

Abstract
The aim of this critical review is to reach a global consensus regarding the introduction of follow-on versions of nonbiological complex drugs (NBCD). A nonbiological complex drug is a medicinal product, not being a biological medicine, where the active substance is not a homo-molecular structure, but consists of different (closely related and often nanoparticulate) structures that cannot be isolated and fully quantitated, characterized and/or described by state of the art physicochemical analytical means and where the clinical meaning of the differences is not known. The composition, quality and in vivo performance of NBCD are highly dependent on manufacturing processes of both the active ingredient as well as in most cases the formulation. The challenges posed by the development of follow-on versions of NBCD are illustrated in this paper by discussing the ‘families’ of liposomes, iron–carbohydrate (‘iron–sugar’) drugs and glatiramoids. It is proposed that the same principles for the marketing authorization of copies of NBCD as for biosimilars be used: the need for animal and/or clinical data and the need to show similarity in quality, safety and efficacy. The regulatory approach of NBCD will have to take into consideration the specific characteristics of the drugs, their formulation and manufacturing process and the resulting critical attributes to achieve their desired quality, safety and efficacy. As with the biosimilars, for the NBCD product, family-specific methods should be evaluated and applied where scientifically proven, including sophisticated quality methods, pharmacodynamic markers and animal models. Concerning substitution and interchangeability of NBCD, it is also advisable to take biosimilars as an example, i.e. (1) substitution without the involvement of a healthcare professional should be discouraged to ensure traceability of the treatment of individual patients, (2) keep an individual patient on a specific treatment if the patient is doing well and only switch if unavoidable and (3) monitor the safety and efficacy of the new product if switching occurs.
KEY WORDS: biosimilars, generics, nonbiological complex drugs, regulatory guidance, substitution
INTRODUCTION
This critical review paper is the outcome of discussions on the science base of the regulatory process for nonbiological complex drugs (NBCD) and the emerging regulatory guidance documents. The paper summarizes the general understanding reached during the discussions with 25 scientific experts from industry, academia and regulatory bodies in a workshop at the FIP Centennial Congress in Amsterdam (2012). A draft document was circulated before and discussed during the meeting. In addition, the topic was discussed during a EUFEPS Regulatory Science Network workshop in October 2012 on ‘Complex Systems, science serving clinical needs’. The aim of this paper is to further reach a global consensus regarding the introduction of follow-on versions of NBCD among all stakeholders: innovators, follow-on manufacturers and regulators. This could for example be achieved by an ICH process. For terminology used in this paper, we refer to the paper ‘Different Pharmaceutical Products Needs Similar Terminology’ published concurrently (1).
A nonbiological complex drug is a medicinal product, not being a biological medicine, where the active substance is not a homo-molecular structure, but consists of different (closely related and often nanoparticulate) structures that cannot be isolated and fully quantitated, characterized and/or described by physicochemical analytical means. It is also unknown which structural elements might impact the therapeutic performance. The composition, quality and in vivo performance of NBCD are highly dependent on manufacturing processes of both the active ingredient as well as the formulation. These products raise specific challenges during the development, manufacturing, clinical testing and quality control by the originator as well as the follow-on producer. Comparability and interchangeability may need to be established during the entire life cycle due to variations caused by scaling up or process improvements and certainly during the development of a follow-on version.
This paper is a follow-up of the paper ‘The therapeutic equivalence of complex drugs’ published in 2011 (2). There is also a recently published report on the New York Academy of Sciences workshop on scientific considerations for complex drugs in the light of established regulatory guidance also addressing the NBCD (3). The current paper should be considered as an interim report because the debate about follow-on versions of complex drugs is ongoing. This paper is based on the discussion during a number of conferences (AAPS 2011, DIA Europe 2012, NYAS 2012 and FIP 2012) discussing complex drugs. It also gives a state of the art update on recently published insights on the topics that are important to be considered when aiming at a globally accepted regulatory pathway for marketing authorization of NBCD copies with a specific emphasis on their nanoparticle properties and the specific challenges related to interchange and substitution (therapeutic equivalence).
It is now generally acknowledged that the standard development programme applied to demonstrate therapeutic equivalence for small, well-characterized molecules does not apply to complex drugs. The classical generic approval was successful for many well-defined, small, low molecular weight drugs where the analytical testing fully characterized the product and showed pharmaceutical sameness to the reference list drug. Together with a proof of bioequivalence to the reference list product, this information led to the submission of an abbreviated approval protocol with a waiver for efficacy and safety studies, leading to interchange and substitution of the products. The new regulatory paradigms developed for biologicals may serve as an example how to regulate NBCD. Since 2006, the European Medicines Agency (EMA) has introduced a series of guidance documents and the Food and Drug Administration (FDA) recently issued three draft guidance documents on biosimilars providing general rules regarding the establishment of similarity to an innovator’s biological medicinal product.
NBCD share a number of characteristic features with biologicals: the structure cannot be fully defined by physicochemical analysis, and the biological and clinical characteristics are highly dependent on the production process. Examples of complex products in our previous communication were the families of the liposomal drugs, glatiramoids, iron–sugar complexes and low molecular weight heparins (LMWH). But more families of drugs or drug products may be considered complex products, i. e. groups of nanomedicines. In the current report, the first three categories will be discussed, whereas the LMWH, being by nature biological products regardless of their legal classification in various regions, do not fall within the new scope of the working group, which compared to the 2010 discussion/article, is now limited to non-biological complex drugs, i.e. NBCD and thus are not considered for this paper. Therefore, there will be an update on typical representatives like liposomes, iron-carbohydrate (‘iron–sugar’) drugs and glatiramoids.
LIPOSOMAL DRUGS
Liposomes are vesicles with (phospho)lipid bilayer membranes that can carry drugs. The safety and efficacy of these liposomal drugs depend on their lipid composition, size, charge, the rigidity of the bilayer and their production process (4). Clinical work on new liposomal formulations of a variety of drugs is thriving, and as of the end of 2012, over 550 clinical trials are registered using the search term ‘liposomes’ under www.clinicaltrials.gov.
The discussion on the regulatory aspects of the introduction of follow-on versions of liposome products focuses on those containing doxorubicin (Doxil®/Caelix®). Recently, the innovator published findings showing that doxorubicin liposomes with a different lipid composition differed in their antitumor activity in animals although they had similar pharmacokinetic (PK) profiles (5). A plea was made for clinical studies to show therapeutic equivalence between original and follow-on versions of liposomal drugs (6).
The FDA published a draft guidance paper on follow-on versions of doxorubicin hydrochloride liposomes (7). The applicant of a follow-on version needs to show that the physicochemical characteristics of the follow-on version are equivalent to the originator’s product. In addition, in vitro studies are needed measuring particle size distribution and doxorubicin release characteristics in different media and a clinical bioequivalence study measuring both free and encapsulated doxorubicin. Clinical efficacy and safety studies are not necessary.
An interesting regulatory precedent was the temporary permission (until 4 February 2013) from the FDA for the use of Lipodox™ because of the shortage of supply of Doxil™. Lipodox™ is marketed in India but had no market approval in the USA or in Europe. In 2013, the FDA approved the first follow-on liposome containing doxorubicin through an Abbreviated New Drug Application pathway.
Also, the EMA issued a reflection paper on the data requirements for intravenous liposomal products with reference to an innovator liposomal product (8). The EMA also considers requesting a physicochemical comparison between innovator and copy as well as a bioequivalence study with pharmacokinetic comparisons in patients of free drug and drug encapsulated by the liposomes. The necessity of a clinical efficacy trial is decided upon on a case-by-case basis. So far, no follow-on versions of liposomal formulations have been approved by the EMA.
COMPLEX IRON (SUGAR) NANOPARTICLE PRODUCTS
An iron–sugar nanoparticle consists of a polynuclear iron(III) hydroxide core surrounded by sugar molecules. Apart from the size, the reduction potential of the iron(III/II) and the strength of the interaction between the iron core and the surrounding sugar define the product’s quality, safety, efficacy and immunogenicity profile. The totality of the physicochemical properties of iron nanoparticle products defines the (only partially understood) bio-interference including their ability to interact with physiological acceptors (transferrin, ferritin, enzymes, specific cells like monocytes, etc.) (9). The iron nanoparticles are defined by proprietary manufacturing processes, which complicates the development of comparable or even interchangeable follow-on products (10). The pharmacokinetic parameters including the dissociation of the complex in plasma and biodistribution of the product into different tissues as well as the kinetics of the release of the active ingredient (iron) and the uptake into the physiological iron metabolism pathways may be profoundly different between products. Differences in efficacy and safety between originator iron sucrose and formerly authorized similar preparations have been published (11–15). These data raise concerns about interchangeability of such products based on both efficacy and safety. Patients treated in a recent study with an iron sucrose similar product required on average 34% more iron compared to patients treated with the originator product (14) (Fig. 1).
Fig. 1.
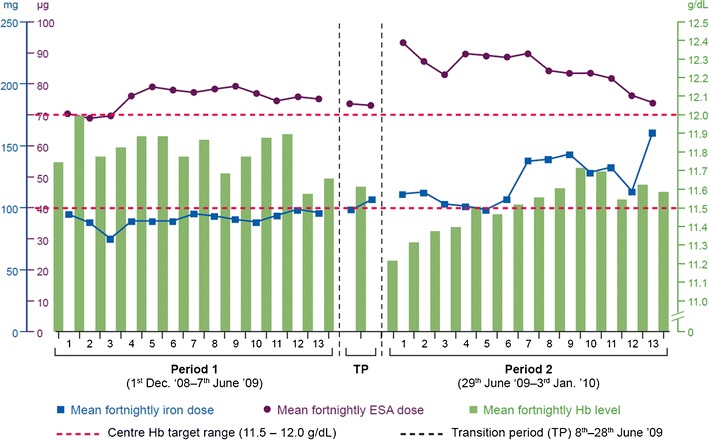
Observational study comparing two subsequent 27-week periods in 75 stable haemodialysis (HD) patients who had received IV iron weekly and an erythropoiesis-stimulating agent (ESA; darbepoetin alfa) once every 2 weeks. Patients received the originator product Venofer® in the period 1 and an iron sucrose similar in period 2. Based on the study by Rottembourg et al. (14)
Given the most common use of chronic intravenous (IV) iron treatment for anemia in haemodialysis patients and knowing that the excretion of iron in man is limited and not well regulated, concern must be raised regarding what potential harmful effects this excess iron may cause (9,16).
Existing quality requirements in pharmacopoeias are also not able to correlate with nonclinical toxicity (oxidative stress) testing in rats (17). This proves that such complex, nonbiological product cannot be fully identified/characterized in vitro. Biologically meaningful differences may appear after injection. Therefore, also an illness like chronic kidney disease may further modify PK-pharmacodynamic (PD), indicating the inappropriateness to check for bioequivalence in healthy volunteers.
Both the FDA and EMA acknowledge that nanoparticle (colloidal) iron (sucrose) preparations cannot be authorized by the so far, well-established (classical) generic approval paradigm for small molecules, which is based on the sameness of the product shown by physicochemical full characterization and a bioequivalence in healthy volunteers (18,19). Both agencies assess the comparability of ‘iron-sucrose-similar’ preparations to a reference product by either focusing on the kinetics of drug clearance and uptake from plasma (FDA) in healthy subjects or on the bio-disposition in target organs and tissues (EMA). Both authorities recognize that simple analysis of drug kinetics in plasma together with demonstrated pharmaceutical equivalence is not sufficient for full therapeutic (efficacy and safety) assessment of these products. Additional clinical testing with suitable patients and a brand name-based post-marketing surveillance are necessary to assess safety, efficacy and comparability of the similar to the original product. The need to also characterize the nanoparticle properties by qualitative and quantitative in vitro testing is recognized in the FDA draft guidance on IV iron carbohydrate complexes. Almost identical recommendations exist for iron sucrose (March 2012) (19), ferumoxytol (December 2012) and sodium ferric gluconate complex (June 2013). This draft guidance requests an in vitro comparison of particle morphology (atomic force microscopy) and diameter sizing (dynamic light scattering) from several batches of the products. Although FDA has proposed methods to better characterize the product physicochemically, the methods are not yet fully established and validated, but reflect FDA’s current thinking. In contrast, the EMA sets priority on a nonclinical equivalence testing approach, including the comparative in vivo assessment of cellular damage by labile (reactive) iron (18). The FDA is reconsidering their approval tools for sodium ferric gluconate because of general concerns. They have solicited a contractor for a prospective 3-year study combining both the current FDA approach and the EMA approach in the (re-) evaluation of Nulecit™ (follow-on) with Ferrlecit™ (originator reference product) requesting nonclinical combined with clinical PK-PD evaluation and toxicity testing (11). It will be interesting to see if the proposed modified ‘generic approach’ (FDA) or the concept of similarity (EMA) for this class of NBCD follow-on versions will detect noticeable structural differences that impact therapeutic comparability and therapeutic equivalence with substitution and interchange. These results will certainly assist an overarching approval process (9).
GLATIRAMOIDS
Glatiramoids comprise a family of synthetic copolymer mixtures comprising the four amino acids, l-glutamic acid, l-alanine, l-lysine and l-tyrosine, in a defined molar ratio. The prototype is glatiramer acetate (Copaxone®), a mixture of hundreds of thousands polypeptide sequences with immune-modulating activity authorized for the treatment of multiple sclerosis. The composition of glatiramer acetate is highly dependent on the manufacturing process. Minor changes in this process can produce altered entities which are likely to significantly affect the safety and efficacy of the product (20).
In addition to its inherent compositional complexity, glatiramer acetate comprises a nano-sized polypeptide mixture with molecules and molecular structures ranging from 1.5 to 550 nm in size; some of them to be deemed proteins because of their size (21). Due to these sizes, Copaxone® is considered to be a colloidal solution. Its biological activities are related to cytokine induction and its immunogenicity that is a key parameter of the quality, safety and efficacy of the drug and very sensitive to any modification of chemical and physical characteristics.
A number of follow-on glatiramoids have been developed that are similar in certain respects to—but not the same as—the original. However, the similarity is restricted to so-called drug’s ‘broad characteristics’ such as molecular weight distribution of polypeptide components in the mixture or amino acid content. Significant difference between Copaxone® and the follow-on glatiramoids were shown using highly sensitive specific procedures developed by the innovator, as well as state-of-the-art analytical and biological methods. Differences were found in primary structure, hydrophobic properties, higher order structure, identification, epitope mapping and potency tests demonstrated by bioassays, measuring effect on monocytes, cytotoxicity and cytokine secretion (22), as well as in a different gene expression pattern for a number of genes that are related to the immune modulatory activity of Copaxone® and are considered to be key elements in its therapeutic effect (23). The clinical implications of the differences observed between Copaxone® and purported generic versions are as yet unclear; therefore, the only meaningful way to evaluate the efficacy and safety ramifications of differences observed between two glatiramoids is by performing comparative clinical studies in patients with multiple sclerosis (Figs. 2 and 3).
Fig. 2.
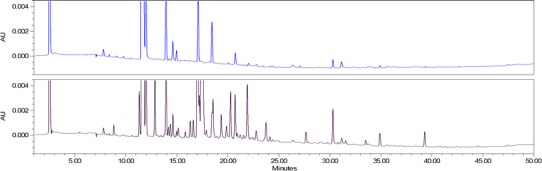
Comparative peptide mapping of originator product Copaxone (blue, upper trace) and a follow-on glatiramoid (black, lower trace) showing clear indication of differences in the primary structure of the drug amino acid sequences (22)
Fig. 3.
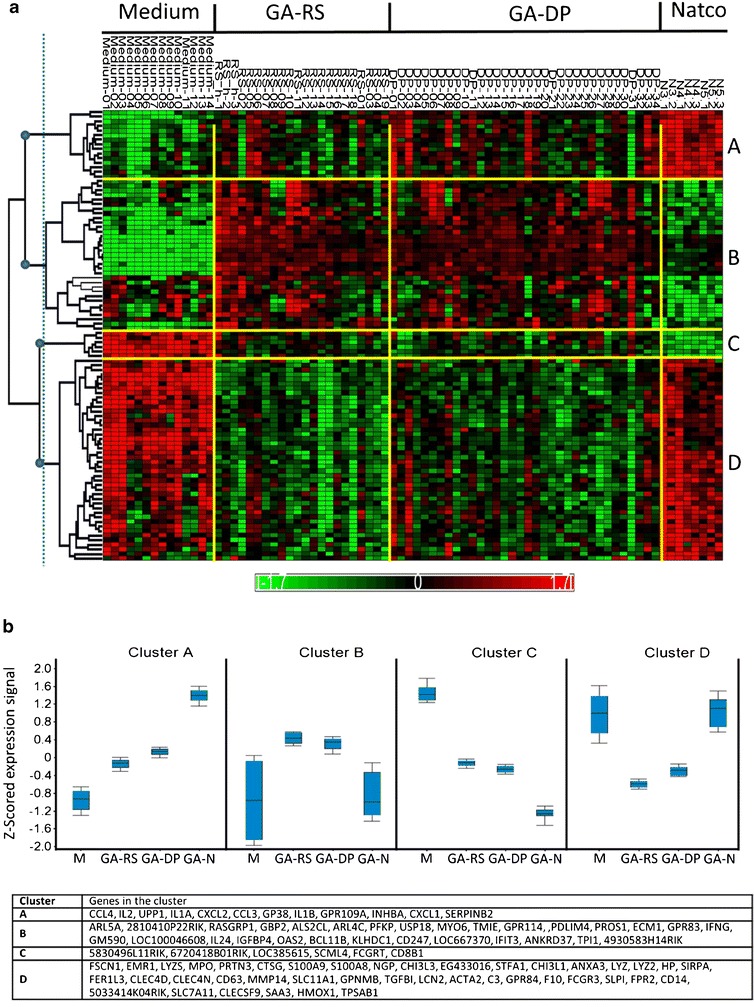
Gene-wise hierarchical clustering of 98 genes with FDR-adjusted p value <0.05 and fold change $1.3 between GA reference standard and eight follow-on Natco lots (23). a Heat map showing clustering results with samples (columns) ordered by sample name and genes (rows) ordered by hierarchical clustering. Four major gene clusters are marked A–D on the right; their root nodes are marked with blue circles on the dendrogram on the left. Yellow bars separate between treatment groups and clusters. b Average expression profile per cluster. For each gene, the mean expression signal per treatment group was calculated. The distribution of these values across genes in each cluster is shown as a box plot. GA-DP glatiramer acetate drug product, GA-N Natco lots, GA-RS glatiramer acetate reference standard, M medium
Apparently, this opinion is shared by the regulatory authorities. Recently, the FDA refused to approve a supplemental new drug application for a different formulation of Copaxone® that used the very same amount of the same active substance in half the injection volume, holding that a clinical trial would be necessary to demonstrate that the change in the formulation (the reduced injection volume) would not compromise the effectiveness of the product. Although not a biological drug as defined by the FDA/EMA, Copaxone® is at least as complicated to characterize as biological medicinal products.
The European Union’s regulatory pathway for biosimilars was released by the EMA in 2005 (24). The companies submitting applications for biosimilars must show that their follow-on products are closely related to reference medicines and do not have any meaningful differences in quality, safety and efficacy. Similar criteria were listed in the recent FDA draft guidelines on biosimilars (25). It will, however, be difficult for a follow-on glatiramoid to comply with these criteria because of inability of complete structural characterization with the existing, even advanced, analytical technology, the impossibility to use conventional PK to demonstrate bioequivalence, the lack of PD markers in predicting the clinical outcomes and the complex and not fully elucidated immune modulatory mode of action. The only way to establish whether a new glatiramoid or its formulation has similar quality/safety and effectiveness of the product is to establish therapeutic equivalence.
CONCLUSION
The regulatory guidelines developed by EMA and FDA discriminate between biological medicinal products and small molecules. The biologicals have their special regulatory position because they are a family of complex molecules which are difficult if not impossible to fully characterize by physicochemical analytical methods. In particular, the EMA has developed a comprehensive regulatory system that has resulted in successful applications for marketing authorization of biosimilars.
For the category of NBCD described in this paper, no dedicated regulatory pathway for follow-on versions has been set up. On a case by case basis, FDA followed the 505(j) route for some follow-on NBCD products. It remains to be seen if the 505(j) route is the most appropriate approval pathway. Occasionally, the FDA has used the guidance document process to develop and communicate regulatory approval requirements for complex drugs. The EMA position is discussed later in the ‘Conclusion’ section.
As reflected by the publication of a steady stream of (draft) guidance documents or reflection papers, there is an increasing awareness also among regulators of the need to adapt standard approval tools for NBCD by a stepwise approach to assess similarity/comparability up to therapeutic equivalence allowing then substitution and interchange between follow-on and reference product. In line with that, the European Directorate for the Quality of Medicines has created a new working party on nonbiological complexes indicating that for the full characterization of such complex products and in contrast to small drugs, new approaches for characterization and identification have to be adopted. Also, FDA considers this concept when looking into new guidance for IV iron nanoparticle products and the solicitation of a new prospective study to evaluate its existing approval tools compared to EMA thinking as mentioned in the part on complex IV iron nanoparticle above (11).
The legal basis for the biosimilar pathway designed by the EMA is in the Human Code, the EU legislation concerning public health. In article 12, it is stated that the follow-on pathway is not allowed for biological products and the marketing authorization request for these products needs to contain animal or clinical data. In the general biosimilar guidelines published by the EMA, the concept of similarity was introduced (24). And to become authorized as a biosimilar in the EU, the applicant needs to show similarity in quality, safety and efficacy.
So, the marketing authorization for a biosimilar needs a full quality dossier comparable with that of an original biological product. In addition, it should contain a comprehensive comparison in physicochemical and other in vitro characteristics showing no clinically relevant differences between biosimilar and control. The full toxicity program does not need to be repeated for a biosimilar, and animal PK and PD studies may be used to demonstrate similarity with the original. To show clinical similarity, it is not necessary to present equivalent efficacy in all hard clinical endpoints for every indications for which the original product is registered. It is allowed to use a biological endpoint that is sensitive to show possible differences in clinical activity between products. Extrapolation to other indication(s) based on this data is allowed, if it can be justified on scientific grounds such as similar mode of action and the same pathogenesis of the different disease conditions.
It is logical to use the same principles for the marketing authorization of copies of NBCD: the need for animal and/or clinical data and the need to show similarity in quality, safety and efficacy. As the NBCD, also the biosimilars are a diverse group of products with a variety in complexity and clinical use for which it is impossible to design a universal regulatory pathway. Therefore, the EMA has published a number of product-specific guidelines. For some products like insulin and filgrastim, a clinical study showing similarity in PD markers in healthy volunteers and/or patients suffices. In others, a clinical endpoint in patients is requested.
The regulatory approach of NBCD should be comparable and will have to take into consideration the specific characteristics of the drugs or their formulation and the resulting critical attributes to achieve their desired quality, safety and efficacy. As with the biosimilars, for the NBCD product family (e.g. liposomes, iron–sugar, glatiramoids), specific methods should be evaluated and applied where scientifically proven, including PD markers and animal models.
The EMA has stated that the marketing authorization as a biosimilar does not imply that the product is interchangeable with the original. This complies with the FDA position that interchangeability of biosimilars needs to be shown in specific studies. The EMA has also discouraged substitution without the involvement of a healthcare professional to ensure traceability of the treatment of individual patients in case of a product-specific safety issue. There is good evidence that such approaches should also apply to NBCD, and Table I gives an overview of the dossier requirements to meet for the different categories of NBCD discussed in this document.
Table I.
Dossier Requirements for NBCD Follow-on Versions
| Follow-on pathway | Additional NBCD similar requirements | ||||||
|---|---|---|---|---|---|---|---|
| Reference product specs (pharmaceutical equivalence) | Clinical PK (bio equivalence) | Extended physicochemical characterizationb | Biological in vitro quality/similarity tests | In vivo toxicity (animal studies) | Extended clinical PK/(PD) equivalence | Clinical studies (safety, efficacy, therapeutic equivalence) | |
| Doxorubicin liposomesa | + | + | + | ? | ? | + | ? |
| Glatiramoids | + | − | + | + | + | − | + |
| Iron–sucrose | + | + | + | − | + | + | + |
+ ‘required’, − ‘not-required’, ? case by case or differences between EMA and FDA regulations
aThe liposome group is quite heterogeneous. Therefore, attention is focused on follow-on versions of the doxorubicin family (i.c. Doxil/Caelix) which is presently under discussion
bFull characterization/quality assessment through all available, relevant analytical in vitro techniques is required
Certainly, other drug products will be recognized as NBCD in addition to the products discussed in the sections above such as, e.g. parenteral emulsions/micelles.
A group where more NBCD candidates will be found is the group of nanomedicines. Examples are polymer–drug conjugates, polymeric nanoparticles, polymeric micelles and iron oxide nanoparticles for diagnostic purposes. It is also expected that these will include older products that are in fact colloidal nanoparticulate systems and will be reclassified as nanoparticles and NBCD or nanosimilars (9).
The authors of this critical review encourage further scientific discussion and multidisciplinary research between experts in the different fields from academia, industry and regulatory bodies including consensus discussions with all stakeholders on an international level to develop meaningful guidance towards the definition of NBCD and the development of any follow-on version.
ACKNOWLEDGMENTS
This paper was written within the framework of the Non Biological Complex Drugs Working Group, hosted at TI Pharma (the Dutch Top Institute Pharma). The NBCD Working Group is currently supported by Vifor Pharma International Inc., Teva Pharmaceutical Industries Ltd. and Sanofi-Aventis Groupe. The authors acknowledge the valuable input of the participants during the discussions at the AAPS, FIP, DIA, EUFEPS and other conferences.
REFERENCES
- 1.Crommelin DJA., de Vlieger JSB, Weinstein V, Mühlebach S, Shah VP, Schellekens H. Different pharmaceutical products need similar terminology. 2013. doi:10.1208/s12248-013-9532-0. [DOI] [PMC free article] [PubMed]
- 2.Schellekens H, Klinger E, Mühlebach S, Brin JF, Storm G, Crommelin DJA. The therapeutic equivalence of complex drugs. Regul Toxicol Pharmacol. 2011;59:176–83. doi: 10.1016/j.yrtph.2010.09.021. [DOI] [PubMed] [Google Scholar]
- 3.Holloway C, Mueller-Berghaus J, Lima BS, Lee SL, Wyatt JS, Nicholas JM, et al. Scientific considerations for complex drugs in light of established and emerging regulatory guidance. Ann N Y Acad Sci. 2012;1276:26–36. doi: 10.1111/j.1749-6632.2012.06811.x. [DOI] [PubMed] [Google Scholar]
- 4.Chang HI, Yeh MK. Clinical development of liposome-based drugs: formulation, characterization, and therapeutic efficacy. Int J Nanomed. 2012;7:49–60. doi: 10.2147/IJN.S26766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mamidi RNVS, Weng S, Stellar S, Wang C, Yu N, Huang T, et al. Pharmacokinetics, efficacy and toxicity of different pegylated liposomal doxorubicin formulations in preclinical models: is a conventional bioequivalence approach sufficient to ensure therapeutic equivalence of pegylated liposomal doxorubicin products? Cancer Chemother Pharmacol. 2010;66:1173–84. doi: 10.1007/s00280-010-1406-x. [DOI] [PubMed] [Google Scholar]
- 6.Barenholz Y. Doxil®—the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160:117–34. doi: 10.1016/j.jconrel.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 7.FDA. Draft guidance on doxorubicin hydrochloride, February 2010, http://www.fda.gov/downloads/Drugs/…/Guidances/UCM199635.pdf. Accessed 3 Dec 2012.
- 8.EMA Committee for Medicinal Products for Human Use CHMP. Draft reflection paper on the data requirements for intravenous liposomal products developed with reference to an innovator liposomal product, 2011. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/07/WC500109479.pdf. Accessed 3 Dec 2012.
- 9.Ehmann F, Saka-Kato K, Duncan R, Hernan Perez de la Ossa D, Pita R, Vidal J-M, et al. Next generation nanomedicines and nanosimilars: EU regulators’ initiatives relating to the development and evaluation of nanomedicines. Nanomedicine. 2013;8:849–56. doi: 10.2217/nnm.13.68. [DOI] [PubMed] [Google Scholar]
- 10.Borchard G, Flühmann B, Mühlebach S. Nanoparticle iron medicinal products—requirements for approval of intended copies of non-biological complex drugs (NBCD) and the importance of clinical comparative studies. Reg Tox Pharm. 2012;64:324–8. doi: 10.1016/j.yrtph.2012.08.009. [DOI] [PubMed] [Google Scholar]
- 11.FDA. Therapeutic equivalence of generic iron complex products (solicitation for a study on Na ferric gluconate; Nulecit (follow-on upon 505j approval) vs Ferrlecit (originator). https://www.fbo.gov/index?s=opportunity&mode=form&id=592788989854da145c8e7b6d103c898d&tab=core&tabmode=list&. Accessed 30 June 2013.
- 12.Martin-Malo A, Merino A, Carracedo J, Alvarez-Lara MA, Ojeda R, Soriano S, et al. Effects of intravenous iron on mononuclear cells during haemodialysis session. Nephrol Dial Transplant. 2012;27:2465–71. doi: 10.1093/ndt/gfr711. [DOI] [PubMed] [Google Scholar]
- 13.Lee ES, Park BR, Kim JS, Lee JJ, Lee IS. Comparison of adverse event profile of intravenous iron sucrose and iron sucrose similar in postpartum and gynecologic operative patients. Curr Med Res Opin. 2013;29:141–7. doi: 10.1185/03007995.2012.760444. [DOI] [PubMed] [Google Scholar]
- 14.Rottembourg J, Kadri A, Leonard E, et al. Do two intravenous iron sucrose preparations have the same efficacy? Nephrol Dial Transplant. 2011;26:3262–7. doi: 10.1093/ndt/gfr024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stein J, Chow AK. Clinical case reports raise doubts about the therapeutic equivalence of an iron sucrose similar preparation compared with iron sucrose originator. Curr Med Res Opin. 2012;28:241–3. doi: 10.1185/03007995.2011.651527. [DOI] [PubMed] [Google Scholar]
- 16.Ghoti H, Rachmilewitz E, Simon-Lopez R, et al. Evidence for tissue iron overload in long-term hemodialysis patients and the impact of withdrawing parenteral iron. Eur J Hematol. 2012;89:87–93. doi: 10.1111/j.1600-0609.2012.01783.x. [DOI] [PubMed] [Google Scholar]
- 17.Toblli JE, Cao G, Oliveri L, Angerosa M. Comparison of oxidative stress and inflammation induced by different intravenous iron sucrose preparation in a rat model. Inflamm Allergy Drug Targets. 2012;11:66–78. doi: 10.2174/187152812798889358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.EMA Committee for Medicinal Products for Human Use (CHMP). Reflection paper on non-clinical studies for generic nanoparticle iron medicinal product applications 17 March 2011, EMA/CHMP/SWP/100094/2011. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/04/WC500105048.pdf. Accessed 3 Dec 2012.
- 19.FDA. Draft guidance on iron sucrose bioequivalence 2012 (March). http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM297630.pdf Accessed 3 Dec 2012.
- 20.Varkony H, Weinstein V, Klinger E, Sterling J, Cooperman H, Komlosh T, et al. The glatiramoid class of immunomodulator drugs. Expert Opin Pharmacother. 2009;10(4). [DOI] [PubMed]
- 21.Citizen Petition submitted by Teva to the FDA, June 4, 2012 http://www.regulations.gov/contentStreamer?objectId=0900006481031791&disposition=attachment&contentType=pdf. Accessed 3 Dec 2012.
- 22.Nicholas JM. Complex drugs and biologics: scientific and regulatory challenges for follow-on products. Drug Inf J. 2012;46(197).
- 23.Bakshi S, Chalifa-Caspi V, Plaschkes I, Perevozkin I, Gurevich M, Schwartz R. Gene expression analysis reveals functional pathways of glatiramer acetate activation. Expert Opin Ther Targets. 2013;17(4):351–62. doi: 10.1517/14728222.2013.778829. [DOI] [PubMed] [Google Scholar]
- 24.European Medicines Agency. Guideline on similar biological medicinal products. London, UK, 30 October 2005. www.emea.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003517.pdf. Accessed 3 Dec 2012.
- 25.CDER. Guidance for industry: scientific considerations for demonstrating biosimilarity to a reference product. US Food and Drug Administration: 9 February 2012. www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf. Accessed 3 Dec 2012.