

Abstract
In recent years, “nutri-epigenetics,” which focuses on the influence of dietary agents on epigenetic mechanism(s), has emerged as an exciting novel area in epigenetics research. Targeting of aberrant epigenetic modifications has gained considerable attention in cancer chemoprevention research because, unlike genetic changes, epigenetic alterations are reversible and occur during early carcinogenesis. Aberrant epigenetic mechanisms, such as promoter DNA methylation, histone modifications, and miRNA-mediated post-transcriptional alterations, can silence critical tumor suppressor genes, such as transcription factors, cell cycle regulators, nuclear receptors, signal transducers, and apoptosis-inducing and DNA repair gene products, and ultimately contribute to carcinogenesis. In an effort to identify and develop anticancer agents which cause minimal harm to normal cells while effectively killing cancer cells, a number of naturally occurring phytochemicals in food and medicinal plants have been investigated. This review highlights the potential role of plant-derived phytochemicals in targeting epigenetic alterations that occur during carcinogenesis, by modulating the activity or expression of DNA methyltransferases, histone modifying enzymes, and miRNAs. We present in detail the epigenetic mode of action of various phytochemicals and discuss their potential as safe and clinically useful chemopreventive strategies.
KEY WORDS: cancer chemoprevention, dietary agents, DNA methylation, epigenetics, histone modification, microRNA
INTRODUCTION
Epigenetics refers to the study of a set of reversible yet heritable changes in gene expression or cell phenotype that occur without any alterations in DNA sequence (1). Due to their reversible nature and early incidence during the process of cancer development, epigenetic modifications have taken the center stage as promising drug targets for cancer prevention. The major epigenetic mechanisms for regulation of gene expression are DNA methylation, alterations in the chromatin structure by post-translational modification of histones and some non-histone proteins, and miRNAs which can either degrade mRNAs or modulate their translation process (2). These epigenetic processes are essential in the regulation of normal functions of the cell during all stages, including development and differentiation (3), and facilitate adaptation to environmental changes, such as nutritional variation or exposure to cigarette smoke, chemicals, radiation, and hormones (4). However, alterations of epigenetic targets may also lead to various diseases, including cancer (5).
DNA Methylation
DNA methylation is catalyzed by DNA methyltransferase (DNMT) enzymes that transfer methyl groups from S-adenosyl-L-methionine (SAM) to a cytosine present next to guanine, known as CpG, forming 5-methylcytosine-guanine dimers. In mammalian cells, the DNMT1 enzyme maintains methylation of DNA during replication, whereas DNMT3a and DNMT3b methylate previously unmethylated DNA sequences and therefore may play an important role in the genesis of cancer (6). Sequences exist in DNA that are rich in CG repeats, known as CpG islands; these repetitive sequences are heavily methylated and act to maintain chromosomal stability (7,8). CpG within the promoter regions of a gene are generally unmethylated. Increased methylation of CpG within gene promoters can lead to transcriptional silencing of tumor suppressors; whereas global DNA hypomethylation in CpG islands can lead to the genomic instability commonly observed in malignant cells (9,10).
Histone Modifications
Gene expression is also regulated epigenetically by post-translational modification of histone proteins, including acetylation, methylation, phosphorylation, ubiquitinylation, sumoylation, and ADP ribosylation. Histone acetylation and methylation are the most common post-translational modifications of histone proteins associated with carcinogenesis (11). Histone acetylation is catalyzed by a class of enzymes known as histone acetyltransferases (HATs), whereas histone deacetylation is catalyzed by histone deacetylases (HDACs). For example, HATs transfer acetyl groups onto the ε-amino group of lysine (K); HDACs remove these acetyl groups. Histone acetylation leads to an open chromatin structure and enables transcription factors to bind to DNA, whereas deacetylation leads to chromatin condensation and transcriptional repression. To date, 18 HDACs and 25 HATs proteins have been identified. HDACs are divided into four classes—I, II, III and IV—based on homology, size, sub-cellular expression, and number of enzymatic domains (12). Class I HDACs consists of HDACs 1, 2, 3, and 8; class II HDACs are comprised of HDACs 4, 5, 6, 7, 9, and10; class IV HDACs have a single member—HDAC11. Class III HDACs are structurally unrelated to the other classes and require NAD+ as a cofactor. This group consists of seven members named sirtuins 1–7 (13). HATs have also been divided into multiple classes—GNAT (hGCN5 and PCAF), MYST (MYST and Tip60), p300/CBP (p300/CBP), SRC (SRC-1), and TAFII250 (TAFII250)—based on structure, homology, and histone specificity (14). Histone methylation occurs at various lysine and arginine residues. Methylation of lysine on histones may either activate or repress gene expression, depending on the location of the lysine residue methylated; for example, methylation at H3K4, H3K36, and H3K79 generally leads to transcriptionally active chromatin, whereas H3K9, H3K27, and H4K20 methylation lead to transcriptional inactivation. Lysine residue methylation on histones is catalyzed by histone lysine methyltransferase (HMTs) enzymes. Like lysine acetylation, lysine methylation is not limited to histone proteins, as several non-histone proteins are also subject to methylation (15).
MicroRNAs
miRNA are small non-coding RNAs of 20–22 nucleotides that inhibit gene expression at the posttranscriptional level to regulate key biological processes, and may also be altered in many diseases, including cancer (16). miRNAs are generated by a complex protein system, which consists of proteins from the Argonaute family, polymerase II-dependent transcription, and Drosha and Dicer ribonucleases from RNA precursor structures (17). miRNAs regulate the translation processes, either by imperfect base-pairing to the mRNA or by affecting mRNA stability. Each miRNA controls several genes within related pathways and are implicated in the initiation and progression of cancer, as they are often deregulated during carcinogenesis due to alterations at the genetic and epigenetic level and defects in their processing (18).
Recent studies have provided convincing evidence that some natural dietary agents, including phytochemicals, have the ability to reverse epigenetic changes before they accumulate and cause disease, such as cancer (19). Because of their ability to alter DNA methylation, histone modifications, and miRNA expression, dietary phytochemicals possess chemopreventive potential. In this review, we highlight several dietary polyphenols which have epigenomic-altering ability (Fig. 1).
Fig. 1.
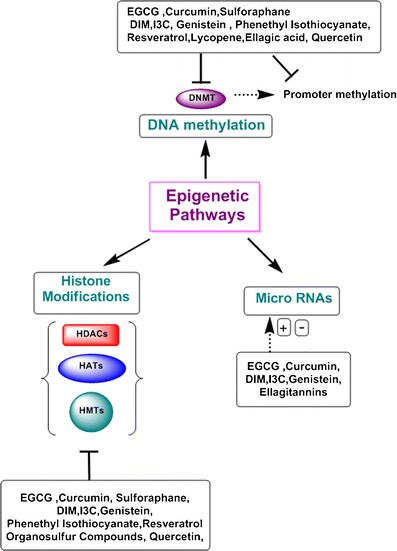
Epigenetic pathways affected by plant phytochemicals. Numerous pathways are deregulated in cancer cells. Major epigenetic mechanisms that regulate gene expression are DNA methylation, alterations in the chromatin structure by post-translational modification of histones, and miRNAs which can either degrade mRNAs or modulate their translation process. Plant phytochemicals have shown to affect epigenetic pathways.  demonstrates regulation;
demonstrates regulation;  demonstrates inhibition
demonstrates inhibition
DIETARY AGENTS AS EPIGENETIC MODULATORS
Tea Polyphenols
Tea, the most widely consumed beverage in the world after water, is consumed in a variety of styles, such as green, black and oolong tea. Tea polyphenols, namely the catechins found in abundance in green tea, have been shown to reduce the risk of various diseases, including cancer. (−)-Epicatechin (EC), (−)-epicatechin-3-gallate (ECG), (−)-epigallocatechin (EGC), and (−)-epigalocatechin-3-gallate (EGCG) are four major catechins present in green tea; EGCG constitutes more than 50% of the total catechin isolated from green tea and has been widely studied for its anticancer effects. Green tea polyphenols have been shown to exert anticancer effects by a number of different mechanisms, including the inhibition of cellular proliferation by induction of cell cycle arrest and apoptosis, and the suppression of oxidative stress, angiogenesis, invasion, and metastasis (20).
Epigenetic aberrations, such as polycomb group (PcG) protein-dependent histone methylation, class-I HDACs dependent deacetylation of histones and some non-histone proteins, and ubiquitination, drive chromatin compaction, and have been shown to reduce tumor suppressor function and increase cancer cell survival; evidence suggests that green tea polyphenols exert their anticancer effects by correcting these epigenetic alterations in cancer cells. Studies with structural analogues of EGCG suggested that D- and B-ring structures of tea polyphenols were important to the inhibition of DNMT activity; molecular modeling supported this conclusion, and suggested that EGCG can form hydrogen bonds with Pro(1223), Glu(1265), Cys(1225), Ser(1229), and Arg(1309) within the catalytic pocket of DNMT. All green tea catechins have been found to inhibit DNMT1 to various extents, with IC50 values ranging from 210 to 470 nM. In vitro analysis has shown that catechol-containing polyphenols inhibit DNMTs by (a) increasing the formation of the potent noncompetitive inhibitor, S-adenosyl-L-homocysteine (SAM), caused by o-methylation of SAM by the catechol-O-methyltransferase enzyme; (b) directly inhibiting the activity of dihydrofolate reductase enzyme, disrupting the folate cycle and increasing SAM levels; and (c) directly inhibiting DNMTs, independent of methylation status. Inhibition of DNMTs ultimately results in DNA hypomethylation and re-expression of proto-oncogenes or other repressed genes (21).
Inhibition of DNA methylation by a commonly consumed dietary constituent was first demonstrated in the human esophageal cancer cell line, KYSE510, a cell line exhibiting loss of tumor suppressor gene p16, RARβ, MGMT, and hMLH1 mRNA expression due to promoter hypermethylation. Treatment of KYSE510 with 5–50 μM EGCG for 6 days led to a reversal of hypermethylation and re-expression of tumor suppressor gene mRNA and protein, similar in effect to classical DNMT inhibitors, 5-aza-2′-deoxycytidine (5-Aza-dC) and zebularine. Similar results were also observed in HT-29 human colon cancer cells and PC-3 prostate cancer cells (22). Treatment of LNCaP human prostate cancer cells with 1–10 μg/ml of GTP for 1–7 days caused a dose- and time-dependent re-expression of GSTP1, a gene silenced in number of cancers by hypermethylation of its promoter. Re-expression of GSTP1 was correlated with inhibition of DNMT1 enzyme activity. GTP treatment also decreased mRNA and protein levels of MBD1, MBD4, and MeCP2, and caused decreased association of MBD2 with accessible Sp1 binding sites, leading to increased binding and transcriptional activation of the GSTP1 gene. GTP treatment did not cause global changes in hypomethylation pattern, but rather promoted the maintenance of genomic integrity. Unlike exposure of cells to 5-Aza-dC, which caused activation of both GSTP1 and S100P, GTP treatment did not cause activation of pro-metastatic gene S100P, demonstrating specificity of GTP to re-express only silenced tumor suppressor genes, thus maintaining genomic stability. GTP treatment also concomitantly decreased the mRNA and protein levels of class-I HDACs 1–3 and increased acetylation of histone H3 and H4 (23). Exposure of human skin cancer A431 cells to EGCG decreased global DNA methylation in a dose-dependent manner, and also decreased 5-methylcytosine, DNMT1, 3a and 3b mRNA and protein levels, H3 at Lysine 9 methylation, and enzymatic activity of DNMTs and HDACs, and increased levels of Histone H3 at Lysine 9 and 14 and Histone H4 at lysine 5, 12, and 16 acetylation in these cells. Additionally, EGCG treatment re-expressed tumor suppressor genes p16 and p21 mRNA and protein, which had been epigenetically silenced in these cells (24). In MCF-7 breast cancer cells, EGCG treatment led to progressive demethylation of the hTERT promoter, including its E2F-1 binding sites, resulting in increased binding of E2F-1, a potent inhibitor, to the hTERT promoter, and further downregulated hTERT by causing hypoacetylation of histone H3K9. The epigenetic regulation of hTERT by EGCG may be a cell specific phenomenon, as treatment of HL60 promyelocytic leukemia cells did not demonstrate an effect similar to the MCF-7 breast cancer cells (25), whereas similar mechanisms of hTERT and cell proliferation inhibition were described in MCF-7 (ER+) and MDA-MB-231 (ER−) breast cancer cells (26). In azoxymethane-Apc Min/+ mouse model of intestinal cancer, mice supplemented with 0.6% (w/v) solution of GTP as the sole source of fluid for 8 weeks showed a significant decrease in methylation of 24 CpG sites within the promoter region of RXR alpha gene, a significantly increase in the protein levels of RXR alpha, and demonstrated that downregulation of RXR alpha is independent of β-catenin expression and is an early event in colon carcinogenesis (27). Treatment of human prostate cancer LNCaP cells (harboring wild-type p53) and PC-3 cells (lacking p53) with 10–80 μg/ml of GTP for 24 h resulted in a dose-dependent inhibition of class I HDAC enzyme activity and enhanced proteasomal degradation of class I HDAC proteins. GTP treatment caused an accumulation of acetylated histone H3 in total cellular chromatin, resulting in increased accessibility to bind with the promoter sequences of p21/waf1 and Bax consistent with cell cycle arrest and induction of apoptosis in both cell lines irrespective of their p53 status (28). GTPs and the major constituent, EGCG, activated p53 in LNCaP human prostate cancer cells by increasing its acetylation at K373 and K382 residues on its C terminus by inhibiting class I HDACs in a dose- and time-dependent manner; discontinuation of GTP or EGCG treatment resulted in loss of p53 acetylation. This GTP or EGCG treatment also resulted in enhanced binding of acetylated p53 on the promoters of p21/waf1 and Bax and increased expression of p21/waf1 and Bax mRNA and protein, leading to cell cycle arrest and apoptosis (29).
The polycomb group (PcG) proteins enhance cell survival by epigenetically regulating gene expression. Increased levels and enhanced activity of PcG proteins lead to increased methylation and reduced acetylation of the histones associated with tumor suppressor genes, causing reduced tumor suppressor activity and increased cell proliferation and survival. Increased expression of two key PcG proteins, Bmi-1 and EZH2, was found in both immortalized keratinocytes and skin cancer cell lines; EGCG treatment of SCC-13 skin cancer cells reduced Bmi-1 and EZH2 protein levels, and global reduction in H3K27 trimethylation was associated with decreased survival. Reduced expression of PcG proteins was also associated with reduced expression of key cell cycle progression proteins (i.e. cyclins, cdks) and increased expression of cell cycle inhibitory proteins (i.e., p21, p27). EGCG treatment also induced apoptosis, as indicated by increased caspase-9, -8, and -3 and PARP cleavage and ratio of Bax/Bcl-xL. This study concluded that GTPs reduce cell survival in skin tumors by influencing PcG-mediated epigenetic regulatory mechanisms (30). It was later reported that reduction in PcG protein level after GTP is associated with their increased ubiquitination and can be blocked by proteasome inhibitors.
Green tea polyphenols have ability to modify the expression of miRNAs in various human cancers. Treatment of human hepatocellular carcinoma HepG2 cells with EGCG altered their miRNAs expression. Thirteen miRNAs were upregulated and 48 downregulated in cells treated with EGCG as compared to untreated cells; RAS, Bcl2, E2F, TGFBR2, and c-Kit are target genes of these upregulated miRNA; HOX family proteins, including PTEN, SMAD, MCL1, SLC16A1, TTK, PRPS1, ZNF513, and SNX19 are target genes of the downregulated miRNA. Treatment with ECGC downregulated Bcl-2 protein expression; transfection with anti-miR-16 inhibitor suppressed miR-16 expression, and counteracted the EGCG effects on Bcl-2 downregulation and induced apoptosis in these cells (31). Treatment of breast cancer MCF-7 cells with Polyphenon-60 significantly altered the expression of 23 miRNAs, including down-regulation of miR-21 and miR-27, which were initially over-expressed in these cancer cells. EGCG treatment induced apoptosis in hepatocellular carcinoma HepG2 cells by increasing miRNA-16 expression leading to decrease in Bcl-2 protein levels. In a xenograft mouse model utilizing prostate cancer LNCaP cells, EGCG treatment repressed the transcriptional activation of androgen receptor (AR), which correlated with significant decrease in androgen-regulated miRNA-21 and upregulation of tumor suppressor, miRNA-330, and inhibited tumor growth (32). The results obtained for miRNA profiling suggests that EGCG may exert its biologic functions through modulation of miRNA expression.
Curcumin
Turmeric is a popular Indian spice associated with cancer prevention and other multiple health benefits. Its major active ingredient, curcumin, has been reported to affect multiple intracellular signaling pathways involved in proliferation, invasion, survival, apoptosis, and inflammation (33). Molecular docking studies demonstrate interaction of curcumin with DNMT1 have suggested that curcumin may inhibit DNMT1 enzymatic activity by covalently blocking its catalytic site by binding to the thiol group of C1226 amino acid. This same study found that Leukemia MV4-11 cells showed global DNA hypomethylation after treatment with curcumin; however, sequence-specific demethylation at the promoter region of epigenetically silenced genes with curcumin was not demonstrated (34). Nrf2, a master regulator of cellular antioxidant defense systems, has been shown to be epigenetically silenced during the progression of prostate tumorigenesis in TRAMP mice. Curcumin treatment of TRAMP C1 cells led to demethylation of the first 5 CpGs within the promoter region of the Nrf2 gene and re-expression of both Nrf2 mRNA and protein, and enhanced expression of a major downstream target gene, NQO1, an anti-oxidative enzyme; this mechanism may be, in part, responsible for the chemopreventive effect of curcumin (35). Curcumin treatment also restored expression of Neurog1, another cancer-related gene silenced by promoter hypermethylation, in prostate cancer LNCaP cells, by demethylating the first 14 CpG sites within its promoter; curcumin also significantly decreased MeCP2 binding to the promoter of Neurog1. Treatment with curcumin increased HDAC1, 4, 5, and 8 levels, but decreased HDAC3. HDAC activity, H3K27me3 levels, binding at the Neurog1 promoter region was decreased after treatment, suggesting ability of curcumin to re-express yet another gene silenced by epigenetic modification in cancer (36). Treatment of SiHa squamous cervical cancer and HeLa adenocarcinoma cervical cancer cell lines with 20 μM curcumin caused demethylation of the promoter and reactivation of RARβ2 gene; the extent of demethylation increased with treatment time from 3 to 6 days in SiHa cells, whereas demethylation in HeLa cells was noted after 6 days treatment (37).
Curcumin is also a potential modulator of histones due to its ability to modulate the activity of HATs and HDACs enzymes. Marcu et al. (38) demonstrated that curcumin is a selective inhibitor of CBP/p300, indicating that alpha-beta unsaturated carbonyl groups in the side chain of curcumin function as Michael reaction sites and are required for activity as HAT inhibitor. Curcumin also promoted proteasome-dependent degradation of CPB/p300 proteins without any effect on PCAF or GCN5 in cells. In addition, acetyltransferase activity of purified p300 was assessed using either histone H3 or p53 as substrate (39). Curcumin effectively blocked the HDAC inhibitor MS-275, inducing histone hyperacetylation in both PC3-M and peripheral blood lymphocytes cells (40). Curcumin treatment inhibited HDAC1, HDAC3, and p300/CBP in Raji cells, which led to decreased activity of NF-κB and Notch1 and caused significant inhibition of cell proliferation. The effect of curcumin on HDACs and HATs was more pronounced and was partially due to increased proteasomal degradation, as protection from degradation by MG-132 could be partially reversed with curcumin treatment (41). Liu et al. also confirmed that curcumin was able to inhibit the expression of class I HDACs and increase expression of Ac-histone H4 in Raji cells (42). Bora-Tatar et al. used HeLa nuclear extract in a fluorometric assay and performed molecular docking for human HDAC8 enzyme; this group reported that curcumin more potently inhibited HDACs as compared to sodium butyrate, a well-known HDAC inhibitor (43). Curcumin treatment in human medulloblastoma DAOY, D283 Med, and D341 cells resulted in cell cycle arrest and apoptosis, accompanied by reduced HDAC4 expression and activity and increased tubulin acetylation, which eventually caused mitotic catastrophe. Treatment with curcumin also reduced the growth of medulloblastoma xenografts tumors in Smo/Smo transgenic medulloblastoma mouse model, significantly increasing mouse survival (44). In human breast cancers, overexpression of enhancer of zeste homolog 2 (EZH2) gene indicates poor prognosis; curcumin treatment of MDA-MB-435 breast cancer cells led to the downregulation of EZH2 expression in a dose- and time-dependent manner, which also correlated with decreased cell proliferation. Curcumin induces downregulation of EZH2 through activation of MAPK, c-Jun NH2-terminal kinase, ERK, and p38, leading to anti-proliferative effects (45).
Curcumin has been shown to modulate miRNA expression in cancer cells. Curcumin modified the expression of 29 miRNA in human pancreatic carcinoma BxPC-3 cells, including miRNA-22 upregulation, leading to suppressed expression of its target genes, Sp1 and ESR1 (46). Curcumin and its derivative, CFD, sensitized human pancreatic cancer cell lines MIAPaCa-E, MIAPaCa-M, and BxPC-3 to gemcitabine by inactivating miR-21, leading to inhibition of NF-κB, COX-2, and their downstream target molecules and reactivation of miR-200b and miR-200c (47). Curcumin treatment significantly downregulated expression of miR-186* in A549/DDP multidrug-resistant human lung adenocarcinoma cells and induced apoptosis (48). Treatment of MCF-7 breast cancer cells with curcumin caused downregulation of Bcl-2 and increased expression of miR-15a and miR-16; silencing of miR-15a and miR-16 by specific inhibitors restored Bcl-2 expression, suggesting that reduced Bcl-2 expression in breast cancer cells after curcumin treatment is due to curcumin ability to upregulate the expression of miR-15a and miR-16 (49). A dose-dependent decrease in miR-21 promoter activity and expression following curcumin treatment was inferred due to reduced binding of AP-1 to the promoter and induction of the tumor suppressor gene, programmed cell death protein 4 (Pdcd4), which is a miR-21 target and is overexpressed in RKO and HCT116 human colon cancer cells, promoting invasion and metastasis. Curcumin also stabilized the expression of the tumor suppressor Pdcd4 in colorectal cancer (50).
Sulforaphane
Another bioactive phytochemical sulforaphane (SFN), widely present in broccoli, sprouts, cabbage and kale, has been widely studied for its anticancer activity. SFN has been shown to enhance xenobiotic metabolism, and induce cell cycle arrest and apoptosis in various human cancer cells (51). Like many other plant polyphenols, SFN can also modify epigenetic events in cancer cells. In human colon cancer CaCo-2 cells, SFN treatment resulted in the downregulation of DNMT1 activity (52). In breast cancer MCF-7 and MDA-MB-231 cells, SFN treatment decreased DNMT1 and DNMT3a activity and caused site-specific CpG demethylation within the first exon of the hTERT gene, facilitating CTCF binding associated with hTERT repression, and leading to the inhibition of the catalytic regulatory subunit of telomerase. SFN treatment increased acetylation of histone H3 at K9 and histone H4 and decreased the histone H3 trimethylation at K9 and K27, respectively in a dose-dependent manner. This increased acetylation and reduced trimethylation of histones facilitated and enhanced the binding of hTERT repressor proteins such as MAD1 and CTCF to the regulatory region of hTERT, causing apoptosis (53). SFN treatment decreased HDAC activity and increased the activity of a β-catenin-responsive reporter (TOPflash) in a dose-dependent manner in human embryonic kidney 293 cells. Though β-catenin or HDAC protein levels were not altered, HDAC activity was diminished, and corresponded with increased acetylation of both global and localized histones and increased acetylated histones bound to the p21 promoter. These findings were also observed in human colorectal cancer HCT116 cells (54). In BPH-1 benign prostate hyperplasia and in LNCaP and PC-3 prostate cancer cells, SFN treatment also inhibited HDAC activity, and subsequently increased the levels of acetylated histones and their binding on the promoters of p21 and Bax genes, induced p21 expression, increased tubulin acetylation in prostate cancer cells, leading to cell cycle arrest and caspase-dependent apoptosis. SFN-induced cytotoxicity was reversed by HDAC6 overexpression (55). SFN exposure to MDA-MB-231, MDA-MB-468, MCF-7, and T47D human breast cancer cell lines caused inhibition of HDAC, increased acetylation of histones, decreased expression of ER, EGFR and HER-2 receptors, and inducted cell cycle arrest and apoptosis (56).
A few, in vivo studies using animal models have suggested that SFN can reduce HDAC activity and increase acetylation of histone proteins. In APC Min/C mice, SFN treatment reduced tumor formation through increased global histone acetylation, increased association of acetylated histone H3 on the promoters of p21 and Bax genes, and increased expression of Bax protein. Wild-type C57BL/6 JC/C mice receiving a single oral dose of 10 μM SFN demonstrated significant inhibition of HDAC activity within the colonic mucosa, with a concomitant yet transient increase in the acetylation levels of Histones H3 and H4 (57). Consumption of 7.5 μM SFN per mouse for 21 days resulted in 40% reduced growth in PC-3 tumor xenograft in nude mice; this correlated with a significant decrease in HDAC activity, increase in global histone acetylation, and increase in the Bax expression in tumors and mononuclear blood cells in these animals (58). A pilot study consisting of 3 human subjects fed 68 g of broccoli sprouts, which are rich in SFN, demonstrated significant inhibition of HDAC activity and increased acetylation of histone H3 and H4 in their peripheral blood mononuclear cells within 3 - 6 h after intake (58).
Indole-3-Carbinol [I3C] and Diindolylmethane [DIM]
Indole-3-carbinol (I3C) is a hydrolyzed product of the phytochemical glucosinolate, present in cruciferous vegetables, including broccoli, cabbage, cauliflower, mustard, and radish. Conversion of glucosinolate to I3C is catalyzed by an enzyme present in these plants called myrosinase; acidic pH in the stomach converts I3C to diindolylmethane (DIM). Both I3C and DIM have been reported to induce apoptosis in many cancer cell lines from solid tumors of different organs by affecting number of kinases and nuclear receptor-mediated signaling (59). A study with human colon cancer HT-29, SW620, RKO, LS174T, and HCT-116 cell lines and in tumor xenografts showed that DIM induced proteosomal-mediated degradation of class I HDACs (1–3, 8) with no effect on class II HDAC proteins. Class I HDAC degradation relieved transcriptional inhibition and increased expression of the cyclin-dependent kinase inhibitors p21/waf1 and p27/Kip1, and caused the cells to arrest in G (2) phase of the cell cycle. This study also found an increased DNA damage associated with the degradation of class I HDAC and induction of cell apoptosis after DIM treatment (60). DIM treatment of gemcitabine-resistant human pancreatic cancer cells MiaPaCa-2, Panc-1, and Aspc-1 resulted in the upregulation of miR-let-7b, miR-let-7e, miR-200b, and miR-200c, and was correlated with the upregulation of the epithelial cell marker, E-cadherin, and downregulation of mesenchymal markers, ZEB1 and vimentin (61). Treatment of pancreatic cells with DIM reduced their invasion capability, which was due to DIM ability to regulate miRNA-146, which in turn reduced expression of EGFR, MTA-2, IRAK-1 and reduced activation of NF-κB pathway in these cells (62). DIM treatment has also been shown to downregulate CDK2, CDK4, and Cdc25A expression, leading to cell cycle arrest, by modulating miR-21 in both estrogen-dependent MCF-7 and ER-negative p53 mutant MDA-MB-468 human breast cancer cells; therefore, regardless of estrogen-dependence and p53 status, DIM can cause cell cycle arrest in breast cancer cells by regulating miR-21. This study also demonstrated inhibition of human breast tumor development by DIM in an in vivo xenograft model (63).
Genistein and Soy Isoflavones
Epidemiological studies have shown that a soy rich diet decreases the incidence of some human cancers, including breast and prostate. Genistein, the active component found in soy, exerts its cancer preventive effects by targeting various pathways relevant in the development of cancer (64). A number of studies have demonstrated that genistein regulates the transcription of various genes by affecting different epigenetic events. Genistein reactivated DNA methylation-silenced p16INK4a, RARβ, and MGMT genes in human esophageal squamous KYSE510 carcinoma cells by inhibiting DNMT activity in a dose-dependent manner, also inhibiting cell growth. Reactivation of RARβ by the reversal of DNA hypermethylation by genistein was also observed in human prostate cancer LNCaP and PC3 cells (65). Treatment of human breast cancer MDA-MB-468 cells with low concentrations of genistein restored the expression of GSTP1 gene by partially demethylating its promoter (66). In human prostate cancer PC-3, DU-145, and LNCaP cell lines, in which promoters of GSTP1 and EPHB2 are strongly methylated, treatment with soy isoflavones genistein and daidzein caused demethylation of these promoters and increased protein expression (67). Similar results were demonstrated with BRCA1, GSTP1 and EPHB2 promoters and protein expression in human prostate cancer DU145 and PC-3 cell lines after treatment with genistein or daidzein (68).
Wnt signaling pathway plays an important role both in normal epithelial regeneration and tumorigenesis in the human colon. Treatment of human colon cancer SW1116 cells with genistein or soy induced WNT5a gene expression by decreasing the methylation of CpG islands on its promoter and causing cell growth inhibition by inhibiting cell proliferation in a time-dependent manner (69). A tumor suppressor gene, BTG3, which is downregulated in renal cancer due to promoter hypermethylation was demethylated in renal cell carcinoma A498, ACHN and HEK-293 cells by inhibition of DNMT activity and MBD2 after treatment with genistein. Genistein treatment significantly decreased promoter methylation, reactivated BTG3 expression, increased levels of acetylated histone H3 and H4, H3K4me2, H3K4me3, and RNA polymerase II, decreased DNA methyl transferase and methyl-binding domain protein 2 activity, and increased HAT activity in prostate cancer cells (70). Results on effect of genistein treatment on DNA methylation are inconsistent; while in vitro studies in cancer cells have shown inhibition of DNMT activity and DNA methylation, in vivo studies have suggested otherwise. A randomized, double-blind trial conducted on premenopausal healthy women (n = 34) to determine the effect isoflavones, including genistein, daidzein, and glycitein taken daily (40 or 140 mg) through one menstrual cycle, on the methylation status of p16, RASSF1A, RARb2, ER, and CCND2 genes known to be silenced due to hypemethylation of their promoters in breast cancer. The RARb2 and CCND2 methylation was found increased in intraductal specimens after treatment which correlated with genistein concentration in the serum (71).
Genistein also possesses histone modifying activity and was shown to induce the expression of p21/waf1/cip1 and p16INK4a tumor suppressor genes in human prostate cancer cells by epigenetic mechanisms involving active chromatin modification, including upregulation of the expression of HATs (72). Genistein, daidzein and equol (a daidzein metabolite) have been shown to stimulate ER-mediated histone acetylation through modulating HAT activity and co-activator activity of ER (73). Treatment of LNCaP and PC-3 prostate cancer cells with genistein activated several aberrantly silenced tumor suppressor genes with unmethylated promoters such as PTEN, CYLD, p53, and FOXO3a by post-translation modification of histone H3K9. PTEN and CYLD genes were reactivated through induction of a substantial remodeling of the heterochromatic domains by demethylation and acetylation of H3K9 and caused the inhibition of PI3K/Akt signaling pathway. Genistein also increased acetylated H3K9 in p53 and FOXO3a via downregulation of histone deacetylase SIRT1 (74). Following genistein treatment, ubiquitination of AR protein in LNCaP cells was increased, due to a decrease in chaperone activity, inhibition of HDAC6, a Hsp90 deacetylase, and enhanced acetylation of Hsp90 (75)
Soy isoflavones may also have the ability to modulate miRNAs. A recent study identified a set of 53 differentially regulated genes by comparing the miRNA profile of untreated and genistein treated UL-3A and UL-3B cells developed from an ovarian cancer patient. Genistein caused upregulation of ERα and ERβ at both the mRNA and proteins level, and reduced the migration potential and invasiveness of genistein treated cells compared to untreated cells. Unfortunately, this study did not elaborate or attempt to characterize the role of miRNAs in the induction of ERα and ERβ (76). In gemcitabine-resistant human pancreatic cancer cell lines MiaPaCa-2, Panc-1, and Aspc-1, there was a positive correlation between miRNA-200 and the mesenchymal markers including ZEB1, slug, and vimentin; both were downregulated after treatment with genistein. Genistein treatment in these cells also reversed their EMT transition (61). In prostate cancer, LNCaP and PC-3 cells upregulation of miRNA-1296 and accumulation of cells in the S phase of the cell cycle were observed after treatment with genistein. The miRNA-1296 upregulation lead to a significant decrease in mRNA and protein levels of its target gene minichromosome maintenance gene [MCM-2] (77). Genistein also caused suppression of uveal melanoma C918 cells growth by inhibiting ZBTB10 gene via downregulation of miRNA-27 (78).
Phenethyl Isothiocyanate
Cruciferous vegetables, like wasabi, horseradish, mustard, radish, brussel sprouts, watercress, nasturtiums, and capers, are also rich in isothiocyanate, a chemical group formed by the substitution of oxygen with sulfur, which provides the characteristic flavors of these vegetables. Phenyl isothiocyanate (PEITC) is one of the most studied isothiocynate for its anticancer activity. PEITC has been reported to induce apoptosis and cell cycle arrest in a number of cancer cell types (79). PEITC demethylated the hypermethylated promoter of the GSTP1 gene, reactivating GSTP1 in androgen-dependent and independent prostate cancer cells. PEITC also inhibited HDAC levels and activity and caused selective changes in the histone acetylation and methylation patterns. This dual PEITC action was more effective than the pharmacological inhibitors of DNMT and HDACs (80). Allyl isothiocyanate treatment of DS19 mouse erythroleukemia cells increased the acetylation of histones with no effect on HDACs (81).
Histone acetylation is virtually undetectable in acute leukemia patients due to high expression and activity of HDACs. Bone marrow cells from acute myeloid leukemia (AML) patients cultured in phenylhexyl isothiocyanate showed significant acetylation of histones, indicating inhibition of HDAC activity by phenylhexyl isothiocyanate (82). Treatment of hepatocellular carcinoma SMMC-7721 cells with phenylhexyl isothiocyanate inhibited cell growth and induced apoptosis, which correlated with increased acetylation of Histone H3 and H4, increased methylation of H3K4, and decreased methylation of H3K9 (83).
PEITC alone, or in combination with other chemopreventive agents, has been reported to inhibit cell proliferation, differentiation, Ras activation, the NF-κB pathway, and angiogenesis, induce apoptosis, reverse p53 function, and prevent the downregulation of miRNAs induced by cigarette smoke. miR-125b, miR-26a, miR-146-pre, let-7a, let-7c, miR-192, miR-222-pre, miR-99, and miR-123 are some of the miRNA downregulated by cigarette smoke; their expression was altered in rats treated with orally administered PEITC for 3 days prior to exposing them to cigarette smoke for 28 consecutive days (84). The effect of PEITC or glucocorticoid budesonide treatment, either alone or in combination, on miRNA expression was analyzed in the mouse liver and lungs. Treatment and subsequent smoke exposure was initiated either directly after birth or after weaning for 2 weeks. PEITC treatment significantly downregulated 9 and upregulated 3 miRNAs in the liver and caused modest effect on miRNA expression in the lungs. Co-treatment significantly upregulated 12 and downregulated 11 miRNAs in comparison to the group treated with only cigarette smoke. These miRNAs expressed differentially were the one associated with genes regulating stress response, protein repair, cell proliferation, and inflammation. No study on the effect of PEITC on miRNA expression in cancer has been reported so far (85).
Resveratrol
Resveratrol, a polyphenol found in red wine, peanuts, and certain berries, possesses antioxidant and anti-inflammatory properties, and has been found to have beneficial effects in cardiovascular disease and cancer (86). Resveratrol exhibits weak DNMT inhibitory activity in MCF7 breast cancer cells and was unable to reverse methylation of several tumor suppressor genes. Resveratrol improved the efficacy of adenosine analogues to inhibit methylation of the promoter of RARβ2 gene; however, resveratrol alone was ineffective. Resveratrol in combination with Vitamin D3 was highly effective in reducing methylation of PTEN promoter and inducing expression of PTEN, downregulating DNMT, and regulating p21 in ER-positive MCF-7 breast cancer cells, but had no notable effects in triple-negative MDA-MB-231 breast cancer cells (87).
Other demonstrated targets of resveratrol are class III HDACs, sirtuin 1 (SIRT1) and p300. Resveratrol activates SIRT1 catalytic core independent of its terminal domains, indicating the existence of a resveratrol binding site within the catalytic core of the enzyme. Low levels of SIRT1 and high levels of survivin were found in mammary tumors of BRCA1 mutant mice; induced expression of BRCA1 led to increased SIRT1 expression by binding of BRCA1 to the promoter of SIRT1. Increased SIRT1 expression in turn inhibited survivin by modulating histone H3K9 levels. Both in vitro and in vivo, resveratrol was able to decrease the acetylation of histone H3K9 by inducing SIRT1 expression and inhibit survivin expression to elicit a profound inhibitory effect on BRCA1 mutant cancer cells, an effective strategy for targeting BRCA1-associated breast cancers (88). Another in vivo study using SIRT1-null mice demonstrated that the number of intestinal polyps induced in Apcmin mutation carrying mice was unaffected by the SIRT1 genotype, but the polyp size was slightly smaller. SIRT1 genotype did not affect the incidence and tumor load of skin papillomas. Topical application of resveratrol to the skin profoundly reduced tumorigenesis, yet the effect was reduced in SIRT1-null mice, suggesting resveratrol requires SIRT1-encoded protein for its protection (89). Resveratrol also prevented epigenetic silencing of tumor suppressor BRCA-1 by the aromatic hydrocarbon receptor in human breast cancer MCF-7 cells by modulating acetylation of H3K9, and H4, association of mono-methylated-H3K9, DNMT1, and methyl-binding domain protein-2 with the promoter of BRCA-1 gene (90).
Resveratrol has also been shown to provide protection from cancer by modifying miRNAs. In human SW480 colon cancer cells, decreased levels of several oncogenic miRNAs, known to target Dicer1, PDCD4, PTEN, and the key effectors of the TGFβ signaling pathway, were found after resveratrol treatment. Treatment significantly caused upregulation of 22 miRNA and downregulated expression of 26 miRNA. Several of the constitutively upregulated miRNA in colon cancer including miR-17, miR-21, miR-25, miR-92a-2 were decreased after treatment with resveratrol. The miR-663 which was found increased possess putative tumor-suppressor functions and targets TGF1 transcript. Components of the TGFβ signaling pathway, including TGFβ RI and RII and SMADs were also reduced suggesting that miR-663 is a target for anticancer activity of resveratrol (88).
Organosulfur Compounds
Allium vegetables, such as garlic, chives, and leeks, have been used to improve immunity and cardiovascular health, for microbial, radiation, and cancer protection, and as hypoglycemic agents in traditional medicine. Risk of stomach and colon cancers is significantly reduced if allium vegetables are consumed regularly. The anticancer activity of these vegetables is attributed to organosulfur compounds, which are released upon processing, and are generated by the decomposition of highly unstable products formed upon conversion of alliin to allicin and other alkyl alkane-thiosulfinates by alliinase. A few of these organosulfur compounds, such as diallyl sulfide [DAS], diallyl disulfide [DADS], and diallyl trisulfide [DATS], have been shown to induce cell cycle arrest and apoptosis, and inhibit cancer growth, angiogenesis and metastasis (91). In in vivo studies, their treatment provides protection from chemically induced cancer of various organs and inhibited tumor growth in xenograft models. DADS and its active metabolite S-allylmercaptocysteine [SAMC] are finally metabolized to allyl mercaptan [AM] and other metabolites (92).
DADS and SAMC has been demonstrated to induce acetylation of histones proteins and inhibition of cell growth in DS19 mouse erythroleukemia cells. Their metabolite AM was a more potent HDAC inhibitor. Direct binding of AM to the catalytic site of HDAC was predicted by performing in silico docking studies and HDACs inhibition potential was confirmed by performing activity assays. DADS treatment increased global acetylation of H3 and H4 histones and increased their binding on the promoter of p21 gene. These events correlated with HDAC inhibition, upregulation of p21 and cell cycle arrest (81). Treatment of human colon cancer Caco-2 cells and human breast cancer T47D cells with SAMC induced histone acetylation where HDAC activity was inhibited with allyl butyrate (93). S-allylmer-captocysteine or allyl isothiocyanate treatment decreased HDACs and HATs in DS19 cells. DADS treatment resulted in hyperacetylation of histones, upregulation p21, arrest of cell cycle and induction of differentiation and apoptosis in a variety of cancer cell lines. Treatment of colon cancer Caco-2 and HT-29 cells with DADS exhibited decreased HDAC and increased acetylation of histones H3 and H4 which correlated with increase expression of p21/Waf1 and cell cycle arrest (93).
Lycopene
Lycopene is present mainly in tomato and tomato products. It is one of the naturally occurring classes of tetra-terpenoids and is a potent antioxidant. In vivo studies using animal cancer models have shown that lycopene can inhibit breast, prostate, and lung tumor growth, but is ineffective in preventing colon, kidney and liver cancers (94). A single dose of 2 μM lycopene was able to partially demethylate the promoter of the GSTP1 tumor suppressor gene in breast cancer cell line MDA-MB-468 cells and increase its mRNA expression. However, RARβ2 gene was not demethylated by lycopene treatment in neither MDA-MB-468 cells nor MCF-7 breast cancer cell lines. Demethylation of RARβ2 and HIN-1 genes in immortalized non-tumorigenic MCF10A fibrocystic breast cells however was observed after 2 weeks of lycopene treatment. The study shows that lycopene has direct DNA demethylating activity (66).
Quercetin
Dietary polyphenols are multi-potent flavonoids with immense potential for the prevention and treatment of cancer (95). Quercetin is one such biflavonoid present predominantly in citrus fruits and buckwheat. In yeast, quercetin activates NAD-dependent histone deacetylase SIRT1. It inhibited the growth of colon cancer RKO cells by reversing the silencing of hypermethylated p16INK4a gene by demethylating its promoter (96). Quercetin treatment inhibited the expression of TNF-induced interferon-gamma-inducible protein 10 [IP-10] and macrophage inflammatory protein 2 [MIP-2] by inhibiting the activity of CBP/p300 and phosphorylation/acetylation of histone H3 on the promoter region of these genes (97). Quercetin treatment in human leukemia HL-60 cells induced FasL-mediated extrinsic apoptosis pathway through caspase-8 activation, Bid cleavage, changes in conformation of Bax and cytochrome c release. Quercetin increased histone H3 acetylation through activation of HAT demonstrating that quercetin induced FasL-related apoptosis by transactivation through activation of c-jun/AP-1 and promotion of histone H3 acetylation in HL-60 cells (98). A recent study reported simultaneous administration of quercetin to DMBA-painted hamsters reduced incidence and burden of tumor, whereas post-treatment of quercetin resulted in a significant tumor growth delay. Quercetin administration caused cell cycle arrest and apoptosis and blocked invasion and angiogenesis. This study found a positive correlation between the inhibition of HDAC1 and DNMT1 by quercetin and its anticancer properties (99).
Ellagitannins
Ellagitannins are polyesters of ellagic acid and a sugar moiety. The presence of these phytochemicals in common food is limited to few fruits and nuts, such as pomegranate, raspberries, strawberries, blackberries, walnuts and almonds. Ellagitannins are widely used in alternative medicine for their antioxidant, radical scavenging, antiviral, antimicrobial, anti-mutagenic, anti-inflammatory, anti-tumor promoting and immunomodulatory properties. Ellagitannins modulate various transcription factors and signaling pathways in their ability to inhibit proliferation and induce apoptosis of cancer cells (100). Treatment of liver cancer HepG2 cells with ellagitannin (BJA3121) isolated from a plant Balanophora japonica, inhibited cell growth and altered the expression of several miRNAs. Ellagitannin treatment, in particular caused increased expression of miR-let-7e, miR-370, miR-373* and miR-526b and decrease expression of let-7a, let-7c, let-7d. These miRNA are correlated with genes involved in cell differentiation and proliferation (101).
Other Dietary Phytochemicals
Other plant phytochemicals which are able to modify epigenetic events and may prove to have potential to be developed as effective chemopreventive and/or therapeutic agent are under investigation. Research study from our group demonstrated that apigenin, a plant flavone has ability to alter histone deacetylation. This study demonstrated that treatment of prostate cancer PC-3 and 22Rv1 cells with 20-40 μM apigenin led to decreased HDAC enzyme activity, decreased protein and mRNA levels of HDAC1 and HDAC3 and resulted in increased global acetylation of histone H3 and H4 and increased localized acetylated histone H3 on the p21/waf1 promoter. Treatment with apigenin also caused increased expression of protein and mRNA of p21/waf1 and BAX and caused cell cycle arrest and induced cell apoptosis in both cancer cells. Study further reported that oral ingestion of apigenin at doses of 20 and 50 μg/mouse/day over an 8-week period resulted in marked reduction in tumor growth, HDAC activity, and HDAC1 and HDAC3 protein expression in PC-3 xenografts of athymic nude mice (102). Other plant flavonoids, including biacalein, cyanidins, rosmarinic acid, silibinin/silymarin, and dihydrocoumarin, are also under investigation for their epigenetic effects in cancer. These compounds are present in the daily food and can effectively cause epigenetic modifications to prevent carcinogenesis and to suppress or delay cancer progression.
SUMMARY, CONCLUSIONS, AND FUTURE DIRECTIONS
Prevention is better than cure, a phrase which holds true for complex and deadly diseases like cancer. As discussed, important cellular functions and cell signaling pathways are deregulated in cancer by epigenetic deregulation of critical tumor suppressor genes by methylation of CpG islands on their promoters, abnormal post-translation modifications of histone and some non-histone proteins by deregulation of acetylation/methylation, and miRNA perturbation. Accumulating evidences indicates that dietary chemopreventive agents can prevent or reverse these epigenetic modifications in cell culture studies and in some animal models of cancer. A concise description of these alterations is shown in Table I.
Table I.
Effect of Dietary Polyphenols on Epigenetic Regulatory Mechanisms
| Phytochemical/Dietary agent (source) | Epigenetic modification(s) | Mechanism(s) | Reference |
|---|---|---|---|
| Green tea polyphenol—epigallocatechin-3-gallate (EGCG) (green tea) | DNA methylation | DNMT inhibitor | (21–32) |
| Histone modifications | Promoter methylation ↓ | ||
| Differential miRNA modulations | SAM, 5mC ↓ | ||
| HDAC activity ↓ | |||
| HAT exp ↑ | |||
| Ac-H3, H3K9Ac, Ac-H4 ↑ | |||
| HMT inhibitor | |||
| BMI-1, SUZ12, EZH2, EED, H3K27me3 ↓ | |||
| miR-16 ↑, miR-21 ↓, miR-27 ↓, miR-330 ↑ | |||
| Curcumin (turmeric) | DNA methylation | DNMT inhibitor | (34–50) |
| Histone modifications | Promoter methylation ↓ 5mC ↓ | ||
| Differential miRNA modulations | MeCP2 binding ↑ | ||
| H3K27me3 ↑ | |||
| HDAC1,HDAC-3,HDAC-8 ↓ | |||
| p300 (HAT) ↓ | |||
| Ac-H3, Ac-H4 ↓ | |||
| miR-22 ↑, miR-186 ↓ | |||
| miR-15a ↑, miR16 ↑ | |||
| Sulforaphane (Broccoli) | DNA methylation | DNMT expression ↓ | (52–58) |
| Histone modifications | Promoter methylation ↓ | ||
| HDAC inhibitor | |||
| Ac-H3 and Ac-H4 ↑ | |||
| Diindolylmethane (DIM) and Indole-3-carbinol (I3C; Cruciferous vegetables—Brassica genus) | Histone modifications | Class I HDACs degradation ↑ | (60–63) |
| Differential miRNA modulations | HDAC-1,2,3 expression ↓ | ||
| miR-let-7b, miR-146, miR- let-7e, miR-200b, and miR-200c,miR-21 ↑ | |||
| Genistein (soy beans) | DNA methylation | DNMT inhibitor | (61,65–78) |
| Histone modifications | Promoter methylation ↓ | ||
| Differential miRNA modulations | HDAC exp ↓ | ||
| HAT exp ↑ | |||
| Ac-H3, Ac-H4, Ac-H3K9 ↑ | |||
| H3K9me2 ↑ | |||
| miR-200 ↓, miR1296 ↑, miR-27a ↓ | |||
| Phenethyl Isothiocyanate (cruciferous vegetables) | DNA methylation | GSTP1 Promoter methylation ↓ | (80–85) |
| Histone modifications | H3 and H4 acetylation ↑, HDAC ↓ | ||
| Resveratrol (Grapes) | DNA methylation | DNMT inhibitor | (87–90) |
| Histone modifications | MBD2 recruitment ↓ | ||
| Promoter methylation ↓ | |||
| MTA1/NuRD corepressor complex ↓ | |||
| Organosulfur Compounds (Allium vegetables such as garlic) | Histone modifications | H3 and H4 acetylation ↑ | (81,92,93) |
| HDAC ↓ | |||
| Lycopene (Tomatoes) | DNA methylation | Promoter methylation ↓ | (66) |
| Quercetin (Citrous fruits and Buck wheat) | DNA methylation | DNMT inhibitor | (96–99) |
| Histone modifications | Promoter methylation ↓ | ||
| Ac-H3 ↑ | |||
| HDAC-1 ↓ | |||
| Ellagitannins (Berries) | Differential miRNA modulations | miR-let-7e, miR-370, miR-373* and miR-526b ↑ let-7a, let-7c, let-7d ↓ | (101) |
↓ downregulation, ↑ upregulation
Future research should be directed on the translation of effect of these dietary phytochemicals to pre-clinical models of cancers and in humans. As some of the effects of these phytochemicals appear to be cell type or organ specific therefore, understanding the mechanism(s) of these differences is a key in designing a personalized regimen for cancer prevention and/or cure. Furthermore, epigenetic defects may eventually lead to genetic defects which are not reversible, appropriate time of interventions using dietary chemopreventive agents which might be effective in slowing down cancer progression may be critical. Hence, it is important to design and perform appropriate experiments needed to address these issues and to analyze the data obtained in an efficient manner. Epigenetic modifications are an integral part of cellular development and differentiation, appropriate exposure time is critical for dietary agent to intervene the epigenetic process. More recent data demonstrate that the combined effect of various phytochemicals may be beneficial than single agent regimen which requires rigorous efforts to determine the exact, dose, duration, and extent of intervention used as a combination approach. Lastly, it is much more challenging and difficult than it seems to develop dietary phytochemicals as effective chemopreventive and/or chemotherapeutic agents despite their widespread recognition and acceptance as evident from a large number of research publications. Although dietary phytochemicals hold great promise in cancer prevention, a number of issues need to be addressed before moving forward to evidence-based clinical trials.
ACKNOWLEDGMENTS
The original work from author’s laboratory outlined in this review was supported by United States Public Health Service Grants RO1CA108512, RO1CA115491 and RO1AT002709. We acknowledge Shyama Prasad Mukherjee (SPM) fellowship provided to GD by the Council of Scientific and Industrial Research (CSIR), India and Fulbright-Nehru Doctoral and Professional Research fellowship provided by United States—India Educational Foundation (USIEF) for her work in the United States. MAB is supported by NIH 5T32DK007316 Ruth L. Kirschstein Pre-Doctoral Fellowship through the Metabolism Training Program. We apologize to those investigators whose original work could not be cited owing to the space limitations.
Conflict of Interest
The authors have no competing interest.
ABBREVIATIONS
- Akt
v-akt murine thymoma viral oncogene homolog 1
- AM
Allyl mercaptan
- AP-1
Activator Protein-1
- AR
Androgen receptor
- Bax
BCL2-associated X protein
- Bcl2
B-cell CLL/lymphoma 2
- Bcl-xL
B-cell lymphoma-extra large
- Bmi-1
B-cell-specific Moloney murine leukemia virus integration site 1
- BRCA1
Breast cancer 1, early onset
- CBP
CREB-binding protein
- CCND2
Cyclin D2
- Cdc25A
Cell division cycle 25 homolog A
- Cdk
Cyclin-dependent kinase
- c-Kit
v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog
- COMT
Catechol-O-methyltransferase
- COX-2
Cyclooxygenase-2
- CYLD
Cylindromatosis (turban tumor syndrome)
- DADS
Diallyl disulfide
- DAS
Diallyl sulfide
- DATS
Diallyl trisulfide
- DHFR
Dihydrofolate reductase
- DMBA
7,12-dimethylbenz(a)anthracene
- DNMT
DNA methyltransferase
- DNMT-3L
DNA (cytosine-5)-methyltransferase 3-like
- E2F
E2F transcription factor
- EC
[−]-epicatechin
- ECG
[−]-epicatechin-3-gallate
- EGC
[−]-epigallocatechin
- EGCG
[−]-epigallocatechin-3-gallate
- EGFR
Epidermal growth factor receptor
- ER
Estrogen receptor
- ERβ
Estrogen receptor beta
- ERBB2
Human epidermal growth factor receptor 2
- ERα
Estrogen receptor alpha
- EZH-2
Enhancer of zeste homolog 2
- FOXO3a
Forkhead box protein O3
- GCN5
SAGA complex histone acetyltransferase catalytic subunit Gcn5
- GSTP1
Glutathione-S-transferase pi 1
- HATs
Histone acetyl transferases
- HDACs
Histone deacetylases
- HER-2
Human epidermal growth factor receptor 2
- HIF-1 α
Hypoxia inducible factor 1, alpha subunit
- HKMTs
Histone lysine methyltransferases
- hMLH1
Human mutL homolog 1
- HOX family proteins
Homeobox family proteins
- HSP90
Heat shock protein 90
- hTERT
Human telomerase reverse transcriptase
- IP-10
TNF-induced interferon-gamma-inducible protein 10
- K
Lysine
- LEF
Lymphoid enhancer factor
- MBD
Methylated DNA binding domain proteins
- MCL1
Induced myeloid leukemia cell differentiation protein Mcl-1
- MCM-2
Minichromosome maintenance gene
- MGMT-O(6)
Methylguanine-DNA methyltransferase
- MIP-2
Macrophage inflammatory protein 2
- miRNA
MicroRNA
- MTA-2
Metastasis associated 1 family member 2
- NF-κB
Nuclear factor kappa-light-chain-enhancer of activated B cells
- Notch1
Notch homolog 1, translocation-associated (Drosophila)
- OSCs
Organosulfur compounds
- p16INK4a
Cyclin-dependent kinase 4 inhibitor A
- p21waf1/cip1
Cyclin-dependent kinase inhibitor 1A
- p53
Tumor protein 53
- PARP
Poly ADP-ribose polymerase
- PCAF
K(lysine) acetyltransferase 2B
- PcG
Polycomb group proteins
- PDCD4
Programmed cell death 4
- PEITC
Phenethyl isothiocyanate
- PRPS1
Phosphoribosyl pyrophosphate synthetase 1
- PTEN
Phosphatase and tensin homolog deleted on chromosome 10
- RARβ2
Retinoic acid receptor, beta 2
- R
Arginine
- RAS
Rat sarcoma transforming oncogene
- RASSF1A
RAS association domain family 1A
- RXR alpha
Retinoid X receptor, alpha
- SAH
S-adenosyl-L-homocysteine
- SAM
S-adenosyl methionine
- SAMC
S-allylmercaptocysteine
- SIRT1
Sirtuin (silent mating type information regulation 2 homolog) 1
- SLC16A1
Solute carrier family 16, member 1
- SNX19
Sorting nexin-19
- Sp1
Transcription Factor Sp1
- TGFBR2
Transforming growth factor, beta receptor II
- TGF-β
Transforming growth factor, beta
- TTK
Phosphotyrosine picked threonine-protein kinase
- VEGF
Vascular endothelial cell growth factor
- ZBTB10
Zinc finger and BTB domain containing 10
- ZEB1
Zinc finger E-box binding homeobox 1
- ZNF513
Zinc finger protein 513
REFERENCES
- 1.Henikoff S, Matzke MA. Exploring and explaining epigenetic effects. Trends Genet. 1997;13:293–295. doi: 10.1016/s0168-9525(97)01219-5. [DOI] [PubMed] [Google Scholar]
- 2.Dehan P, Kustermans G, Guenin S, Horion J, Boniver J, Delvenne P. DNA methylation and cancer diagnosis: new methods and applications. Expert Rev Mol Diagn. 2009;9:651–657. doi: 10.1586/erm.09.53. [DOI] [PubMed] [Google Scholar]
- 3.Illingworth R, Kerr A, Desousa D, Jorgensen H, Ellis P, Stalker J, et al. A novel CpG island set identifies tissue-specific methylation at developmental gene loci. PLoS Biol. 2008;6:e22. doi: 10.1371/journal.pbio.0060022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suter MA, Aagaard-Tillery KM. Environmental influences on epigenetic profiles. Semin Reprod Med. 2009;27:380–390. doi: 10.1055/s-0029-1237426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Issa JP, Kantarjian HM. Targeting DNA methylation. Clin Cancer Res. 2009;15:3938–3946. doi: 10.1158/1078-0432.CCR-08-2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. 2007;8:286–298. doi: 10.1038/nrg2005. [DOI] [PubMed] [Google Scholar]
- 8.Kopelovich L, Crowell JA, Fay JR. The epigenome as a target for cancer chemoprevention. J Natl Cancer Inst. 2003;95:1747–1757. doi: 10.1093/jnci/dig109. [DOI] [PubMed] [Google Scholar]
- 9.Gama-Sosa MA, Slagel VA, Trewyn RW, Oxenhandler R, Kuo KC, Gehrke CW, et al. The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res. 1983;11:6883–6894. doi: 10.1093/nar/11.19.6883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goelz SE, Vogelstein B, Hamilton SR, Feinberg AP. Hypomethylation of DNA from benign and malignant human colon neoplasms. Science. 1985;228:187–190. doi: 10.1126/science.2579435. [DOI] [PubMed] [Google Scholar]
- 11.Fullgrabe J, Kavanagh E, Joseph B. Histone onco-modifications. Oncogene. 2011;30:3391–3403. doi: 10.1038/onc.2011.121. [DOI] [PubMed] [Google Scholar]
- 12.Mottet D, Castronovo V. Histone deacetylases: target enzymes for cancer therapy. Clin Exp Metastasis. 2008;25:183–189. doi: 10.1007/s10585-007-9131-5. [DOI] [PubMed] [Google Scholar]
- 13.Sauve AA, Wolberger C, Schramm VL, Boeke JD. The biochemistry of sirtuins. Annu Rev Biochem. 2006;75:435–465. doi: 10.1146/annurev.biochem.74.082803.133500. [DOI] [PubMed] [Google Scholar]
- 14.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spange S, Wagner T, Heinzel T, Kramer OH. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol. 2009;41:185–198. doi: 10.1016/j.biocel.2008.08.027. [DOI] [PubMed] [Google Scholar]
- 16.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 17.Winter J, Jung S, Keller S, Gregory RI, Diederichs S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol. 2009;11:228–234. doi: 10.1038/ncb0309-228. [DOI] [PubMed] [Google Scholar]
- 18.Brait M, Sidransky D. Cancer epigenetics: above and beyond. Toxicol Mech Methods. 2011;21:275–288. doi: 10.3109/15376516.2011.562671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hardy TM, Tollefsbol TO. Epigenetic diet: impact on the epigenome and cancer. Epigenomics. 2011;3:503–518. doi: 10.2217/epi.11.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Siddiqui IA, Adhami VM, Saleem M, Mukhtar H. Beneficial effects of tea and its polyphenols against prostate cancer. Mol Nutr Food Res. 2006;50:130–143. doi: 10.1002/mnfr.200500113. [DOI] [PubMed] [Google Scholar]
- 21.Lee WJ, Shim JY, Zhu BT. Mechanisms for the inhibition of DNA methyltransferases by tea catechins and bioflavonoids. Mol Pharmacol. 2005;68:1018–1030. doi: 10.1124/mol.104.008367. [DOI] [PubMed] [Google Scholar]
- 22.Fang MZ, Wang Y, Ai N, Hou Z, Sun Y, Lu H, et al. Tea polyphenol (-)-epigallocatechin-3- gallate inhibits DNA methyltransferase and reactivates methylation silenced genes in cancer cell lines. Cancer Res. 2003;63:7563–7570. [PubMed] [Google Scholar]
- 23.Pandey M, Shukla S, Gupta S. Promoter demethylation and chromatin remodeling by green tea polyphenols leads to re-expression of GSTP1 in human prostate cancer cells. Int J Cancer. 2010;126:2520–2533. doi: 10.1002/ijc.24988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nandakumar V, Vaid M, Katiyar SK. (-)-Epigallocatechin-3-gallate reactivates silenced tumor suppressor genes, Cip1/p21 and p16INK4a, by reducing DNA methylation and increasing histones acetylation in human skin cancer cells. Carcinogenesis. 2011;32:537–544. doi: 10.1093/carcin/bgq285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berletch JB, Liu C, Love WK, Andrews LG, Katiyar SK, Tollefsbol TO. Epigenetic and genetic mechanisms contribute to telomerase inhibition by EGCG. J Cell Biochem. 2008;103:509–519. doi: 10.1002/jcb.21417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meeran SM, Patel SN, Chan TH, Tollefsbol TO. A novel prodrug of epigallocatechin-3-gallate: differential epigenetic hTERT repression in human breast cancer cells. Cancer Prev Res (Phila) 2001;4:1243–1254. doi: 10.1158/1940-6207.CAPR-11-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Volate SR, Muga SJ, Issa AY, Nitcheva D, Smith T, Wargovich MJ. Epigenetic modulation of the retinoid X receptor alpha by green tea in the azoxymethane-Apc Min/+ mouse model of intestinal cancer. Mol Carcinog. 2009;48:920–933. doi: 10.1002/mc.20542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thakur VS, Gupta K, Gupta S. Green tea polyphenols causes cell cycle arrest and apoptosis in prostate cancer cells by suppressing class I histone deacetylases. Carcinogenesis. 2012;33:377–384. doi: 10.1093/carcin/bgr277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thakur VS, Gupta K, Gupta S. Green tea polyphenols increase p53 transcriptional activity and acetylation by suppressing class I histone deacetylases. Int J Oncol. 2012;41:353–361. doi: 10.3892/ijo.2012.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Balasubramanian S, Adhikary G, Eckert RL. The Bmi-1 polycomb protein antagonizes the (-)-epigallocatechin-3-gallate-dependent suppression of skin cancer cell survival. Carcinogenesis. 2010;31:496–503. doi: 10.1093/carcin/bgp314. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 31.Tsang WP, Kwok TT. Epigallocatechin gallate up-regulation of miR-16 and induction of apoptosis in human cancer cells. J Nutr Biochem. 2010;21:140–146. doi: 10.1016/j.jnutbio.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 32.Fix LN, Shah M, Efferth T, Farwell MA, Zhang B. MicroRNA expression profile of MCF-7 human breast cancer cells and the effect of green tea polyphenon-60. Cancer Genomics Proteomics. 2010;7:261–277. [PubMed] [Google Scholar]
- 33.Kunnumakkara AB, Anand P, Aggarwal BB. Curcumin inhibits proliferation, invasion, angiogenesis and metastasis of different cancers through interaction with multiple cell signaling proteins. Cancer Lett. 2008;269:199–225. doi: 10.1016/j.canlet.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 34.Liu Z, Xie Z, Jones W, Pavlovicz RE, Liu S, Yu J, et al. Curcumin is a potent DNA hypomethylation agent. Bioorg Med Chem Lett. 2009;19:706–709. doi: 10.1016/j.bmcl.2008.12.041. [DOI] [PubMed] [Google Scholar]
- 35.Khor TO, Huang Y, Wu TY, Shu L, Lee J, Kong AN. Pharmacodynamics of curcumin as DNA hypomethylation agent in restoring the expression of Nrf2 via promoter CpGs demethylation. Biochem Pharmacol. 2011;82:1073–1078. doi: 10.1016/j.bcp.2011.07.065. [DOI] [PubMed] [Google Scholar]
- 36.Shu L, Khor TO, Lee JH, Boyanapalli SS, Huang Y, Wu TY et al. Epigenetic CpG demethylation of the promoter and reactivation of the expression of neurog1 by curcumin in prostate LNCaP cells. AAPS J. 2011:606–14. [DOI] [PMC free article] [PubMed]
- 37.Jha AK, Nikbakht M, Parashar G, Shrivastava A, Capalash N, Kaur J. Reversal of hypermethylation and reactivation of the RARbeta2 gene by natural compounds in cervical cancer cell lines. Folia Biol (Praha) 2010;56:195–200. doi: 10.14712/fb2010056050195. [DOI] [PubMed] [Google Scholar]
- 38.Marcu MG, Jung YJ, Lee S, Chung EJ, Lee MJ, Trepel J, et al. Curcumin is an inhibitor of p300 histone acetylatransferase. Med Chem. 2006;2:169–174. doi: 10.2174/157340606776056133. [DOI] [PubMed] [Google Scholar]
- 39.Balasubramanyam K, Varier RA, Altaf M, Swaminathan V, Siddappa NB, Ranga U, et al. Curcumin, a novel p300/CREB-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase dependent chromatin transcription. J Biol Chem. 2004;279:51163–51171. doi: 10.1074/jbc.M409024200. [DOI] [PubMed] [Google Scholar]
- 40.Kang J, Chen J, Shi Y, Jia J, Zhang Y. Curcumin-induced histone hypoacetylation: the role of reactive oxygen species. Biochem Pharmacol. 2005;69:1205–1213. doi: 10.1016/j.bcp.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 41.Chen Y, Shu W, Chen W, Wu Q, Liu H, Cui G. Curcumin, both histone deacetylase and p300/CBP-specific inhibitor, represses the activity of nuclear factor kappa B and Notch-1 in Raji cells. Basic Clin Pharmacol Toxicol. 2007;101:427–433. doi: 10.1111/j.1742-7843.2007.00142.x. [DOI] [PubMed] [Google Scholar]
- 42.Liu HL, Chen Y, Cui GH, Zhou JF. Curcumin, a potent anti-tumor reagent, is a novel histone deacetylase inhibitor regulating B-NHL cell line Raji proliferation. Acta Pharmacol Sin. 2005;26(5):603–609. doi: 10.1111/j.1745-7254.2005.00081.x. [DOI] [PubMed] [Google Scholar]
- 43.Bora-Tatar G, Dayangac-Erden D, Demir AS, Dalkara S, Yelekci K, Erdem-Yurter H. Molecular modifications on carboxylic acid derivatives as potent histone deacetylase inhibitors: activity and docking studies. Bioorg Med Chem. 2009;17:5219–5228. doi: 10.1016/j.bmc.2009.05.042. [DOI] [PubMed] [Google Scholar]
- 44.Lee SJ, Krauthauser C, Maduskuie V, Fawcett PT, Olson JM, Rajasekaran SA. Curcumin-induced HDAC inhibition and attenuation of medulloblastoma growth in vitro and in vivo. BMC Cancer. 2011;11:144. doi: 10.1186/1471-2407-11-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hua WF, Fu YS, Liao YJ, Xia WJ, Chen YC, Zeng YX, et al. Curcumin induces down- regulation of EZH2 expression through the MAPK pathway in MDA-MB-435 human breast cancer cells. Eur J Pharmacol. 2010;637:16–21. doi: 10.1016/j.ejphar.2010.03.051. [DOI] [PubMed] [Google Scholar]
- 46.Sun M, Estrov Z, Ji Y, Coombes KR, Harris DH, Kurzrock R. Curcumin (diferuloylmethane) alters the expression profiles of microRNAs in human pancreatic cancer cells. Mol Cancer Ther. 2008;7:464–473. doi: 10.1158/1535-7163.MCT-07-2272. [DOI] [PubMed] [Google Scholar]
- 47.Ali S, Ahmad A, Banerjee S, Padhye S, Dominiak K, Schaffert JM, et al. Gemcitabine sensitivity can be induced in pancreatic cancer cells through modulation of miR-200 and miR-21 expression by curcumin or its analogue CDF. Cancer Res. 2010;70:3606–3617. doi: 10.1158/0008-5472.CAN-09-4598. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 48.Zhang J, Zhang T, Ti X, Shi J, Wu C, Ren X, et al. Curcumin promotes apoptosis in A549/DDP multidrugresistant human lung adenocarcinoma cells through an miRNA signaling pathway. Biochem Biophys Res Commun. 2010;399:1–6. doi: 10.1016/j.bbrc.2010.07.013. [DOI] [PubMed] [Google Scholar]
- 49.Yang J, Cao Y, Sun J, Zhang Y. Curcumin reduces the expression of Bcl-2 by upregulating miR-15a and miR-16 in MCF-7 cells. Med Oncol. 2010;27:1114–1118. doi: 10.1007/s12032-009-9344-3. [DOI] [PubMed] [Google Scholar]
- 50.Mudduluru G, George-William JN, Muppala S, Asangani IA, Regalla K, Nelson LD, et al. Curcumin regulates miR-21 expression and inhibits invasion and metastasis in colorectal cancer. Biosci Rep. 2011;31:185–197. doi: 10.1042/BSR20100065. [DOI] [PubMed] [Google Scholar]
- 51.Clarke JD, Dashwood RH, Ho E. Multi-targeted prevention of cancer by sulforaphane. Cancer Lett. 2008;269:291–304. doi: 10.1016/j.canlet.2008.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Traka M, Gasper AV, Smith JA, Hawkey CJ, Bao Y, Mithen RF. Transcriptome analysis of human colon caco-2 cells exposed to sulforaphane. J Nutr. 2005;135:1865–1872. doi: 10.1093/jn/135.8.1865. [DOI] [PubMed] [Google Scholar]
- 53.Meeran SM, Patel SN, Tollefsbol TO. Sulforaphane causes epigenetic repression of hTERT expression in human breast cancer cell lines. PLoS One. 2010;5(7):e11457. doi: 10.1371/journal.pone.0011457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Myzak MC, Karplus PA, Chung FL, Dashwood RH. A novel mechanism of chemoprotection by sulforaphane: inhibition of histone deacetylase. Cancer Res. 2004;64:5767–5774. doi: 10.1158/0008-5472.CAN-04-1326. [DOI] [PubMed] [Google Scholar]
- 55.Myzak MC, Hardin K, Wang R, Dashwood RH, Ho E. Sulforaphane inhibits histone deacetylase activity in BPH-1, LnCaP and PC-3 prostate epithelial cells. Carcinogenesis. 2006;27:811–819. doi: 10.1093/carcin/bgi265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pledgie-Tracy A, Sobolewski MD, Davidson NE. Sulforaphane induces cell type-specific apoptosis in human breast cancer cell lines. Mol Cancer Ther. 2007;6:1013–1021. doi: 10.1158/1535-7163.MCT-06-0494. [DOI] [PubMed] [Google Scholar]
- 57.Myzak MC, Dashwood WM, Orner GA, Ho E, Dashwood RH. Sulforaphane inhibits histone deacetylase in vivo and suppresses tumorigenesis in Apc-minus mice. FASEB J. 2006;20:506–508. doi: 10.1096/fj.05-4785fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Myzak MC, Tong P, Dashwood WM, Dashwood RH, Ho E. Sulforaphane retards the growth of human PC-3 xenografts and inhibits HDAC activity in human subjects. Exp Biol Med (Maywood) 2007;232:227–234. [PMC free article] [PubMed] [Google Scholar]
- 59.Banerjee S, Kong D, Wang Z, Bao B, Hillman GG, Sarkar FH. Attenuation of multi-targeted proliferation-linked signaling by 3,30-diindolylmethane (DIM): from bench to clinic. Mutat Res. 2011;728:47–66. doi: 10.1016/j.mrrev.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li Y, Li X, Guo B. Chemopreventive agent 3,30-diindolylmethane selectively induces proteasomal degradation of class I histone deacetylases. Cancer Res. 2010;70:646–654. doi: 10.1158/0008-5472.CAN-09-1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li Y, Vandenboom TG, 2nd, Kong D, Wang Z, Ali S, Philip PA, et al. Up-regulation of miR- 200 and let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer Res. 2009;69:6704–6712. doi: 10.1158/0008-5472.CAN-09-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li Y, Vandenboom TG, Wang Z, Ali S, Philip PA, Sarkar FH. miR-146a suppresses invasion of pancreatic cancer cells. Cancer Res. 2010;70:1486–1495. doi: 10.1158/0008-5472.CAN-09-2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jin Y, Zou X, Feng X. 3,30-Diindolylmethane negatively regulates Cdc25A and induces a G2/M arrest by modulation of microRNA 21 in human breast cancer cells. Anticancer Drugs. 2010;21:814–822. doi: 10.1097/CAD.0b013e32833e53ea. [DOI] [PubMed] [Google Scholar]
- 64.Banerjee S, Li Y, Wang Z, Sarkar FH. Multi-targeted therapy of cancer by genistein. Cancer Lett. 2008;269:226–242. doi: 10.1016/j.canlet.2008.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fang MZ, Chen D, Sun Y, Jin Z. Reversal of hypermethylation and reactivation of p16INK4a, RARbeta, and MGMT genes by genistein and other isoflavones from soy. Clin Cancer Res. 2005;11(19 Pt 1):7033–7041. doi: 10.1158/1078-0432.CCR-05-0406. [DOI] [PubMed] [Google Scholar]
- 66.King-Batoon A, Leszczynska JM, Klein CB. Modulation of gene methylation by genistein or lycopene in breast cancer cells. Environ Mol Mutagen. 2008;49:36–45. doi: 10.1002/em.20363. [DOI] [PubMed] [Google Scholar]
- 67.Vardi A, Bosviel R, Rabiau N, Adjakly M, Satih S, Dechelotte P, et al. Soy phytoestrogens modify DNA methylation of GSTP1, RASSF1A, EPH2 and BRCA1 promoter in prostate cancer cells. In Vivo. 2010;24:393–400. [PubMed] [Google Scholar]
- 68.Adjakly M, Bosviel R, Rabiau N, Boiteux JP, Bignon YJ, Guy L, et al. DNA methylation and soy phytoestrogens: quantitative study in DU-145 and PC-3 human prostate cancer cell lines. Epigenomics. 2011;3:795–803. doi: 10.2217/epi.11.103. [DOI] [PubMed] [Google Scholar]
- 69.Wang Z, Chen H. Genistein increases gene expression by demethylation of WNT5a promoter in colon cancer cell line SW1116. Anticancer Res. 2010;30:4537–4545. [PubMed] [Google Scholar]
- 70.Majid S, Dar AA, Shahryari V, Hirata H, Ahmad A, Saini S, et al. Genistein reverses hypermethylation and induces active histone modifications in tumor suppressor gene B-cell translocation gene 3 in prostate cancer. Cancer. 2010;116:66–76. doi: 10.1002/cncr.24662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Qin W, Zhu W, Shi H, Hewett JE, Ruhlen RL, Macdonald RS, et al. Soy isoflavones have an antiestrogenic effect and alter mammary promoter hypermethylation in healthy premenopausal women. Nutr Cancer. 2009;61:238–244. doi: 10.1080/01635580802404196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Majid S, Kikuno N, Nelles J, Noonan E, Tanaka Y, Kawamoto K, et al. Genistein induces the p21WAF1/CIP1 and p16INK4a tumor suppressor genes in prostate cancer cells by epigenetic mechanisms involving active chromatin modification. Cancer Res. 2008;68:2736–2744. doi: 10.1158/0008-5472.CAN-07-2290. [DOI] [PubMed] [Google Scholar]
- 73.Hong T, Nakagawa T, Pan W, Kim MY, Kraus WL, Ikehara T, et al. Isoflavones stimulate estrogen receptor-mediated core histone acetylation. Biochem Biophys Res Commun. 2004;317:259–264. doi: 10.1016/j.bbrc.2004.03.041. [DOI] [PubMed] [Google Scholar]
- 74.Kikuno N, Shiina H, Urakami S, Kawamoto K, Hirata H, Tanaka Y, et al. Genistein mediated histone acetylation and demethylation activates tumor suppressor genes in prostate cancer cells. Int J Cancer. 2008;123:552–560. doi: 10.1002/ijc.23590. [DOI] [PubMed] [Google Scholar]
- 75.Basak S, Pookot D, Noonan EJ, Dahiya R. Genistein down-regulates androgen receptor by modulating HDAC6-Hsp90 chaperone function. Mol Cancer Ther. 2008;7:3195–3202. doi: 10.1158/1535-7163.MCT-08-0617. [DOI] [PubMed] [Google Scholar]
- 76.Parker LP, Taylor DD, Kesterson J, Metzinger DS, Gercel-Taylor C. Modulation of microRNA associated with ovarian cancer cells by genistein. Eur J Gynaecol Oncol. 2009;30:616–621. [PubMed] [Google Scholar]
- 77.Majid S, Dar AA, Saini S, Chen Y, Shahryari V, Liu J, et al. Regulation of minichromosome maintenance gene family by microRNA-1296 and genistein in prostate cancer. Cancer Res. 2010;70:2809–2818. doi: 10.1158/0008-5472.CAN-09-4176. [DOI] [PubMed] [Google Scholar]
- 78.Sun Q, Cong R, Yan H, Gu H, Zeng Y, Liu N, et al. Genistein inhibits growth of human uveal melanoma cells and affects microRNA-27a and target gene expression. Oncol Rep. 2009;22:563–567. doi: 10.3892/or_00000472. [DOI] [PubMed] [Google Scholar]
- 79.Cheung KL, Kong AN. Molecular targets of dietary phenethyl isothiocyanate and sulforaphane for cancer chemoprevention. AAPS J. 2010;12:87–97. doi: 10.1208/s12248-009-9162-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang LG, Beklemisheva A, Liu XM, Ferrari AC, Feng J, Chiao JW. Dual action on promoter demethylation and chromatin by an isothiocyanate restored GSTP1 silenced in prostate cancer. Mol Carcinog. 2007;46:24–31. doi: 10.1002/mc.20258. [DOI] [PubMed] [Google Scholar]
- 81.Lea MA, Randolph VM, Lee JE, des Bordes C. Induction of histone acetylation in mouse erythroleukemia cells by some organosulfur compounds including allyl isothiocyanate. Int J Cancer. 2001;92:784–789. doi: 10.1002/ijc.1277. [DOI] [PubMed] [Google Scholar]
- 82.Xiao L, Huang Y, Zhen R, Chiao JW, Liu D, Ma X. Deficient histone acetylation in acute leukemia and the correction by an isothiocyanate. Acta Haematol. 2010;123:71–76. doi: 10.1159/000264628. [DOI] [PubMed] [Google Scholar]
- 83.Huang YQ, Ma XD, Lai YD, Wang XZ, Chiao JW, Liu DL. Phenylhexyl isothiocyanate (PHI) regulates histone methylation and acetylation and induces apoptosis in SMMC-7721 cells. Zhonghua Gan Zang Bing Za Zhi. 2010;18:209–212. doi: 10.3760/cma.j.issn.1007-3418.2010.03.014. [DOI] [PubMed] [Google Scholar]
- 84.Izzotti A, Calin GA, Steele VE, Cartiglia C, Longobardi M, Croce CM, et al. Chemoprevention of cigarette smoke-induced alterations of microRNA expression in rat lungs. Cancer Prev Res (Phila) 2010;3:62–72. doi: 10.1158/1940-6207.CAPR-09-0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Izzotti A, Larghero P, Cartiglia C, Longobardi M, Pfeffer U, Steele VE, et al. Modulation of microRNA expression by budesonide, phenethyl isothiocyanate and cigarette smoke in mouse liver and lung. Carcinogenesis. 2010;31:894–901. doi: 10.1093/carcin/bgq037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Savouret JF, Quesne M. Resveratrol and cancer: a review. Biomed Pharmacother. 2002;56:84–87. doi: 10.1016/s0753-3322(01)00158-5. [DOI] [PubMed] [Google Scholar]
- 87.Stefanska B, Rudnicka K, Bednarek A, Fabianowska-Majewska K. Hypomethylation and induction of retinoic acid receptor beta 2 by concurrent action of adenosine analogues and natural compounds in breast cancer cells. Eur J Pharmacol. 2010;638:47–53. doi: 10.1016/j.ejphar.2010.04.032. [DOI] [PubMed] [Google Scholar]
- 88.Tili E, Michaille JJ, Alder H, Volinia S, Delmas D, Latruffe N, et al. Resveratrol modulates the levels of microRNAs targeting genes encoding tumor-suppressors and effectors of TGF signaling pathway in SW480 cells. Biochem Pharmacol. 2010;80:2057–2065. doi: 10.1016/j.bcp.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang RH, Zheng Y, Kim HS, Xu X, Cao L, Luhasen T, et al. Interplay among BRCA1, SIRT1, and surviving during BRCA1-associated tumorigenesis. Mol Cell. 2008;32:11–20. doi: 10.1016/j.molcel.2008.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Papoutsis AJ, Lamore SD, Wondrak GT, Selmin OI, Romagnolo DF. Resveratrol prevents epigenetic silencing of BRCA-1 by the aromatic hydrocarbon receptor in human breast cancer cells. J Nutr. 2010;140:1607–1614. doi: 10.3945/jn.110.123422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ariga T, Seki T. Antithrombotic and anticancer effects of garlic-derived sulfur compounds: a review. Biofactors. 2006;26:93–103. doi: 10.1002/biof.5520260201. [DOI] [PubMed] [Google Scholar]
- 92.Lea MA, Randolph VM, Patel M. Increased acetylation of histones induced by diallyl disulfide and structurally related molecules. Int J Oncol. 1999;15:347–352. doi: 10.3892/ijo.15.2.347. [DOI] [PubMed] [Google Scholar]
- 93.Druesne N, Pagniez A, Mayeur C, Thomas M, Cherbuy C, Duee PH, et al. Diallyl disulfide (DADS) increases histone acetylation and p21(waf1/cip1) expression in human colon tumor cell lines. Carcinogenesis. 2004;25:1227–1236. doi: 10.1093/carcin/bgh123. [DOI] [PubMed] [Google Scholar]
- 94.Giovannucci E. Tomatoes, tomato-based products, lycopene, and cancer: review of the epidemiologic literature. J Natl Cancer Inst. 1999;91:317–331. doi: 10.1093/jnci/91.4.317. [DOI] [PubMed] [Google Scholar]
- 95.Gibellini L, Pinti M, Nasi M, Montagna JP, De Biasi S, Roat E, et al. Quercetin and cancer chemoprevention. Evid Based Complement Alternat Med. 2011;2011:591356. doi: 10.1093/ecam/neq053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tan S, Wang C, Lu C, Zhao B, Cui Y, Shi X, et al. Quercetin is able to demethylate the p16INK4a gene promoter. Chemotherapy. 2009;55:6–10. doi: 10.1159/000166383. [DOI] [PubMed] [Google Scholar]
- 97.Ruiz PA, Braune A, Holzlwimmer G, Quintanilla-Fend L, Haller D. Quercetin inhibits TNF- induced NF-kappaB transcription factor recruitment to proinflammatory gene promoters in murine intestinal epithelial cells. J Nutr. 2007;137:1208–1215. doi: 10.1093/jn/137.5.1208. [DOI] [PubMed] [Google Scholar]
- 98.Lee WJ, Chen YR, Tseng TH. Quercetin induces FasL-related apoptosis, in part, through promotion of histone H3 acetylation in human leukemia HL-60 cells. Oncol Rep. 2011;25:583–591. doi: 10.3892/or.2010.1097. [DOI] [PubMed] [Google Scholar]
- 99.Priyadarsini RV, Vinothini G, Murugan RS, Manikandan P, Nagini S, et al. The flavonoid quercetin modulates the hallmark capabilities of hamster buccal pouch tumors. Nutr Cancer. 2011;63:218–226. doi: 10.1080/01635581.2011.523503. [DOI] [PubMed] [Google Scholar]
- 100.Heber D. Multi-targeted therapy of cancer by ellagitannins. Cancer Lett. 2008;269:262–268. doi: 10.1016/j.canlet.2008.03.043. [DOI] [PubMed] [Google Scholar]
- 101.Wen XY, Wu SY, Li ZQ, Liu ZQ, Zhang JJ, Wang GF, et al. Ellagitannin (BJA3121), an anti-proliferative natural polyphenol compound, can regulate the expression of miRNAs in HepG2 cancer cells. Phytother Res. 2009;23:778–784. doi: 10.1002/ptr.2616. [DOI] [PubMed] [Google Scholar]
- 102.Pandey M, Kaur P, Shukla S, Abbas A, Fu P, Gupta S. Plant flavone apigenin inhibits HDAC and remodels chromatin to induce growth arrest and apoptosis in human prostate cancer cells: in vitro and in vivo study. Mol Carcinog. 2012;51:952–962. doi: 10.1002/mc.20866. [DOI] [PMC free article] [PubMed] [Google Scholar]