

Abstract
Polycomb repressive complex 2 (PRC2) is an essential chromatin-modifying enzyme that implements gene silencing. PRC2 methylates histone H3 on lysine-27 and is conserved from plants to flies to humans. In Drosophila melanogaster, PRC2 contains four core subunits: E(Z), SU(Z)12, ESC, and NURF55. E(Z) bears a SET domain that houses the enzyme active site. However, PRC2 activity depends upon critical inputs from SU(Z)12 and ESC. The stimulatory mechanisms are not understood. We present here functional dissection of the SU(Z)12 subunit. SU(Z)12 contains two highly conserved domains: an ∼140-amino-acid VEFS domain and a Cys2-His2 zinc finger (ZnF). Analysis of recombinant PRC2 bearing VEFS domain alterations, including some modeled after leukemia mutations, identifies distinct elements needed for SU(Z)12 assembly with E(Z) and stimulation of histone methyltransferase. The results define an extensive VEFS subdomain that organizes the SU(Z)12-E(Z) interface. Although the SU(Z)12 ZnF is not needed for methyltransferase in vitro, genetic rescue assays show that the ZnF is required in vivo. Chromatin immunoprecipitations reveal that this ZnF facilitates PRC2 binding to a genomic target. This study defines functionally critical SU(Z)12 elements, including key determinants of SU(Z)12-E(Z) communication. Together with recent findings, this illuminates PRC2 modulation by conserved inputs from its noncatalytic subunits.
INTRODUCTION
Polycomb repressive complex 2 (PRC2) is a chromatin-modifying enzyme widely deployed in metazoans for gene silencing. Its signature activity is to methylate histone H3 on lysine-27 (K27) (1–4). PRC2 can methylate substrates bearing zero, one, or two preexisting methyl groups, with the trimethylated product (H3-K27me3) viewed as the main output that operates in silencing. Polycomb repressors, including subunits of PRC2, were originally defined in Drosophila melanogaster, where they play critical roles in the spatial control of Hox gene expression (5, 6). The broad conservation of PRC2 among single-celled eukaryotes, fungal species, plants, and animals (7–9) underscores its role as an evolutionarily ancient cog of the chromatin regulatory machinery. Commensurate with its widespread distribution, PRC2 functions in many vital biological processes, including fundamental cell fate decisions, the transcriptional control circuitry of both embryonic and adult stem cells, and cancer epigenetics (reviewed in references 7, 10–12, and 13).
PRC2 contains four core subunits, defined as the set of stoichiometric partners obtained upon purification from fly or human cells (1–4). In Drosophila, the subunits are E(Z), SU(Z)12, ESC and NURF55. E(Z) is the catalytic subunit and its SET domain houses the active site that performs histone methylation. However, unlike other examples of SET domain methyltransferases, such as SUV39H or DIM-5 (14, 15), E(Z) is enzymatically inactive on its own (4). To attain robust activity, E(Z) must assemble with two critical noncatalytic partners, SU(Z)12 and ESC (16–19). In contrast, the NURF55 subunit appears dispensable for in vitro PRC2 methyltransferase (17, 18), and its in vivo contribution to PRC2 function is not yet established (20, 21). A key issue in understanding how PRC2 works, then, is to determine how the SU(Z)12 and ESC subunits interface with E(Z), modulate its enzymatic efficiency, and impact its functional output in vivo.
Mammalian PRC2 contains the same four core subunits as in Drosophila, but there is greater diversity of mammalian PRC2 family complexes due to multiple expressed subunit variants (reviewed in references 22, 23, and 24). Multiple genes encode mammalian E(Z) (EZH2 and EZH1), NURF55 (RbAp48, RbAp46), and multiple forms of the ESC homolog (EED) are produced by alternative translation start sites (25, 26). Thus, it is simpler to study PRC2 in Drosophila with its much smaller set of variants. Here, all subunits are encoded by a single gene except for the ESC/ESCL pair, which appear functionally similar but predominate at different times of fly development (27–29).
The SU(Z)12 protein contains two discrete domains that display marked evolutionary conservation (Fig. 1A). The ∼140-amino-acid VEFS domain was originally identified by high similarity among plant, fly, and human versions of SU(Z)12 (30). Removal of the entire VEFS domain by in-frame deletion results in failure of PRC2 assembly (17), suggesting that it contains binding determinants for E(Z). The second highly conserved domain is a single potential Cys2-His2 zinc finger. This ZnF does not appear to supply DNA-binding capacity in vitro (30), and its molecular role and requirement in SU(Z)12 function have not been defined. Besides these two domains, the N-terminal half of SU(Z)12 contains moderately conserved subregions interspersed with more divergent regions. The location of a partial loss-of-function mutation (30), G274D, provides the main evidence to date that this region has function in vivo. This N-terminal region contains a binding site for the NURF55 subunit (31), but the in vivo requirement for this site has not been assessed. There is also no atomic structure yet for either SU(Z)12 in its entirety or for the VEFS or ZnF domains in isolation.
Fig 1.

SU(Z)12 VEFS domain mutations with differential effects upon PRC2 assembly or enzyme activity. (A) Domain organization of SU(Z)12. Three main functional domains are shown, with their percent identities between fly and human. NBE denotes the NURF55-binding element (31). G274D is a missense mutation corresponding to the fly allele Su(z)122 (30). (B) Amino acid sequence of the central portion of the VEFS domain, with the Drosophila melanogaster sequence (residues 542 to 605) on top and sequences from selected animal species aligned below. Residues mutated in T-cell or myeloid leukemia subtypes (45, 46) are underlined, and W581 is highlighted in gray and asterisked. Missense mutations analyzed here are depicted above, and the extent of an in-frame deletion is shown below. (C) Assembly of recombinant PRC2 bearing indicated VEFS domain mutations. Complexes were purified via Flag-ESC. WT denotes the wild-type four-subunit complex. (D) HMTase activities of PRC2 bearing indicated VEFS domain mutations, using HeLa polynucleosomes (left panel) or recombinant Drosophila H3/H4 tetramers (right panel) as substrates. Amido black staining to visualize histones in each reaction is shown below. For quantitation, histone bands were excised and subjected to scintillation counting. The wild type was set to 100%, and the activity of mutants was derived by averaging results from the 20 and 50 nM PRC2 samples.
Here we analyze recombinant PRC2 complexes in vitro and transgenic versions of SU(Z)12 in vivo to investigate the conserved SU(Z)12 domains. Dissection of the VEFS domain reveals distinct functional modules. Residues spanning an extensive C-terminal portion of this domain are required for stable SU(Z)12 binding to E(Z). Other VEFS residues, located primarily in an adjacent more N-terminal region, are not required for PRC2 complex assembly but are needed for full enzymatic function. This includes a pair of consecutive EK residues, conserved from fungi to humans. In contrast, we found that the ZnF is dispensable for PRC2 activity in vitro but is nevertheless required for SU(Z)12 function during fly development. Chromatin immunoprecipitations imply that the ZnF contributes to SU(Z)12 chromatin targeting. Similarly, the N-terminal half of SU(Z)12 is not needed for enzyme activity in vitro but is required for genetic rescue. Collectively, these results define discrete elements within the VEFS domain that supply the SU(Z)12-E(Z) interface and methyltransferase stimulation, as well as elements outside the VEFS domain that are functionally critical in vivo.
MATERIALS AND METHODS
Expression and purification of recombinant PRC2 complexes.
Baculovirus expression of recombinant proteins in insect Sf9 cells was performed using the Bac-to-Bac system (Invitrogen). Anti-FLAG immunoaffinity purification of complexes was performed as described previously (17, 32), with washes of resin-bound PRC2 containing up to 1.2 M KCl. Mutant complexes were prepared in parallel with a wild-type control and purified at least twice independently for each complex. Assembly of complexes was assessed by Coomassie staining after sodium dodecyl sulfate (SDS)-gel electrophoresis.
Baculovirus constructs and site-directed mutagenesis.
Full-length cDNAs encoding FLAG-ESC, E(Z), SU(Z)12, and NURF55 inserted into pFastBac1 were described previously (4, 17). Site-directed SU(Z)12 missense mutations were generated by using a QuikChange mutagenesis kit (Agilent Technologies). Hemagglutinin (HA)-tagged SU(Z)12 and N-terminally or C-terminally truncated derivatives were generated by tailed PCR strategies. In-frame deletions were generated using a PCR strategy as described previously (33). Mutations and deletions were sequenced to confirm intact SU(Z)12 coding region.
Histone methyltransferase assays and substrates.
Histone methyltransferase (HMTase) assays were performed as described previously (17) and were repeated at least twice using independent preparations of mutant and wild-type complexes purified in parallel. Histone content was tracked by amido black staining. HMTase reactions using 3H-labeled S-adenosylmethionine ([3H]SAM) were performed for 1 h at 30°C. Quantitation of HMTase activity, including kinetic analysis (see Fig. 2C), was determined by scintillation counting of excised histone bands. Vmax and Km were determined by nonlinear regression analysis using the Michaelis-Menten model supplied by Prism version 6.0 software (GraphPad Software, La Jolla, CA). For detection of mono-, di-, and trimethylated products (see Fig. 2B), reactions were performed for 18 h using nonradioactive SAM. Polynucleosome substrate, consisting of 8- to 12-mers purified from HeLa cells, was prepared as described previously (17) and used at 60 ng/μl. H3/H4 tetramers were prepared after coexpression of Drosophila histone H3 and H4 in Escherichia coli (34) and used at 50 ng/μl. Mononucleosomes (kindly provided by Karim-Jean Armache and Bob Kingston) were prepared using recombinant, unmodified Xenopus histones and the 601 nucleosome positioning sequence as described previously (35). HMTase stimulation by K27me3 peptide (Fig. 2A) was determined using recombinant mononucleosomes and 40 μM histone H3(15-34) peptide, either unmodified or bearing K27me3.
Fig 2.
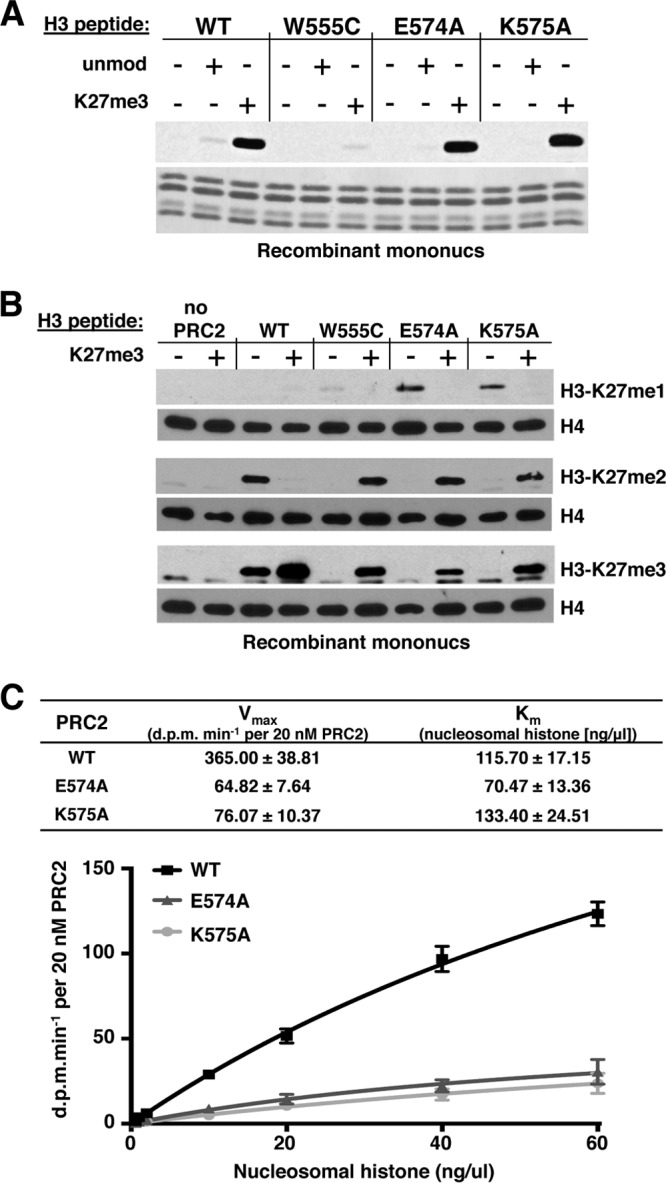
Characterization of altered HMTase activities of VEFS missense mutants. (A) HMTase activities of PRC2 bearing indicated SU(Z)12 VEFS mutations, using recombinant mononucleosomes as substrate. Reactions were performed in the presence (+) or absence (−) of 40 μM histone H3(15-34) peptide either unmodified or trimethylated at K27. Histones were visualized by amido black staining (bottom). (B) Western blots to detect accumulation of K27me1, K27me2, and K27me3 products after 18-h HMTase reactions using mononucleosomes and indicated forms of PRC2. Histone loading was tracked by Western blotting detection of histone H4. Reactions contained (+) or lacked (−) 40 μM histone H3(15-34) peptide bearing K27me3. (C) Kinetic analyses of E574A and K575A HMTase activities. Reactions were performed with wild-type or mutant forms of PRC2 using various amounts of HeLa polynucleosome substrate. Vmax and Km values were determined by nonlinear regression analysis (see Materials and Methods). Error bars show the standard errors of the mean determined from three independent experiments.
Antibodies, Western blots, and coimmunoprecipitations.
Embryo extracts for Western blot analysis were prepared as described previously (17, 36). Proteins were fractionated on 8 to 10% SDS-PAGE gels and transferred to Protran (Schleicher & Schuell). Blots performed on embryo extracts were incubated with anti-HA antibody (Cell Signaling) at 1:200 and with antitubulin (Sigma) at 1:1,000 as a control for lane loading. Blots performed on S2 cell extracts were incubated with anti-FLAG antibodies (M5; Sigma) at 1:3,000 or with anti-SU(Z)12 antibodies (4) at 1:250. Blots to detect H3-K27 methylation status (see Fig. 2B) were performed with anti-K27me1 (1:500; Epigentek), anti-K27me2 (1:500; Millipore), and anti-K27me3 (1:500; Millipore) rabbit polyclonal antibodies and were reprobed with an anti-H4 monoclonal antibody (1:1,000; Thermo Scientific) as a loading control. Immunoprecipitations were performed as described previously (29, 37) using 15 μl of anti-E(Z) (38) for embryo extracts or 30 μl of anti-FLAG M2 beads (Sigma) for S2 cell extracts.
S2 cell transfections and ChIP assays.
Constructs were built, using the pAc5.1 vector (Invitrogen), to express SU(Z)12 transgenes from an actin promoter. Aliquots of 2 × 106 Drosophila S2 cells were transfected with 2 μg of pAc5.1 empty vector, pAc5.1-Flag-SU(Z)12-WT, or pAc5.1-Flag-SU(Z)12-ΔZnF using Fugene HD-6 reagent (Roche). Cells were harvested 3 days after transfection for Western blot analysis and chromatin immunoprecipitation (ChIP). ChIP was performed as described previously (39, 40). Briefly, cells were treated with 1% formaldehyde for 1 h at 24°C, lysed, and chromatin was sheared to ∼500 bp using a Sonic Dismembrator 500 (Fisher). Ten percent of the input material was saved at −80°C. The remainder was diluted 1:10 and precleared in 50 μl of protein A-agarose beads (Roche) for 1 h at 4°C. A total of 30 μl of anti-Flag M2 beads (Sigma) was added to the precleared extracts, and immunoprecipitation was performed overnight at 4°C. After washing, elution, and cross-link reversal, DNA was purified by using a QIAquick PCR purification kit (Qiagen). Quantitative real-time PCR was performed using Platinum SYBR green qPCR Supermix-UDG (Invitrogen) as described previously (40). Primer pairs used to amplify DNA from the PRE region upstream of the Hox gene Ubx were as described previously (40). Quantitative PCR was performed in duplicate for each sample from four independent transfection experiments, with the four averages displayed in Fig. 6D.
Fig 6.

SU(Z)12 lacking its zinc finger shows diminished chromatin binding. (A) Map depicts the Ubx transcription start region and a PRE located ∼25 kb upstream within the bxd regulatory region. Fragments 1, 2, and 3 represent amplicons used in ChIP assays. Fragments 1 and 2, located within the PRE, have been described (40). Fragment 3, located 2 kb downstream of the PRE, provides an off-target, negative control. (B) Western blots on transfected S2 cells demonstrate that the wild type and SU(Z)12ΔZnF accumulate to equivalent levels. “Int control” is an ∼40-kDa endogenous S2 cell protein that cross-reacts with anti-FLAG antibody and serves here as a loading control. (C) Coimmunoprecipitation assays on S2 cells transfected in parallel with indicated constructs. Immunoprecipitations were performed with anti-E(Z), and exogenous SU(Z)12 was detected by Western blotting with anti-HA. Input lanes contain one-sixth the amount of extract used for the immunoprecipitations. (D) ChIP assays to detect association of SU(Z)12 with Ubx PRE DNA in fly S2 cells. Bar graphs depict Q-PCR ChIP signals obtained with anti-FLAG antibodies on samples from S2 cells transfected with empty vector or vectors that express wild-type or ZnF-deleted SU(Z)12. Signals at two PRE subregions (PRE1 and PRE2) and an off-target control region, defined in panel A, are shown. Error bars show standard errors of the mean determined from four independent transfection experiments. For the ZnF mutant versus wild-type comparisons, Student's t test yielded a P of ≈0.05 for PRE1 and a P of ≈0.03 for PRE2.
Generation of SU(Z)12 transgenic lines and assay for genetic rescue.
A 5.8-kb genomic fragment encompassing the Su(z)12 gene, including ∼0.7 kb upstream of the translation start site (3L, 19911150 to 19916900 [Flybase]), was PCR amplified and inserted into the P-element transformation vector pCaSper4. After embryo microinjections, transgenic flies were selected by w+ eye color, and independent lines were established. Three lines with inserts on chromosome 2 were made homozygous and tested for rescue, as described below. For constructs bearing the Su(z)12 promoter fused to full-length Su(z)12 cDNA (see Fig. 5A), the 5.8-kb genomic fragment was modified by (i) inserting an HA tag at the translation start site, (ii) replacing the genomic segment downstream of a unique exon 1 StuI site with the corresponding cDNA segment, and (iii) relocating the Su(z)12 transgene to the patt-B vector, designed for phiC31-targeted integration (41). PhiC31-mediated transgenic lines were generated using the VK37 host strain, with integration at a chromosome 2 landing site at cytological location 22A (41). Inserts containing wild-type, ZnF mutant (see Fig. 5) and ΔN mutant (see Fig. 7) versions of SU(Z)12 were made homozygous and tested for rescue as follows, using the Su(z)123- and Su(z)124-null alleles. Both alleles contain premature stop codons upstream of the ZnF (30). Transgenic males were mated to w; Su(z)123 h th e/TM6C, Sb Tb e females. F1 w; [22A:w+, Su(z)12]/+; Su(z)123 h th e/+ male progeny were selected as w+, nonstubble adults and crossed to w; Su(z)124 h th e/TM6C, Sb Tb e females. Rescue is indicated by the survival of w+, nonstubble, ebony flies. The rescued flies displayed the recessive hairy (h) and thread (th) phenotypes, confirming that they carried the Su(z)123/Su(z)124-null heteroallelic combination. ΔN rescue of the Su(z)125 hypomorph was tested by mating w; [22A:w+, Su(z)12ΔN]/CyO; +/TM6C, Sb Tb e males to w; Su(z)125/TM6C, Sb Tb e females and then mating the w; [22A:w+, Su(z)12ΔN]/+; Su(z)125/TM6C, Sb Tb e male progeny to w; Su(z)123/TM6C, Sb Tb e females. Rescue is indicated by the survival of w+, nonstubble flies.
Fig 5.
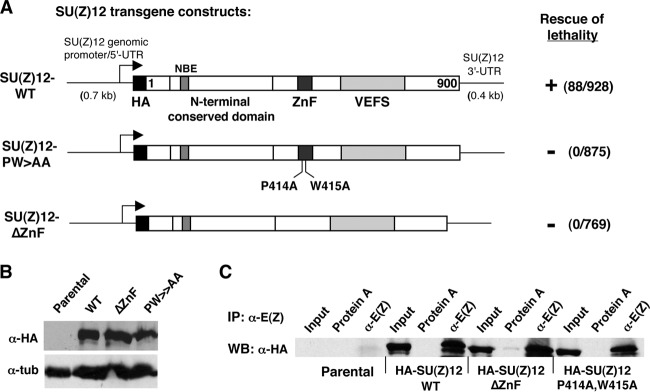
Tests for in vivo function of the SU(Z)12 zinc finger by transgene rescue. (A) Transgene constructs to express wild-type (WT) or indicated zinc finger mutant forms of SU(Z)12. Functional domains are indicated. HA denotes a single epitope tag at the N terminus of the coding region. The arrow represents the transcription start site. The ability to rescue Su(z)123/Su(z)124-null animals to adulthood is indicated at the right. The number of rescued adults observed relative to the total number of adult progeny scored is indicated. (B) Western blots with anti-HA antibodies to detect accumulation of transgenic SU(Z)12 proteins in embryo extracts. “Parental” indicates nontransgenic fly line used as the recipient for transgene insertions. Antitubulin was used as a loading control. (C) Coimmunoprecipitation assays on embryo extracts from control (parental) and indicated transgenic lines. Immunoprecipitations were performed with anti-E(Z), and associated transgenic SU(Z)12 was detected by Western blotting with anti-HA.
Fig 7.

Analysis of PRC2 lacking the N-terminal half of SU(Z)12. (A) Assembly of recombinant PRC2 bearing SU(Z)12ΔN, lacking amino acids 1 to 402. Diagram at top shows domain content of SU(Z)12ΔN. (B) HMTase activity of PRC2 bearing SU(Z)12ΔN is comparable to the wild type. Assays used HeLa polynucleosomes (top) or recombinant histone H3/H4 tetramers (bottom) as a substrate. (C) Western blotting with anti-HA antibodies to detect indicated transgenic SU(Z)12 proteins in embryo extracts. Total protein amounts loaded per lane are indicated, including 2-fold serial dilutions of wild-type HA-SU(Z)12 extract. Antitubulin was used as a loading control. (D) Coimmunoprecipitation assays on embryo extracts from control (parental) and the indicated transgenic lines. Immunoprecipitations were performed with anti-E(Z), and associated transgenic SU(Z)12 was detected by Western blotting with anti-HA antibody.
RESULTS
VEFS mutations that mimic SU(Z)12 alleles in hematological malignancies inactivate PRC2 by distinct mechanisms.
Recently, oncogenic mutations in human subunits of PRC2 have been described from analysis of patients with different types of leukemias (42, 43). Although most reported links between PRC2 and cancer entail overexpression or gain of function of PRC2 subunits, T-cell and myeloid leukemias have instead been associated with PRC2 loss-of-function (44–46). In addition to nonsense and frameshift mutations, several of the apparent inactivating changes are missense mutations that alter EZH2, SU(Z)12, or EED (45, 46). Notably, all of the SU(Z)12 missense alleles map to within the VEFS domain, which highlights its key contributions to function. However, the mechanistic basis of PRC2 disruption by these oncogenic mutations has not been fully addressed. To investigate molecular consequences, we generated corresponding VEFS substitutions in fly SU(Z)12 and tested their impacts upon PRC2 assembly and methyltransferase activity.
Figure 1B highlights three residues located in the central portion of the VEFS domain (underlined) that are altered in leukemias (45, 46). Each of these residues—W555, E574, and N582—is conserved among the animal species shown. Figures 1C and D show that mutations of these residues have differential effects upon PRC2 assembly and activity. Of the three substitutions, N582Y is the only SU(Z)12 mutant that fails to generate a four-subunit complex (Fig. 1C, left panel). Instead, only E(Z) is coenriched with Flag-ESC, indicating that SU(Z)12-N582Y binding to E(Z) is disrupted. This assembly failure is not due to reduced accumulation or instability of N582Y protein in Sf9 cells since Western blot analysis verifies comparable levels of wild-type and mutant proteins in the coinfection samples (not shown).
Histone methyltransferase assays, using polynucleosome substrate, yielded distinct outcomes for these mutants (Fig. 1D, left panel). Although both W555C and E574A retain assembly, W555C shows a dramatic decrease in enzyme function, whereas E574A produces a more modest decline in activity of ∼4-fold. Consistent with assembly failure, no activity is seen with N582Y (not shown). Thus, PRC2 is inactivated by N582Y via disassembly and by W555C through faulty stimulation of E(Z) methyltransferase.
The basis of E574A loss of function is less certain from these in vitro tests; assays on mammalian PRC2 bearing SUZ12 substitution at the analogous position (E610) similarly found complex assembly preserved and methyltransferase activity diminished only ∼2-fold (46). We note that the charged residues E574 and its neighbor K575 are invariant in plant, animal, and fungal SU(Z)12 and are thus more fundamentally conserved than either W555 or N582 (see Fig. 3A). This implies functional importance that contrasts with the modest molecular defects described thus far. To gain fuller understanding, we expanded the biochemical tests to include alternative histone substrates, multiplicity of methyl products, and kinetic analysis (Figs. 1D and 2).
Fig 3.

Elements of the SU(Z)12 VEFS domain required for the interface with E(Z). (A) Sequence alignments of the VEFS domain segment spanning residues 544 to 652 in Drosophila (top sequence). Locations of residues required for PRC2 enzyme stimulation and for binding of SU(Z)12 to E(Z) are indicated at top. Key missense mutations and clustered alanine substitutions described in the present study and previous work (17, 49) are shown. Positions of conserved hydrophobic residues are highlighted in dark green (conserved among animals, plants, and fungi) or light green (conserved among animals and plants). The absolutely conserved EK residues are indicated in blue and red. Predicted alpha-helical content is displayed at bottom, as determined by the RaptorX structure prediction program (77). The alpha-helices shown reflect positions with >50% helical probability. (B) Assembly of recombinant PRC2 bearing indicated SU(Z)12 clustered alanine substitutions (left panel) or C-terminal deletions (right panel). Asterisks indicate detection of progressively truncated SU(Z)12. (C) Subunit architecture of fly PRC2, adapted from the structure of human PRC2 determined by electron microscopy (50). Contours of the four core subunits are displayed and locations of key domains discussed here are indicated. The asterisk denotes location of the K27me3-binding site on ESC (47, 48). AEBP2, which is included in the human PRC2 structure (50), is omitted here for simplicity.
Figure 1D (right panel) shows that E574A and K575A display more severe loss of HMTase when tested on recombinant H3/H4 tetramers produced in E. coli. A key difference between the substrates used here is that the polynucleosomes, purified from HeLa cells, bear preexisting modifications that are devoid from the tetramers. Since histone H3 tails with preinstalled K27me3 can stimulate PRC2 HMTase (47, 48), the more robust activity of the EK motif mutants on polynucleosomes could reflect retention of this stimulatory mechanism, which operates through ESC. To directly test this, we performed HMTase assays using unmodified mononucleosome substrate with added H3 tail peptides bearing or lacking K27me3. Figure 2A shows that E574A and K575A are stimulated by K27me3 peptide, akin to the wild type, whereas W555C is only very weakly stimulated.
To address whether the VEFS mutations differentially impact the generation of K27me1, K27me2, or K27me3, we performed Western blots on the HMTase reaction products (Fig. 2B). These assays used recombinant mononucleosomes and conditions as in Fig. 2A, except the SAM methyl donor was nonradioactive and reactions were incubated for 18 h to maximize detection of each accumulated methyl product. Without added K27me3 peptide, wild-type PRC2 proceeds to generate K27me2 and K27me3, whereas in all three mutants, only the first reaction product, K27me1, was detected. Upon peptide stimulation, all enzyme activities were boosted, with the wild type yielding primarily K27me3 and the mutants producing readily detectable levels of K27me2 and K27me3. Thus, despite reduced activities, all three mutants retain capacity to perform the three successive PRC2 methylation reactions.
Taken together, the in vitro results indicate that W555C enzyme function is severely reduced, whereas the EK mutants retain more partial function. To further investigate the EK mutant defects, we performed kinetic analysis to determine whether substrate binding (Km) or catalytic turnover (Vmax) is altered (Fig. 2C). Using HeLa polynucleosomes as substrate, we found that wild-type PRC2 displays a Vmax of ∼400 dpm/min/20 nM enzyme complex, an observation consistent with the range of Vmax values reported for fly PRC2 on polynucleosome substrates (49). However, both E574A and K575A mutant complexes show substantially reduced Vmax, whereas Km appears not to be significantly altered (Fig. 2C). This suggests that the EK motif of the SU(Z)12 VEFS domain contributes to the intrinsic catalytic efficiency of PRC2, presumably by impacting, directly or indirectly, the E(Z) SET domain (see Discussion).
Determinants within the SU(Z)12 VEFS domain of the interface with E(Z).
We were intrigued that a single amino acid change, N582Y, could so dramatically disrupt SU(Z)12 binding to E(Z) (Fig. 1C), since this could identify a key substituent of the SU(Z)12-E(Z) interface. However, this nonconservative change introduces a bulky side chain, so its properties might not accurately reflect normal contributions of this residue or region. Instead, this mutation could disrupt E(Z) binding by creating steric conflict. We noted that the immediate vicinity has a high percentage of large aliphatic and aromatic residues (Fig. 1B), which could reflect a binding mode that features hydrophobic contacts. Thus, to address whether this VEFS subregion contains determinants for E(Z) binding, we mutated several conserved hydrophobes, singly and in combination. We also generated an in-frame deletion (Δ578-603) that removes N582 and many of the nearby large hydrophobes.
Figure 1C (right panel) shows PRC2 assembly results with SU(Z)12 bearing these VEFS alterations. Significantly, the single W581A mutation, immediately adjacent to N582, is sufficient to disrupt PRC2 assembly (lane 2). Two other lesions that perturb W581, a double substitution (lane 4) and Δ578-603 (lane 6), also disrupt assembly. In contrast, substitutions for the hydrophobic residues V585, F590, and V591 have no discernible impact. Western blot analysis confirmed that all mutant proteins accumulated in infected Sf9 cells (data not shown), indicating that assembly failure cannot be attributed to protein instability.
Consistent with W581 supplying a key determinant for E(Z) binding, this residue is absolutely conserved in SU(Z)12 VEFS domains from fungi to plants to humans (Fig. 3A). There is also conservation of numerous other hydrophobic residues extending C terminally as far as L645 (Fig. 3A, highlighted in green). Consequently, we tested PRC2 assembly of three clustered alanine mutants that alter conserved hydrophobes dispersed in this region (Fig. 3A, bold brackets). Each of these alterations disrupts PRC2 assembly, ranging from complete (mutant FILH) to partial (mutant MFLD) disassembly (Fig. 3B, left panel). To delimit the extent of the VEFS domain required for assembly, we also tested a series of C-terminal truncations (Fig. 3B, right panel). We found that robust assembly requires VEFS residues extending to between 655 and 682. Thus, a large VEFS subdomain, spanning ∼100 residues from V570 to as far as 682, organizes the SU(Z)12 interface with E(Z). Consistent with this, the structure of human PRC2 recently derived by electron microscopy (50) places the VEFS domain in close proximity to portions of E(Z) (Fig. 3C). This interface appears functionally distinct from the VEFS subregion, spanning D546 to W555 (Fig. 3A), that contains residues implicated in E(Z) methyltransferase stimulation rather than stable association of PRC2 subunits (17, 49; see Discussion).
The SU(Z)12 zinc finger is not required for PRC2 histone methyltransferase in vitro.
The Cys2-His2 zinc finger (ZnF) of SU(Z)12 is conserved from fungi to plants and animals (Fig. 4A). Nevertheless, a molecular role for this zinc finger has not been described and its functional requirement in PRC2 has not been determined. In a previous study of fly PRC2 (17), a pair of SU(Z)12 ZnF residues conserved among plants and animals, E406 and L426, were mutated and tested for PRC2 assembly and activity. These mutants behaved indistinguishably from the wild type, suggesting that either these substitutions were ineffective in disrupting the ZnF or that the ZnF is dispensable for function in vitro.
Fig 4.

Properties of recombinant PRC2 complexes bearing mutations or deletion of the SU(Z)12 zinc finger. (A) Amino acid sequence alignment of the SU(Z)12 zinc finger from a wide array of organisms. Sequence from Drosophila melanogaster is shown on top line. The defining Cys and His residues (dark gray outline) are absolutely conserved, and other conserved hydrophobic residues are highlighted in light gray. The bracket on top delimits the region of the fly zinc finger tested here by missense mutations. Thalassiosira pseudonana (asterisk) is a species of diatoms. (B) Assembly of recombinant PRC2 bearing indicated zinc finger mutations or in-frame deletion (ΔZnF). (C) HMTase activities of PRC2 bearing the indicated zinc finger mutations or in-frame deletion, as determined using HeLa polynucleosomes (top) or recombinant H3/H4 tetramers as substrate (bottom). Histones were visualized by amido black staining.
To more definitively assess ZnF function, we constructed three additional SU(Z)12 mutants. The C413A/C416A double mutant eliminates presumed zinc-binding residues, which should more fully disable the ZnF. In addition, we constructed the double mutant P414A/W415A, which alters one of several conserved large hydrophobic residues (Fig. 4A, highlighted in light gray). To guarantee ZnF disruption, we also generated an in-frame deletion (ΔZnF) that eliminates the entire domain spanning residues 406 to 441. Figure 4B shows that all of the ZnF mutants assemble into four-subunit complexes. We do observe a subtle but reproducible decline in the level of associated NURF55 compared to the wild type. However, when ΔZnF complex was affinity purified under less stringent wash conditions, with an ionic strength (0.3 M KCl) closer to the physiological level, stoichiometric assembly of all four subunits was obtained (data not shown). HMTase assays revealed that robust activity is retained for all three ZnF mutants on both polynucleosome and H3/H4 tetramer substrates (Fig. 4C). Western blots confirmed that the ΔZnF complex is similar to the wild type in capacity to produce mono-, di-, and trimethylated H3-K27 products (data not shown). We conclude that the SU(Z)12 ZnF is dispensable for intrinsic enzyme activity of PRC2 and, overall, there is little discernible defect after ZnF disruption in vitro.
In vivo requirement for the SU(Z)12 zinc finger.
To definitively assess whether the ZnF is required for SU(Z)12 function, we sought to test the genetic activity of transgenic SU(Z)12 constructs bearing ZnF mutation or deletion. Since Su(z)12 genetic rescue has not previously been reported in Drosophila, we first defined a 5.8-kb genomic DNA fragment that rescues lethality of Su(z)12-null animals. All three of the tested transgene inserts rescued Su(z)12-null animals to adult viability. Consistent with robust rescue, the surviving P[Su(z)12+]/+; Su(z)123/Su(z)124 adults lacked overt segmental transformations that would indicate partial loss of Polycomb silencing. Thus, this relatively compact genomic fragment, with <1 kb of upstream DNA, is sufficient to provide Su(z)12+ function.
To test Su(z)12 constructs with an impaired ZnF, we wanted to avoid the inherent variability due to random P element integration. Consequently, we switched to a phiC31-based vector (41) and targeted integration to a single genomic location for further transgene assays. Figure 5A (top) shows design of the wild-type Su(z)12 transgene used. This construct contains the same amount of upstream DNA as the initial rescue construct but differs in two respects: (i) the coding region is supplied by cDNA rather than a genomic segment and (ii) an HA epitope tag is inserted at the N terminus. Similar to the genomic construct, this hybrid genomic/cDNA transgene rescues Su(z)123/Su(z)124-null animals to adulthood at a frequency consistent with robust genetic function (Fig. 5A).
We next generated transgenes identical to SU(Z)12-WT but with the ZnF either fully deleted or bearing the P414A/W415A double substitution (Fig. 5A). These ZnF mutant transgenes were inserted at the same integration site, and the transgenic lines were tested for accumulation of HA-tagged SU(Z)12. As shown in Fig. 5B, anti-HA Western blots on embryo extracts revealed ΔZnF and P414A/W415A protein expression at levels comparable to the wild-type control. To address assembly with a PRC2 partner, we performed coimmunoprecipitations on embryo extracts using anti-E(Z) antibodies. These coimmunoprecipitations demonstrated that the ZnF mutants retain association with E(Z) in vivo, as seen with the wild-type HA-tagged SU(Z)12 tested in parallel (Fig. 5C). Nevertheless, we found that both the ΔZnF and the P414A/W415A transgenes failed to rescue the lethality of Su(z)123/Su(z)124-null animals, indicating that the ZnF is required for in vivo function. Adult survivors were never observed among many hundreds of progeny scored (Fig. 5A). We noted that the P414A/W415A rescue crosses yielded substantial numbers of dead pupae, which were not observed with ΔZnF rescue crosses or control crosses lacking transgene. Dissection of several P414A/W415A pupae that reached the pharate stage revealed extra sex comb phenotypes, as seen previously with Su(z)12 hypomorphs (30). Since Su(z)12-null animals die prior to the third larval stage (30), these findings imply that the ΔZnF mutant has little or no genetic function, whereas P414A/W415A may retain partial function.
Zinc finger removal diminishes SU(Z)12 chromatin association.
We hypothesized that the failure of the ZnF mutants to supply SU(Z)12 genetic rescue might reflect defects in binding and/or targeting to chromatin sites. This would not be readily addressed by in vitro assays (Fig. 4), which feature PRC2 binding to substrates by mass action rather than physiological targeting mechanisms. To address chromatin binding properties of SU(Z)12ΔZnF, we pursued chromatin immunoprecipitation (ChIP) assays to track association at a well-characterized target, the Polycomb response element (PRE) located ∼25 kb upstream of the Hox gene Ubx (Fig. 6A). We initially attempted ChIPs to track transgenic HA-SU(Z)12 (Fig. 5A) at this Ubx PRE in larval wing discs. We and others have described ChIP detection of endogenous PcG proteins at this target in wing discs (40, 51, 52). However, we were unable to reproducibly detect wild-type transgenic HA-SU(Z)12 at this PRE by anti-HA ChIP, which may reflect differences in antibody properties. Consequently, we retooled the SU(Z)12 constructs, replacing the HA tags with Flag tags and shifting to a vector that delivers transient expression in cultured fly S2 cells rather than germ line transformation. The same Ubx PRE is an established, functional PcG target in S2 cells (40, 52).
We generated constructs to express wild-type and ΔZnF versions of Flag-SU(Z)12 under constitutive control. Figure 6B illustrates that equivalent levels of wild-type and ΔZnF proteins are obtained in S2 cells after parallel transfection with these constructs. To assess whether these Flag-tagged SU(Z)12 proteins could assemble with PRC2 partners in S2 cells, we performed coimmunoprecipitations with anti-Flag antibodies. As seen with embryo extracts (Fig. 5C), SU(Z)12-ΔZnF is capable of E(Z) association in S2 cell extracts in a manner similar to the wild type (Fig. 6C). We then performed ChIP assays on S2 cells transfected with these constructs or with empty vector. Anti-Flag ChIPs detected SU(Z)12-WT binding to regions within the Ubx PRE (denoted PRE1 and PRE2) but much less association with an off-target control region located 2 kb away (Fig. 6D). These assays revealed that binding of SU(Z)12-ΔZnF to the PRE is significantly reduced compared to SU(Z)12-WT. Figure 6D depicts a compilation of ChIP data from four independent transfection experiments; the diminished ΔZnF signal, compared to the wild type, was reproducible for both PRE fragments in all four assays, and the magnitude of this differential ranged from 2- to 4-fold. These results suggest that the SU(Z)12 ZnF contributes to the chromatin binding capacity of PRC2.
Tests for functional contributions of the SU(Z)12 N-terminal half.
The N-terminal half of SU(Z)12 contains interspersed elements of moderate conservation, as well as a binding site for the NURF55 subunit (31). To address contributions of this N-terminal half, we tested a truncated SU(Z)12 containing residues 403 to 900 (termed ΔN). Figure 7A shows that SU(Z)12ΔN assembles stably with E(Z) and ESC but that NURF55 association is dramatically reduced. A weak band is visible at the NURF55 position and Western blot analysis indicates that, although some NURF55 remains associated, it is significantly reduced compared to wild-type PRC2 (data not shown). Despite this subunit loss, PRC2 bearing SU(Z)12ΔN retains enzyme activity at a level similar to that of the wild type (Fig. 7B). Moreover, Western blots revealed that PRC2 bearing SU(Z)12ΔN retains capacity to produce the H3-K27me1, H3-K27me2, and H3-K27me3 reaction products (data not shown). These results are consistent with previous findings that trimeric fly PRC2 solely lacking NURF55 retains robust histone methyltransferase (17, 18).
To address the in vivo requirement for the SU(Z)12 N-terminal half, we generated an HA-tagged SU(Z)12ΔN transgene that parallels HA-SU(Z)12-WT (Fig. 5A). After phiC31-mediated integration at the same chromosomal site used above, the ΔN transgene was tested for rescue of lethality in Su(z)123/Su(z)124-null animals. Rescue did not occur, with zero Su(z)12-null animals surviving to adulthood out of 1,020 total progeny scored. Western blots were performed to evaluate whether this reflected failure of SU(Z)12ΔN accumulation versus failure to function in vivo. Figure 7C shows that SU(Z)12ΔN protein of the expected size is readily detected in embryo extracts (right lane), at a level approximately 2- to 3-fold lower than wild-type HA-SU(Z)12. Coimmunoprecipitations showed that this SU(Z)12ΔN associates with E(Z) in embryo extracts (Fig. 7D), suggesting that, as in vitro (Fig. 7A), it retains ability to assemble into a multimeric complex. Since the 2-fold reduction of Su(z)12 dosage in null or deficiency heterozygotes does not compromise viability, this failure to rescue Su(z)12-null animals seems more likely due to functional defects of SU(Z)12ΔN complexes than to the marginally reduced level of SU(Z)12. To further address this, we repeated the genetic rescue tests using the hypomorphic Su(z)125 allele. This allele retains substantial partial function, since Su(z)125/null animals survive through late pupal stages to form uneclosed pharate adults (30). If the failure of ΔN to rescue SU(Z)12-null animals was simply due to marginally lower accumulation, we reasoned that it might retain the ability to rescue this Su(z)12 hypomorph from pharate to surviving adults. Crosses to test SU(Z)12ΔN rescue of both Su(z)125/Su(z)123 and Su(z)125/Su(z)124 animals were performed (see Materials and Methods). No rescue was observed after scoring 229 and 287 total progeny, respectively, whereas robust rescue was obtained with HA-SU(Z)12-WT transgene control (127/563 and 72/348 [survivors/total progeny scored], respectively). Taken together, these findings imply that the N-terminal half of SU(Z)12 is not needed to form a multimeric PRC2 with robust methyltransferase but that it is nevertheless critically required for gene silencing in vivo.
DISCUSSION
Cooperative action and inputs of subunits within PRC2.
A critical aspect of PRC2 design is that the catalytic subunit, E(Z), is not configured for efficient histone methylation on its own. Studies on worm, fly, and mammalian PRC2 have all found that the E(Z)/EZH2 subunit must be partnered with, at a minimum, two other noncatalytic subunits to attain robust methyltransferase (16–19). Structural and mutational studies have begun to shed light on how these SU(Z)12 and ESC/EED subunits modulate PRC2 enzyme function. EED contains a binding pocket for H3-K27me3 (47, 48). Mutational disruption of this aromatic cage diminishes PRC2 activity, thereby defining at least one site on EED that functions in enzyme stimulation. This positive mode of PRC2 modulation is suggested to maintain K27me3 marks in local chromatin during cell cycle progression (47, 53; reviewed in reference 24).
Recently, another mode of positive PRC2 modulation, this time acting through SU(Z)12, was described (49). The study reported that PRC2 responds to local nucleosome density via binding affinity for the histone H3 peptide spanning residues 35 to 42. When H3(35-42) peptide is added, PRC2 enzyme activity was found to be stimulated up to 30-fold in vitro (49). Intriguingly, the H3(35-42) peptide binding site maps within the VEFS domain, in close proximity to elements analyzed here (Fig. 3A; see below). Our findings that VEFS mutants retain stimulation by K27me3 (Fig. 2A), together with previous results (49), suggest that the stimulatory mechanisms operating through SU(Z)12 versus through ESC/EED are independent.
SU(Z)12 has also been shown to mediate inhibitory responses to local chromatin state (31, 54). In this case, PRC2 enzyme activity is dampened by nucleosome substrates bearing H3-K4me3 or H3-K36me2/3. Since these inhibitory K4 and K36 methyl marks are associated with active transcription, this could restrain PRC2 in regions where activation predominates over silencing. The C-terminal portion of SU(Z)12, encompassing the VEFS domain, is implicated in this inhibitory mechanism as well (31). Together, these multiple inputs are thought to fine-tune PRC2 activity to adjust local amounts of H3-K27me3 (55).
VEFS domain elements for stimulating histone methyltransferase.
Earlier work established that the SU(Z)12 VEFS domain is needed for binding to E(Z) and assembly of PRC2 (17). In addition, missense mutations targeted at conserved, acidic residues revealed a VEFS subregion that stimulates PRC2 HMTase. Specifically, the VEFS mutations D546A and E550A substantially reduced HMTase while preserving robust PRC2 assembly (17). These findings suggested that separable modules for E(Z) stimulation and E(Z) contact might exist within the VEFS domain. The recently defined binding site for stimulatory H3(35-42) peptide (49) maps precisely to this same subregion (Fig. 3A). Indeed, a triple point mutant, E548A/E550A/D552A, reduces H3(35-42) binding to PRC2 and eliminates HMTase stimulation by this peptide or by dense polynucleosome arrays (49). Thus, there is an HMTase stimulatory module at VEFS residues 546 to 552 implicated in sensing local nucleosome density. The effects of the nearby W555C mutation, which dramatically reduces HMTase while preserving overall PRC2 assembly (Fig. 1C and D), may be explained by impact on this stimulatory module. In addition, we find that the highly conserved EK motif (Fig. 3A) is needed for optimal HMTase but not PRC2 assembly (Fig. 1C and D and Fig. 2). Further work is needed to determine the mechanistic relationship between the stimulatory module in the 546-552 region (17, 49) and this EK motif located 25 residues more C-terminal.
The VEFS domain supplies a key interface with E(Z).
Our results indicate that an extensive C-terminal region of the VEFS domain is critical for stable SU(Z)12-E(Z) interaction and PRC2 assembly (Fig. 3). Initial clues were provided by mutations W581A and N582Y, either of which disrupts SU(Z)12 assembly into PRC2 (Fig. 1C). The dramatic consequence of removing the conserved large hydrophobe, W581, suggests that hydrophobic interactions contribute significantly to organization of the SU(Z)12-E(Z) interface. Indeed, clustered substitutions that disrupt conserved hydrophobes V570, F603, or F620 each impair SU(Z)12 assembly into PRC2 (Fig. 3B). We note that this ∼100-amino-acid region is heavily predicted to favor alpha-helical conformation and that the conserved hydrophobes tend to be spaced periodically, often with a 3- to 4-amino-acid or a 7- to 8-amino-acid separation (Fig. 3A). Thus, this C-terminal VEFS region may form a multi-helix bundle or other helical arrangement anchored by hydrophobic contacts on potential amphipathic helices. We speculate that this helical VEFS subregion supplies the interface with E(Z). Derivation of a high-resolution structure should illuminate how this interface is organized.
Juxtaposing cogs for PRC2 stimulation.
How does SU(Z)12 stimulation of PRC2 histone methyltransferase work? Kinetic analysis of the EK mutants (Fig. 2C), and previous work (49), indicate that VEFS elements impact Vmax of assembled PRC2, suggesting that they influence the efficiency of catalytic methyl transfer. Thus, at some level, stimulatory inputs must be conveyed to the SET domain active site in E(Z). Given the arrangement of VEFS functional elements (Fig. 3A), we suggest that the module for HMTase stimulation communicates through the adjacent E(Z) interface to impact the enzyme active site. How this works in molecular detail is not known. Although allosteric mechanisms have been presumed, there is little information yet on how PRC2 conformations might change in response to subunit contacts or binding of chromatin features. Importantly, the electron microscopy (EM)-derived structure of human PRC2 (50) provides a key architectural framework to consider potential models. This structure positions the SU(Z)12 VEFS domain in close proximity to the EZH2 SET domain (Fig. 3C), a finding consistent with direct molecular communication between VEFS and the catalytic core of PRC2. Thus, we can envision direct and indirect mechanisms by which the VEFS domain could impact the active site. In a direct mechanism, VEFS elements could physically contact and optimize the SAM-binding or lysine substrate-binding pockets of the SET domain. Precedent for direct modes of SET domain optimization is provided by other SET proteins, such as DIM-5 and SET7/9, where POST-SET elements help create a functionally efficient lysine-binding pocket (56). In DIM-5 this occurs through coordination of a zinc cluster (15), and in SET7/9 the POST-SET furnishes an interacting alpha-helix that stabilizes this pocket (57). An instructive example is also provided by the MLL1 histone methyltransferase which, like PRC2, relies upon noncatalytic subunits to attain robust activity (58). Here, it appears that the lysine-binding channel adopts an unproductive “open” conformation in unassembled MLL1, and binding of partner subunits, including WDR5 and RbBP5, induces closure and optimization of this site (59, 60). Alternatively, an indirect stimulatory mechanism might operate. In this scenario, the VEFS domain could contact a remote E(Z) surface, outside of the SET domain, and optimization could occur via conformational changes propagated through E(Z). To reveal how the E(Z) SET domain is reconfigured and optimized by VEFS inputs will require data on high-resolution structure and dynamics of the critical interacting elements.
Taken together, the biochemical and functional data suggest that the VEFS domain is an integrator that senses local chromatin state and conveys this information to control PRC2 catalytic efficiency. This key regulatory role highlights the VEFS interface with E(Z)/EZH2 as a potential target for PRC2 modulation by small molecules. Since recently described EZH2 inhibitors work via competition with the methyl donor, SAM (61, 62), they very likely bind directly to the SET domain SAM-binding pocket. With ∼50 SET domain proteins in humans (63), this raises concerns about potential off-target effects in clinical applications. An alternative strategy might be to target the SU(Z)12-EZH2 interface, whose architecture should be unique among SET domain methyltransferases. These considerations are underscored by the likelihood that aberrant PRC2 function is a driver in certain leukemias (42, 43, 61, 62) and possibly in many other types of cancer (10, 12).
Contribution of the SU(Z)12 zinc finger.
Our results indicate that the SU(Z)12 zinc finger is not needed for intrinsic histone methyltransferase (Fig. 4). These findings are broadly consistent with recent reports on fly and mammalian PRC2, suggesting that the minimal region of SU(Z)12 needed for HMTase is a C-terminal portion that spans just the VEFS domain. In one study, on mouse PRC2, a “VEFS-only” form of SU(Z)12 can partner with EZH2-EED to create HMTase (31). However, this minimal PRC2 is described as having poor enzyme activity and so does not function as well as the intact complex. In another study on fly PRC2, a VEFS-only portion of SU(Z)12 (residues 507 to 630), along with E(Z) and ESC, produced robust histone methyltransferase (49), although the stability of this minimal complex may appear compromised. Thus, although further work should solidify whether the VEFS domain is quantitatively sufficient for full HMTase, all studies agree that the SU(Z)12 ZnF is dispensable for in vitro PRC2 activity.
The enigma of a highly conserved ZnF that contributes little to PRC2 in vitro prompted us to perform in vivo assays for function. Indeed, we found that the SU(Z)12 ZnF is needed for genetic activity in Drosophila transgene experiments (Fig. 5A). Despite stable accumulation and retained ability to bind E(Z) in embryos and cultured fly cells (Fig. 5B and C and Fig. 6B and C), SU(Z)12ΔZnF exhibited a strong loss of function in genetic rescue tests. The main molecular defect that we detected was revealed by ChIP assays, which suggest that the ZnF is needed for optimal PRC2 binding to chromatin targets (Fig. 6D).
In mechanistic terms, how might the SU(Z)12 ZnF contribute to chromatin binding by PRC2? One possibility is that the ZnF interacts with extrinsic factors that guide PRC2 to chromatin sites. There are numerous candidate factors to consider. In Drosophila, the main factor implicated in PRC2 genomic targeting is the DNA-binding protein PHO (52, 64). However, direct interactions between SU(Z)12 and PHO have not been reported. Furthermore, the molecular basis for PHO-PRC2 interaction is unresolved since PHO can bind in pairwise tests to E(Z) or ESC (52) but does not stably interact with intact recombinant PRC2 (65). Alternatively, the SU(Z)12 ZnF could mediate interactions with PCL, which is implicated in PRC2 chromatin binding in fly and mammalian cells (66–68) or with JARID2, which influences PRC2 targeting in ES cells (69–71) and associates with PRC2 purified from Drosophila (72). Finally, we note that PRC2 structure (50) positions the SU(Z)12 ZnF in close proximity to the DNA-binding domain of AEBP2. AEBP2 is a DNA-binding zinc finger protein, originally copurified with PRC2 from HeLa cells (1), that has been suggested to help target mammalian PRC2 to chromatin sites (16, 73). AEBP2 was included in the recombinant PRC2 used for structure determination since it helped stabilize the holocomplex (50). Thus, the SU(Z)12 ZnF could aid PRC2 targeting through direct interactions with a bona fide DNA-binding factor. Functional studies of the Drosophila AEBP2 homolog, known as Jing (74), will be needed to address this hypothesis.
Contribution of the SU(Z)12 N-terminal half: implications for the PRC2 bottom lobe.
The EM-derived structure of human PRC2 (50) positions the core subunits in two distinct territories: a top lobe that contains E(Z), EED, and the C-terminal half of SU(Z)12 and a bottom lobe that contains the N-terminal half of SU(Z)12 and NURF55 (Fig. 3C). The “business end” of PRC2, then, resides in the top lobe which harbors the catalytic SET domain and all known sites on EED/ESC and SU(Z)12 that impact HMTase activity. Consistent with this layout, we found that a PRC2 complex missing the N-terminal half of SU(Z)12 and NURF55, corresponding essentially to an isolated top lobe, retained HMTase comparable to the wild type in vitro (Fig. 7B). This finding prompts further questions: how critical are this half of SU(Z)12 and NURF55 to PRC2 function? What does the bottom lobe do? Using genetic tests, we found that SU(Z)12ΔN is severely compromised for function in vivo, since it failed to rescue either SU(Z)12-null animals or hypomorphs. The SU(Z)12 N-terminal half contains a NURF55-binding site (Fig. 1A) (31), and the loss of NURF55 from the SU(Z)12ΔN complex (Fig. 7A) suggests that much of the SU(Z)12ΔN loss of function can be attributed to dislodging of NURF55. However, the functional contribution of NURF55 to fly PRC2 silencing is not yet clear (20, 21). Furthermore, a recent Neurospora study implies that NURF55 is not required for HMTase in vivo but is needed for PRC2 binding to a subset of genomic sites (75). Defining its precise in vivo role in PRC2 is technically nontrivial since NURF55 functions in many different chromatin complexes (55, 76). To further address this, genetic and molecular analysis of SU(Z)12 altered by surgical removal of its NURF55-binding site should be useful.
ACKNOWLEDGMENTS
We are grateful to Rick Jones for providing anti-E(Z) antibodies, to Jürg Müller for supplying Su(z)12 mutant fly stocks, and to Karim-Jean Armache and Bob Kingston for supplying an aliquot of recombinant mononucleosomes. We thank Aidan Peterson, Arpan Ghosh, and Mark Murphy for helpful discussions.
This study was supported by National Institutes of Health grant GM49850 to J.A.S.
Footnotes
Published ahead of print 7 October 2013
REFERENCES
- 1. Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. 2002. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 298:1039–1043 [DOI] [PubMed] [Google Scholar]
- 2. Czermin B, Melfi R, McCabe D, Seitz V, Imhof A, Pirrotta V. 2002. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell 111:185–196 [DOI] [PubMed] [Google Scholar]
- 3. Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. 2002. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 16:2893–2905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Muller J, Hart CM, Francis NJ, Vargas ML, Sengupta A, Wild B, Miller EL, O'Connor MB, Kingston RE, Simon JA. 2002. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell 111:197–208 [DOI] [PubMed] [Google Scholar]
- 5. Lewis EB. 1978. A gene complex controlling segmentation in Drosophila. Nature 276:565–570 [DOI] [PubMed] [Google Scholar]
- 6. Struhl G. 1981. A gene product required for correct initiation of segmental determination in Drosophila. Nature 293:36–41 [DOI] [PubMed] [Google Scholar]
- 7. Sawarkar R, Paro R. 2010. Interpretation of developmental signaling at chromatin: the Polycomb perspective. Dev. Cell 19:651–661 [DOI] [PubMed] [Google Scholar]
- 8. Shaver S, Casas-Mollano JA, Cerny RL, Cerutti H. 2010. Origin of the polycomb repressive complex 2 and gene silencing by an E(z) homolog in the unicellular alga Chlamydomonas. Epigenetics 5:301–312 [DOI] [PubMed] [Google Scholar]
- 9. Whitcomb SJ, Basu A, Allis CD, Bernstein E. 2007. Polycomb Group proteins: an evolutionary perspective. Trends Genet. 23:494–502 [DOI] [PubMed] [Google Scholar]
- 10. Bracken AP, Helin K. 2009. Polycomb group proteins: navigators of lineage pathways led astray in cancer. Nat. Rev. Cancer 9:773–784 [DOI] [PubMed] [Google Scholar]
- 11. Mills AA. 2010. Throwing the cancer switch: reciprocal roles of polycomb and trithorax proteins. Nat. Rev. Cancer 10:669–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Simon JA, Lange CA. 2008. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat. Res. 647:21–29 [DOI] [PubMed] [Google Scholar]
- 13. Surface LE, Thornton SR, Boyer LA. 2010. Polycomb group proteins set the stage for early lineage commitment. Cell Stem Cell 7:288–298 [DOI] [PubMed] [Google Scholar]
- 14. Rea S, Eisenhaber F, O'Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD, Jenuwein T. 2000. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406:593–599 [DOI] [PubMed] [Google Scholar]
- 15. Zhang X, Yang Z, Khan SI, Horton JR, Tamaru H, Selker EU, Cheng X. 2003. Structural basis for the product specificity of histone lysine methyltransferases. Mol. Cell 12:177–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cao R, Zhang Y. 2004. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol. Cell 15:57–67 [DOI] [PubMed] [Google Scholar]
- 17. Ketel CS, Andersen EF, Vargas ML, Suh J, Strome S, Simon JA. 2005. Subunit contributions to histone methyltransferase activities of fly and worm polycomb group complexes. Mol. Cell. Biol. 25:6857–6868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nekrasov M, Wild B, Muller J. 2005. Nucleosome binding and histone methyltransferase activity of Drosophila PRC2. EMBO Rep. 6:348–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pasini D, Bracken AP, Jensen MR, Lazzerini Denchi E, Helin K. 2004. Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J. 23:4061–4071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Anderson AE, Karandikar UC, Pepple KL, Chen Z, Bergmann A, Mardon G. 2011. The enhancer of trithorax and polycomb gene Caf1/p55 is essential for cell survival and patterning in Drosophila development. Development 138:1957–1966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wen P, Quan Z, Xi R. 2012. The biological function of the WD40 repeat-containing protein p55/Caf1 in Drosophila. Dev. Dyn. 241:455–464 [DOI] [PubMed] [Google Scholar]
- 22. Margueron R, Reinberg D. 2011. The Polycomb complex PRC2 and its mark in life. Nature 469:343–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Simon JA, Kingston RE. 2009. Mechanisms of polycomb gene silencing: knowns and unknowns. Nat. Rev. Mol. Cell. Biol. 10:697–708 [DOI] [PubMed] [Google Scholar]
- 24. Simon JA, Kingston RE. 2013. Occupying chromatin: Polycomb mechanisms for getting to genomic targets, stopping transcriptional traffic, and staying put. Mol. Cell 49:808–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kuzmichev A, Jenuwein T, Tempst P, Reinberg D. 2004. Different EZH2-containing complexes target methylation of histone H1 or nucleosomal histone H3. Mol. Cell 14:183–193 [DOI] [PubMed] [Google Scholar]
- 26. Margueron R, Li G, Sarma K, Blais A, Zavadil J, Woodcock CL, Dynlacht BD, Reinberg D. 2008. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol. Cell 32:503–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kurzhals RL, Tie F, Stratton CA, Harte PJ. 2008. Drosophila ESC-like can substitute for ESC and becomes required for Polycomb silencing if ESC is absent. Dev. Biol. 313:293–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ohno K, McCabe D, Czermin B, Imhof A, Pirrotta V. 2008. ESC, ESCL, and their roles in Polycomb group mechanisms. Mech. Dev. 125:527–541 [DOI] [PubMed] [Google Scholar]
- 29. Wang L, Jahren N, Vargas ML, Andersen EF, Benes J, Zhang J, Miller EL, Jones RS, Simon JA. 2006. Alternative ESC and ESC-like subunits of a polycomb group histone methyltransferase complex are differentially deployed during Drosophila development. Mol. Cell. Biol. 26:2637–2647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Birve A, Sengupta AK, Beuchle D, Larsson J, Kennison JA, Rasmuson-Lestander A, Muller J. 2001. Su(z)12, a novel Drosophila Polycomb group gene that is conserved in vertebrates and plants. Development 128:3371–3379 [DOI] [PubMed] [Google Scholar]
- 31. Schmitges FW, Prusty AB, Faty M, Stutzer A, Lingaraju GM, Aiwazian J, Sack R, Hess D, Li L, Zhou S, Bunker RD, Wirth U, Bouwmeester T, Bauer A, Ly-Hartig N, Zhao K, Chan H, Gu J, Gut H, Fischle W, Muller J, Thoma NH. 2011. Histone methylation by PRC2 is inhibited by active chromatin marks. Mol. Cell 42:330–341 [DOI] [PubMed] [Google Scholar]
- 32. Joshi P, Carrington EA, Wang L, Ketel CS, Miller EL, Jones RS, Simon JA. 2008. Dominant alleles identify SET domain residues required for histone methyltransferase of Polycomb repressive complex 2. J. Biol. Chem. 283:27757–27766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Makarova O, Kamberov E, Margolis B. 2000. Generation of deletion and point mutations with one primer in a single cloning step. Biotechniques 29:970–972 [DOI] [PubMed] [Google Scholar]
- 34. Levenstein ME, Kadonaga JT. 2002. Biochemical analysis of chromatin containing recombinant Drosophila core histones. J. Biol. Chem. 277:8749–8754 [DOI] [PubMed] [Google Scholar]
- 35. Dyer PN, Edayathumangalam RS, White CL, Bao Y, Chakravarthy S, Muthurajan UM, Luger K. 2004. Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods Enzymol. 375:23–44 [DOI] [PubMed] [Google Scholar]
- 36. Ng J, Hart CM, Morgan K, Simon JA. 2000. A Drosophila ESC-E(Z) protein complex is distinct from other polycomb group complexes and contains covalently modified ESC. Mol. Cell. Biol. 20:3069–3078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jones CA, Ng J, Peterson AJ, Morgan K, Simon J, Jones RS. 1998. The Drosophila esc and E(z) proteins are direct partners in polycomb group-mediated repression. Mol. Cell. Biol. 18:2825–2834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Carrington EA, Jones RS. 1996. The Drosophila Enhancer of zeste gene encodes a chromosomal protein: examination of wild-type and mutant protein distribution. Development 122:4073–4083 [DOI] [PubMed] [Google Scholar]
- 39. Ratnaparkhi GS, Duong HA, Courey AJ. 2008. Dorsal interacting protein 3 potentiates activation by Drosophila Rel homology domain proteins. Dev. Comp. Immunol. 32:1290–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang L, Jahren N, Miller EL, Ketel CS, Mallin DR, Simon JA. 2010. Comparative analysis of chromatin binding by Sex Comb on Midleg (SCM) and other polycomb group repressors at a Drosophila Hox gene. Mol. Cell. Biol. 30:2584–2593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Groth AC, Fish M, Nusse R, Calos MP. 2004. Construction of transgenic Drosophila by using the site-specific integrase from phage phiC31. Genetics 166:1775–1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. McCabe MT, Graves AP, Ganji G, Diaz E, Halsey WS, Jiang Y, Smitheman KN, Ott HM, Pappalardi MB, Allen KE, Chen SB, Della Pietra A, III, Dul E, Hughes AM, Gilbert SA, Thrall SH, Tummino PJ, Kruger RG, Brandt M, Schwartz B, Creasy CL. 2012. Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc. Natl. Acad. Sci. U. S. A. 109:2989–2994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sneeringer CJ, Scott MP, Kuntz KW, Knutson SK, Pollock RM, Richon VM, Copeland RA. 2010. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc. Natl. Acad. Sci. U. S. A. 107:20980–20985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, Waghorn K, Zoi K, Ross FM, Reiter A, Hochhaus A, Drexler HG, Duncombe A, Cervantes F, Oscier D, Boultwood J, Grand FH, Cross NC. 2010. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat. Genet. 42:722–726 [DOI] [PubMed] [Google Scholar]
- 45. Ntziachristos P, Tsirigos A, Van Vlierberghe P, Nedjic J, Trimarchi T, Flaherty MS, Ferres-Marco D, da Ros V, Tang Z, Siegle J, Asp P, Hadler M, Rigo I, De Keersmaecker K, Patel J, Huynh T, Utro F, Poglio S, Samon JB, Paietta E, Racevskis J, Rowe JM, Rabadan R, Levine RL, Brown S, Pflumio F, Dominguez M, Ferrando A, Aifantis I. 2012. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat. Med. 18:298–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Score J, Hidalgo-Curtis C, Jones AV, Winkelmann N, Skinner A, Ward D, Zoi K, Ernst T, Stegelmann F, Dohner K, Chase A, Cross NC. 2012. Inactivation of polycomb repressive complex 2 components in myeloproliferative and myelodysplastic/myeloproliferative neoplasms. Blood 119:1208–1213 [DOI] [PubMed] [Google Scholar]
- 47. Margueron R, Justin N, Ohno K, Sharpe ML, Son J, Drury WJ, III, Voigt P, Martin SR, Taylor WR, De Marco V, Pirrotta V, Reinberg D, Gamblin SJ. 2009. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 461:762–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xu C, Bian C, Yang W, Galka M, Ouyang H, Chen C, Qiu W, Liu H, Jones AE, MacKenzie F, Pan P, Li SS, Wang H, Min J. 2010. Binding of different histone marks differentially regulates the activity and specificity of polycomb repressive complex 2 (PRC2). Proc. Natl. Acad. Sci. U. S. A. 107:19266–19271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yuan W, Wu T, Fu H, Dai C, Wu H, Liu N, Li X, Xu M, Zhang Z, Niu T, Han Z, Chai J, Zhou XJ, Gao S, Zhu B. 2012. Dense chromatin activates Polycomb repressive complex 2 to regulate H3 lysine 27 methylation. Science 337:971–975 [DOI] [PubMed] [Google Scholar]
- 50. Ciferri C., G. C. Lander, A Maiolica, F Herzog, R Aebersold, E Nogales. 2012. Molecular architecture of human polycomb repressive complex 2. elife 1:e00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Papp B, Muller J. 2006. Histone trimethylation and the maintenance of transcriptional ON and OFF states by trxG and PcG proteins. Genes Dev. 20:2041–2054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang L, Brown JL, Cao R, Zhang Y, Kassis JA, Jones RS. 2004. Hierarchical recruitment of polycomb group silencing complexes. Mol. Cell 14:637–646 [DOI] [PubMed] [Google Scholar]
- 53. Hansen KH, Bracken AP, Pasini D, Dietrich N, Gehani SS, Monrad A, Rappsilber J, Lerdrup M, Helin K. 2008. A model for transmission of the H3K27me3 epigenetic mark. Nat. Cell Biol. 10:1291–1300 [DOI] [PubMed] [Google Scholar]
- 54. Yuan W, Xu M, Huang C, Liu N, Chen S, Zhu B. 2011. H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. J. Biol. Chem. 286:7983–7989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. O'Meara MM, Simon JA. 2012. Inner workings and regulatory inputs that control Polycomb repressive complex 2. Chromosoma 121:221–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cheng X, Zhang X. 2007. Structural dynamics of protein lysine methylation and demethylation. Mutat. Res. 618:102–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Xiao B, Jing C, Wilson JR, Walker PA, Vasisht N, Kelly G, Howell S, Taylor IA, Blackburn GM, Gamblin SJ. 2003. Structure and catalytic mechanism of the human histone methyltransferase SET7/9. Nature 421:652–656 [DOI] [PubMed] [Google Scholar]
- 58. Dou Y, Milne TA, Ruthenburg AJ, Lee S, Lee JW, Verdine GL, Allis CD, Roeder RG. 2006. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat. Struct. Mol. Biol. 13:713–719 [DOI] [PubMed] [Google Scholar]
- 59. Avdic V, Zhang P, Lanouette S, Groulx A, Tremblay V, Brunzelle J, Couture JF. 2011. Structural and biochemical insights into MLL1 core complex assembly. Structure 19:101–108 [DOI] [PubMed] [Google Scholar]
- 60. Southall SM, Wong PS, Odho Z, Roe SM, Wilson JR. 2009. Structural basis for the requirement of additional factors for MLL1 SET domain activity and recognition of epigenetic marks. Mol. Cell 33:181–191 [DOI] [PubMed] [Google Scholar]
- 61. Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, Sacks JD, Raimondi A, Majer CR, Song J, Scott MP, Jin L, Smith JJ, Olhava EJ, Chesworth R, Moyer MP, Richon VM, Copeland RA, Keilhack H, Pollock RM, Kuntz KW. 2012. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat. Chem. Biol. 8:890–896 [DOI] [PubMed] [Google Scholar]
- 62. McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A, III, Diaz E, LaFrance LV, Mellinger M, Duquenne C, Tian X, Kruger RG, McHugh CF, Brandt M, Miller WH, Dhanak D, Verma SK, Tummino PJ, Creasy CL. 2012. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492:108–112 [DOI] [PubMed] [Google Scholar]
- 63. Copeland RA, Solomon ME, Richon VM. 2009. Protein methyltransferases as a target class for drug discovery. Nat. Rev. Drug Discov. 8:724–732 [DOI] [PubMed] [Google Scholar]
- 64. Oktaba K, Gutierrez L, Gagneur J, Girardot C, Sengupta AK, Furlong EE, Muller J. 2008. Dynamic regulation by polycomb group protein complexes controls pattern formation and the cell cycle in Drosophila. Dev. Cell 15:877–889 [DOI] [PubMed] [Google Scholar]
- 65. Klymenko T, Papp B, Fischle W, Kocher T, Schelder M, Fritsch C, Wild B, Wilm M, Muller J. 2006. A Polycomb group protein complex with sequence-specific DNA-binding and selective methyl-lysine-binding activities. Genes Dev. 20:1110–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Casanova M, Preissner T, Cerase A, Poot R, Yamada D, Li X, Appanah R, Bezstarosti K, Demmers J, Koseki H, Brockdorff N. 2011. Polycomblike 2 facilitates the recruitment of PRC2 Polycomb group complexes to the inactive X chromosome and to target loci in embryonic stem cells. Development 138:1471–1482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hunkapiller J, Shen Y, Diaz A, Cagney G, McCleary D, Ramalho-Santos M, Krogan N, Ren B, Song JS, Reiter JF. 2012. Polycomb-like 3 promotes polycomb repressive complex 2 binding to CpG islands and embryonic stem cell self-renewal. PLoS Genet. 8:e1002576. 10.1371/journal.pgen.1002576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Savla U, Benes J, Zhang J, Jones RS. 2008. Recruitment of Drosophila Polycomb-group proteins by Polycomblike, a component of a novel protein complex in larvae. Development 135:813–817 [DOI] [PubMed] [Google Scholar]
- 69. Li G, Margueron R, Ku M, Chambon P, Bernstein BE, Reinberg D. 2010. Jarid2 and PRC2, partners in regulating gene expression. Genes Dev. 24:368–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pasini D, Cloos PA, Walfridsson J, Olsson L, Bukowski JP, Johansen JV, Bak M, Tommerup N, Rappsilber J, Helin K. 2010. JARID2 regulates binding of the Polycomb repressive complex 2 to target genes in ES cells. Nature 464:306–310 [DOI] [PubMed] [Google Scholar]
- 71. Peng JC, Valouev A, Swigut T, Zhang J, Zhao Y, Sidow A, Wysocka J. 2009. Jarid2/Jumonji coordinates control of PRC2 enzymatic activity and target gene occupancy in pluripotent cells. Cell 139:1290–1302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Herz HM, Mohan M, Garrett AS, Miller C, Casto D, Zhang Y, Seidel C, Haug JS, Florens L, Washburn MP, Yamaguchi M, Shiekhattar R, Shilatifard A. 2012. Polycomb repressive complex 2-dependent and -independent functions of Jarid2 in transcriptional regulation in Drosophila. Mol. Cell. Biol. 32:1683–1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kim H, Kang K, Kim J. 2009. AEBP2 as a potential targeting protein for Polycomb repression complex PRC2. Nucleic Acids Res. 37:2940–2950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Culi J, Aroca P, Modolell J, Mann RS. 2006. jing is required for wing development and to establish the proximo-distal axis of the leg in Drosophila melanogaster. Genetics 173:255–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Jamieson K, Rountree MR, Lewis ZA, Stajich JE, Selker EU. 2013. Regional control of histone H3 lysine 27 methylation in Neurospora. Proc. Natl. Acad. Sci. U. S. A. 110:6027–6032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Suganuma T, Pattenden SG, Workman JL. 2008. Diverse functions of WD40 repeat proteins in histone recognition. Genes Dev. 22:1265–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kallberg M, Wang H, Wang S, Peng J, Wang Z, Lu H, Xu J. 2012. Template-based protein structure modeling using the RaptorX web server. Nat. Protoc. 7:1511–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]