

Abstract
Trimethylated histone H3 lysine 4 (H3K4) and H3K27 generally mark transcriptionally active and repressive chromatins, respectively. In most cell types, these two modifications are mutually exclusive, and this segregation is crucial for the regulation of gene expression. However, how this anticorrelation is achieved has not been fully understood. Here, we show that removal of the H3K27 trimethyl mark facilitates recruitment of SET1-like H3K4 methyltransferase complexes to their target genes by eliciting a novel interaction between histone H3 and two common subunits, WDR5 and RBBP5, of SET1-like complexes. Consistent with this result, H3K27 trimethylation destabilizes interactions of H3 with SET1-like complexes and antagonizes their ability to carry out H3K4 trimethylation of peptide (H3 residues 1 to 36), histone octamer, and mononucleosome substrates. Altogether, our studies reveal that H3K27 trimethylation of histone H3 represses a previously unrecognized interaction between H3 and SET1-like complexes. This provides an important mechanism that directs the anticorrelation between H3K4 and H3K27 trimethylation.
INTRODUCTION
Trimethylation of histone H3 at lysine 4 (H3K4me3) is a mark for active chromatin that counters the repressive chromatin milieu imposed by H3K9 and H3K27 methylation in higher eukaryotes (1). H3K4 trimethylation is primarily carried out by the vertebrate H3K4 methyltransferases SET1A/SET1B (SET1A/B) and MLL1 to MLL4, which form a family of related SET1-like complexes (2, 3). Each complex contains a unique H3K4 methyltransferase enzyme, complex-specific subunits, and a common subcomplex, consisting of RBBP5, ASH2L, WDR5, and DPY-30, that facilitates the H3K4 methyltransferase activity of SET1-like complexes (2, 3). We previously identified the first mammalian SET1-like complexes containing MLL3 and MLL4 (MLL3/4), which play essential roles in transactivation by retinoic acid receptor (RAR) and multiple other transcription factors (3–5). Unique subunits in these two complexes include PA1, PTIP, the H3K27 demethylase UTX, and the transcriptional coactivator ASC-2 (3, 4, 6–9). These complexes were originally named ASCOM (for ASC-2 complex)-MLL3 and ASCOM-MLL4 (4, 5).
WDR5 was earlier reported to bind to K4-dimethylated H3 (10) and, in subsequent structural studies, to interact not only with dimethylated H3K4, by virtue of both water-mediated hydrogen bonding and the altered hydrophilicity of the modified K4, but also with the second arginine residue (H3R2) of H3 (11–14). The arginine methyltransferase PRMT6 catalyzes H3R2 dimethylation in vitro and controls global levels of H3R2 dimethylation in vivo (15, 16). Of note, and as a paradigm for the present study, H3R2 methylation by PRMT6 is blocked by the presence of the H3K4 trimethyl mark, whereas the H3R2 methyl mark interferes with binding of WDR5 to the H3 tail and thus prevents H3K4 trimethylation by SET1-like complexes (15, 16).
It is well established that, in general, H3K4 trimethylation is inversely correlated with H3K27 trimethylation throughout the genome in most vertebrate cell types (1), although these marks may also coexist in discrete or overlapping regions of individual genes (17–21). Our findings in this report reveal that H3K27 trimethylation of histone H3 inhibits a strong, but previously unrecognized, interaction between H3 and SET1-like complexes, thus making H3K27 demethylation a prerequisite for efficient H3K4 trimethylation by SET1-like complexes. In addition, H3K4 trimethylation has been shown to inhibit H3K27 trimethylation by the PRC2 complex (22). Taken together, these results reveal the molecular basis underlying the important anticorrelation between H3K4 and H3K27 trimethylation.
MATERIALS AND METHODS
Plasmids, H3 peptides, and methylated H3.
pGEX4T-1 vectors expressing the following were generated by PCR and verified by sequencing: histone H3 residues 1 to 25 [H3(1–25)], 1 to 36 [H3(1–36)], 1 to 57 [H3(1–57)], 5 to 36 [H3(5–36)], 5 to 57 [H3(5–57)], 10 to 31 [H3(10–31)], and 10 to 36 [H3(10–36)]; H3 residues 10 to 36 with the point mutations R26A and K27F, respectively; H3 residues 1 to 57 bearing the point mutations R2F [H3(1–57)(R2F)], K4F, and K27F [H3(1–57)(K27F)], respectively; and pcDNA3 expressing WDR5 with the point mutation S91F. The cDNA sequences of UTX in pCS2 hUTX (where hUTX is human UTX) or its mutant H1146A (UTX-H1146A) (23) were mutated to be resistant to small interfering RNA targeting UTX (si-UTX) (24). Histone H3 peptides corresponding to residues 1 to 20 or 1 to 36 were chemically synthesized. Semisynthetic full-length H3 with trimethylated K27 (K27me3) was prepared via expressed protein ligation as described previously (25). Briefly a synthetic peptide α-thioester corresponding to residues 1 to 28 of H3 and containing the K27me3 modification was ligated to the remainder of H3 (containing an Ala29Cys mutation) prepared through recombinant means. Following ligation, the ligation product was desulfurized to convert the Cys29 ligation back to the native alanine.
RT/qRT-PCR analysis.
Reverse transcription/quantitative reverse transcription-PCRs (RT/qRT-PCRs) were performed as described previously (5). For retinoic acid (RA) treatment, cells were incubated with 0.1 μM RA for 12 h before harvest. The primers for human p21 were 5′-ACT TCC TCC TCC CCA CTT GT-3′ and 5′-AGG TGA GGG GAC TCC AAA GT-3′. The primers for cyclophilin A (control) were 5′-GTC TCC TTC GAG CTG TTT GC-3′ and 5′-GAT GCC AGG ACC TGT ATG CT-3′. The primers for RAR-β2 were 5′-GGT CCT CTG ACT GAC CTT GT-3′ and 5′-GGA AAC ATG TGA GGC TTG CT-3′. The primers for PDCD4 (for programmed cell death 4) were 5′-ATG ATG TGG AGG AGG TGG ATG TG-3′ and 5′-CCA ATG CTA AGG ATA CTG CCA AC-3′. The primers for IGFBP2 (for insulin-like growth factor binding protein 2) were 5′-CAG AGA CTC GAG CAC AGC AC-3′ and 5′-GAT GAC CGG GGT TTA AAG GT-3′. The primers for BTG2 (for B-cell translocation gene 2) were 5′-CCT GGG CAG AGA GTG AAA AG-3′ and 5′-CCT TCC ATC CTA ACC CCA AT-3′.
ChIP analysis.
Chromatin immunoprecipitation (ChIP) assays were performed as described previously (5). For RA treatment in nonkinetic ChIP analyses, cells were incubated with 0.1 μM RA for 2 h before cross-linking. The antibodies included anti-H3K27me3 antibody (ab6002; Abcam), anti-H3K4me3 antibody (ab8580; Abcam), anti-H3 antibody (ab1791; Abcam), anti-UTX antibody (a gift from K. Helin), and anti-Flag antibody (F-3165; Sigma). The antibodies against ASC-2, MLL3, and RBBP5 were as described previously (5). The ChIP primers for the p21 RA response element (RARE) were 5′-TGT TTC AGG CAC AGA AAG GAG GCA-3′ and 5′-TCC GCT CCC ATC TAC CTC ACA C-3′. The primers for +1 region of p21 were 5′-GGT GTG AGC AGC TGC CGA AGT-3′ and 5′-TCG GTG CCT CGG CGA ATC C-3′. The primers for RAR-β2 were 5′-CTG CTG GGA GTT TTT AAG C-3′ and 5′-GGC AAA GAA TAG ACC CTC C-3′. The primers for +1 region of PDCD4 were 5′-CTG GTT CCC TCT TTC TGA GC-3′ and 5′-GCG AAT CTT CCT CAC AGG AT-3′.
Purification of the human ASCOM, SET1, and MLL1 complexes.
ASCOM was immunopurified from HeLa that stably express Flag-PA1 using anti-Flag M2 affinity resin and eluted in buffer containing 100 μg/ml of 3×FLAG peptide (Sigma) as described previously (26). Human SET1 and reconstituted MLL1 complexes were purified as described previously (26, 27).
Histone methyltransferase assay.
One microgram of H3(1–20) or H3(1–36) peptide was mixed with 10 to 20 ng of the human SET1 complex or 100 ng of reconstituted MLL1 complex in reaction buffer (10 mM HEPES, pH 7.8, 30 mM KCl, 2.5 mM dithiothreitol [DTT], 2.5 mM EDTA) along with 1 μCi of S-adenosyl-l-[methyl-3H] (3H-SAM) (GE Healthcare) and 50 μM cold SAM (BioLab). After incubation at 30 οC for 60 min, the samples were resolved in 15% SDS-PAGE followed by either autoradiography or immunoblotting. The Quantity One program (Bio-Rad) was used for quantitative analysis of H3K4 methylation levels.
Peptide pulldown assays.
C-terminal biotinylated histone H3 peptides were immobilized on Dynabeads MyOne Streptavidin C1 (Invitrogen) and subsequently incubated either with in vitro-translated mouse WDR5 or human RBBP5 or with a purified ASCOM or SET1 complex in binding buffer (150 mM NaCl, 50 mM Tris [pH 8.0], 1 mM DTT, 0.1% NP-40, protease inhibitors) for 30 min at 4°C in a rotator. Interacting proteins were pulled down with Dynabeads using a DynaMag-Spin magnet (Invitrogen) and washed four times with 1 ml of binding buffer and once with 1 ml of binding buffer containing 300 mM NaCl. Bound proteins were analyzed on 15% SDS-PAGE gels for Coomassie staining or immunoblotting with anti-Flag and other indicated antibodies.
Mononucleosome binding and methylation assays.
Mononucleosomes were assembled on a biotinylated 601 sequence fragment (197 bp) (28) by salt dialysis (29) with either an unmodified histone-containing octamer or an H3K27me3-containing octamer (29). The quality and quantity of mononucleosomes were determined by gel electrophoresis. Mononucleosomes were mixed with Streptavidin M280 beads (Invitrogen) in 10 mM HEPES-KOH (pH 7.9), 0.2 mM EDTA, 10% glycerol, 300 mM KCl, and 0.2% NP-40 buffer. After samples were washed, the immobilized mononucleosomes were incubated with the purified ASCOM complex for 3 h at 4οC in a rotator. The binding of ASCOM with mononucleosomes was analyzed by immunoblotting with WDR5 and RBBP5 antibodies (Bethyl Laboratories, Inc.). For the histone methyltransferase assays, 30-μl reaction mixtures contained mononucleosomes, SET1C, ASCOM or reconstituted MLL1 complex, and 50 μM SAM in reaction buffer (50 μM MgCl2, 10 mM HEPES [pH 7.8], 50 mM KCl, 2.5 mM DTT, 0.25 mM EDTA). After an overnight incubation at 30°C, reactions were terminated by addition of 10 μl of 5× SDS sample buffer and analyzed by immunoblotting.
MTT assays.
For in vitro cytotoxicity assays, HEK293 cells transfected with a short hairpin RNA (shRNA) vector for scrambled UTX or si-UTX were treated with an increasing concentration of RA for 48 h and subjected to a standard MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] assay (30).
Statistical analysis.
All values are presented as means ± standard errors of the means (SEM). Differences between groups were analyzed by a two-tailed Student's t test.
RESULTS
Identification of a novel SET1-like complex binding site in H3 that is sensitive to K27 trimethylation.
In searching for the mechanistic basis underlying the general anticorrelation between H3K4 and H3K27 trimethylation, we considered the possibility that the SET1-like complexes make an additional histone H3 contact that is blocked by a trimethyl mark at H3K27. In this regard, glutathione S-transferase (GST)–H3(1–57) and GST-H3(1–36) fusion proteins bound an independently expressed WDR5 with a much higher affinity than GST-H3(1–25) (Fig. 1A). Consistent with these results, an affinity-purified ASCOM, isolated from HEK293 cells that stably express Flag-tagged PA1, also showed stronger binding to GST-H3(1–57) and GST-H3(1–36) than to GST-H3(1–25) (Fig. 1A). Importantly, GST-H3(5–57), which is devoid of the R2 and K4 residues that constitute the reported interaction interface for WDR5 (11–14), still showed a strong interaction with WDR5 (Fig. 1B). Further deletional analysis revealed that these secondary interactions with WDR5 were still conserved with H3 residues 10 to 31 (Fig. 1B). Together, these results suggest that an additional, independent WDR5-interacting domain is contained in histone H3 residues 10 to 31. Consistent with the results of the GST pulldown assays, WDR5 showed much stronger binding to an unmodified H3(1–36) peptide than to an unmodified H3(1–20) peptide (Fig. 1C). As reported earlier (10), the H3(1–20) peptide interacted with WDR5 most strongly when K4 was dimethylated but failed to bind the “control” RBBP5 subunit that, like WDR5, contains WD40 repeats (Fig. 1C). Unexpectedly, however, RBBP5 did bind the H3(1–36) peptide, and this interaction was markedly enhanced by K4 trimethylation (Fig. 1C). Related to and consistent with these results, RBBP5 did not interact with GST-H3(1–25) but showed very weak interactions with GST-H3(1–36) and much stronger interactions with GST-H3(1–57) (Fig. 1A and B). Overall, these results demonstrate that H3 interacts with both WDR5 and RBBP5 not only through the previously reported WDR5 interface consisting of H3R2 and dimethylated H3K4 (11–14) but also through a second interaction domain present within the H3 tail region containing residues 10 to 31.
Fig 1.
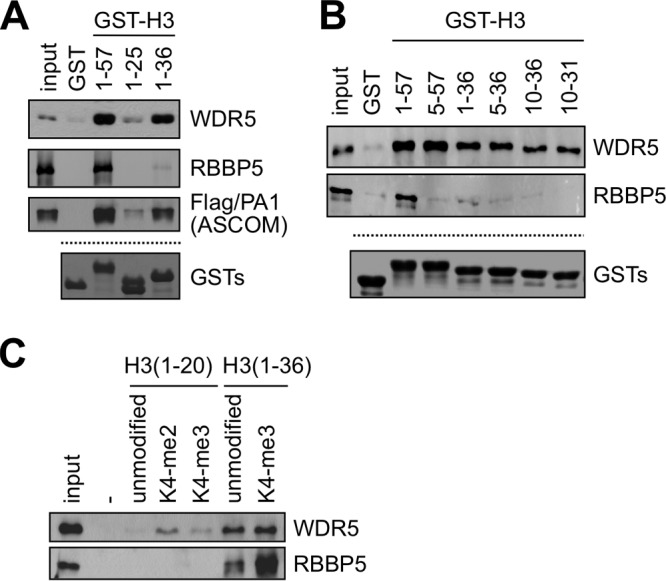
Histone H3 contains dual interaction interfaces for WDR5 and RBBP5. (A and B) GST pulldown assay with the indicated GST fusions and either in vitro translated Flag-tagged WDR5, Flag-RBBP5, or purified ASCOM complex with Flag-PA1. WDR5, RBBP5, and ASCOM were detected by immunoblotting with anti-Flag antibody. Coomassie blue staining of GST fusions is shown as a loading control. (C) Immobilized peptide pulldown assay using the indicated H3 peptides with either in vitro translated Flag-WDR5 or Flag-RBBP5. WDR5 and RBBP5 were detected by immunoblotting with anti-Flag antibody. All inputs are 5%. K4-me2, K4-dimethylated H3.
Next, we tested whether the newly identified WDR5- and RBBP5-interacting domain in H3 is sensitive to H3K27 trimethylation. GST-H3(1–57) readily bound the WDR5 and RBBP5 subunits of SET1-like complexes, as shown above, whereas it showed no interactions with either the isolated PA1 or the isolated ASH2L subunit (Fig. 2A). GST-H3(1–57) also interacted with the affinity-purified ASCOM from HEK293 cells (Fig. 2A). As an initial step to further analyze effects of H3 methylation on binding, we sought to mimic the bulky trimethylated state by introducing phenylalanine substitutions for R2, K4, and K27 into GST-H3(1–57) as described previously (31). The R2F mutation, which is thought to mimic the dimethylation of H3R2 by PRMT6 (15, 16), resulted in moderately decreased binding of GST-H3(1–57) to WDR5, RBBP5, and ASCOM (Fig. 2A). In contrast, the K4F mutation resulted in no significant changes in binding affinity. This is likely because the increase in the binding affinity of dimethylated H3K4 for WDR5 (10) and of trimethylated H3K4 for RBBP5 (Fig. 1C) cannot be recapitulated by the phenylalanine substitution. Strikingly, GST-H3(1–57)(K27F) was as inefficient as GST-H3(1–57)(R2F) in binding WDR5, RBBP5, and ASCOM (Fig. 2A). In addition, the strong binding of GST-H3(10–36) to WDR5 was abolished by the K27F mutation (Fig. 2B). Together, these results raised the possibility that H3K27 trimethylation inhibits the interactions of the second H3 domain that interacts with WDR5 and RBBP5. To directly test this, we used H3(1–36) peptides that were either unmodified or trimethylated at K4 or K27 or at both K4 and K27. K27 trimethylation of the H3(1–36) peptide significantly reduced its interaction with the isolated WDR5 and RBBP5 proteins and with the affinity-purified ASCOM (monitored by the presence of Flag-tagged PA1 and RBBP5) and SET1 (monitored by the presence of Flag-tagged CFP1, a SET1 complex-specific subunit, and RBBP5) complexes, whereas K4 trimethylation of the H3(1–36) peptide modestly enhanced these interactions (Fig. 2C). Interestingly, concomitant trimethylation at H3K4 and H3K27, which may mimic the bivalent chromatin in embryonic stem cells (17–19), also strongly blocked these interactions (Fig. 2C). To further test these interactions using more physiologically relevant substrates, we reconstituted mononucleosomes with either unmodified H3 or a semisynthetic K27-trimethylated H3. Interestingly, the ASCOM complex (which behaved similarly to the SET1 complex in binding to H3 peptides) clearly distinguished between these mononucleosomes, showing reduced binding (based on analysis of its WDR5 and RBBP5 subunits) to mononucleosomes containing trimethylated K27 (Fig. 2D). Similar results were also obtained with the SET1 complex (data not shown). Altogether, these results reveal that interactions of histone H3 with WDR5 and RBBP5 and with the parental MLL3/4 (ASCOM) and SET1 complexes are enhanced by H3K4 trimethylation but weakened by H3K27 trimethylation. As WDR5 and RBBP5 are common components of all SET1-like complexes, the MLL1 and MLL2 complexes are expected to respond similarly to H3K4 and H3K27 trimethylation.
Fig 2.
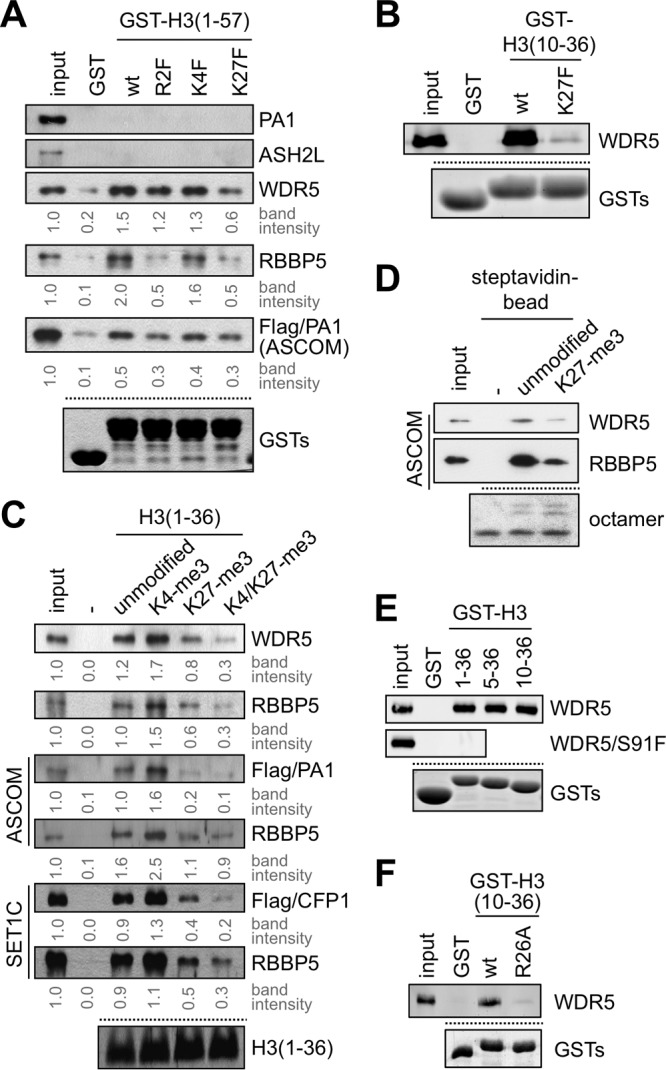
H3K27 trimethylation impairs the interaction of H3 and WDR5/RBBP5. (A, B, E, and F) GST pulldown assay with GST fusions to indicated H3 fragments and either in vitro translated Flag-PA1, Flag-ASH2L, Flag-WDR5, Flag-WDR5/S91F, or Flag-RBBP5 or purified ASCOM complex with Flag-tagged PA1. PA1, ASH2L, WDR5, RBBP5, and ASCOM were detected by immunoblotting with anti-Flag antibody. Coomassie blue staining of GST fusions is shown as a loading control. (C) Immobilized peptide pulldown assay using indicated H3 peptides either with in vitro translated Flag-WDR5 or Flag-RBBP5 or with purified ASCOM (via Flag-PA1 or RBBP5) or purified SET1 complex (via Flag-CFP1 or RBBP5). WDR5, RBBP5, PA1, and CFP1 were detected by immunoblotting with anti-Flag or RBBP5 antibody. Coomassie blue staining of H3 peptides is shown as a loading control. (D) Binding assays of ASCOM (monitored by anti-WDR5 and anti-RBBP5 immunoblotting) to immobilized reconstituted mononucleosomes containing either unmodified or K27 trimethylated H3. Coomassie blue staining of octamers in mononucleosomes is shown as a loading control. Similar results were also obtained with the SET1 complex (data not shown). Input in panel D is 10%, and all other inputs are 5%. wt, wild type.
Given that WDR5 has been shown to directly interact with the A1 (weakly) and R2 (strongly) residues of H3 (reviewed in references 32 and 33) and that H3K27 is also preceded by A25 and R26, we postulated that WDR5 might utilize a similar strategy to recognize both the H3R2 interface and the second interface located within H3 residues 10 to 31. In favor of this possibility, mutation of WDR5 S91 to phenylalanine, which has been shown to impair the interaction of WDR5 with H3R2 (14), also disrupted the interactions of WDR5 with GST fusions containing H3(1–36), H3(5–36), and H3(10–36) (Fig. 2E; also data not shown). Moreover, and importantly, the mutation of H3R26 to alanine disrupted the interaction of GST-H3(10–36) with WDR5 (Fig. 2F). Overall, these results suggest that the second WDR5 interaction domain located within H3 residues 10 to 31 is potentially similar to the WDR5 interaction interface in the extreme N terminus of H3 involving H3R2.
H3K27 trimethylation interferes with H3K4 trimethylation by SET1-like complexes.
The above results suggested that the anticorrelation between H3K4 and H3K27 trimethylation is established, at least in part, by an “active” mechanism in which the H3K27 trimethyl mark blocks the interaction between SET1-like complexes and histone H3, leading to inhibition of SET1-like complex-mediated H3K4 trimethylation. To test this, we monitored H3K4 methylation of the H3(1–36) peptide by the affinity-purified human SET1 complex. Interestingly, and consistent with the binding results, the K27 trimethylated H3(1–36) peptide was a much poorer substrate for the SET1 complex than the unmodified H3(1–36) peptide, especially at shorter incubation periods (Fig. 3A and B). Also consistent with the binding results shown in Fig. 1C, the H3(1–36) peptide was a much better substrate than the H3(1–20) peptide (Fig. 3A). Similar to what was observed for the SET1 complex, H3K27 trimethylation also inhibited H3K4 methylation of the H3(1–36) peptide by both a reconstituted MLL1 complex (Fig. 3C) and the affinity-purified ASCOM (data not shown). Interestingly, immunoblot analysis with mono-, di-, and trimethylated H3K4-specific antibodies revealed that H3K27 trimethylation severely impaired H3K4 trimethylation of the H3(1–36) peptide by the human SET1 complex, while it affected the levels of H3K4 dimethylation relatively less significantly (Fig. 3D). Similar results were observed when corresponding histone octamers, which are thought to be dissociated into H2A:H2B dimers and (H3:H4)2 tetramers under low-salt conditions, were employed as substrates (Fig. 3E). In contrast, and most importantly, H3K27 trimethylation resulted in both a dramatic reduction in H3K4 trimethylation and significant reductions in H3K4 monomethylation and dimethylation by the SET1 complex on the more physiological mononucleosome substrate (Fig. 3F). Similarly, when histone octamers and ASCOM were employed, we observed a dramatic reduction in both H3K4 monomethylation and trimethylation (Fig. 3G). Overall, our results demonstrate that the H3K27 trimethyl mark, through its effect on WDR5/RBBP5-mediated binding and function of SET1/MLL complexes, is a crucial player in achieving the anticorrelation between H3K4 and H3K27 trimethylation.
Fig 3.

H3K27 trimethylation attenuates H3K4 trimethylation. (A to C) Human SET1-complex- and MLL1 complex (MLL1 com)-dependent H3K4 methylation is inhibited by H3K27 trimethylation in vitro. H3K4 methylation assays with human SET1/MLL1 complexes on H3(1–20) and H3(1–36) peptides with or without the indicated trimethyl modifications at K4 or K27 are shown. (D) Immunoblot analysis of H3K4 methylation states upon incubation of the indicated H3(1–36) peptides with the human SET1 complex. (E to G) H3K4 methylation of unmodified and H3K27 trimethylated histone octamers (E and G) and mononucleosomes (F) by the human SET1 complex (E and F) or ASCOM (G). H3K4me1, H3 monomethylated at K4; α, anti.
UTX is required for H3K4 trimethylation by ASCOM.
Our results suggest that removal of the H3K27 trimethyl mark is a prerequisite for efficient H3K4 trimethylation by SET1-like complexes. This leads to the prediction that H3K27 demethylases cooperate with SET1-like complexes to activate transcription of target genes by triggering the formation of transcriptionally active chromatin marked by H3K4 trimethylation. It is notable that whereas other SET1-like complexes have not been found to contain any H3K27 demethylases as integral subunits, with the identity of the H3K27 demethylases required for their functions being unknown, ASCOM does contain the H3K27 demethylase UTX as an integral subunit (7, 8). Thus, we examined the role of UTX in transactivation of five direct RAR target genes that included p21, RAR-β2, PDCD4 (for programmed cell death 4), IGFBP2 (for insulin-like growth factor binding protein 2), and BTG2 (for B-cell translocation gene 2) (5, 34–36). Notably, the knockdown of UTX by si-UTX RNA (24) profoundly compromised RA-mediated upregulation of expression of all five genes (Fig. 4A to E). Expression of these genes was rescued by reexpression of an si-UTX-resistant, wild-type UTX but not by an enzymatically inactive mutant form (UTX-H1146A) of si-UTX-resistant UTX (Fig. 4A to E). These results suggest that the H3K27 demethylase activity of UTX is required for the transcriptional activation of RAR target genes by ASCOM. In further support of the physiological functionality of our findings, we assessed the antiproliferative activity of RA in the presence of si-UTX, and in MTT assays RA-dependent antiproliferation of HEK293 cells was significantly impaired in the presence of si-UTX (Fig. 5).
Fig 4.

The catalytic activity of UTX is required for RA-induced transactivation. qRT-PCR analysis of RA-dependent transcription of p21 (A), RAR-β2 (B), PDCD4 (C), IGFBP2 (D), and BTG2 (E) in HEK293 cells transfected with scrambled siRNA (NC) or siRNA against UTX (si-UTX) and either wild-type (UTX-wt) or catalytically inactive (UTX-m) forms of si-UTX-resistant UTX. *, P < 0.05; ***, P < 0.001.
Fig 5.
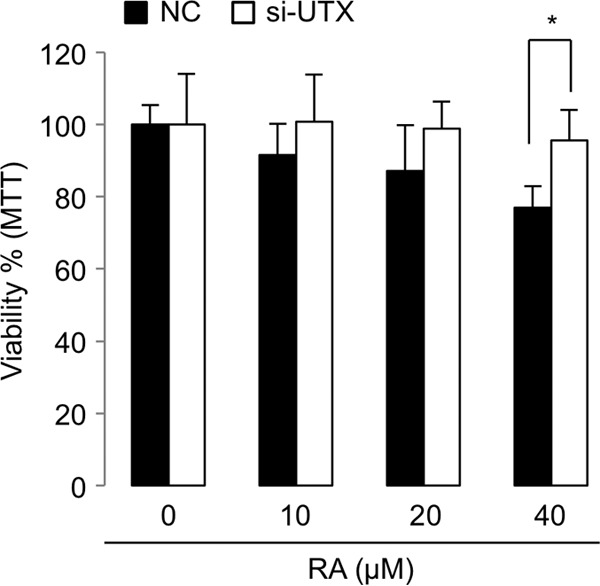
MTT assays with HEK293 cells. Treatment of cells with an increasing amount of RA for 40 h resulted in a reduction in cell proliferation, but knockdown of UTX by si-UTX interfered with this antiproliferative action of RA. *, P < 0.05.
To monitor a possible effect of UTX loss on the assembly of ASCOM per se, ASCOM was immunopurified from HEK293 cells that stably express Flag-PA1 using an anti-Flag antibody. The formation of ASCOM, as determined by coimmunoprecipitation of ASC-2 and RBBP5 with Flag-PA1, was not affected by downregulation of UTX (Fig. 6A). These results suggest that UTX is dispensable for the assembly of ASCOM. In a further analysis of the basis of the UTX requirement, RA treatment resulted in a significant reduction of H3K27 trimethylation and a strong induction of H3K4 trimethylation on both the RARE and the transcription start site (+1) regions of the p21 gene in cells expressing a scrambled control siRNA (Fig. 6B and C). Intriguingly, the knockdown of UTX increased the basal level of H3K27 trimethylation on the +1 and the RARE regions of the p21 gene and attenuated the RA-dependent suppression of H3K27 trimethylation (Fig. 6B). Moreover, the UTX knockdown markedly blunted the induction of H3K4 trimethylation by RA on both the +1 and RARE regions of the p21 gene (Fig. 6C). These results suggest that UTX is required not only for removal of H3K27 trimethyl marks but also for efficient H3K4 trimethylation by ASCOM and raise the interesting possibility that the UTX-mediated H3K27 demethylation is needed for the stable recruitment of ASCOM to its target genes. In support of this idea, the knockdown of UTX markedly reduced the RA-dependent recruitment of the RBBP5 component of ASCOM to the +1 region of the p21 gene (Fig. 6D), indicating that UTX is important for recruiting ASCOM to the p21 initiation site. Control (rescue) experiments with wild-type UTX and the catalytically inactive UTX-H1146A mutant (23) showed, first, that both exhibited robust intracellular associations with RBBP5 (Fig. 6E) indicative of incorporation into ASCOM and, second, that wild-type UTX restored efficient recruitment of RBBP5 to the +1 region of p21 upon RA treatment while mutant UTX-H1146A was not as effective as wild-type UTX (Fig. 6F). Similar results were also obtained with the +1 regions of two RA target genes, RAR-β2 and PDCD4 (Fig. 7). Interestingly, the knockdown of UTX did not affect recruitment of RBBP5 to the p21 RARE region (Fig. 6D), consistent with our results showing that UTX is dispensable for the assembly of ASCOM (Fig. 6A) and, especially, that ASCOM (and UTX) is recruited to RAR target genes via interactions between ASC-2 and RAR (5). Indeed, UTX, together with other subunits of ASCOM, was recruited to p21, RAR-β2, and PDCD4 in an RA-dependent manner in HEK293 cells (Figs. 6, 7, and 8). Together, these results clearly demonstrate that the UTX H3K27 demethylase activity (i.e., removal of H3K27 trimethyl marks) is required for RAR-mediated transactivation by facilitating recruitment of ASCOM to the +1 region of RAR target genes upon RA treatment.
Fig 6.

Removal of H3K27 trimethylation is required for RA-induced transactivation. (A) Coimmunoprecipitation assays with the indicated antibodies using HEK293 cells transfected with scrambled siRNA (NC) or si-UTX. (B to D) ChIP showing levels of H3K27 trimethylation (B), H3K4 trimethylation (C), and RBBP5 occupancy (D) of the transcription start site (+1) and RARE regions of p21 in HEK293 cells transfected with scrambled siRNA (NC) or si-UTX. (E) Coimmunoprecipitation assays with the indicated antibodies using HEK293/Flag-PA1 cells transfected with si-UTX and either the wild-type (UTX-wt) or catalytically inactive (UTX-m) form of si-UTX-resistant UTX. (F) ChIP showing that RA-dependent recruitment of RBBP5 to the +1 region of p21 in HEK293 cells transfected with si-UTX is restored more readily by the wild-type UTX (UTX-wt) form than by the catalytically inactive (UTX-m) form of si-UTX-resistant UTX. In contrast, both forms of UTX were readily recruited to the +1 region of p21. WB, Western blotting. *, P < 0.05; **, P < 0.01.
Fig 7.

UTX is also required for the transition between an H3K27me3-repressed state and an H3K4me3-activated state of additional RAR target genes, RAR-β2 (A to D) and PDCD4 (E to H). ChIP analysis was performed for levels of H3K27 (A and E) and H3K4 trimethylation (B and F) and for RBBP5 occupancy (C and G) using HEK293/Flag-PA1 cells transfected with scrambled siRNA (NC) or si-UTX. (D and H) ChIP analyses showing levels of RBBP5 occupancy of RAR-β2 (D) and PDCD4 (H) in HEK293 cells transfected with si-UTX and either wild-type (UTX-wt) or catalytically inactive (UTX-m) forms of si-UTX-resistant UTX. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Fig 8.

Removal of H3K27 trimethyl marks precedes H3K4 trimethylation. (A) ChIP showing RA-dependent recruitment of UTX along with other subunits of ASCOM-MLL3/MLL4 to the RAR-β2 promoter in HEK293 cells that stably express Flag-tagged PA1 (HEK293/Flag-PA1 cells). (B to H) ChIP showing recruitment of ASC-2 (B), RBBP5 (C), MLL4 (D), MLL3 (E), and UTX (F) as well as H3K27 trimethylation (G) and H3K4 trimethylation (H) levels at the transcription start site (+1) and RARE regions of p21 in HEK293 cells following RA treatment for 0, 0.5, 1, 2, and 4 h. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
H3K27 demethylation precedes H3K4 trimethylation by ASCOM.
To further test our model that H3K27 demethylation is a prerequisite for H3K4 trimethylation by SET1-like complexes, we focused on the p21 gene. In contrast to the RAR-β2 gene in which the RARE is located only 93 bp upstream of the transcription start site, the p21 gene contains an RARE that is well separated (located 1,186 bp upstream) from the transcription start site. This has allowed us to independently monitor (by ChIP) the initial RA-induced recruitment of ASCOM to the RARE and the subsequent roles of ASCOM in modifying nucleosomes situated closer to the +1 region. Our results revealed the following: (i) that ASC-2, MLL3/4, UTX, and RBBP5 are enriched in the RARE region by 30 min post-RA treatment (Fig. 8B to F); (ii) that the induced level of ASC-2 at the RARE region decreases after 30 min and, interestingly, that it is not enriched in the +1 region (Fig. 8B), indicating that ASC-2 is needed only for initial recruitment of ASCOM to the RARE, likely via the previously defined RA-dependent interactions between ASC-2 and RAR (5); (iii) that MLL3/4, UTX, and RBBP5 show significant enrichment at the +1 region only after 1 to 2 h of RA treatment (Fig. 8C to F), thus lagging by at least 30 min their recruitment to the RARE region and suggesting that initial recruitment of ASCOM to the RARE region is followed by subsequent enrichment of ASCOM at the +1 region; (iv) that trimethylated H3K27 levels at the RARE region begin to decrease at 30 min after RA treatment, whereas trimethylated H3K27 levels at the +1 region decrease only later at 1 h after RA treatment (Fig. 8G); and (v) that, in contrast, trimethylated H3K4 levels at the RARE region begin to increase 1 h after RA treatment, whereas trimethylated H3K4 levels at the +1 region begin to increase 2 h after RA treatment (Fig. 8H). Taken together, these results strongly suggest that H3K27 demethylation by UTX precedes H3K4 trimethylation by ASCOM at both the RARE and +1 regions of the p21 gene and that these changes occur first in the RARE region and subsequently in the +1 region. Of note, an earlier analysis of RA-induced Hox genes indicated that UTX recruitment and the resulting H3K27 demethylation were concomitant with H3K4 methylation (37). However, the use in that study of much longer time points (0, 18, and 24 h) than those used here (0, 0.5, 1, 2, and 4 h) precluded a clear determination, in contrast to the current analysis, of the exact order of ASCOM-UTX recruitment, H3K27 demethylation, and H3K4 methylation events.
DISCUSSION
Our study has identified a novel SET1-like complex-interacting domain in the H3 tail that involves both WDR5 and RBBP5 subunits. This strong interaction domain within H3 residues 10 to 31 has remained undetected to date, most likely because all of the previous studies employed shorter H3 peptides that lack this region. Although the precise nature of this new interface awaits further investigation, our results indicate that it involves H3R26 and is disrupted by H3K27 trimethylation and thereby may play a critical role in establishing the anticorrelation between H3K4 and H3K27 trimethylation (Fig. 9). This interface might serve as an important interaction surface that facilitates binding of SET1-like complexes to H3, at least prior to the H3K4 dimethylation that further stabilizes the interaction between WDR5 and H3R2 (10–14) (Fig. 1C). In this regard, our findings have significant implications for the interpretation of recent results indicating the presence in MLL/SET1 polypeptides of an H3R2-like WDR5 interaction (WIN) motif that interacts with WDR5 in a manner competitive with H3K4 monomethylated and dimethylated H3 (38–40). Thus, the second H3-WDR5/RBBP5 interface may facilitate H3–SET1-like complex interactions that allow more flexibility in the potentially dynamic interactions of WDR5 with MLL/SET1.
Fig 9.

Model for the interplay between H3K4 and H3K27 trimethylation. WDR5/RBBP5 subcomplex binds to either R2 or R26, and the R26 binding is inhibited by methylation of adjacent K27. It remains to be determined whether R2 and R26 are selectively or randomly recognized by WDR5 and RBBP5. It is also unclear whether the WDR5/RBBP5 complex recognizes R2 and R26 on the same H3 tail or on two different H3 tails. It also remains to be tested whether H3K4 trimethylation occurs in cis (i.e., on the same histone H3 molecule that interacts with WDR5/RBBP5 through H3R26) or in trans (on the other H3 molecule that lacks the interaction of WDR5/RBBP5 with H3R26). Recruitment of SET1-like complexes occurs through interactions between specific subunits of SET1-like complexes and their target transcription factors (step i). For ASCOM-MLL3/MLL4, this provides UTX and results in removal of H3K27 trimethyl marks, while JMJD3 or an unidentified H3K27 demethylase may work for other SET1-like complexes (step ii). The loss of H3K27 trimethyl marks then allows H3K4 trimethylation, which further stabilizes the interaction between SET1-like complexes and their target genes and may also provide a means to propagate SET1-like complexes and H3K4 methylation through adjacent regions of activated genes (step iii).
The H3K27 methylation-sensitive nature of this second interface, along with the stabilizing effect of H3K4 dimethylation on H3R2-WDR5 interactions (10–14), provides critical new insights into the molecular basis of recruitment of SET1-like complexes to their target genes. We along with others have shown that the initial tethering of SET1-like complexes to their target genes involves direct interactions between subunits of SET1-like complexes and transcription factors (3, 5, 41, 42), which are often signal dependent. This is exemplified by the RA-induced recruitment of ASCOM-MLL3 and ASCOM-MLL4 to RAREs through the RA-dependent interaction between RAR and ASC-2 (5) (Fig. 9, step i). Importantly, this initial recruitment event brings the H3K27 demethylase UTX to RAREs. The removal of the H3K27 trimethyl mark by UTX may in turn elicit strong interactions between ASCOM and the newly defined WDR5- and RBBP5-interacting interface in H3 (Fig. 9, step ii), which subsequently leads to efficient H3K4 trimethylation and transcriptional activation of the RAR target genes (Fig. 9, step iii).
Interestingly, on H3 peptide and histone octamer substrates, SET1 complex-mediated H3K4 monomethylation and dimethylation are less affected than H3K4 trimethylation by the H3K27 trimethyl mark (Fig. 3D and E). Although the molecular basis for this is not understood, it may simply be that trimethylation uniquely requires stronger (stabilizing) interactions, mediated by the newly defined interface, between the H3 tail and the catalytic core of SET1-like complexes. Thus, it is possible that the initial tethering of ASCOM to its target genes through transcription factors (e.g., RAR for the RAR target genes) could be sufficient for ASCOM to carry out H3K4 monomethylation and dimethylation, while further stabilization through interactions of WDR5 and RBBP5 with dimethylated H3K4 (10) and the second interface might be required for H3K4 trimethylation (Fig. 9). On the other hand, we note that these considerations apply mainly to effects of H3K27 trimethyl marks on H3K4 methylation of nonphysiological (nonnucleosomal) histone substrates and that with nucleosomal substrates there are pronounced effects of the H3K27 trimethyl mark not only on H3K4 trimethylation but also on monomethylation and dimethylation (Fig. 3F). Thus, the combined H3 peptide and mononucleosome substrate analyses indicate that additional constraints to H3K4 methylation are imposed by the nucleosomal structure and thereby elicit requirements for the second H3-WDR5/SET1C interaction not only for H3K4 trimethylation but also for efficient monomethylation and dimethylation. These results also have important implications for current studies, mostly using short peptide substrates, on the mechanism of action of H3K4 methyltransferases (reviewed in references 32 and 33). Interestingly, even with an octamer substrate, both H3K4 monomethylation and trimethylation by ASCOM are affected by H3K27me3 (Fig. 3G).
WDR5 likely utilizes a similar mode of interaction toward H3R2 and H3R26 (and possibly toward H3R8 as well). First, all three arginine residues are preceded by alanine, similar to the AR-containing WIN motif in MLL/SET proteins (38–40). Second, mutation of WDR5 S91 to phenylalanine disrupts the interaction of WDR5 not only with H3R2 (14) but also with the newly defined, second WDR5 interaction domain in H3 (Fig. 2E). The second interface is also disrupted by the mutation of H3R26 to alanine (Fig. 2F). Third, similar to demethylation of H3R2 (15, 16), demethylation of H3K9 (43) and H3K27 (this study) facilitates H3K4 trimethylation. However, it remains to be determined whether WDR5 and RBBP5 bind nonselectively to either H3R2 or H3R26 or whether they preferentially bind one site. We favor the latter possibility, given our in vitro results showing that RBBP5 may require additional H3 residues between amino acids 36 and 57 for stronger interactions (Fig. 1A and B). However, it is also possible that the weak interaction of RBBP5 with GST-H3(1-36) relative to the strong interaction with GST-H3(1-57) results from an arbitrary structural impact of fusion. Thus, the GST fusion in GST-H3(1-36) may interfere with the second WDR5/RBBP5 interaction domain in H3, restricting its interaction with RBBP5, while this interference is structurally relieved in GST-H3(1-57). This notion is supported by the strong interaction of H3(1-36) peptide with RBBP5 (Fig. 1C). Intriguingly, the newly described interactions of RBBP5 with the H3(1-36) peptide were significantly enhanced by the H3K4 trimethyl group (Fig. 1C). However, it remains to be determined whether RBBP5, like WDR5 (10), binds the H3K4 dimethylated H3 tail more strongly than the H3K4 trimethylated H3 tail. It is also possible that the stabilization of ASCOM-H3 interactions through di- or trimethylated H3K4 facilitates propagation of H3K4 trimethylation to downstream regions of genes (e.g., by elongating SET1-like complexes) or transmission of the identical H3K4 methylation pattern to the cognate H3 locus on the opposite DNA strand during DNA replication. Future studies should also delineate whether H3K4 trimethylation occurs in cis (i.e., on the same histone H3 molecule that interacts with WDR5/RBBP5 through H3R26) or in trans (on the other H3 molecule that lacks the interaction of WDR5/RBBP5 with H3R26), as recently described (20, 21, 44–47). Importantly, our results are compatible with both possibilities (Fig. 9).
Because H3K4 trimethylation is a hallmark of the transcriptional activation function of SET1-like complexes (1, 2), our results suggest that removal of H3K27 trimethyl marks is a critical step for optimal H3K4 trimethylation and transcriptional activation by SET1-like complexes in vivo. Our demonstration of the effects of UTX loss on RAR target gene induction in HEK293 cells clearly supports this notion. Thus, the knockdown of UTX disrupts the transition between the two H3 methylation states on RAR target genes by preventing not only the efficient removal of the H3K27me3 mark but also the deposition of the H3K4me3 mark (Fig. 6 and 7). As a consequence, RA-dependent induction of RAR target genes is significantly impaired (Fig. 4). Importantly, removal of the H3K27me3 marks could be a general feature that precedes the stable recruitment of all SET1-like complexes to their target genes and subsequent H3K4 trimethylation. Therefore, it is possible that while UTX functions as an intrinsic partner of ASCOM-MLL3 and ASCOM-MLL4, the JMJD3 H3K27 demethylase (6) or other yet-to-be revealed H3K27 demethylases are required for the H3K4 trimethylation activity of the SET1A/B-, MLL1-, or MLL2-containing SET1-like complexes that lack UTX. In support of this idea, JMJD3 was shown to be associated with the core subcomplex of SET1-like complexes (48).
Our demonstration of an inhibitory role of H3K27 trimethylation on H3K4 trimethylation by SET1-like complexes is also relevant to the formation and/or maintenance of the bivalent chromatin domains in embryonic stem cells (17–21). The lack of H3K4me3 and H3K27me3 on individual histones in HeLa cells (49) and the inhibition by dual H3K4me3 marks of nucleosomal H3K27 methylation by PRC2 (22) had suggested that H3K27me3 and H3K4me3 may not coexist on individual nucleosomes. However, a recent elegant study has documented the joint presence of asymmetrically positioned (different H3 tails) H3K4me3 and H3K27me3 marks on individual nucleosomes (20). Using reconstituted nucleosomes, this study further showed that nucleosomes with one H3K4me3 mark, but not nucleosomes with two H3K4me3 marks, could be H3K27 methylated by PRC2. Whereas these studies (20, 22) indicated cis inhibition of H3K37 methylation by H3K4me3, our results indicate reciprocal cis inhibition of H4K3 methylation by H3K27me3 as well as an underlying mechanism involving H3K27me3-mediated inhibition of H3-WDR5/RBBP5 interactions and corresponding SET1-like complex binding (Fig. 9). Although the regulatory factors leading to the generation of the bivalent marks are less clear (reviewed in reference 21), these complementary cis-inhibitory mechanisms may thus contribute to the formation and/or maintenance of the asymmetric bivalent H3K4-H3K27 methylation state by restricting symmetric H3K4-H3K27 methylation events. However, if SET1-like complexes are able to methylate nucleosomes with a single H3K27me3 mark, just as PRC2 can methylate nucleosomes with a single H3K4me3 mark (20), these fundamental reactions can then be regulated by other factors that may act primarily at the level of recruitment (21).
Interestingly, the H3K9 demethylase JMJD2B recently was reported to be associated with the MLL3/4 complex, at least in MCF7 cells, and H3K9 demethylation has also been shown to be a prerequisite for H3K4 methylation (43). Therefore, MLL3/4 complexes appear to be capable of containing or associating with two distinct demethylases, UTX and JMJD2B, although it remains to be determined if this is a general feature in different cell types. In addition, H3K4 methylation has been shown to stimulate H3K27 demethylase activity through binding to the plant homeodomain (PHD) of JHDM1D (50). Overall, these results suggest a highly dynamic interplay among H3K4, H3K9, and H3K27 methylation events.
In summary, we have shown that interactions between H3 and SET1-like complexes involve an additional interface that is directly inhibited by H3K27 trimethylation, providing a critical molecular basis for the anticorrelation between H3K4 and H3K27 trimethylation. Moreover, this second interface and the previously described interface involving H3R2 and dimethylated H3K4 (10–14) appear to be coregulated through a dynamic interplay involving methylation of both H3K4 and H3K27, which sets the stage for future investigations.
ACKNOWLEDGMENTS
We thank Kristian Helin, Kai Ge, and Jaehoon Kim for reagents.
This work was supported by a March of Dimes grant (to S.-K.L.), National Institutes of Health grants DK064678 (to J.W.L.), NS054941 (to S.-K.L.), DK071900 (to R.G.R.), and CA148354 (to T.W.M. and R.G.R.), and grants from Basic Science Research Program (2012R1A1A1001749) and Bio and Medical Technology Development Program (2012M3A9C6050508) of the National Research Foundation funded by the Republic of South Korea (MEST), National R&D Program for Cancer Control, Ministry of Health and Welfare, Republic of Korea (1220120) (to S.L.), and the World Class University program through the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology (R31-10105) (to J.W.L.).
Footnotes
Published ahead of print 14 October 2013
REFERENCES
- 1. Shilatifard A. 2006. Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annu. Rev. Biochem. 75:243–269 [DOI] [PubMed] [Google Scholar]
- 2. Shilatifard A. 2008. Molecular implementation and physiological roles for histone H3 lysine 4 (H3K4) methylation. Curr. Opin. Cell Biol. 20:341–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lee S, Roeder RG, Lee JW. 2009. Roles of histone H3-lysine 4 methyltransferase complexes, in NR-mediated gene transcription. Prog. Mol. Biol. Transl. Sci. 87:343–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Goo YH, Sohn YC, Kim DH, Kim SW, Kang MJ, Jung DJ, Kwak E, Barlev NA, Berger SL, Chow VT, Roeder RG, Azorsa DO, Meltzer PS, Suh PG, Song EJ, Lee KJ, Lee YC, Lee JW. 2003. Activating signal cointegrator 2 belongs to a novel steady-state complex that contains a subset of trithorax group proteins. Mol. Cell. Biol. 23:140–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lee S, Lee DK, Dou Y, Lee J, Lee B, Kwak E, Kong YY, Lee SK, Roeder RG, Lee JW. 2006. Coactivator as a target gene specificity determinant for histone H3 lysine 4 methyltransferases. Proc. Natl. Acad. Sci. U. S. A. 103:15392–15397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Swigut T, Wysocka J. 2007. H3K27 demethylases, at long last. Cell 131:29–32 [DOI] [PubMed] [Google Scholar]
- 7. Cho YW, Hong T, Hong S, Guo H, Yu H, Kim D, Guszczynski T, Dressler GR, Copeland TD, Kalkum M, Ge K. 2007. PTIP associates with MLL3- and MLL4-containing histone H3 lysine 4 methyltransferase complex. J. Biol. Chem. 282:20395–20406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Issaeva I, Zonis Y, Rozovskaia T, Orlovsky K, Croce CM, Nakamura T, Mazo A, Eisenbach L, Canaani E. 2007. Knockdown of ALR (MLL2) reveals ALR target genes and leads to alterations in cell adhesion and growth. Mol. Cell. Biol. 27:1889–1903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lee J, Kim DH, Lee S, Yang QH, Lee DK, Lee SK, Roeder RG, Lee JW. 2009. A tumor suppressive coactivator complex of p53 containing ASC-2 and histone H3-lysine-4 methyltransferase MLL3 or its paralogue MLL4. Proc. Natl. Acad. Sci. U. S. A. 106:8513–8518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wysocka J, Swigut T, Milne TA, Dou Y, Zhang X, Burlingame AL, Roeder RG, Brivanlou AH, Allis CD. 2005. WDR5 associates with histone H3 methylated at K4 and is essential for H3 K4 methylation and vertebrate development. Cell 121:859–872 [DOI] [PubMed] [Google Scholar]
- 11. Schuetz A, Allali-Hassani A, Martín F, Loppnau P, Vedadi M, Bochkarev A, Plotnikov AN, Arrowsmith CH, Min J. 2006. Structural basis for molecular recognition and presentation of histone H3 by WDR5. EMBO J. 25:4245–4252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Couture JF, Collazo E, Trievel RC. 2006. Molecular recognition of histone H3 by the WD40 protein WDR5. Nat. Struct. Mol. Biol. 13:698–703 [DOI] [PubMed] [Google Scholar]
- 13. Ruthenburg AJ, Wang W, Graybosch DM, Li H, Allis CD, Patel DJ, Verdine GL. 2006. Histone H3 recognition and presentation by the WDR5 module of the MLL1 complex. Nat. Struct. Mol. Biol. 13:704–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Han Z, Guo L, Wang H, Shen Y, Deng XW, Chai J. 2006. Structural basis for the specific recognition of methylated histone H3 lysine 4 by the WD-40 protein WDR5. Mol. Cell 22:137–144 [DOI] [PubMed] [Google Scholar]
- 15. Hyllus D, Stein C, Schnabel K, Schiltz E, Imhof A, Dou Y, Hsieh J, Bauer UM. 2007. PRMT6-mediated methylation of R2 in histone H3 antagonizes H3 K4 trimethylation. Genes Dev. 21:3369–3380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guccione E, Bassi C, Casadio F, Martinato F, Cesaroni M, Schuchlautz H, Lüscher B, Amati B. 2007. Methylation of histone H3R2 by PRMT6 and H3K4 by an MLL complex are mutually exclusive. Nature 449:933–937 [DOI] [PubMed] [Google Scholar]
- 17. Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, Jaenisch R, Wagschal A, Feil R, Schreiber SL, Lander ES. 2006. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125:315–326 [DOI] [PubMed] [Google Scholar]
- 18. Pan G, Tian S, Nie J, Yang C, Ruotti V, Wei H, Jonsdottir GA, Stewart R, Thomson JA. 2007. Whole-genome analysis of histone H3 lysine 4 and lysine 27 methylation in human embryonic stem cells. Cell Stem Cell 1:299–312 [DOI] [PubMed] [Google Scholar]
- 19. Zhao XD, Han X, Chew JL, Liu J, Chiu KP, Choo A, Orlov YL, Sung WK, Shahab A, Kuznetsov VA, Bourque G, Oh S, Ruan Y, Ng HH, Wei CL. 2007. Whole-genome mapping of histone H3 Lys4 and 27 trimethylations reveals distinct genomic compartments in human embryonic stem cells. Cell Stem Cell 1:286–298 [DOI] [PubMed] [Google Scholar]
- 20. Voigt P, LeRoy G, Drury WJ, III, Zee BM, Son J, Beck DB, Young NL, Garcia BA, Reinberg D. 2012. Asymmetrically modified nucleosomes. Cell 151:181–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Voigt P, Tee WW, Reinberg D. 2013. A double take on bivalent promoters. Genes Dev. 27:1318–1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schmitges FW, Prusty AB, Faty M, Stützer A, Lingaraju GM, Aiwazian J, Sack R, Hess D, Li L, Zhou S, Bunker RD, Wirth U, Bouwmeester T, Bauer A, Ly-Hartig N, Zhao K, Chan H, Gu J, Gut H, Fischle W, Müller J, Thomä NH. 2011. Histone methylation by PRC2 is inhibited by active chromatin marks. Mol. Cell 42:330–341 [DOI] [PubMed] [Google Scholar]
- 23. Hong S, Cho YW, Yu LR, Yu H, Veenstra TD, Ge K. 2007. Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc. Natl. Acad. Sci. U. S. A. 104:18439–18444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Agger K, Cloos PA, Christensen J, Pasini D, Rose S, Rappsilber J, Issaeva I, Canaani E, Salcini AE, Helin K. 2007. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 449:731–734 [DOI] [PubMed] [Google Scholar]
- 25. Muir TW. 2003. Semisynthesis of proteins by expressed protein ligation. Annu. Rev. Biochem. 72:249–289 [DOI] [PubMed] [Google Scholar]
- 26. Tang Z, Chen WY, Shimada M, Nguyen UT, Kim J, Sun XJ, Sengoku T, McGinty RK, Fernandez JP, Muir TW, Roeder RG. 2013. SET1 and p300 act synergistically, through coupled histone modifications, in transcriptional activation by p53. Cell 154:297–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dou Y, Milne TA, Ruthenburg AJ, Lee S, Lee JW, Verdine GL, Allis CD, Roeder RG. 2006. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat. Struct. Mol. Biol. 13:713–719 [DOI] [PubMed] [Google Scholar]
- 28. Lowary PT, Widom J. 1998. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 276:19–42 [DOI] [PubMed] [Google Scholar]
- 29. Simon MD, Chu F, Racki LR, de la Cruz CC, Burlingame AL, Panning B, Narlikar GJ, Shokat KM. 2007. The site-specific installation of methyl-lysine analogs into recombinant histones. Cell 128:1003–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Plumb JA, Milroy R, Kaye SB. 1989. Effects of the pH dependence of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide-formazan absorption on chemosensitivity determined by a novel tetrazolium-based assay. Cancer Res. 49:4435–4440 [PubMed] [Google Scholar]
- 31. Huq MD, Tsai NP, Khan SA, Wei LN. 2007. Lysine trimethylation of retinoic acid receptor-alpha: a novel means to regulate receptor function. Mol. Cell. Proteomics 6:677–688 [DOI] [PubMed] [Google Scholar]
- 32. Cosgrove MS, Patel A. 2010. Mixed lineage leukemia: a structure-function perspective of the MLL1 protein. FEBS J. 277:1832–1842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Trievel RC, Shilatifard A. 2009. WDR5, a complexed protein. Nat. Struct. Mol. Biol. 16:678–680 [DOI] [PubMed] [Google Scholar]
- 34. Afonja O, Juste D, Das S, Matsuhashi S, Samuels HH. 2004. Induction of PDCD4 tumor suppressor gene expression by RAR agonists, antiestrogen and HER-2/neu antagonist in breast cancer cells. Evidence for a role in apoptosis. Oncogene 23:8135–8145 [DOI] [PubMed] [Google Scholar]
- 35. Passeri D, Marcucci A, Rizzo G, Billi M, Panigada M, Leonardi L, Tirone F, Grignani F. 2006. Btg2 enhances retinoic acid-induced differentiation by modulating histone H4 methylation and acetylation. Mol. Cell. Biol. 26:5023–5032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schmid C, Schläpfer I, Waldvogel M, Meier PJ, Schwander J, Böni-Schnetzler M, Zapf J, Froesch ER. 1992. Differential regulation of insulin-like growth factor binding protein (IGFBP)-2 mRNA in liver and bone cells by insulin and retinoic acid in vitro. FEBS Lett. 303:205–209 [DOI] [PubMed] [Google Scholar]
- 37. Lee MG, Villa R, Trojer P, Norman J, Yan KP, Reinberg D, Di Croce L, Shiekhattar R. 2007. Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science 318:447–450 [DOI] [PubMed] [Google Scholar]
- 38. Patel A, Dharmarajan V, Cosgrove MS. 2008. Structure of WDR5 bound to mixed lineage leukemia protein-1 peptide. J. Biol. Chem. 283:32158–32161 [DOI] [PubMed] [Google Scholar]
- 39. Patel A, Vought VE, Dharmarajan V, Cosgrove MS. 2008. A conserved arginine-containing motif crucial for the assembly and enzymatic activity of the mixed lineage leukemia protein-1 core complex. J. Biol. Chem. 283:32162–32175 [DOI] [PubMed] [Google Scholar]
- 40. Song JJ, Kingston RE. 2008. WDR5 interacts with mixed lineage leukemia (MLL) protein via the histone H3-binding pocket. J. Biol. Chem. 283:35258–35264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Patel SR, Kim D, Levitan I, Dressler GR. 2007. The BRCT-domain containing protein PTIP links PAX2 to a histone H3, lysine 4 methyltransferase complex. Dev. Cell 13:580–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tyagi S, Chabes AL, Wysocka J, Herr W. 2007. E2F activation of S phase promoters via association with HCF-1 and the MLL family of histone H3K4 methyltransferases. Mol. Cell 27:107–119 [DOI] [PubMed] [Google Scholar]
- 43. Shi L, Sun L, Li Q, Liang J, Yu W, Yi X, Yang X, Li Y, Han X, Zhang Y, Xuan C, Yao Z, Shang Y. 2011. Histone demethylase JMJD2B coordinates H3K4/H3K9 methylation and promotes hormonally responsive breast carcinogenesis. Proc. Natl. Acad. Sci. U. S. A. 108:7541–7546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Garcia BA, Pesavento JJ, Mizzen CA, Kelleher NL. 2007. Pervasive combinatorial modification of histone H3 in human cells. Nat. Methods 4:487–489 [DOI] [PubMed] [Google Scholar]
- 45. Taverna SD, Ueberheide BM, Liu Y, Tackett AJ, Diaz RL, Shabanowitz J, Chait BT, Hunt DF, Allis CD. 2007. Long-distance combinatorial linkage between methylation and acetylation on histone H3 N termini. Proc. Natl. Acad. Sci. U. S. A. 104:2086–2091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xu C, Bian C, Yang W, Galka M, Ouyang H, Chen C, Qiu W, Liu H, Jones AE, MacKenzie F, Pan P, Li SS, Wang H, Min J. 2010. Binding of different histone marks differentially regulates the activity and specificity of polycomb repressive complex 2 (PRC2). Proc. Natl. Acad. Sci. U. S. A. 107:19266–19271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Margueron R, Justin N, Ohno K, Sharpe ML, Son J, Drury WJ, 3rd, Voigt P, Martin SR, Taylor WR, De Marco V, Pirrotta V, Reinberg D, Gamblin SJ. 2009. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 461:762–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. De Santa F, Totaro MG, Prosperini E, Notarbartolo S, Testa G, Natoli G. 2007. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell 130:1083–1094 [DOI] [PubMed] [Google Scholar]
- 49. Young NL, DiMaggio PA, Plazas-Mayorca MD, Baliban RC, Floudas CA, Garcia BA. 2009. High throughput characterization of combinatorial histone codes. Mol. Cell. Proteomics 8:2266–2284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Horton JR, Upadhyay AK, Qi HH, Zhang X, Shi Y, Cheng X. 2010. Enzymatic and structural insights for substrate specificity of a family of jumonji histone lysine demethylases. Nat. Struct. Mol. Biol. 17:38–43 [DOI] [PMC free article] [PubMed] [Google Scholar]