

Abstract
The role of RhoA in promoting directed cell migration has been complicated by studies showing that it is activated both in the front and the rear of migrating cells. We report here that the RhoA-specific guanine nucleotide exchange factor Syx is required for the polarity of actively migrating brain and breast tumor cells. This function of Syx is mediated by the selective activation of the RhoA downstream effector Dia1, the subsequent reorganization of microtubules, and the downregulation of focal adhesions and actin stress fibers. The data argue that directed cell migration requires the precise spatiotemporal regulation of Dia1 and ROCK activities in the cell. The recruitment of Syx to the cell membrane and the subsequent selective activation of Dia1 signaling, coupled with the suppression of ROCK and activation of cofilin-mediated actin reorganization, plays a key role in establishing cell polarity during directed cell migration.
INTRODUCTION
Rho GTPases (including RhoA, Rac1, and Cdc42) are key mediators of cytoskeletal dynamics (1). In concert with adhesion receptors and polarity complexes, Rho GTPases regulate apicobasal polarity and epithelial cell adhesion, or front-end polarity and directed cell migration (2–4). Rac1 and Cdc42 activities are associated with actin reorganization and membrane protrusion at the leading cell edge, promoting either directed cell migration or cell-cell contact (1, 2, 4, 5). RhoA is linked with either a strengthening or weakening of intercellular junctions, through mechanisms that likely involve the antagonistic functions of its effectors (6–8). In polarized cell migration, RhoA function was originally thought to be restricted to the retraction of the trailing end (9, 10). However, biosensor studies indicated that activated RhoA is also found at the leading edges of migrating cells (11–14). Therefore, RhoA effects toward front-rear polarity and directed cell migration cannot be explained by selective RhoA activation at the front or the rear of migrating cells. An alternative possibility is that the precise spatiotemporal activation of RhoA coupled with the selective activation of distinct downstream effectors, such as the Rho associated kinases (ROCK1 and -2) or the formin family member Diaphanous homologue 1 (mDia or DIAP1; referred to herein as Dia1), accounts for RhoA effects on cell polarity and directed cell migration. However, no selective mechanism for the activation of one versus another Rho effector has been reported to date, nor has the mechanism of the functional antagonism between ROCK and Dia1 been elucidated.
RhoA binds to and is activated by guanine nucleotide exchange factors (GEFs). The RhoA-specific (15, 16) synectin-binding RhoA exchange factor (Syx; also known as TECH or PLEKHG5) is involved in endothelial cell migration (17, 18), as well as endothelial cell junction integrity, barrier function, and vascular leakiness (8). Syx localizes to the cell membrane through its interaction with members of the Crumbs polarity complex (8, 18, 19). In the present study, we show that Syx is required for the polarity of actively migrating brain and breast cancer cells. Our data support a model where the precise spatiotemporal activation of RhoA by Syx, its selective coupling to Dia1, and the suppression of ROCK are required for proper polarization of actively migrating tumor cells.
MATERIALS AND METHODS
Cell culture, transfection, lentivirus production, and infections.
U251 cells were cultured in Dulbecco modified Eagle medium (DMEM; Cellgro) containing 10% fetal bovine serum (FBS), an additional 2 mM l-glutamine, and 1% nonessential amino acids; Hs578T and HeLa cells were cultured in DMEM–10% FBS. U251 and HeLa cells were transfected with Lipofectamine 2000 (Invitrogen) or TransIT-HeLaMonster (Mirus), respectively, according to the manufacturer's instructions.
Lentiviral vectors (pLKO) encoding a nontarget shRNA sequence, along with human specific shRNA targeting Syx and Dia1, were purchased from Open Biosystems (Syx shRNA1, TRCN0000130291; Syx shRNA2, TRCN0000128190; Dia1 shRNA1, NM_005219.2-2523s1c1; Dia1 shRNA2, NM_005219.2-2557s1c1). Virus was produced using Virapower lentivirus packaging mix according to the manufacturer's protocol (Invitrogen), and cells were infected as described previously (20). Infected cells were selected with 5 μg of puromycin (Sigma-Aldrich)/ml for 48 h.
Antibodies, constructs, and reagents.
The following antibodies were used: mouse anti-Syx (KIAA0720, 5A9; Novus); mouse antivinculin, rabbit anti-Golim4, rabbit antiactin, and rabbit anticofilin (Sigma-Aldrich); mouse anti-GFP (Invitrogen); rat anti-α-tubulin and rabbit anti-Glu-tubulin (Millipore); mouse anti-EB1, anti-Dia1, and mouse anti-Mupp1 (BD Bioscience); rabbit anti-MYPT1 pThr853 (Unites States Biological); rabbit anti-MYPT1 and antiphosphocofilin (Ser3) (Cell Signaling); and mouse anti-RhoA and rabbit anti-APC (Santa Cruz).
pEYFP-Syx and pEYFP-Syx-ΔPBM have been previously described (8). pEYFP-mDia1ΔN3-Syx(C) was generated by a two-step subcloning procedure: GFP-mDia1ΔN3 (21) was initially subcloned into pEYFP-C1 (Clontech) using the BglII and SalI restriction sites; the C-terminal 100 amino acids of murine Syx were then PCR amplified and subcloned in frame into pEYFP-mDia1ΔN3. pEYFP-SyxN596A and pEYFP-Syx-ΔDH (lacking amino acids 406 to 596 of murine Syx) were generated using a QuikChange multisite-directed mutagenesis kit (Stratagene). All constructs were sequenced to ensure absence of mutations.
ROCK inhibitors Y-27632 (Sigma) and H1152 (Calbiochem) were dissolved in sterile water to 10 mM. Nocodazole (Sigma-Aldrich) was dissolved in dimethyl sulfoxide to a stock concentration of 10 mM.
Immunofluorescence, immunoblotting, and quantitative reverse transcription-PCR (RT-PCR).
For immunofluorescence, the cells were fixed with either methanol or 3% paraformaldehyde as previously described (22, 23). Samples were probed with the respective primary antibodies, followed by incubation with highly cross-absorbed Alexa Fluor (488 and 594) secondary antibodies (Invitrogen). Images were acquired with a Zeiss LSM 510 META confocal laser-scanning microscope, using a Plan-Neofluar 40×, 1.3-numerical-aperture (NA) objective oil immersion objective or a Plan-Apochromat 63×, 1.4-NA oil immersion objective.
For immunoblotting, cell lysates were prepared using radioimmunoprecipitation assay buffer, resolved using SDS-PAGE, transferred to nitrocellulose membrane, probed with primary and secondary antibodies, and detected with enhanced chemiluminescence (GE Healthcare).
For quantitative RT-PCR (qRT-PCR), total RNA was isolated using miRCURY RNA isolation kit (Exiqon). Equal amounts of RNA were converted to cDNA using a high-capacity cDNA reverse transcriptase kit (Applied Biosystems) according to the manufacturer's instructions. TaqMan FAST Universal PCR master mix (Applied Biosystems) was used for qPCRs (triplicate for each sample). The data were collected and analyzed by an Applied Biosystems Prism 7900 sequence detector and Sequence Detection System software (Applied Biosystems). The samples were probed for Syx (catalog no. Hs00299154_m1; Applied Biosystems) or Dia1 (catalog no. Hs00946556_m1; Applied Biosystems) and normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH; catalog no. Hs99999905_m1; Applied Biosystems) or 18S (catalog no. Hs99999901_s1; Applied Biosystems). The mRNA abundance was calculated using the ΔΔCT method (24).
RhoA activity assay.
The level of active RhoA was determined using rhotekin pulldown assays as previously described (25). Briefly, HeLa cells were transfected with the respective constructs and cleared cell lysates were incubated with reconstituted glutathione S-transferase (GST)-fused rhotekin-RBD protein beads (Cytoskeleton) to precipitate GTP-bound RhoA. Active, GTP-bound RhoA, as well as total RhoA, was visualized by SDS-PAGE and Western blotting with a RhoA-specific monoclonal antibody (26C4; Santa Cruz Biotechnology).
Scratch-wound, Transwell, and xCELLigence migration assays.
For scratch-wound assays, U251 cells were plated and cultured at confluence on cover glass for 24 h. Scratching the cell monolayer with a P200 pipette tip induced a wound. At 5 hours after wound formation, the cells were fixed and stained for immunofluorescence analysis.
HeLa cell migration was determined using Transwell migration chambers (8-μm pore size; Becton Dickinson). Cells were serum starved overnight before seeding at a density of 5 × 104 cells per chamber in serum-free medium. Cells were allowed to attach for 3 h, and then 5% FBS was added to the lower chamber as a chemoattractant. Cells that migrated to the underside of the migration chambers after 12 h at 37°C in 5% CO2 were stained with crystal violet and counted, as described previously (25). The data from several experiments were expressed as percent control and represent the means ± the standard deviations (SD) of three independent determinations performed in triplicate.
The xCELLigence system (Roche Applied Science) was used to monitor Transwell cell migration of U251 and Hs578T cells, according to the manufacturer's instructions. Briefly, cells were serum starved for 1 h before seeding onto a CIM plate 16 at a density of 3 × 104 cells/well in serum-free medium. In the lower chamber, 10% FBS-containing medium or serum-free medium were used as a chemoattractant or control, respectively. Cell migration was monitored for 10 h. Each condition was performed in quadruplicate.
RESULTS
Syx regulates microtubules, focal adhesions, and static cell polarity.
Directed cell migration and increased invasion are major factors in the poor prognosis of many tumors, especially human glioblastoma multiforme (GBM), a predominantly lethal brain tumor. Although Syx is expressed primarily in endothelial cells, we found it to be widely expressed in a panel of conventional human GBM cell lines, as well as in several human GBM tumors transplanted serially in mice (Fig. 1A and B). The level of Syx expression in U251 cells, a classic cellular model of human GBM, was comparable to its expression in human umbilical vein endothelial cells (HUVECs) (Fig. 1A). Knockdown of syx by each of two nonoverlapping shRNAs markedly reduced Syx expression (Fig. 1C) and drastically affected U251 cell morphology (Fig. 1D and E). The elongated morphology of U251 cells expressing nontarget shRNA was replaced by a flattened, apolar phenotype upon the expression of syx shRNAs. Syx depleted cells exhibited a significant increase in the number and size of focal adhesions (visualized by vinculin staining) and a redistribution of actin filaments to stress fibers and circumferential actin bundles (Fig. 1D). Syx depletion also resulted in the dramatic reorganization of microtubules, which retracted from the cell membrane and lost the ability to coalesce in bundles (Fig. 1E, arrows). These results collectively suggest that the ability of U251 to polarize in the absence of exogenously manipulated extracellular matrix (ECM) or directional cues depends upon Syx.
Fig 1.

Syx is necessary to maintain U251 static cell polarity. (A) Western blot analysis of Syx expression in a panel of conventional glioblastoma multiforme (GBM) cancer cell lines. HUVECs are included as a positive control. (B) Western blot analysis of Syx expression in human GBM tumors serially propagated in the flanks of mice. (C) For the left panel, a quantitative real-time PCR analysis of Syx mRNA levels was performed in cells expressing control shRNA versus either of two nonoverlapping syx shRNAs, normalized to GAPDH (glyceraldehyde-3-phosphate dehydrogenase) mRNA levels. The right panels presents the results of a Western blot analysis showing the efficiency of Syx protein depletion in U251 cells expressing the same shRNAs. (D) Immunofluorescence staining of vinculin (green) and f-actin (phalloidin; red) in U251 cells expressing control shRNA versus either of two nonoverlapping syx shRNAs. Note the progressive cell rounding and loss of front-rear polarity from control shRNA expressing cells to Syx shRNA2-expressing cells. (E) As in panel D, cells expressing indicated shRNAs were stained for α-tubulin. Arrows point to examples of unbundled microtubules.
The establishment of front-end polarity and active migration of tumor cells require Syx.
To assess the ability of tumor cells to migrate directionally in the absence of Syx, we used scratch-wound and Transwell migration assays. Upon wounding a confluent cell monolayer, cells at the wound's edge extend lamellipodia, polarize, and move directionally to fill in the wound. This behavior is clearly evident in leading-edge U251 cells expressing control shRNA (Fig. 2A), which extend lamellipodia in the direction of the wound. In contrast, Syx-depleted U251 cells exhibit an apolar phenotype and lack a polarized lamellipodium distribution toward the wound (Fig. 2A). Consistent with a polarity defect, the ability of U251 cells to reorganize their Golgi apparatus in front of the nucleus and toward the wound is effectively blocked by Syx depletion with either of two different shRNAs (Fig. 2B). Next, we utilized the xCELLigence (Roche Applied Science) Transwell assay, which quantitates cell migration over time, to examine the ability of U251 cells to migrate toward a gradient of FBS, used here as a chemoattractant. Figure 2C shows that over the course of 10 h, Syx-depleted cells were unable to migrate directionally toward the chemoattractant cue.
Fig 2.

Syx is required for tumor cell polarization and directional cell migration. (A) Immunofluorescence staining of vinculin and f-actin (phalloidin) at the leading edge of a wounded U251 monolayer. Cells were fixed and processed for staining 5 h after wounding. (B) As in panel A, cells were fixed and stained for Golgi integral membrane protein 4 (golim4) and α-tubulin 5 h after wounding was induced. Note the complete loss of directional Golgi staining in Syx-depleted cells. (C) xCELLigence Transwell migration assay measuring the real-time migration pattern of control shRNA versus syx shRNA expressing U251 cells toward the FBS chemoattractant. (D) qRT-PCR analysis of Syx mRNA levels in breast cancer cell lines, normalized to respective 18S rRNA levels. The results were then calculated as a percentage of 18S-normalized Syx mRNA in U251 cells. (E) Western blot analysis showing the expression level of Syx protein in Hs578T cells compared to U251. (F) qRT-PCR analysis of Syx mRNA levels in Hs578T cells expressing control shRNA versus either of two nonoverlapping syx shRNAs, normalized to GAPDH mRNA levels. (G) As in panel C, the directional migration of syx shRNA expressing Hs578T cells toward a gradient of FBS was impaired.
To determine whether Syx acts similarly in other cells, we assessed Syx mRNA expression in several breast cancer cell lines (Fig. 2D) and selected Hs578T cells for further evaluation given their ability to migrate and their comparable levels of Syx expression to U251 cells (Fig. 2E). Depletion of Syx in Hs578T cells (Fig. 2F) also resulted in a flattened, apolar cell phenotype (not shown) and blocked Hs578T directed cell migration in the xCELLigence assay (Fig. 2G). Combined, the data show that the establishment of front-end polarity and the active migration of tumor cells require Syx.
Syx membrane localization and exchange activity are required for cell polarity and migration.
Depletion of Syx did not change overall RhoA activity, as measured by a whole-cell RhoA activity assay, in normally cultured U251 or HeLa cells (data not shown). This is not entirely surprising, given the reported cross talk between RhoGEFs. For example, depletion of p114RhoGEF also fails to reduce overall RhoA levels, due to a compensatory activation of GEFH1, a microtubule-associated RhoGEF (26). Although we have not formally tested this hypothesis, the induction of focal adhesion and stress fiber formation and the reorganization of the microtubule network upon Syx depletion suggest the potential involvement of other locally acting RhoGEFs, and possibly GEFH1. Nonetheless, a local effect of Syx on RhoA activity could still be important to the establishment of Syx-dependent front-end polarity and active migration of tumor cells. To test whether Syx promotes cell migration by locally activating RhoA at the leading edges of migrating tumor cells, we utilized a panel of yellow fluorescent protein (YFP)- or V5-tagged Syx mutants either lacking the C-terminal PDZ binding motif (Syx-ΔPBM), carrying a point mutation that disrupts GEF activity (Syx-N596A), or lacking the entire DH catalytic domain (Syx-ΔDH) (Fig. 3A). Previous studies have established that the recruitment of Syx to the cell membrane requires interaction with members of the Crumbs polarity complex via its PDZ binding motif (8, 18). First, we assessed the ability of membrane or Rho uncoupled Syx mutants to rescue the morphology and polarity of Syx-depleted U251 cells. Ectopically expressed murine YFP-Syx accumulated at leading cell edges and rescued the polarized morphology of U251 cells expressing syx shRNA (Fig. 3B). In contrast, Syx-ΔPBM failed to localize to the cell periphery or to reverse the apolar phenotype of Syx-depleted cells. Consistent with its inability to activate RhoA (Fig. 3C), ectopic expression of the Syx-N596A mutant also failed to reverse the apolar phenotype of Syx-depleted cells, despite its localization at cell edges (Fig. 3B).
Fig 3.

Localization and GEF activity of Syx are required for Syx effects on cell polarity and migration. (A) Schematic domain structures of murine pEYFP-Syx, pEYFP-Syx-ΔPBM, pEYFP-Syx-N596A, and pEYFP-Syx-ΔDH. DH, Dbl homology; PH, pleckstrin homology; PDZ-BM, PDZ-binding motif. (B) Immunofluorescence experiment comparing the morphology of Syx-depleted U251 cells expressing pEYFP-Syx, pEYFP-Syx-ΔPBM, or pEYFP-Syx-N596A. The framed regions are shown separately to highlight the localization of expressed constructs; arrows indicate pEYFP-Syx or pEYFP-Syx-N596A that localized at cell edges. Fixed cells were stained for YFP and α-tubulin. (C) Western blot analysis of active and total RhoA from lysates of HeLa cells expressing the indicated v5-tagged constructs. (D) Transwell migration of control shRNA versus syx shRNA expressing HeLa cells toward the FBS chemoattractant. Migration data are expressed as the % control (mean ± the SD; n = 9). Western blot analysis shows the knockdown efficiency of the shRNAs. (E) Transwell migration of HeLa cells transiently expressing pEYFP, pEYFP-Syx, pEYFP-Syx-ΔPBM, or pEYFP-Syx-ΔDH. Migration data are expressed as the % control (mean ± the SD; n = 9). Western blot analysis of active RhoA in lysates of HeLa cells expressing the indicated constructs.
We also assessed the effect of ectopically expressing Syx mutants in HeLa cells, which express endogenous Syx and migrate toward serum (FBS) in a Syx-dependent manner (Fig. 3D). Ectopic expression of YFP-Syx in HeLa cells increased cell migration by 2.5-fold, whereas the expression of either the membrane-uncoupled (ΔPBM) or the catalytically inactive (ΔDH) Syx mutants failed to significantly affect cell migration (Fig. 3D). Importantly, Rho activity assays verified that Syx-ΔDH is unable to induce RhoA activation, while Syx-ΔPBM induces RhoA to a similar extent as wild-type Syx (Fig. 3E). Combined, the data indicate that while Syx is not a major contributor to the overall cellular RhoA activity, both its membrane localization and exchange activity are required for cell polarity and directed cell migration.
Dia1 depletion and microtubule depolymerization phenocopy Syx depletion defects.
Since the GEF activity of Syx was required for its effects on cell polarity and migration, we sought to assess the involvement of two key Rho downstream effectors: ROCK and Dia1. Pharmacological inhibition of ROCK in U251 cells induced a multipolar, stellate cell morphology, with a significant reduction in actin stress fiber organization (Fig. 4A). Similar phenotypic changes were also observed in cells treated with a second ROCK inhibitor (data not shown). In contrast, silencing dia1 with specific shRNAs (Fig. 4B) resulted in flattened, apolar cells, accompanied by increased size and number of focal adhesions and actin reorganization (Fig. 4A), phenotypes that were indistinguishable from those observed upon Syx depletion. In agreement with the phenotypic changes that are opposite those of ROCK suppression, depletion of either Syx or Dia1 resulted in increased phosphorylation of the myosin-binding subunit of myosin phosphatase (MYPT) at T853 (Fig. 4C), a direct measure of ROCK activation (27). Phosphorylation of cofilin at S3 (an indirect target of ROCK activation [for a review, see reference 28]) was also increased in Syx- or Dia1-depleted cells (Fig. 4C). S3 phosphorylation inactivates cofilin's actin severing activity (29, 30), which may account, at least in part, for the increased presence of actin stress fibers in Syx and Dia1-depleted cells. As expected, a dose-dependent reduction in cofilin S3 phosphorylation was observed in U251 cells treated with the ROCK inhibitor (Fig. 4D).
Fig 4.

Dia1 opposes ROCK and phenocopies Syx in U251 cell morphology. (A) Immunofluorescence experiment comparing the morphology of U251 cells expressing control versus either of two nonoverlapping dia1 shRNAs, or treated with ROCK inhibitor (Y-27632, 10 μM, 12 h). Fixed cells were stained for vinculin and f-actin (phalloidin) or for α-tubulin. ROCK inhibition resulted in a stellate, multipolar cell morphology, whereas dia1 shRNA expression induced a well-spread, apolar phenotype. (B) Western blot analysis showing the efficiency of Dia1 protein depletion in U251 cells expressing the dia1 shRNAs. (C) Western blot analysis of phosphoT853-MYPT, total MYPT, phosphoS3-cofilin, and total cofilin in U251 cells expressing control-, syx-, or dia1-specific shRNAs. (D) Western blot analysis of phosphoS3-cofilin in U251 cells treated overnight with increasing amounts of ROCK inhibitor (Y-27632). (E) U251 cells were treated with nocodazole (10 μM, 5 h), fixed, and stained for vinculin and f-actin (phalloidin) or for α-tubulin. Nocodazole treatment increased focal adhesions and induced an apolar cell morphology.
A close inspection of microtubules in U251 cells revealed an interesting dichotomy (Fig. 4A): similar to Syx-depleted cells, Dia1 knockdown induced retraction of microtubules from the cell membrane and blocked microtubules from coalescing into bundles, whereas in contrast, the suppression of ROCK induced pronounced microtubule bundling. Normal U251 cells exhibited an intermediate phenotype, suggesting that spatial regulation of ROCK and Dia1 activities is critical for proper cell polarization. Combined, the data suggest that Dia1 opposes ROCK activity and mediates the effects of Syx on cell polarity and migration.
Dia1 has been previously linked to directed cell migration (31–33), as well as to the turnover of focal adhesions (31). Interestingly, microtubules have long been associated with focal adhesion turnover (34–36). To examine the potential role of microtubules in Syx function, we initially treated U251 cells with the microtubule depolymerizing agent nocodazole. Treated cells largely phenocopied the Syx and Dia1 depletion-induced morphological changes, suggesting that microtubules are involved in Syx function (Fig. 4E).
Dia1 activation, stable microtubule capture and bundling, and suppression of ROCK mediate cell polarization.
Dia1 is thought to recruit adenomatous polyposis coli (APC) to the cell's leading edge, where APC interacts with the microtubule end-capping protein EB1 to promote microtubule capture in a process that is regulated by glycogen synthase kinase 3β(GSK3β) (33, 37, 38). To test the hypothesis that Syx affects Dia1-dependent microtubule capture and bundling, we first examined control and Syx-depleted cells for EB1 localization. We observed an overall increase in EB1 staining in Syx-depleted cells, suggesting increased levels of microtubule plus ends (Fig. 5A). Importantly, clusters of EB1 puncta could be readily seen in control cells, where they associated with the tips of microtubule bundles at the cell's edge (Fig. 5A). In contrast, in cells expressing syx shRNA, EB1 was not found in clusters and was not associated with microtubule bundles.
Fig 5.

Syx-targeted constitutively active Dia1 promotes microtubule bundling and multipolar cell morphology. (A) Immunofluorescence staining of the microtubule plus-end capping protein EB1 and α-tubulin in U251 cells expressing control versus syx shRNA. In control cells, large clusters of EB1 puncta are found associated with microtubule bundles at the cell periphery (arrows), whereas Syx-depleted cells lack both large microtubule bundles and the associated EB1 clusters at their tips. (B) Schematic domain structures of murine Syx, Dia1, Dia1ΔN3 (a constitutively active mutant lacking the auto-inhibitory Rho binding domain), and Dia1ΔN3-Syx(C) (conjugation of the last 100 amino acids of Syx to the activated Dia1 mutant). DH, Dbl homology; PH, pleckstrin homology; PDZ-BM, PDZ-binding motif; FH, formin homology. (C) Immunofluorescence staining of Syx-depleted (syx shRNA2) U251 cells expressing pEYFP-Syx, pEYFP-Dia1ΔN3, or pEYFP-Dia1ΔN3-Syx(C). The framed regions are shown separately to highlight the localization of expressed constructs; arrows indicate pEYFP-Syx or pEYFP-Dia1ΔN3-Syx(C) that localized and induced microtubule bundles at cell edges. Fixed cells were stained for YFP and α-tubulin. (D) Immunofluorescence staining of Syx-depleted (syx shRNA1) U251 cells transiently expressing YFP-Dia1ΔN3-Syx(C), which induces microtubule bundling and a multipolar cell morphology similar to that seen with ROCK inhibition in Fig. 4A.
Next, we assessed whether activation of Dia1 specifically at membrane complexes where Syx is targeted can rescue the Syx depletion defects in microtubule dynamics, cell morphology, and polarity. We assumed that endogenous Syx acts by locally activating RhoA, which then associates with the autoinhibitory Rho-binding domain of Dia1 (39), resulting in Dia1 activation. Thus, we generated a yellow fluorescent protein (YFP)-tagged fusion construct (Fig. 5B) consisting of a constitutively active Dia1 fragment (ΔN3; lacking the autoinhibitory Rho-binding domain) fused to a C-terminal Syx fragment containing the PDZ binding motif [syx(C)] responsible for membrane recruitment of Syx. The fusion protein, YFP-Dia1(ΔN3)-syx(C), is expected to promote Dia1-induced signaling events selectively at Syx-targeted membrane complexes. Transient expression of YFP-Dia1(ΔN3)-syx(C) in Syx-depleted U251 cells caused pronounced microtubule bundling and a dramatic phenotypic change from an apolar phenotype to a multipolar, stellate cell morphology (Fig. 5C and D). Importantly, this phenotypic reversion was not observed in cells expressing YFP-Dia1(ΔN3), indicating that proper localization, in addition to Dia1 activation, is required for microtubule bundling and cell polarization (Fig. 5C).
To examine the mechanism of Dia1 action, we then assessed microtubule capture at the cell periphery. Consistent with microtubule capture, the phenotypic change induced by ectopic expression of YFP-Dia1(ΔN3)-syx(C) was accompanied by the presence of EB1 clusters at the tips of microtubule bundles, colocalizing at cell protrusions with the Dia1 fusion protein (Fig. 6A). Microtubule capture at the cell periphery is thought to induce microtubule stabilization and promote cargo delivery to the cell's leading edge (40). In agreement, U251 cells expressing the YFP-Dia1(ΔN3)-syx(C) fusion protein exhibited increased levels of detyrosinated (stabilized) α-tubulin (also known as Glu-tubulin) associated mainly with microtubule bundles (Fig. 6B), compared to surrounding Syx-depleted cells. Furthermore, endogenous APC largely colocalized with the fusion protein at cell protrusions associated with microtubule bundles (Fig. 6C). Combined the data argue that Syx selectively couples RhoA activation to Dia1 signaling at the cell membrane, inducing microtubule capture and bundling although the local recruitment of APC and the clustering of EB1.
Fig 6.
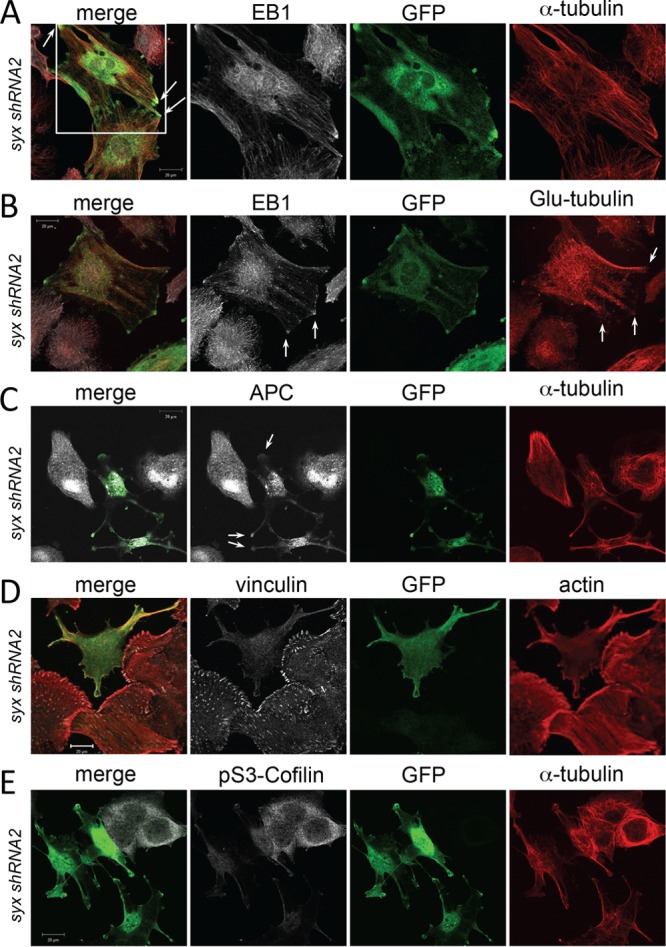
Syx-dependent Dia1 activation induces microtubule capture and stabilization and functional suppression of ROCK signaling. (A) Immunofluorescence staining of Syx-depleted (syx shRNA2) U251 cells transiently expressing YFP-Dia1ΔN3-Syx(C). Note the colocalization (arrows) of large EB1 clusters and microtubule bundles at the tips of YFP-Dia1ΔN3-Syx(C)-expressing cells. (B) Immunofluorescence staining of Syx-depleted U251 cells showing colocalization (arrows) of ectopically expressed YFP-Dia1ΔN3-Syx(C) with EB1 clusters associated with the tips of stable (detyrosinated) Glu-tubulin microtubule bundles. (C) Immunofluorescence staining of Syx-depleted U251 cells shows colocalization of YFP-Dia1ΔN3-Syx(C) with APC at the tips of cells (arrows). (D) Immunofluorescence staining of vinculin and actin in Syx-depleted U251 cells transiently expressing YFP-Dia1ΔN3-Syx(C). Focal adhesions and actin stress fibers are dramatically reduced in cells expressing YFP-Dia1ΔN3-Syx(C). (E) Immunofluorescence staining of cofilin phosphorylated at Ser 3 (pS3-Cofilin), an indirect measure of ROCK activity, in Syx-depleted cells transiently expressing YFP-Dia1ΔN3-Syx(C). pS3-Cofilin is markedly reduced in cells expressing YFP-Dia1ΔN3-Syx(C).
Given the antagonistic relationship of Dia1 and ROCK activities on microtubules we also determined the effect of the YFP-Dia1(ΔN3)-syx(C) fusion protein on focal adhesion size, stress fiber formation, and cofilin S3 phosphorylation, all indirect measures of ROCK activity. U251 cells expressing the fusion protein exhibited fewer and smaller focal adhesions and decreased number of stress fibers (Fig. 6D), as well as significantly reduced levels of phosphorylated cofilin at S3 (Fig. 6E), suggesting that membrane-localized Dia1 activity suppresses ROCK activity in these cells and promotes actin severing and reorganization.
DISCUSSION
Here, we uncover a novel mechanism that mediates microtubule-dependent cell polarization and directed cell migration of human brain and breast cancer cells. The key component of this pathway is the RhoA-specific GEF Syx, which selectively couples RhoA activation at leading cell edges with Dia1 signaling. Syx depletion in cancer cells impairs cell polarity and blocks directed cell migration. These effects are accompanied by lack of microtubule capture at the cell periphery and deficient microtubule bundling and stabilization. The effects of Syx depletion are phenocopied by suppressing Dia1 function and are rescued by selectively activating Dia1 at the cell membrane of cells lacking Syx. Importantly, the data indicate that localization of Syx to the cell membrane, Syx GEF activity toward Rho, and subsequent localized activation of Dia1 signaling are all essential for the establishment of front-rear polarity and the directed migration of human cancer cells.
An important question regarding the role of Syx in cell migration is the molecular mechanism by which it couples Rho activation to Dia signaling. Previous studies have shown that the localization of Syx to the cell membrane depends on the presence of the PDZ binding motif (8, 17, 18). The PDZ binding motif of Syx mediates association with members of the Crumbs polarity complex (8, 18), including PATJ and Pals1, which have been implicated in the directed migration of epithelial cells (41). The selectivity of Syx for Dia1 activation also requires the PDZ binding motif, as Syx constructs lacking this domain induce actin stress fibers, a classic ROCK-mediated response, when expressed in epithelial cells (8). Syx and Dia1 colocalize at the leading edges of U251 cells; however, Dia1 membrane localization is not affected by Syx depletion (data not shown). The mechanism of Dia1 recruitment to the leading edges of migrating cells is currently unclear. In turn, Dia1 is thought to recruit APC and EB1 to the leading edge, thus stabilizing microtubules (33). Interestingly, the polarity protein Scribble also modulates microtubule capture at the front of migrating cells via a Cdc42- and APC-dependent pathway (37, 42). Therefore, it is possible that two or more polarity complexes cooperate to properly localize and activate Cdc42, Rho, APC, and Dia during directed cell migration. Interestingly, depletion of RhoC but not RhoA resulted in breast and prostate cancer cell spreading similar to that seen in U251 cells upon Syx depletion (43). This effect of RhoC was attributed to downstream activation of the Dia-related formin FMNL3. Therefore, one possibility is that Syx couples selectively with RhoC at the leading edges of migrating cells. Another possibility is that Syx activates RhoA, but additional interactions of the Syx-activated RhoA may direct its selectivity toward Dia. For example, recent studies have argued that the effector specificity of RhoA toward rhotekin, another Rho effector, is induced upon interaction with S100A4 (44). As with Dia, the recruitment of ROCK to the plasma membrane is unaffected by Syx depletion (unpublished observations). ROCK binds several membrane-associated proteins, such as p120 catenin, Shroom 3, and p114RhoGEF (26, 45, 46), suggesting that its recruitment to the cell membrane is highly regulated and that its activation is also dependent on its immediate associated protein complex. The data suggest that Syx does not affect the localization of the Rho effectors per se but rather their activation. Indeed, the formation of select signaling modules at the plasma membrane may explain the specificity of Syx for Dia activation and its inability to activate ROCK.
Syx depletion has profound effects on microtubule organization. Microtubule capture, bundling, and stabilization are likely key events in cell polarization during migration. Our observation that the two Rho effectors, Dia1 and ROCK, exhibit opposite effects on microtubules suggests that their differential activation within the cell, and their antagonistic cross talk can provide the means for cell polarization. In agreement, depletion of either Syx or Dia1 increased ROCK activity, whereas expression of the YFP-Dia1(ΔN3)-syx(C) fusion protein suppressed cofilin S3 phosphorylation and phenocopied the effects of ROCK inhibition on microtubule bundling, actin organization, and cell morphology.
Based on these observations, we propose that directed cell migration requires the precise spatiotemporal regulation of Dia1 and ROCK activities in the cell. The recruitment of Syx to the cell membrane and the subsequent selective and localized activation of Dia1 signaling, coupled with the suppression of ROCK and activation of cofilin-mediated actin reorganization plays a key role in establishing cell polarity during directed cell migration. In support of this, depletion of either Syx or Dia1 promotes an apolar cell phenotype, while Syx-directed Dia1 activation or suppression of ROCK induces a multipolar cell morphology (see the model in Fig. 7). The model implies that regulation of Syx membrane recruitment and subsequent signaling via the Syx/RhoA/Dia1 pathway is critical for tumor cell migration, the major contributor in the dissemination and poor outcome of GBM and metastatic tumors. Thus, elucidation of this process could lead to the identification of novel targets for therapeutic intervention in aggressive invasive cancer.
Fig 7.
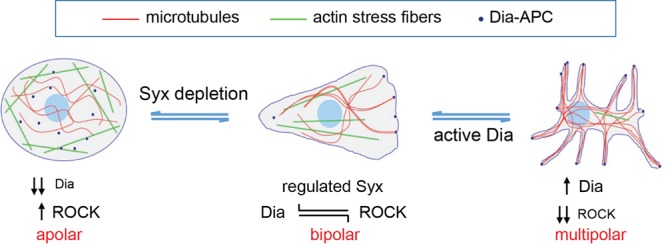
Model of Syx function. Proposed model in which locally regulated Syx activation signals through Dia to induce microtubule capture and bundling, suppress ROCK, establish front-rear (bipolar) cell polarity, and promote directed cell migration; Depletion of either Syx or Dia1 results in an apolar phenotype, while constitutive Syx/Dia1 activation or inhibition of ROCK results in a multipolar phenotype. The model suggests that the precise spatiotemporal activation and subsequent cross talk of downstream Rho effectors, rather than RhoA activation itself, is crucial for the establishment of cell polarization during migration.
ACKNOWLEDGMENTS
This study was supported by National Institutes of Health grants R01 NS069753 and R21 NS070117 (P.Z.A.).
We thank Arie Horowitz (Cleveland Clinic Foundation), Shuh Narumiya (Kyoto University), and Mark McNiven, Jeff Salisbury, and Brian Necela (Mayo Clinic) for providing reagents, technical support, and critical comments.
Footnotes
Published ahead of print 14 October 2013
REFERENCES
- 1. Hall A. 1998. Rho GTPases and the actin cytoskeleton. Science 279:509–514 [DOI] [PubMed] [Google Scholar]
- 2. Nelson WJ. 2009. Remodeling epithelial cell organization: transitions between front-rear and apical-basal polarity. Cold Spring Harbor Perspect. Biol. 1:a000513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fukata M, Nakagawa M, Kaibuchi K. 2003. Roles of Rho-family GTPases in cell polarisation and directional migration. Curr. Opin. Cell Biol. 15:590–597 [DOI] [PubMed] [Google Scholar]
- 4. Iden S, Collard JG. 2008. Crosstalk between small GTPases and polarity proteins in cell polarization. Nat. Rev. Mol. Cell. Biol. 9:846–859 [DOI] [PubMed] [Google Scholar]
- 5. Braga V. 2000. The crossroads between cell-cell adhesion and motility. Nat. Cell Biol. 2:E182–E184 [DOI] [PubMed] [Google Scholar]
- 6. Sahai E, Marshall CJ. 2002. ROCK and Dia have opposing effects on adherens junctions downstream of Rho. Nat. Cell Biol. 4:408–415 [DOI] [PubMed] [Google Scholar]
- 7. Gavard J, Patel V, Gutkind JS. 2008. Angiopoietin-1 prevents VEGF-induced endothelial permeability by sequestering Src through mDia. Dev. Cell 14:25–36 [DOI] [PubMed] [Google Scholar]
- 8. Ngok SP, Geyer R, Liu M, Kourtidis A, Agrawal S, Wu C, Seerapu HR, Lewis-Tuffin LJ, Moodie KL, Huveldt D, Marx R, Baraban JM, Storz P, Horowitz A, Anastasiadis PZ. 2012. VEGF and Angiopoietin-1 exert opposing effects on cell junctions by regulating the Rho GEF Syx. J. Cell Biol. 199:1103–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. 2003. Cell migration: integrating signals from front to back. Science 302:1704–1709 [DOI] [PubMed] [Google Scholar]
- 10. Raftopoulou M, Hall A. 2004. Cell migration: Rho GTPases lead the way. Dev. Biol. 265:23–32 [DOI] [PubMed] [Google Scholar]
- 11. Pertz O, Hodgson L, Klemke RL, Hahn KM. 2006. Spatiotemporal dynamics of RhoA activity in migrating cells. Nature 440:1069–1072 [DOI] [PubMed] [Google Scholar]
- 12. Kurokawa K, Matsuda M. 2005. Localized RhoA activation as a requirement for the induction of membrane ruffling. Mol. Biol. Cell 16:4294–4303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Machacek M, Hodgson L, Welch C, Elliott H, Pertz O, Nalbant P, Abell A, Johnson GL, Hahn KM, Danuser G. 2009. Coordination of Rho GTPase activities during cell protrusion. Nature 461:99–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kardash E, Reichman-Fried M, Maitre JL, Boldajipour B, Papusheva E, Messerschmidt EM, Heisenberg CP, Raz E. 2010. A role for Rho GTPases and cell-cell adhesion in single-cell motility in vivo. Nat. Cell Biol. 12:47–53 [DOI] [PubMed] [Google Scholar]
- 15. De Toledo M, Coulon V, Schmidt S, Fort P, Blangy A. 2001. The gene for a new brain specific RhoA exchange factor maps to the highly unstable chromosomal region 1p36.2-1p36.3. Oncogene 20:7307–7317 [DOI] [PubMed] [Google Scholar]
- 16. Marx R, Henderson J, Wang J, Baraban JM. 2005. Tech: a RhoA GEF selectively expressed in hippocampal and cortical neurons. J. Neurochem. 92:850–858 [DOI] [PubMed] [Google Scholar]
- 17. Liu M, Horowitz A. 2006. A PDZ-binding motif as a critical determinant of Rho guanine exchange factor function and cell phenotype. Mol. Biol. Cell 17:1880–1887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ernkvist M, Luna Persson N, Audebert S, Lecine P, Sinha I, Liu M, Schlueter M, Horowitz A, Aase K, Weide T, Borg JP, Majumdar A, Holmgren L. 2009. The Amot/Patj/Syx signaling complex spatially controls RhoA GTPase activity in migrating endothelial cells. Blood 113:244–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Estevez MA, Henderson JA, Ahn D, Zhu XR, Poschmann G, Lubbert H, Marx R, Baraban JM. 2008. The neuronal RhoA GEF, Tech, interacts with the synaptic multi-PDZ-domain-containing protein, MUPP1. J. Neurochem. 106:1287–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Soto E, Yanagisawa M, Marlow LA, Copland JA, Perez EA, Anastasiadis PZ. 2008. p120 catenin induces opposing effects on tumor cell growth depending on E-cadherin expression. J. Cell Biol. 183:737–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ishizaki T, Morishima Y, Okamoto M, Furuyashiki T, Kato T, Narumiya S. 2001. Coordination of microtubules and the actin cytoskeleton by the Rho effector mDia1. Nat. Cell Biol. 3:8–14 [DOI] [PubMed] [Google Scholar]
- 22. Thoreson MA, Anastasiadis PZ, Daniel JM, Ireton RC, Wheelock MJ, Johnson KR, Hummingbird DK, Reynolds AB. 2000. Selective uncoupling of p120(ctn) from E-cadherin disrupts strong adhesion. J. Cell Biol. 148:189–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yanagisawa M, Kaverina IN, Wang A, Fujita Y, Reynolds AB, Anastasiadis PZ. 2004. A novel interaction between kinesin and p120 modulates p120 localization and function. J. Biol. Chem. 279:9512–9521 [DOI] [PubMed] [Google Scholar]
- 24. Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408 [DOI] [PubMed] [Google Scholar]
- 25. Yanagisawa M, Anastasiadis PZ. 2006. p120 catenin is essential for mesenchymal cadherin-mediated regulation of cell motility and invasiveness. J. Cell Biol. 174:1087–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Terry SJ, Zihni C, Elbediwy A, Vitiello E, Leefa Chong San IV, Balda MS, Matter K. 2011. Spatially restricted activation of RhoA signaling at epithelial junctions by p114RhoGEF drives junction formation and morphogenesis. Nat. Cell Biol. 13:159–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Birukova AA, Smurova K, Birukov KG, Kaibuchi K, Garcia JG, Verin AD. 2004. Role of Rho GTPases in thrombin-induced lung vascular endothelial cells barrier dysfunction. Microvasc. Res. 67:64–77 [DOI] [PubMed] [Google Scholar]
- 28. Amano M, Fukata Y, Kaibuchi K. 2000. Regulation and functions of Rho-associated kinase. Exp. Cell Res. 261:44–51 [DOI] [PubMed] [Google Scholar]
- 29. Arber S, Barbayannis FA, Hanser H, Schneider C, Stanyon CA, Bernard O, Caroni P. 1998. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature 393:805–809 [DOI] [PubMed] [Google Scholar]
- 30. Yang N, Higuchi O, Ohashi K, Nagata K, Wada A, Kangawa K, Nishida E, Mizuno K. 1998. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature 393:809–812 [DOI] [PubMed] [Google Scholar]
- 31. Yamana N, Arakawa Y, Nishino T, Kurokawa K, Tanji M, Itoh RE, Monypenny J, Ishizaki T, Bito H, Nozaki K, Hashimoto N, Matsuda M, Narumiya S. 2006. The Rho-mDia1 pathway regulates cell polarity and focal adhesion turnover in migrating cells through mobilizing Apc and c-Src. Mol. Cell. Biol. 26:6844–6858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Goulimari P, Kitzing TM, Knieling H, Brandt DT, Offermanns S, Grosse R. 2005. Gα12/13 is essential for directed cell migration and localized Rho-Dia1 function. J. Biol. Chem. 280:42242–42251 [DOI] [PubMed] [Google Scholar]
- 33. Wen Y, Eng CH, Schmoranzer J, Cabrera-Poch N, Morris EJ, Chen M, Wallar BJ, Alberts AS, Gundersen GG. 2004. EB1 and APC bind to mDia to stabilize microtubules downstream of Rho and promote cell migration. Nat. Cell Biol. 6:820–830 [DOI] [PubMed] [Google Scholar]
- 34. Kaverina I, Krylyshkina O, Small JV. 2002. Regulation of substrate adhesion dynamics during cell motility. Int. J. Biochem. Cell Biol. 34:746–761 [DOI] [PubMed] [Google Scholar]
- 35. Wittmann T, Waterman-Storer CM. 2001. Cell motility: can Rho GTPases and microtubules point the way? J. Cell Sci. 114:3795–3803 [DOI] [PubMed] [Google Scholar]
- 36. Palazzo AF, Gundersen GG. 2002. Microtubule-actin cross-talk at focal adhesions. Sci. STKE 2002:PE31. [DOI] [PubMed] [Google Scholar]
- 37. Etienne-Manneville S, Hall A. 2003. Cdc42 regulates GSK-3β and adenomatous polyposis coli to control cell polarity. Nature 421:753–756 [DOI] [PubMed] [Google Scholar]
- 38. Eng CH, Huckaba TM, Gundersen GG. 2006. The formin mDia regulates GSK3β through novel PKCs to promote microtubule stabilization but not MTOC reorientation in migrating fibroblasts. Mol. Biol. Cell 17:5004–5016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Watanabe N, Kato T, Fujita A, Ishizaki T, Narumiya S. 1999. Cooperation between mDia1 and ROCK in Rho-induced actin reorganization. Nat. Cell Biol. 1:136–143 [DOI] [PubMed] [Google Scholar]
- 40. Palazzo AF, Cook TA, Alberts AS, Gundersen GG. 2001. mDia mediates Rho-regulated formation and orientation of stable microtubules. Nat. Cell Biol. 3:723–729 [DOI] [PubMed] [Google Scholar]
- 41. Shin K, Wang Q, Margolis B. 2007. PATJ regulates directional migration of mammalian epithelial cells. EMBO Rep. 8:158–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Osmani N, Vitale N, Borg JP, Etienne-Manneville S. 2006. Scrib controls Cdc42 localization and activity to promote cell polarization during astrocyte migration. Curr. Biol. 16:2395–2405 [DOI] [PubMed] [Google Scholar]
- 43. Vega FM, Fruhwirth G, Ng T, Ridley AJ. 2011. RhoA and RhoC have distinct roles in migration and invasion by acting through different targets. J. Cell Biol. 193:655–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen M, Bresnick AR, O'Connor KL. 2013. Coupling S100A4 to rhotekin alters Rho signaling output in breast cancer cells. Oncogene 32:3754–3764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nishimura T, Takeichi M. 2008. Shroom3-mediated recruitment of Rho kinases to the apical cell junctions regulates epithelial and neuroepithelial planar remodeling. Development 135:1493–1502 [DOI] [PubMed] [Google Scholar]
- 46. Smith AL, Dohn MR, Brown MV, Reynolds AB. 2012. Association of Rho-associated protein kinase 1 with E-cadherin complexes is mediated by p120-catenin. Mol. Biol. Cell 23:99–110 [DOI] [PMC free article] [PubMed] [Google Scholar]