

Abstract
Micelles are attractive delivery systems for hydrophobic drugs due to their small size and the ease of application. However, the limited drug loading capacity and the intrinsic poor stability of drug-loaded formulations represent two major issues for some micellar systems. In this study, we designed and synthesized a micelle-forming PEG-lipopeptide conjugate with two Fmoc groups located at the interfacial region, and two oleoyl chains as the hydrophobic core. The significance of Fmoc groups as a broadly applicable drug-interactive motif that enhances the carrier–drug interaction was examined using eight model drugs of diverse structures. Compared with an analogue without carrying a Fmoc motif, PEG5000-(Fmoc-OA)2 demonstrated a lower value of critical micelle concentration and three-fold increases of loading capacity for paclitaxel (PTX). These micelles showed tubular structures and small particle sizes (∼70 nm), which can be lyophilized and readily reconstituted with water without significant changes in particle sizes. Fluorescence quenching study illustrated the Fmoc/PTX π–π stacking contributes to the carrier/PTX interaction, and drug-release study demonstrated a much slower kinetics than Taxol, a clinically used PTX formulation. PTX/PEG5000-(Fmoc-OA)2 mixed micelles exhibited higher levels of cytotoxicity than Taxol in several cancer cell lines and more potent inhibitory effects on tumor growth than Taxol in a syngeneic murine breast cancer model (4T1.2). We have further shown that seven other drugs can be effectively formulated in PEG5000-(Fmoc-OA)2 micelles. Our study suggests that micelle-forming PEG-lipopeptide surfactants with interfacial Fmoc motifs may represent a promising formulation platform for a broad range of drugs with diverse structures.
Electronic supplementary material
The online version of this article (doi:10.1208/s12248-013-9536-9) contains supplementary material, which is available to authorized users.
KEY WORDS: drug-interactive motif, micelle, paclitaxel, slow release
INTRODUCTION
Drug discovery and development is an extremely time-consuming and costly process, during which only very few candidates survive the screening process and eventually become clinical drugs. Many promising candidates are eliminated due to poor solubility and bioavailability (1,2). Drug formulations have gained increasing attention as a strategy to maximize the success of drug development process and to improve the performance of existing drugs (3).
Micelles are an attractive delivery system due to the ease of application and their small particle sizes, which allow effective passive targeting to tissues with leaky vasculature such as tumors and inflammatory tissues based on the enhanced penetration and retention (EPR) effect (4–6). However, there are several issues with conventional micellar systems such as low drug loading capacity and limited colloidal stability, which limit their successful in vivo applications (7). Incorporation of drugs into hydrophobic core of typical micelle formulations is largely based on hydrophobic–hydrophobic interactions. While these micellar formulations are effective in formulating very few drugs that are highly hydrophobic or lipophilic, they have limited effectiveness in formulating many drugs that are only moderately hydrophobic.
Studies from Park’s group have shown that inclusion of hydrotropic motifs into the hydrophobic domain of polymeric micelles significantly improves the compatibility of the core-forming blocks with the drugs that are not entirely hydrophobic/lipophilic (8,9). Hydrotropes are small molecular amphiphiles that increase the aqueous solubility of poorly soluble agents. This strategy has led to significant improvement in both drug loading capacity and the colloidal stability of drug-loaded micelles. The study by Yoo et al. showed that covalent coupling of a drug molecule (doxorubicin, DOX) into the hydrophobic domain of polymeric micelles resulted in an improved system for loading of the same drug, e.g., DOX (10,11). These studies highlight the benefit of introducing additional structural variables to the traditional polymeric micellar systems.
The idea of including an additional drug-interactive domain in lipidic micellar systems has not been studied before. However, the importance of such approach has been suggested by recent studies with several “unconventional” pegylated surfactants with vitamin E, embelin, or farnesylthiosalicylic acid (FTS, a Ras inhibitor), instead of simple lipids as a hydrophobic domain (12–16). Vitamin E, embelin, and FTS all have an interfacial aromatic ring linked to an acyl chain, which may contribute significantly to the improved formulation properties over the surfactants with simple lipid chains. We hypothesized that expansion of the interfacial region to purposely include motifs that have drug interaction potential could result in further improved lipidic micellar systems. We have experimentally demonstrated that such motif can be identified via a screening process and then incorporated into the interfacial region of a pegylated surfactant to add a drug interactive functionality. With JP4-039, a mitochondria-targeted antioxidant featuring a peptide derivative carrying a nitroxide group, we have shown that such approach is both feasible and significant. We have found that 9-fluorenylmethoxycarbonyl (Fmoc) moiety, a functional group that is routinely used for amino acid protection, was the best among several motifs examined (17).
The aim of the present study is to examine the broad applicability of our new micellar system in formulating different drugs of diverse structures. The potential of the new formulation in delivery of paclitaxel to tumor cells was also investigated in vitro and in vivo.
MATERIALS AND METHODS
Materials
Paclitaxel (PTX, 98%) was purchased from AK Scientific, Inc. (CA, USA). α-Fmoc-ε-Boc-lysine, di-Boc-lysine, N,N′-dicyclohexylcarbodiimide (DCC), N-hydroxysuccinimide (NHS), trifluoroacetic acid (TFA), and triethylamine (TEA) were obtained from Acros Organic (NJ, USA), and oleic acid (OA) was from Alfa Aesar (MA, USA). Monomethoxy PEG5000, 4-dimethylaminopyridine (DMAP), ninhydrin, and other unspecified chemicals were all purchased from Sigma-Aldrich (MO, USA). Dulbecco’s phosphate-buffered saline (DPBS), Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), and 100× penicillin–streptomycin solution were purchased from Invitrogen (NY, USA). All solvents used in this study were HPLC grade.
Cell Culture
PC-3 and DU145 are two androgen-independent human prostate cancer cell lines and were obtained from ATCC (VA, USA). 4T1-2 is a mouse metastatic breast cancer cell line and was kindly provided by Dr. Zhaoyang You at University of Pittsburgh School of Medicine. All cells were cultured at 37°C in DMEM containing 10% FBS and 1% penicillin–streptomycin in a humidified environment with 5% CO2.
Synthesis of PEG5000-Lys-(α-Fmoc-ε-oleoyl lysine)2 (PEG5000-(Fmoc-OA)2)
The synthetic routes for PEG5000-(Fmoc-OA)2 and PEG5000-di-oleoyl lysine (PEG5000-OA2) were depicted in Scheme 1.
Scheme 1.
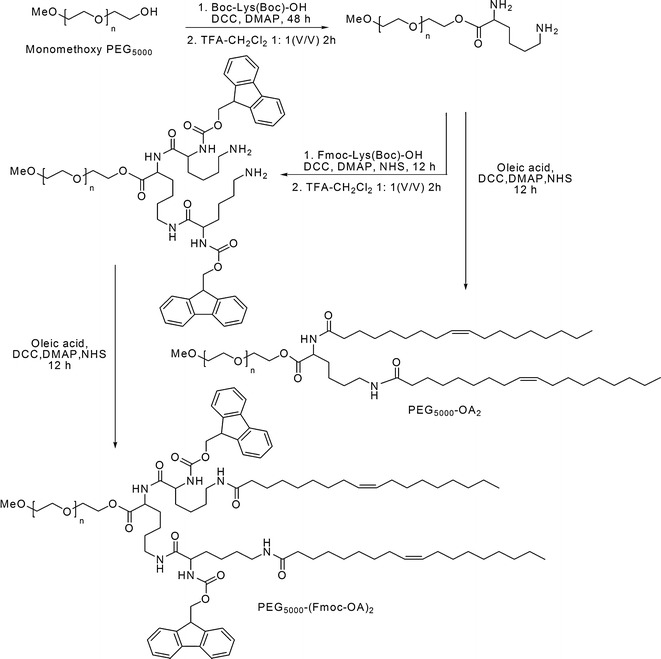
Synthetic route of PEG5000-(Fmoc-OA)2 and PEG5000-OA2
Monomethoxy PEG5000 (1 eq.) was dissolved in CH2Cl2 and mixed with di-Boc-lysine (1.5 eq.), DCC (1.8 eq.), and DMAP (0.3 eq.). After stirring at room temperature for 24 h, another portion of di-Boc-lysine (1.5 eq.) and DCC (1.8 eq.) was added into reaction mixture and the reaction was allowed for another 24 h. White solid precipitate was then removed by filtration, and the filtrate was added into 10-fold volume of cold ethyl ether to precipitate the PEG derivative, followed by three washes with cold ethanol and ether. The obtained PEG5000-di-Boc-lysine ester was dissolved in CH2Cl2/TFA (1:1, v/v) at the concentration of 0.3 g/mL, and stirred for 2 h at room temperature to remove the Boc moiety. After removal of most of the solvent, the PEG derivative was precipitated in cold ether and washed three times with cold ethanol and ether.
α-Fmoc-ε-Boc-lysine (3 eq.), NHS (3.6 eq.), DCC (4 eq.), and DMAP (0.8 eq.) were dissolved in CH2Cl2 and activated at 37°C for 4 h, followed by mixing with PEG5000-di-NH2-lysine (1 eq.) and TEA (3 eq.). The reaction was allowed at 37°C overnight until completion as indicated by the negative results in the ninhydrin tests. The white solid precipitate was removed by filtration, and the PEG derivative was precipitated in cold ether and washed by cold ethanol and ether. The PEG5000-Lys-(α-Fmoc-ε-Boc lysine)2 obtained was dissolved in CH2Cl2/TFA (1:1, v/v) and stirred for 2 h at room temperature to remove the Boc moiety. The resulting PEG5000-Lys-(α-Fmoc-ε-NH2 lysine)2 was then purified by cold ether and ethanol precipitation.
The NH2-terminated PEG5000-Lys-(α-Fmoc-ε-NH2 lysine)2 (1 eq.) was conjugated with oleic acid (OA, 3 eq.) that was pre-activated with NHS (3.6 eq.), DCC (4 eq.), and DMAP (0.8 eq.) for 4 h at 37°C. The conjugating reaction was allowed overnight until negative results were shown in the ninhydrin tests. After filtration and precipitation, the obtained PEG5000-Lys-(α-Fmoc-ε-oleoyl lysine)2 (PEG5000-(Fmoc-OA)2) was purified via three washes with cold ethanol and ether.
Synthesis of PEG5000-OA2
The PEG5000-di-NH2-lysine was synthesized as described above and the di-NH2-terminated PEG was reacted with OA (3 eq.) that was pre-activated with NHS (3.6 eq.), DCC (4 eq.), and DMAP (0.8 eq.) for 4 h at 37°C. The reaction was completed as indicated by the negative results in the ninhydrin tests. After filtration and precipitation, the obtained PEG5000-OA2 was purified by three washes with cold ethanol and ether.
Preparation and Characterization of Drug-Loaded Micelles
All of the drugs were dissolved in chloroform (10 mg/mL) and were mixed with PEG5000-(Fmoc-OA)2 (100 mg/mL in chloroform) at various carrier/drug molar ratios. The organic solvent was removed by a stream of nitrogen to generate a thin film at the bottom of a glass tube, and the trace amount of solvent was further removed under vacuum for 2 h. DPBS was added to hydrate and suspend the thin film to form drug-loaded micelles as clear solution, followed by filtration through 220 nm PVDF syringe filter to remove any unincorporated drug. The drug-free micelles were prepared via a same procedure without the addition of drug. Drug-free or drug-loaded PEG5000-OA2 micelles were also similarly prepared as described above.
The diameter and size distribution of the micelles were examined via a Malvern Zeta Nanosizer. The morphology of both drug-free and drug-loaded micelles was observed by transmission electron microscopy (TEM) after negative staining. Drug in micelles was extracted by methanol and detected by Waters Alliance 2695–2998 high-performance liquid chromatography (HPLC) system with a Lichrospher® 100 RP-18 column (250 × 4.6 mm) equipped with a UV detector at 227 nm at room temperature and a mobile phase of 80:20 (v/v) of methanol/water at the flow rate of 0.8 mL/min. The drug loading capacity and efficiency were calculated according to the formula listed below:
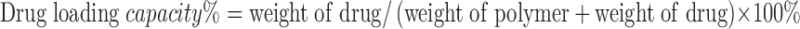 |
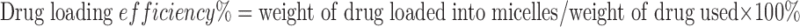 |
Determination of the Critical Micelle Concentration
The fluorescence probe pyrene was employed to measure the critical micelle concentrations of PEG5000-(Fmoc-OA)2 and PEG5000-OA2 micelles. Various amounts of the conjugates in chloroform were mixed with pyrene (4 × 10−5 M in chloroform). The organic solvent was completely removed, and 2 mL of DPBS was added to each tube to form the micelles with the conjugate concentrations ranging from 1 × 10−4 to 0.5 mg/mL and a final pyrene concentration of 6 × 10−7 M. The fluorescence intensity of each sample was detected at 334/390 nm (excitation/emission) via Synergy H1 Hybrid Multi-Mode Microplate Reader (Winooski, VT), and the critical micelle concentration (CMC) value was determined from the threshold concentration, where the sharp increase in pyrene fluorescence intensity is observed.
Fluorescence Quenching Study
PTX-loaded PEG5000-(Fmoc-OA)2 micelles and cholesterol (Chol)/PEG5000-(Fmoc-OA)2 were prepared according to the procedure described above, and the carrier concentration was kept at 1.5 mg/mL in all of the samples. The fluorescence intensity of the samples at the wavelength of 300 ∼ 460 nm was recorded with an excitation wavelength at 270 nm by using a Synergy H1 Hybrid Multi-Mode Microplate Reader.
Changes of Particle Sizes Before and After Lyophilization/Reconstitution
One milliliter of PTX-loaded PEG5000-(Fmoc-OA)2 micelles (PTX concentration at 1 mg/mL) was prepared according to the procedure described above. The micelle solution was frozen at −80°C and then lyophilized overnight to obtain white powder. The obtained powder was suspended in 1 mL of distilled water to reconstitute the micelle solution. Particle sizes of the micelles before and after lyophilization/reconstitution were measured by dynamic light scattering method using a Zetasizer (Malvern).
In Vitro Drug Release
Two milliliters of PTX/PEG5000-(Fmoc-OA)2 or PTX/PEG5000-OA2 mixed micelles, or Taxol formulation (6 mg/mL PTX in Cremophor EL/ethanol 1:1,diluted to 1 mg PTX/mL with DPBS) was placed in a dialysis tube (MWCO 12 kDa, Spectrum Laboratories) that was incubated in 200 mL DPBS (pH = 7.4) containing 0.5% (w/v) Tween 80 at 37°C with gentle shaking. The concentration of PTX remaining in the dialysis bags at scheduled times was similarly detected by HPLC as described above.
In Vitro Cytotoxicity
4T1.2 (1000 cells/well), PC-3 (3000 cells/well), or DU145 (2000 cells/well) cells were seeded in 96-well plates and incubated in DMEM containing 10% FBS and 1% streptomycin-penicillin at 37°C for 24 h. PTX/PEG5000-(Fmoc-OA)2 mixed micelles or Taxol formulation were added to cells in triplicate at the PTX concentrations from 6.25 to 200 ng/mL and cells were further incubated for 72 h. Then 20 μL of 3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT) in DPBS (5 mg/mL) was added to each well. Four hours later, the medium was removed and 150 μL of DMSO was added to each well to solubilize the formazan crystal. The absorbance was measured at a wavelength of 550 nm and a reference wavelength at 630 nm with a microplate reader. Untreated cells were included as a control. Cell viability was calculated according to the following formula:
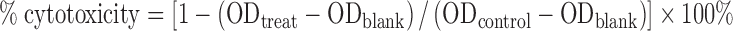 |
Animals
Female BALB/c mice (10 to 12 weeks) were purchased from Charles River (Davis, CA) and were housed under pathogen-free conditions according to AAALAC guidelines. All animal-related experiments were performed in full compliance with institutional guidelines and approved by the Animal Use and Care Administrative Advisory Committee at the University of Pittsburgh.
In Vivo Tumor Inhibition Study
An aggressive syngeneic murine breast cancer model (4T1.2) was employed to evaluate the tumor inhibition effect of PTX-loaded PEG5000-(Fmoc-OA)2 micelles. For establishment of the tumor model, 2 × 105 of 4T1.2 cells in 100 μL of DPBS were inoculated s.c. at the right flank of female BALB/c mice, and treatments were initiated (day 1) when the tumor volume reached ∼50 mm3. Mice were randomly divided into four groups (n = 4) and received i.v. administration of PTX/PEG5000-(Fmoc-OA)2 micelles (10 mg PTX/kg), PTX/PEG5000-(Fmoc-OA)2 micelles (20 mg PTX/kg), Taxol (10 mg PTX/kg), and saline, respectively, on days 1, 3, 5, 7, and 9. Tumor sizes were monitored by a digital caliper and calculated based on the formula (L × W2)/2, where L is the longest and W is the shortest tumor diameters (millimeters). Data were presented as relative tumor volume (the tumor volume at a given time point divided by the tumor volume prior to first treatment). Mice were sacrificed when tumors reached 2000 mm3 or developed ulceration. The change of body weights of all mice was monitored during the entire course of treatment to evaluate the potential toxicity of different formulations.
Statistical Analysis
In all statistical analysis, Student’s t-test was performed between two groups, and the significance level was set at a probability of p < 0.05 or p < 0.01. All results were reported as the means ± standard error unless otherwise indicated.
RESULTS
Synthesis and Characterization of PEG5000-Lipopeptide with Drug-Interactive Motifs
We have previously shown that inclusion of Fmoc motifs into PEG-lipid conjugates at the interfacial region significantly improved the drug-loading capacity and the formulation stability with JP4-039 as a model drug (17). Our data showed that PEG-lipopeptides with two or four Fmoc motifs were more active than the one with one Fmoc motif in formulating JP4-039. This study is focused on the PEG-lipopeptide with two Fmoc motifs due to its relative simplicity. PEG5000 was chosen to construct the hydrophilic motif as the resulting PEG5000-lipopeptide performed better than the counterpart with PEG2000 or PEG1000 in formulating several compounds in a preliminary study (data not shown).
Figure 1 shows the structure of PEG5000-(Fmoc-OA)2, in which two oleic acids and two Fmoc moieties were conjugated to PEG5000 using lysine–(lysine)2 as a bridge. Scheme 1 shows the procedures for the synthesis of both PEG5000-(Fmoc-OA)2 and PEG5000-OA2, a control carrier without Fmoc motifs. 1H NMR spectrum of PEG5000-(Fmoc-OA)2 shows signals at 3.63 ppm attributed to the methylene protons located at the terminal of PEG backbone, the Fmoc proton signals at 7.9–7.3 ppm, the signals attributed to the double bond at the carbon chain of OA at 5.25 ppm, the signals attributed to the methyl of OA at 0.89 ppm, and the carbon chain signals of OA at 1.25–1.05 ppm (Fig. S1). The 1H NMR spectrum for PEG5000-OA2 shows similar proton signals except lack of proton signals for Fmoc (Fig. S2). The molecular weights of the PEG5000-(Fmoc-OA)2 and PEG5000-OA2 conjugates measured by MALDI-TOF Mass Spectrum are close to the theoretical values (Figs. S3 and S4).
Fig. 1.
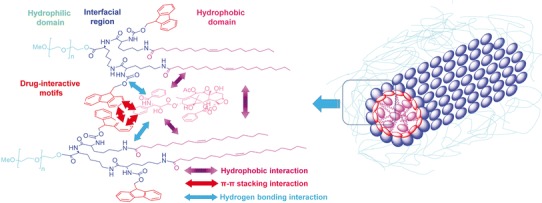
Structure of PEG5000-Lys-(α-Fmoc-ε-oleoyl lysine)2 and the postulated modes of carrier-drug and carrier-carrier interactions
Biophysical Characterization of Drug-Free and PTX-Loaded PEG-Lipopeptide Micelles
As an initial step to examine the general applicability of our PEG-lipopeptide in formulating different drugs of diverse structures, we examined the efficiency of PEG5000-(Fmoc-OA)2 in delivering PTX to tumor cells in vitro and in vivo. PEG5000-OA2 was used as a control formulation.
Both PEG5000-(Fmoc-OA)2 and PEG5000-OA2 readily formed micelles in DPBS. Figure 2 shows the size distribution and TEM images of PEG5000-(Fmoc-OA)2 and PEG5000-OA2 micelles. The size of PEG5000-OA2 is around 20 nm as determined by dynamic light scattering (DLS) (Fig. 2a). TEM images show spherical particles for PEG5000-OA2 micelles (Fig. 2b) and the sizes of the particles on TEM were consistent with that determined by DLS. PEG5000-(Fmoc-OA)2 formed particles of slightly larger size (∼60 nm) as determined by DLS (Fig. 2d). TEM revealed mostly tubular structures (Fig. 2e), suggesting formation of filamentous micelles.
Fig. 2.
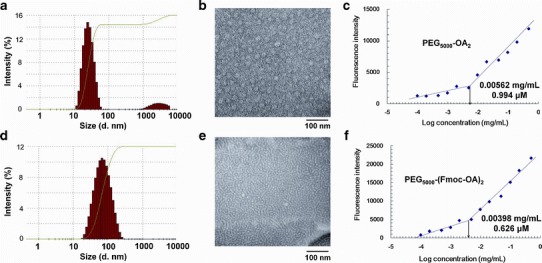
Size distribution of PEG5000-OA2 (a) and PEG5000-(Fmoc-OA)2 (d) measured by DLS, and TEM of PEG5000-OA2 (b) and PEG5000-(Fmoc-OA)2 (e) micelles. The spherical particles with a diameter around 20 nm were observed for PEG5000-OA2 micelles, while filamentous micelles with tubular structure were observed for PEG5000-(Fmoc-OA)2 micelles. CMC measurements of the PEG5000-OA2 (c) and PEG5000-(Fmoc-OA)2 (f) micelles using pyrene as a fluorescence probe. The fluorescence intensity was plotted as a function of logarithmic concentration of micelles
The CMC values of PEG5000-(Fmoc-OA)2 and PEG5000-OA2 micelles were measured using pyrene as a fluorescence probe. As shown in Fig. 2c, f, the CMC of PEG5000-(Fmoc-OA)2 is 0.626 μM, which is lower than those of PEG5000-OA2 (0.994 μM) and many reported surfactants (18–20).
Table I shows the size, drug loading capacity (DLC), and drug loading efficiency (DLE) for PEG5000-(Fmoc-OA)2/PTX mixed micelles in comparison with PEG5000-OA2 formulation. PTX could be formulated in PEG5000-(Fmoc-OA)2 micelles at a carrier/drug molar ratio as low as 0.75/1 although the size of the resulting PTX-loaded micelles stayed stable for only 1.5 h. There was little change in the particle sizes before and after the incorporation of PTX. Under this condition, the DLC and DLE were 15.19% and 56.29%, respectively. An increase in the carrier/drug input ratio was associated with an increase in the DLE and significantly improved colloidal stability of the PEG5000-(Fmoc-OA)2/PTX mixed micelles. The particles stayed stable for more than 20 h at carrier/drug ratios of 5/1 and above.
Table I.
Biophysical Characterization of Drug-Free and PTX-Loaded PEG5000-OA2 and PEG5000-(Fmoc-OA)2 Micelles
| Micelles | Molar ratio | Size (nm) | PDI | DLC (%) | DLE (%) | Stability (h) |
|---|---|---|---|---|---|---|
| PEG5000-OA2 | – | 26.76 ± 0.44 | 0.262 | – | – | – |
| PEG5000-OA2/PTXa | 2.5:1 | 23.52 ± 0.21 | 0.103 | 5.70 | ND | 1 |
| 5:1 | 22.62 ± 0.33 | 0.102 | 2.93 | ND | 1.5 | |
| 7.5:1 | 23.93 ± 0.33 | 0.098 | 1.97 | ND | 3.5 | |
| PEG5000-(Fmoc-OA)2 | – | 58.72 ± 1.06 | 0.220 | – | – | – |
| PEG5000-(Fmoc-OA)2/PTXa | 0.75:1 | 66.26 ± 0.46 | 0.233 | 15.19 | 56.29 | 1.5 |
| 1:1 | 70.28 ± 1.25 | 0.256 | 11.84 | 88.73 | 2 | |
| 2.5:1 | 69.42 ± 1.91 | 0.248 | 5.10 | 80.50 | 3.5 | |
| 5:1 | 73.55 ± 1.26 | 0.258 | 2.62 | 92.95 | 22 | |
| 7.5:1 | 73.13 ± 1.29 | 0.245 | 1.76 | 97.73 | 70 |
PDI polydispersity index, DLC drug loading capacity, DLE drug loading efficiency, ND not determined
aPTX concentration in micelles were kept at 1 mg/mL; drug-free micelle concentration was 20 mg/mL
On the contrary, a minimal carrier/drug ratio of 2.5/1 was needed to load PTX into PEG5000-OA2 micelles and the resulting mixed micelles were only stable for 1 h. The PEG5000-OA2/PTX mixed micelles were also significantly less stable than PEG5000-(Fmoc-OA)2/PTX mixed micelles at other carrier/drug ratios examined.
The above data clearly demonstrated that the inclusion of Fmoc motifs into PEG5000-OA2 micelles significantly improved the PTX loading capacity and the stability of PTX-loaded micelles. To gain insight into the role of Fmoc in the carrier/PTX interaction, we then conducted a fluorescence quenching study with PEG5000-(Fmoc-OA)2. Figure 3 shows the fluorescence spectrum of PEG5000-(Fmoc-OA)2 with a maximum fluorescence at 305 nm upon excitation at a wavelength of 270 nm. This fluorescence spectrum appears to be specific for Fmoc as minimal fluorescence was detected for PEG5000-OA2 (data not shown). Interestingly, the fluorescence intensity was decreased with the addition of PTX, and the extent of fluorescence quenching was correlated to amount of PTX. However, no quenching was observed in the presence of the same amount of cholesterol (Chol), a hydrophobic molecule lacking aromatic rings. It has been reported that intermolecular π–π stacking between aromatic rings can effectively cause fluorescence quenching through energy transfer (21). Our data suggest a role of Fmoc/PTX π–π stacking in the carrier/PTX interaction.
Fig. 3.
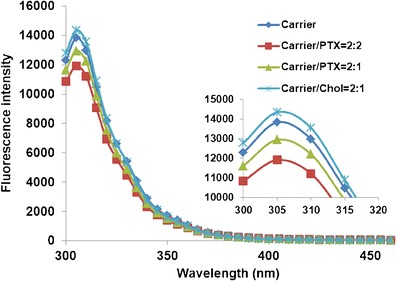
Fluorescence quenching of PEG5000-(Fmoc-OA)2. The concentration of PEG5000-(Fmoc-OA)2 was fixed at 1.5 mg/mL and mixed with PTX and Chol at designated molar ratios in PBS. Intensity of fluorescence emitted between 300 and 460 nm was recorded with excitation wavelength at 270 nm
Figure S5a shows the 1H-NMR spectra of PEG5000-(Fmoc-OA)2/PTX in CDCl3 and in deuterated water, respectively. In CDCl3, signals from both carrier and PTX were clearly observed. In contrast, all of the proton signals of PTX were suppressed in deuterated water, indicating complete entrapment of PTX inside self-assembled micelles in aqueous solution.
We then examined the effect of freezing and lyophilization on the colloidal stability of PEG5000-(Fmoc-OA)2/PTX mixed micelles. As shown in Fig. S6, there were essentially no changes in the size distribution for PEG5000-(Fmoc-OA)2/PTX mixed micelles following lyophilization and reconstitution of the lyophilized power with water.
Release Kinetics of PTX-Loaded Mixed Micelles
The kinetics of PTX release from PTX-loaded PEG5000-(Fmoc-OA)2 micelles was examined via dialysis method and compared to PEG5000-OA2/PTX micelles and Taxol formulation. As shown in Fig. 4, the PTX-loaded PEG5000-(Fmoc-OA)2 micelles showed much enhanced stability. After first 24 h, only 27.88% of formulated PTX was released from PTX/PEG5000-(Fmoc-OA)2 mixed micelles, while 55.90% and 62.88% of PTX was released from PEG5000-OA2/PTX micelles and Taxol formulation, respectively. The T1/2 of PTX release is 19.8 and 14.4 h for PEG5000-OA2/PTX and Taxol formulation, respectively, while only 33% of PTX was released from PEG5000-(Fmoc-OA)2 micelles even after 72 h.
Fig. 4.
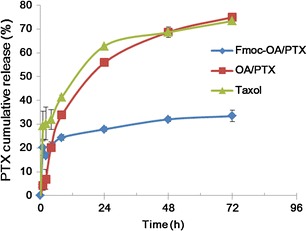
Cumulative PTX release profile from PTX-loaded PEG5000-(Fmoc-OA)2 micelles (Fmoc-OA/PTX), PEG5000-OA2 micelles (OA/PTX) and Taxol. PTX concentration was kept at 1 mg/mL in all the formulations, and DPBS (pH 7.4) containing 0.5% (w/v) Tween 80 was utilized as release medium
In Vitro Cytotoxicity Study
The in vitro cytotoxicity of PTX-loaded PEG5000-(Fmoc-OA)2 micelles was evaluated with three different tumor cell lines, 4T1.2, PC-3, and DU145, and compared to that of Taxol, a clinical PTX formulation. PTX-loaded PEG5000-(Fmoc-OA)2 micelles exhibited higher levels of cytotoxicity than Taxol in all three cell lines tested (Fig. 5a–c). Interestingly, PEG5000-(Fmoc-OA)2 micelles alone showed modest cytotoxicity towards 4T1.2 cells (Fig. 5a) while they showed minimal effect on the growth of prostate cancer cells PC-3 and DU145 (Fig. 5b, c). This might be due to the different proliferation rates of these cell lines. The difference of intracellular esterase activity among these cell lines may also contribute to the different levels of cytotoxicity.
Fig. 5.
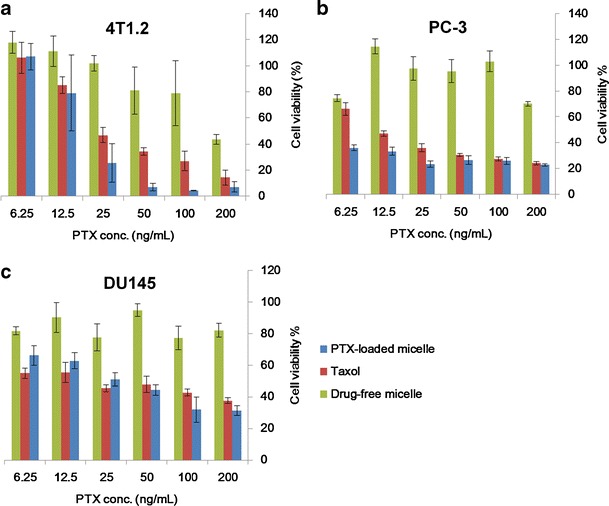
Cytotoxicity of PTX-loaded PEG5000-(Fmoc-OA)2 micelles, drug-free PEG5000-(Fmoc-OA)2 micelles, and Taxol to 4T1.2 mouse breast cancer cell line (a), human prostate cancer cell lines PC-3 (b), and DU145 (c). Cells were treated for 72 h, and cytotoxicity was determined by MTT assay
In Vivo Antitumor Activity
The in vivo therapeutic activity of PTX formulated in PEG5000-(Fmoc-OA)2 micelles was investigated in a syngeneic murine breast cancer model (4T1.2). As shown in Fig. 6, Taxol exhibited modest effects in inhibiting the tumor growth at a PTX dose of 10 mg/kg. At the same PTX dosage, PTX formulated in PEG5000-(Fmoc-OA)2 micelles was more effective than Taxol in inhibiting the tumor growth (p < 0.05). The tumor growth inhibitory effect of PTX/PEG5000-(Fmoc-OA)2 micelles was further enhanced when the PTX dosage was increased to 20 mg/kg (p < 0.01). No apparent weight changes were noticed in all of the treatment groups.
Fig. 6.
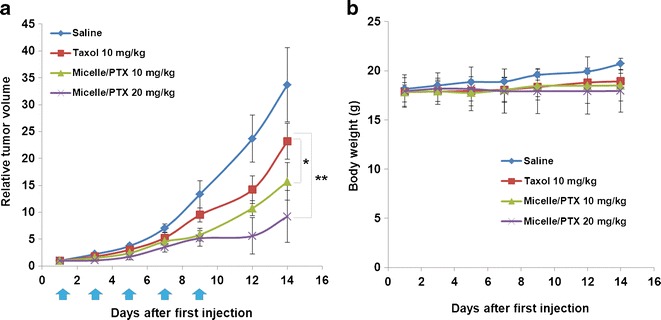
a Tumor inhibitory activity of PTX-loaded PEG5000-(Fmoc-OA)2 micelles. BALB/c mice were inoculated s.c. with 4T1.2 cells (2 × 105 cells/mouse). Five days later, mice received various treatments on days 1, 3, 5, 7, and 9, and tumor growth was monitored and plotted as relative tumor volume. *P < 0.05 (PEG5000-(Fmoc-OA)2/PTX at 10 mg/kg vs. Taxol at 10 mg/kg). **P < 0.01 (PEG5000-(Fmoc-OA)2/PTX at 20 mg/kg vs. Taxol at 10 mg/kg). N = 4. b Changes of body weight in mice in different treatment groups
Effectiveness of PEG5000-(Fmoc-OA)2 Micelles in Formulating Seven Other Drugs of Different Structures
The above study clearly demonstrated that the inclusion of Fmoc motifs into PEG-lipid micelles improved PTX loading capacity and facilitates its delivery to tumor cells in vitro and in vivo. We then went on to test the effectiveness of our improved system in formulating seven additional model drugs of different structures (Fig. 7) to test the general utility of our formulation. These include probucol (cholesterol-lowing drug), niclosamide (antiparasitic agent), JP4-039 (antioxidant), progesterone (female hormone), cyclosporin A (immunosuppressant), nifedipine (Ca2+ channel blocker), and griseofulvin (antifungal agent). The structural features of these compounds were summarized in Table II, and they were loaded into PEG5000-(Fmoc-OA)2 micelles at the concentration of 1 mg drug/mL, respectively, which represents a 71 ∼ 1.7 × 105-fold increase in solubility compared with the original aqueous solubility of each compound. The drug candidates were well encapsulated inside the hydrophobic core of PEG5000-(Fmoc-OA)2 micelles, respectively, as demonstrated by the suppression of proton signals of each compound in deuterated water (Fig. 7a–f).
Fig. 7.
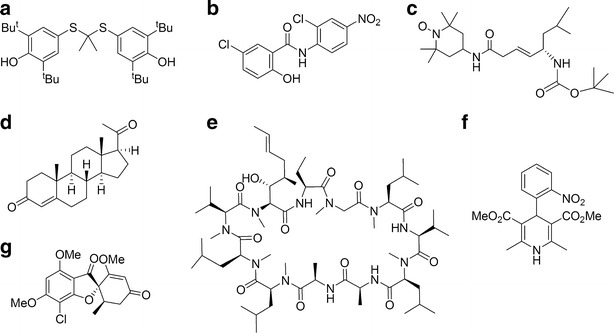
Chemical structure of seven drug candidates effectively formulated in PEG5000-(Fmoc-OA)2 micelles. a Probucol, b niclosamide, c JP4-039, d progesterone, e cyclosporin A, f nifedipine, and g griseofulvin
Table II.
Effectiveness of PEG5000-(Fmoc-OA)2 Micelles in Formulating Drugs of Diverse Structures
| Drug candidate | Biologic activities | Number of aromatic rings | Structural features | Log P | Free drug solubility (μg/mL) | Carrier/drug molar ratioa | DLC (%) | Particle size (nm) |
|---|---|---|---|---|---|---|---|---|
| Probucol | Cholesterol-lowing drug | 2 | Dimerized di-tert-butylthiophenol | 8.08b | 0.006b | 2.5 | 3.15 | 74.22 ± 2.57 |
| Niclosamide | Antiparasitic agent | 2 | Salicylanilide | 4.56b | 0.23b | 2.5 | 2.02 | 81.94 ± 4.37 |
| JP4-039 | Antioxidant | 0 | Peptide | N/A | N/A | 2.5 | 3.13 | 55.90 ± 0.75 |
| Progesterone | Female hormone | 0 | Steroid hormone | 3.87b | 7b | 0.25 | 16.52 | 70.57 ± 0.73 |
| Cyclosporin A | Immunosuppressant | 0 | 11 amino acids cyclic peptide | 3.0b | 6.6b | 2.5 | 7.04 | 72.79 ± 1.62 |
| Nifedipine | Ca2+ channel blocker. | 2 | Nitrophenyl dihydropyridine dicarboxylate | 2.5b | 5b | 0.5 | 9.83 | 63.75 ± 1.22 |
| Griseofulvin | Antifungal agent | 1 | Methoxy benzofuran, cyclohexene-dione | 2.0b | 14b | 2.5 | 2.17 | 66.79 ± 0.96 |
DISCUSSION
We have confirmed and extended our previous work that the inclusion of a drug-interactive motif at the interfacial region of surfactants significantly improves the drug-loading capacity and formulation stability. PEG-lipopeptides were originally developed to formulate JP4-039, a peptide-based antioxidant (17). Data from the present study suggest that our concept is applicable to various types of therapeutic agents of diverse structures.
One of the key findings of our study is the unusual propensity of Fmoc motif in interacting with many types of drugs, ranging from PTX, steroids, to hydrophobic peptide drugs with linear or cyclic configuration (Table II). These agents have one or more aromatic or heterocyclic ring structures or multiple hydrophobic side chain groups if it is a peptide derivative and can form hydrogen bond with other molecules. These features are quite ubiquitous among many drugs and drug candidates, suggesting that this motif may have the utility for a broad spectrum of compounds. These data strongly suggest that α-Fmoc behaves as a “formulation chemophor” or a structural unit capable of interacting with many pharmaceutical agents. The molecular basis for such propensity of interactions is likely due to a combination of the fused aromatic ring structure of fluorenyl group and its carbamate and amide linkages. Fluorenyl group is a compact hydrophobic motif capable of forming hydrophobic π–π interactions with compounds carrying one or more aromatic ring structures. Such interaction is normally stronger than the van der Waals interaction between alkyl chains. The carbamate and other amine linkages in the conjugate are known to facilitate hydrogen bonding interactions, which shall also contribute to both carrier/carrier and carrier/drug interactions (Fig. 1). The importance of Fmoc was supported by the observations that the performance of the PEG-lipopeptides was significantly compromised if it was replaced by other motifs such as Boc (Gao et al., unpublished data).
Similar to a PEG-lipopeptide with four Fmoc motifs (17), PEG5000-(Fmoc-OA)2 formed tubular structures, suggesting the formation of filamentous micelles (Fig. 2e). These structures were well retained following the incorporation of PTX. This is likely due to strong interaction among the Fmoc-containing PEG-lipopeptides. It is known that Fmoc-containing short peptides tend to show strong interaction among themselves to form tubular structures that resulted in formation of hydrogels (22). However, the lipid motif may also contribute to the formation of tubular structures as a PEG5000-Fmoc counterpart without OA chains formed spherical particles in the absence or presence of loaded PTX (data not shown). It has been reported that filamentous polymeric micelles exhibit substantially longer half-life in the blood compared to the spherical counterparts (23). More studies are needed to examine the in vivo pharmacokinetics profiles of our PEG-lipopeptides as well as the mechanisms involved in various modes of self-assemblies.
In addition to examining the general utility of our system in formulating different types of drugs of diverse structures, we further explored its potential in the delivery of PTX to tumor cells. PTX is a first-line therapeutic agent for various types of cancers; however, its clinical application is limited by the toxicity and poor water solubility (24–27). Taxol is an alcohol/Cremophor formulation that can cause local irritation and severe histamine-mediated hypersensitivity reactions (28–30). PEG-derivatized phospholipid has been shown to be able to form simple micellar formulation with PTX, but has limited loading capacity (31–33). We also noticed a low PTX loading capacity with a similar system based on PEG5000-OA2 conjugate (Table I). Incorporation of Fmoc into this system led to a three-fold increase in PTX loading capacity. In addition, the colloidal stability of PTX/PEG5000-(Fmoc-OA)2 mixed micelles was significantly improved compared to that of PTX-loaded PEG5000-OA2 micelles (Table I). This is corroborated by the observation that PTX formulated in PEG5000-(Fmoc-OA)2 micelles also demonstrated a much slower kinetics of PTX release compared to Taxol formulation and PEG5000-OA2 micelles without Fmoc motifs (Fig. 4). These improvements may be attributed to the enhanced interaction between the carrier and PTX. Several mechanisms are likely to be involved in the interactions between the carrier and PTX including hydrophobic/hydrophobic interaction and hydrogen bonding. The Fmoc/PTX π–π stacking shall also contribute significantly to the carrier/PTX interactions as supported by the data from the fluorescence quenching study (Fig. 3). More studies are needed in the future to better understand the mechanism of carrier/PTX interaction.
We have further demonstrated an improved antitumor activity for our micellar PTX compared to Taxol formulation both in vitro and in vivo. At an increased dosage of PTX (20 mg/kg), which is beyond the maximum tolerated dose of Taxol (14), our micelle formulation led to a further enhancement in tumor inhibitory effect with minimal toxicity (Fig. 6). The improved performance is likely due to an enhanced carrier/drug interaction and an improved stability of the PTX micelle formulation in vivo, which presumably lead to an effective delivery of PTX to tumors through the EPR effect.
CONCLUSIONS
We have shown that incorporation of Fmoc motifs into a PEG-lipopeptide conjugate resulted in an improved formulation that is effective in formulating eight model drugs of diverse structures. We have further shown that incorporation of PTX into our new formulation led to improved antitumor activity over Taxol in vitro and in vivo. Our study suggests that micelle-forming PEG-lipopeptide surfactants with interfacial Fmoc motifs may represent a promising drug formulation platform for a broad range of drugs with diverse structures.
Electronic Supplementary Material
(DOCX 18 kb)
(TIFF 1401 kb)
{kind=link}
(TIFF 1412 kb)
{kind=link}
(TIFF 2892 kb)
{kind=link}
(TIFF 2925 kb)
{kind=link}
(TIFF 348 kb)
{kind=link}
(TIFF 9314 kb)
{kind=link}
(TIFF 1521 kb)
{kind=link}
ACKNOWLEDGMENTS
This work was supported in part by NIH grants (R01GM102989, R21CA173887, and R21CA155983) and a DOD grant (BC09603).
Contributor Information
Xiang Gao, Email: xig9@pitt.edu.
Song Li, Email: sol4@pitt.edu.
REFERENCES
- 1.Serajuddin AT. Salt formation to improve drug solubility. Adv. Drug Deliv. Rev. 2007;59:603–616. doi: 10.1016/j.addr.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 2.Löbenberg R, Amidon GL, Vierira M. Solubility as a limiting factor to drug absorption. In: Dressman JB, Lennernäs H, editors. Oral Drug Absorption: Prediction and Assessment. New York: Marcel Dekker; 2000. p. 137. [Google Scholar]
- 3.Matsumura Y. Poly(amino acid) micelle nanocarriers in preclinical and clinical studies. Adv. Drug Deliv. Rev. 2008;60:899–914. doi: 10.1016/j.addr.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 4.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control. Release. 2000;65:271–284. doi: 10.1016/S0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 5.Maeda H. The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Adv. Enzyme Regul. 2001;41:189–207. doi: 10.1016/S0065-2571(00)00013-3. [DOI] [PubMed] [Google Scholar]
- 6.Jain RK. Vascular and interstitial barriers to delivery of therapeutic agents in tumors. Cancer Metastasis Rev. 1990;9:253–266. doi: 10.1007/BF00046364. [DOI] [PubMed] [Google Scholar]
- 7.Gaucher G, Dufresne MH, Sant VP, Kang N, Maysinger D, Leroux JC. Block copolymer micelles: preparation, characterization and application in drug delivery. J Control. Release. 2005;109:169–188. doi: 10.1016/j.jconrel.2005.09.034. [DOI] [PubMed] [Google Scholar]
- 8.Kim JY, Kim S, Papp M, Park K, Pinal R. Hydrotropic solubilization of poorly water-soluble drugs. J Phar Sci. 2010;99:3953–3965. doi: 10.1002/jps.21895. [DOI] [PubMed] [Google Scholar]
- 9.Kim JY, Kim S, Pinal R, Park K. Hydrotropic polymer micelles as versatile vehicles for delivery of poorly water-soluble drugs. J Control. Release. 2011;152:13–20. doi: 10.1016/j.jconrel.2011.02.014. [DOI] [PubMed] [Google Scholar]
- 10.Yoo HS, Park TG. Folate-receptor-targeted delivery of doxorubicin nano-aggregates stabilized by doxorubicin–PEG–folate conjugate. J Control. Release. 2004;100:247–256. doi: 10.1016/j.jconrel.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 11.Yoo HS, Park TG. Folate receptor targeted biodegradable polymeric doxorubicin micelles. J Control. Release. 2004;96:273–283. doi: 10.1016/j.jconrel.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 12.Mi Y, Liu Y, Feng SS. Formulation of Docetaxel by folic acid-conjugated d-α-tocopheryl polyethylene glycol succinate 2000 (Vitamin E TPGS2k) micelles for targeted and synergistic chemotherapy. Biomaterials. 2011;32:4058–4066. doi: 10.1016/j.biomaterials.2011.02.022. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Z, Tan S, Feng SS. Vitamin E TPGS as a molecular biomaterial for drug delivery. Biomaterials. 2012;33:4889–4906. doi: 10.1016/j.biomaterials.2012.03.046. [DOI] [PubMed] [Google Scholar]
- 14.Lu J, Huang Y, Zhao W, Marquez RT, Meng X, Li J, et al. PEG-derivatized embelin as a nanomicellar carrier for delivery of paclitaxel to breast and prostate cancers. Biomaterials. 2013;34:1591–1600. doi: 10.1016/j.biomaterials.2012.10.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang Y, Lu J, Gao X, Li J, Zhao W, Sun M, et al. PEG-derivatized embelin as a dual functional carrier for the delivery of paclitaxel. Bioconjug. Chem. 2012;23:1443–1451. doi: 10.1021/bc3000468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang X, Lu J, Huang Y, Zhao W, Chen Y, Li J, et al. PEG–farnesylthiosalicylate conjugate as a nanomicellar carrier for delivery of paclitaxel. Bioconjug. Chem. 2013;24:464–472. doi: 10.1021/bc300608h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao X, Huang Y, Makhov AM, Epperly M, Lu J, Grab S, et al. Nanoassembly of surfactants with interfacial drug-interactive motifs as tailor-designed drug carriers. Mol. Pharm. 2013;10:187–198. doi: 10.1021/mp300319m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kang N, Leroux JC. Triblock and star-block copolymer of N-(2-hydroxypropyl)methacrylamide or N-vinyl-2-pyrrolidone and d,l-lactide: synthesis and self-assembling properties in water. Polymer. 2004;45:8967–8980. doi: 10.1016/j.polymer.2004.10.081. [DOI] [Google Scholar]
- 19.Lavasanifar A, Samuel J, Kwon GS. The effect of alkyl core structure on micellar properties of poly(ethylene oxide)-block-poly(l-aspartamide) derivatives. Colloids Surf. B Biointerfaces. 2001;22:115–126. doi: 10.1016/S0927-7765(01)00147-3. [DOI] [PubMed] [Google Scholar]
- 20.Kwon GS, Natio M, Yokoyama M, Okano T, Sakurai Y, Kataoka K. Micelles based on AB block copolymers of poly(ethylene oxide) and poly(β benzyl l-aspartate) Langmuir. 1993;9:945–949. doi: 10.1021/la00028a012. [DOI] [Google Scholar]
- 21.Shirai K, Matsuoka M, Fukunishi K. Fluorescence quenching by intermolecular π–π interactions of 2,5-bis(N,N-dialkylamino)-3,6-dicyanopyrazines. Dyes Pigm. 1999;42:95–101. doi: 10.1016/S0143-7208(99)00013-3. [DOI] [Google Scholar]
- 22.Zhou M, Smith AM, Das AK, Hodson NW, Collins RF, Ulijn RV, et al. Self-assembled peptide-based hydrogels as scaffolds for anchorage-dependent cells. Biomaterials. 2009;30:2523–2530. doi: 10.1016/j.biomaterials.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 23.Geng Y, Dalhaimer P, Cai S, Tsai R, Tewari M, Minko T, et al. Shape effects of filaments versus spherical particles in flow and drug delivery. Nat. Nanotechnol. 2007;2:249–255. doi: 10.1038/nnano.2007.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goldspiel BR. Clinical overview of the taxanes. Pharmacotherapy. 1997;17:110S–125S. [PubMed] [Google Scholar]
- 25.Rowinsky EK, Cazenave LA, Donehower RC. Taxol: a novel investigational antimicrotubule agent. J. Natl. Cancer Inst. 1990;82:1247–1259. doi: 10.1093/jnci/82.15.1247. [DOI] [PubMed] [Google Scholar]
- 26.Spencer CM, Faulds D. Paclitaxel: a review of its pharmacodynamic and pharmacokinetic properties and therapeutic potential in the treatment of cancer. Drugs. 1994;48:794–847. doi: 10.2165/00003495-199448050-00009. [DOI] [PubMed] [Google Scholar]
- 27.Sollott SJ, Cheng L, Pauly RR, Jenkins GM, Monticone RE, Kuzuya M, et al. Taxol inhibits neointimal smooth muscle cell accumulation after angioplasty in the rat. J. Clin. Invest. 1995;95:1869–1876. doi: 10.1172/JCI117867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gelderblom H, Verweij J, Nooter K, Sparreboom A. Cremophor EL: the drawbacks and advantages of vehicle selection for drug formulation. Eur. J. Cancer. 2001;37:1590–1598. doi: 10.1016/S0959-8049(01)00171-X. [DOI] [PubMed] [Google Scholar]
- 29.Weiss RB, Donehower RC, Wiernik PH, Ohnuma T, Gralla RJ, Trump DL, et al. Hypersensitivity reactions from taxol. J. Clin. Oncol. 1990;8:1263–1268. doi: 10.1200/JCO.1990.8.7.1263. [DOI] [PubMed] [Google Scholar]
- 30.Kloover JS, den Bakker MA, Gelderblom M, van Meerbeeck JP. Fatal outcome of a hypersensitivity reaction to paclitaxel: a critical review of premedication regimens. Br. J. Cancer. 2004;90:304–305. doi: 10.1038/sj.bjc.6601303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao Z, Lukyanov AN, Singhal A, Torchilin VP. Diacyl-polymer micelles as nanocarriers for poorly soluble anticancer drugs. Nano Lett. 2002;2:979–982. doi: 10.1021/nl025604a. [DOI] [Google Scholar]
- 32.Lukyanov AN, Torchilin VP. Micelles from lipid derivatives of water-soluble polymers as delivery systems for poorly soluble drugs. Adv. Drug Deliv. Rev. 2004;56:1273–1289. doi: 10.1016/j.addr.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 33.Gao Z, Lukyanov AN, Chakilam AR, Torchilin VP. PEG–PE/phosphatidylcholine mixed immunomicelles specifically deliver encapsulated taxol to tumor cells of different origin and promote their efficient killing. J. Drug Target. 2003;11:87–92. doi: 10.1080/1061186031000138623. [DOI] [PubMed] [Google Scholar]
- 34.Jack DB. Handbook of clinical pharmacokinetics data. Houndmills, UK: MacMillan Publisher’s Ltd; 1992.
- 35.Persson EM, Gustafsson AS, Carlsson AS, Nilsson RG, Knutson L, Forsell P, et al. The effects of food on the dissolution of poorly soluble drugs in human and in model small intestinal fluids. Pharm. Res. 2005;22:2141–2151. doi: 10.1007/s11095-005-8192-x. [DOI] [PubMed] [Google Scholar]
- 36.Nendza M, Müller M. Discriminating toxicant classes by mode of action: 3. Substructure indicators. SAR QSAR Environ Res. 2007;18:155–168. doi: 10.1080/10629360601054354. [DOI] [PubMed] [Google Scholar]
- 37.Yang W, de Villiers MM. Effect of 4-sulphonato-calix[n]arenes and cyclodextrins on the solubilization of niclosamide, a poorly water soluble anthelmintic. AAPS J. 2005;7:E241–E248. doi: 10.1208/aapsj070123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alvarez Núñez FA, Yalkowsky SH. Correlation between log P and Clog P for some steroids. J. Pharm. Sci. 1997;86:1187–1189. doi: 10.1021/js970050a. [DOI] [PubMed] [Google Scholar]
- 39.Nandi I, Bateson M, Bari M, Joshi HN. Synergistic effect of PEG-400 and cyclodextrin to enhance solubility of progesterone. AAPS PharmSciTech. 2003;4:1–5. doi: 10.1208/pt040101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang ZQ, Xu J, Pan P, Zhang XN. Preparation of an alternative freeze-dried pH-sensitive cyclosporine A loaded nanoparticles formulation and its pharmacokinetic profile in rats. Pharmazie. 2009;64:26–31. [PubMed] [Google Scholar]
- 41.Novalbos J, Abad-Santos F, Zapater P, Cano-Abad MF, Moradiellos J, Sánchez-García P, et al. Effects of dotarizine and flunarizine on chromaffincell viability and cytosolic Ca2+ Eur. J. Pharmacol. 1999;366:309–317. doi: 10.1016/S0014-2999(98)00916-9. [DOI] [PubMed] [Google Scholar]
- 42.Yang W, de Villiers MM. The solubilization of the poorly water soluble drug nifedipine by water soluble 4-sulphonic calix[n]arenes. Eur. J. Pharm. Biopharm. 2004;58:629–636. doi: 10.1016/j.ejpb.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 43.Mithani SD, Bakatselou V, TenHoor CN, Dressman JB. Estimation of the increase in solubility of drugs as a function of bile salt concentration. Pharm. Res. 1996;13:163–167. doi: 10.1023/A:1016062224568. [DOI] [PubMed] [Google Scholar]
- 44.Gramatté T. Griseofulvin absorption from different sites in the human small intestine. Biopharm. Drug Dispos. 1994;15:747–759. doi: 10.1002/bdd.2510150903. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 18 kb)
(TIFF 1401 kb)
(TIFF 1412 kb)
(TIFF 2892 kb)
(TIFF 2925 kb)
(TIFF 348 kb)
(TIFF 9314 kb)
(TIFF 1521 kb)