table 7S). Among them, isoprenylated flavonoids glyasperin C (C16), licocoumarone (C17), licobenzofuran (C18), and wighteone (C19) might strongly inhibit 1A2, 2C9, 2C19, and 2D6, while cyclization of isoprenyl group (C11–C15) could affect the ligand-protein binding, especially for 2C19 and 2D6. No potent inhibitor of 3A4 was found. If dual side chains were contained in the structure, inhibition to 1A2 could be weakened (C22 and C23). These results were consistent with the experimental values and structure–activity relationship we obtained from of 40 licorice compounds.
DISCUSSION
In this study, we found that a series of licorice compounds, especially isoprenylated flavonoids and arylcoumarins, could significantly inhibit the activities of cytochrome P450 isozymes in vitro. Among them, only the following compounds were prevalent in licorice extract: liquiritigenin (9), isoliquiritigenin (20), glycycoumarin (23), semilicoisoflavone B (24), kumatakenin (25), licoisoflavone A (26), licoricone (27), glycyrol (30), licoflavonol (32), topazolin (33), glycyrin (34), and licoisoflavone B (C15) (Fig. 1 and ESM Fig. 1S). Combined with our previous metabolic and pharmacokinetic studies, aglycones 9, 20, 23, 24, 26, 27, 30, and 32 could be detected in rats plasma after oral intake of licorice water extract, either in their unchanged form or as phase II metabolites (glucuronides or sulfates) (19). They could play the major role in the overall activity of licorice extracts to inhibit P450 enzymes. However, key licorice components for P450 inhibition are not limited to the above compounds. According to the metabolic fates of licorice compounds (19,20), some hydrophilic glycosides could be metabolized into less polar aglycones. For instance, liquiritin (4), liquiritin apioside (3), isoliquiritin (6), and isoliquiritin apioside (5) could all be converted into liquiritigenin (9), isoliquiritigenin (20), and davidigenin (18) after oral administration of licorice water extract (19). Although LWE contained abundant glycosides and few aglycones, the in vivo metabolism of LWE resulted in a high level plasma exposure of liquiritigenin (dual Cmax at 68.4 ± 16 and 78.5 ± 27 ng/mL) and isoliquiritigenin (dual Cmax at 44.7 ± 20 and 55.7 ± 22 ng/mL). In this regard, the glycosides did contribute to the P450 inhibition of licorice despite their weak in vitro activities (ESM Fig. 7SA). Similarly, ononin (7) could also be hydrolyzed to produce formononetin (22), which showed inhibitory effect to CYP 1A2 and 2D6. Saponin is another type of abundant compounds in licorice, especially in the water extract. Although all the tested saponins showed weak in vitro P450 activities, the saccharide chain could be eliminated in vivo to produce glycyrrhetinic acid (ESM Fig. 7SB), which showed moderate inhibition activities against P450 2D6 and 3A4 at 10 μM. As a result, the maximal concentration of glycyrrhetinic acid in rats plasma, after an oral administration of 5 g/kg licorice water extract, could be 14.6 ± 3.3 μg/mL, with a long circulation time (Tmax) of 13.6 ± 5.7 h. This may be the reason why licorice could alter the pharmacokinetics of several chemical drugs, while LWE showed little in vitro inhibition activities on P450 enzymes (8,10–12). On the other hand, kumatakenin (25), topazolin (33), and glycyrin (34) showed low bioavailability according to our metabolic studies and they may not play important role in the P450 inhibition of licorice.
Based on our results, the licorice constituents that may incur high risk of HDI were summarized in Table IV according to their target P450 isozymes. Selected drugs metabolized by P450 isozymes were listed correspondingly (8,38). Many of these medications were commonly used for chronic diseases, such as atorvastatin (for type II diabetes), celecoxib (for osteoarthritis), amitriptyline (antidepressant), and metoprolol (for hypertension). These drugs have potential to interact with licorice, especially when licorice was extensively used as an adjuvant agent, food supplement, and flavoring additive.
Table IV.
High-Risk Licorice Compounds Which Might Affect the Metabolism of Popular Chemical Drugs
| Chemical drug (brand name) | CYP | High-risk licorice compounds (No.) |
|---|---|---|
| amitriptyline (Elavil), haloperidol (Haldol), ondansetron (Zofran), propranolol (Inderal), theophylline (Theo-Dur), verapamil (Calan, Isoptin) (38) | 1A2 | Glycyrol (30), licoflavonol (32), glycycoumarin (23), liquiritigenin (9), semilicoisoflavone B (24), licoisoflavone A (26), licoricone (27) |
| omeprazole (Prilosec), lansoprazole (Prevacid), pantoprazole (Protonix), diazepam (Valium), carisoprodol (Soma), nelfinavir (Viracept) (38) | 2C19 | Glycycoumarin (23), licoricone (27), licoflavonol (32), isoliquiritigenin (20), licoisoflavone A (26), semilicoisoflavone B (24), licoisoflavone B (C15) |
| celecoxib (Celebrex), diclofenac (Voltaren), glipizide (Glucotrol), ibuprofen (Motrin), irbesartan (Avapro), losartan (Cozaar), phenytoin (Dilantin), torsemide (Demadex), warfarin (Coumadin) (8) | 2C9 | Glycyrol (30), glycycoumarin (23), licoisoflavone A (26), licoflavonol (32) |
| flecainide (Tambocor), imipramine (Tofranil), metoprolol (Toprol-XL), ondansetron (Zofran), paroxetine (Paxil), risperidone (Risperdal), tramadol (Ultram), venlafaxine (Effexor) (38) | 2D6 | Licoflavonol (32), semilicoisoflavone B (24), liquiritigenin (9), licoisoflavone A (26), glycycoumarin (23), licoisoflavone B (C15) |
| atorvastatin (Lipitor), ketoconazole (Nizoral), itraconazole (Sporanox), fexofenadine (Allegra), triazolam (Halcion) (8) | 3A4/5 | Licoflavonol (32), isoliquiritigenin (20), semilicoisoflavone B (24), licoisoflavone A (26) |
CONCLUSIONS
Licorice could affect the metabolism of some drugs by interacting with cytochrome P450 enzymes. The likely P450 modulators in licorice were identified in this study by combining their in vitro inhibition and previously published in vivo pharmacokinetic results. We concluded that liquiritigenin (9), isoliquiritigenin (20), together with seven isoprenylated flavonoids and arylcoumarins, viz. glycycoumarin (23), semilicoisoflavone B (24), licoisoflavone A (26), licoricone (27), glycyrol (30), licoflavonol (32), and licoisoflavone B (C15) may be the key compounds responsible for licorice–P450 interactions. Moreover, although flavonoid glycosides and saponins showed weak P450 activities, they could be converted into P450 inhibitors through in vivo metabolism.
Electronic supplementary material
PDF 534 kb
ACKNOWLEDGMENTS
This work was supported by National Natural Science Foundation of China (nos. 81173644 and 81222054), and the Program for New Century Excellent Talents in University from Chinese Ministry of Education (no. NCET-11-0019).
REFERENCES
- 1.United States Pharmacopoeia, USP34-NF29. Rockville: United States Pharmacopoeial Convention; 2011. pp. 1184–5. [Google Scholar]
- 2.Morgan AG, Pacsoo C, McAdam WA. Maintenance therapy: a two year comparison between Caved-S and cimetidine treatment in the prevention of symptomatic gastric ulcer recurrence. Gut. 1985;26:599–602. doi: 10.1136/gut.26.6.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Armanini D, Castello R, Scaroni C, Bonanni G, Faccini G, Pellati D, et al. Treatment of polycystic ovary syndrome with spironolactone plus licorice. Eur J Obstet Gynecol Reprod Biol. 2007;131:61–7. doi: 10.1016/j.ejogrb.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 4.Ferrari P, Sansonnens A, Dick B, Frey FJ. In vivo 11 beta-HSD-2 activity: variability, salt-sensitivity, and effect of licorice. Hypertension. 2001;38:1330–6. doi: 10.1161/hy1101.096112. [DOI] [PubMed] [Google Scholar]
- 5.Datla R, Rao SR, Murthy KJ. Excretion studies of nitrofurantoin and nitrofurantoin with deglycyrrhizinated liquorice. Indian J Physiol Pharmacol. 1981;25:59–63. [PubMed] [Google Scholar]
- 6.Natural Standard Professional Monograph. Licorice (Glycyrrhiza glabra) and DGL (deglycyrrhizinated licorice). http://www.naturalstandard.com. (2012). Accessed 22 Jul 2012.
- 7.Omar HR, Komarova I, El-Ghonemi M. Licorice abuse: time to send a warning message. Ther Adv Endocrinol Metab. 2012;4:125–38. doi: 10.1177/2042018812454322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.MedlinePlus (knowledgebase). National Institutes of Health, Rockville. 2013. http://medlineplus.gov. Accessed 22 Jul 2013.
- 9.Wienkers LC, Heath TG. Predicting in vivo drug interactions from in vitro drug discovery data. Nat Rev Drug Discov. 2005;4:25–33. doi: 10.1038/nrd1851. [DOI] [PubMed] [Google Scholar]
- 10.Guengerich FP. Cytochrome P450 and chemical toxicology. Chem Res Toxicol. 2008;21:70–83. doi: 10.1021/tx700079z. [DOI] [PubMed] [Google Scholar]
- 11.Lin SP, Tsai SY, Hou YC, Chao PDL. Glycyrrhizin and licorice significantly affect the pharmacokinetics of methotrexate in rats. J Agric Food Chem. 2009;57:1854–9. doi: 10.1021/jf8029918. [DOI] [PubMed] [Google Scholar]
- 12.Hou YC, Lin SP, Chao PDL. Liquorice reduced cyclosporine bioavailability by activating P-glycoprotein and CYP 3A. Food Chem. 2012;135:2307–12. doi: 10.1016/j.foodchem.2012.07.061. [DOI] [PubMed] [Google Scholar]
- 13.Methlie P, Husebye EES, Hustad S, Lien EA, Løvås K. Grapefruit juice and licorice increase cortisol availability in patients with Addison’s disease. Eur J Endocrinol. 2011;165:761–9. doi: 10.1530/EJE-11-0518. [DOI] [PubMed] [Google Scholar]
- 14.Jeong HG, You HJ, Park SJ, Moon AR, Chung YC, Kang SK, et al. Hepatoprotective effects of 18β-glycyrrhetinic acid on carbon tetrachloride-induced liver injury: inhibition of cytochrome P450 2E1 expression. Pharmacol Res. 2002;46:221–7. doi: 10.1016/S1043-6618(02)00121-4. [DOI] [PubMed] [Google Scholar]
- 15.Kent UM, Aviram M, Rosenblat M, Hollenberg PF. The licorice root-derived isoflavan glabridin inhibits the activities of human cytochrome P450s 3A4, 2B6, and 2C9. Drug Metab Dispos. 2002;30:709–15. doi: 10.1124/dmd.30.6.709. [DOI] [PubMed] [Google Scholar]
- 16.Tu JH, He YJ, Chen Y, Fan L, Zhang W, Tan ZR, et al. Effect of glycyrrhizin on the activity of CYP3A enzyme in humans. Eur J Clin Pharmacol. 2010;66:805–10. doi: 10.1007/s00228-010-0814-5. [DOI] [PubMed] [Google Scholar]
- 17.Zhao K, Ding M, Cao H, Cao ZX. In vitro metabolism of glycyrrhetinic acid by human and rat liver microsomes and its interactions with six CYP substrates. J Pharm Pharmacol. 2012;64:1445–51. doi: 10.1111/j.2042-7158.2012.01516.x. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Q, Ye M. Chemical analysis of the Chinese herbal medicine Gan-cao (licorice) J Chromatogr A. 2009;1216:1954–69. doi: 10.1016/j.chroma.2008.07.072. [DOI] [PubMed] [Google Scholar]
- 19.Xiang C, Qiao X, Wang Q, Li R, Miao WJ, Guo DA, et al. From single compounds to herbal extract: a strategy to systematically characterize the metabolites of licorice in rats. Drug Metab Dispos. 2012;39:1597–608. doi: 10.1124/dmd.111.038695. [DOI] [PubMed] [Google Scholar]
- 20.Qiao X, Ye M, Xiang C, Wang Q, Liu CF, Miao WJ, et al. Analytical strategy to reveal the in vivo process of multi-component herbal medicine: a pharmacokinetic study of licorice using liquid chromatography coupled with triple quadrupole mass spectrometry. J Chromatogr A. 2012;1258:84–93. doi: 10.1016/j.chroma.2012.08.041. [DOI] [PubMed] [Google Scholar]
- 21.Walsky RL, Obach RS. Validated assays for human cytochrome P450 activities. Drug Metab Dispos. 2004;32:647–60. doi: 10.1124/dmd.32.6.647. [DOI] [PubMed] [Google Scholar]
- 22.Walsky RL, Boldt SE. In vitro cytochrome P450 inhibition and induction. Curr Drug Metab. 2008;9:928–39. doi: 10.2174/138920008786485128. [DOI] [PubMed] [Google Scholar]
- 23.U.S. Food and Drug Administration. Guidance for industry: bioanalytical method validation. 2001. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070107.pdf. Accessed 03 Jan 2012.
- 24.U.S. Food and Drug Administration. Guidance for industry: drug interaction studies—study design, data analysis, and implications for dosing and labeling. 2006. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM072101.pdf. Accessed 3 Jan 2012.
- 25.He F, Bi H, Xie Z, Zuo Z, Li J, Li X, et al. Rapid determination of six metabolites from multiple cytochrome P450 probe substrates in human liver microsome by liquid chromatography/mass spectrometry: application to high-throughput inhibition screening of terpenoids. Rapid Commun Mass Spectrom. 2007;21:635–43. doi: 10.1002/rcm.2881. [DOI] [PubMed] [Google Scholar]
- 26.Kim MJ, Kim H, Cha IJ, Park JS, Shon JH, Liu KH, et al. High-throughput screening of inhibitory potential of nine cytochrome P450 enzymes in vitro using liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom. 2005;19:2651–8. doi: 10.1002/rcm.2110. [DOI] [PubMed] [Google Scholar]
- 27.Kunze KL, Wienkers LC, Thummel KE, Trager WF. Warfarin-fluconazole, I. Inhibition of the human cytochrome P450-dependent metabolism of warfarin by fluconazole: in vitro studies. Drug Metab Dispos. 1996;24:414–21. [PubMed] [Google Scholar]
- 28.Qiu R, Zhang R, Sun J, Jiye A, Hao H, Peng Y, et al. Inhibitory effects of seven components of danshen extract on catalytic activity of cytochrome P450 enzyme in human liver microsomes. Drug Metab Dispos. 2008;36:1308–14. doi: 10.1124/dmd.108.021030. [DOI] [PubMed] [Google Scholar]
- 29.Ekroos M, Sjögren T. Structural basis for ligand promiscuity in cytochrome P450 3A4. Proc Natl Acad Sci U S A. 2006;103:13682–7. doi: 10.1073/pnas.0603236103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou SF, Wang B, Yang LP, Liu LP. Structure, function, regulation and polymorphism and the clinical significance of human cytochrome P450 1A2. Drug Metab Rev. 2010;42:268–354. doi: 10.3109/03602530903286476. [DOI] [PubMed] [Google Scholar]
- 31.Mo SL, Zhou ZW, Yang LP, Wei MQ, Zhou SF. New insights into the structural features and functional relevance of human cytochrome P450 2C9. Part I. Curr Drug Metab. 2009;10:1075–126. doi: 10.2174/138920009790820129. [DOI] [PubMed] [Google Scholar]
- 32.Egnell AC, Eriksson C, Albertson N, Houston B, Boyer S. Generation and evaluation of a CYP2C9 heteroactivation pharmacophore. J Pharmacol Exp Ther. 2003;307:878–87. doi: 10.1124/jpet.103.054999. [DOI] [PubMed] [Google Scholar]
- 33.Reynald RL, Sansen S, Stout CD, Johnson EF. Structural characterization of human cytochrome P450 2C19: active site differences between P450’s 2C8, 2C9 and 2C19. J Biol Chem. 2012;287:44581–91. doi: 10.1074/jbc.M112.424895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hritz J, de Ruiter A, Oostenbrink C. Impact of plasticity and flexibility on docking results for cytochrome P450 2D6: a combined approach of molecular dynamics and ligand docking. J Med Chem. 2008;51:7469–77. doi: 10.1021/jm801005m. [DOI] [PubMed] [Google Scholar]
- 35.Wang A, Savas U, Hsu MH, Stout CD, Johnson EF. Crystal structure of human cytochrome P450 2D6 with prinomastat bound. J Biol Chem. 2012;287:10834–43. doi: 10.1074/jbc.M111.307918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li L, Chang Z, Pan Z, Fu ZQ, Wang X. Modes of heme binding and substrate access for cytochrome P450 CYP74A revealed by crystal structures of allene oxide synthase. Proc Natl Acad Sci U S A. 2008;105:13883–8. doi: 10.1073/pnas.0804099105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sevrioukova IF, Poulos TL. Understanding the mechanism of cytochrome P450 3A4: recent advances and remaining problems. Dalton Trans. 2013;42:3116–26. doi: 10.1039/c2dt31833d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.CYP P450 drug interaction table (database). U.S. Food and Drug Administration, Rockville. 2009. http://www.fda.gov/drugs. Accessed 22 Jul 2013.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDF 534 kb