

Abstract
Tumor necrosis factor alpha (TNF-α) plays a role in apoptosis and proliferation in multiple types of cells, and defects in TNF-α-induced apoptosis are associated with various autoimmune diseases. Here, we show that TRIM27, a tripartite motif (TRIM) protein containing RING finger, B-box, and coiled-coil domains, positively regulates TNF-α-induced apoptosis. Trim27-deficient mice are resistant to TNF-α–d-galactosamine-induced hepatocyte apoptosis. Trim27-deficient mouse embryonic fibroblasts (MEFs) are also resistant to TNF-α–cycloheximide-induced apoptosis. TRIM27 forms a complex with and ubiquitinates the ubiquitin-specific protease USP7, which deubiquitinates receptor-interacting protein 1 (RIP1), resulting in the positive regulation of TNF-α-induced apoptosis. Our findings indicate that the ubiquitination-deubiquitination cascade mediated by the TRIM27-USP7 complex plays an important role in TNF-α-induced apoptosis.
INTRODUCTION
The tripartite motif (TRIM) family of proteins is defined by a set of domains known as the RBCC motif, which consists of the RING (really interesting new gene 1) finger, one or two B-box motifs, and a coiled-coil region (1). The RING finger is a zinc-binding domain with an important role in ubiquitin (Ub) E3 ligase binding to Ub-conjugating enzymes (E2) (2). The B box is structurally related to the RING domain and is required for various biological functions (3), although its roles remain unclear. The coiled-coil region is mainly involved in homo- and heterodimeric interactions for large-complex formation (4). Many TRIM proteins are induced by interferons (IFNs), which are crucial for resistance to pathogens, and a series of recent studies indicates that some TRIM proteins play an important role in the broader immune response (5). For instance, TRIM25 ubiquitinates the caspase recruitment domains (CARD) of RIG-I (retinoic acid inducible gene I), a cytosolic receptor for viral RNAs, and this ubiquitination is required for IFN-β production and NF-κB promoter activation (6). TRIM30α negatively regulates Toll-like receptor (TLR)-mediated NF-κB activation by targeting the adaptors TAB2 (transforming growth factor β-activated kinase 1 [TAK1]-binding protein 2) and TAB3 for degradation, which results in the inhibition of TRAF6 (TNF receptor-associated factor 6) activity and IκBα phosphorylation (7). TRIM56 induces K63-linked polyubiquitination of STING (stimulator of interferon genes), which regulates intracellular DNA-mediated, type I IFN-dependent innate immunity (8). This ubiquitination is required for STING dimerization, TBK1 (TANK [TRAF family member-associated NF-κB activator]-binding kinase 1) recruitment, and IFN-β promoter activation. TRIM23 mediates K27-linked ubiquitination of NEMO (NF-κB essential modulator), which is important for NF-κB activation downstream of RIG-like receptor and TLR3 in response to viral double-stranded RNA (dsRNA) (9).
TRIM27 (also known as the Ret finger protein [RFP]) is one of the TRIM family proteins and was originally found to be fused to the ret proto-oncogene in transformed NIH 3T3 cells (10). TRIM27 plays a role in transcriptional regulation through interaction with retinoblastoma protein (Rb) and the Mi-2β-containing histone deacetylase complex in the nucleus (11, 12). TRIM27 contains a nuclear export sequence and shuttles between the cytoplasm and the nucleus (13). In the cytosol, TRIM27 interacts with the IκB kinase (IKK) family of kinases and with TBK1, which inhibits NF-κB-dependent and IRF3-dependent transcriptional activation (14). This inhibition does not depend on the RING finger of TRIM27, and the mechanism underlying the negative regulation by TRIM27 has not been elucidated.
Tumor necrosis factor alpha (TNF-α) is a potent inflammatory cytokine that induces apoptosis of tumor cells but not normal cells, with the exception of hepatocytes, neural cells, and thymocytes (15). Dysregulation of TNF-α signaling is often correlated with autoimmune diseases, including type 1 diabetes and Crohn's disease (16). Recent studies have shown that ubiquitination plays a crucial role in TNF-α signaling (17). Upon binding to TNF-α, TNF receptor 1 (TNFR1) recruits TRADD (TNFR1-associated death domain protein) (18) and triggers the formation of two distinct complexes, complex I and complex II (19). The membrane-associated complex I, which activates NF-κB, contains TRADD, TRAF2, TRAF5, RIP1 (receptor-interacting protein kinase 1), cIAP1 (cellular inhibitor of apoptosis 1), and cIAP2. Both cIAP1 and cIAP2 are RING finger-containing ubiquitin ligases that catalyze the K63-linked polyubiquitination of RIP1 (20, 21). K63 linked polyubiquitin (poly-Ub) chains recruit TAK1 and IKK complexes by binding to the regulatory subunits TAB2 and NEMO, activating NF-κB to induce a group of cell survival genes (22). Activation of NF-κB via complex I triggers the dissociation of TRADD, RIP1, and TRAF2 from complex I and the formation of cytoplasmic complex II, which contains FADD (Fas-associated death domain protein) and procaspase-8. Procaspase-8 is processed into the mature caspase-8, which cleaves procaspase-3 into the mature effector caspase-3 to initiate apoptosis (19). The balance between complex I-dependent cell survival and complex II-dependent apoptosis is regulated by multiple factors, and TNF-α does not induce apoptosis in most cells. NF-κB activation rapidly induces the expression of several antiapoptotic proteins, including cIAPs and c-FLIP (cellular FLICE-inhibitory protein), which prevent the activation of procaspase-8. NF-κB upregulates the expression of A20, which forms a ubiquitin-editing complex that removes K63-linked poly-Ub chains to induce RIP1 degradation (23, 24). Another deubiquitinating enzyme, CYLD, which is the product of the cylindromatosis tumor suppressor gene CYLD, also specifically cleaves K63-linked poly-Ub chains bound to RIP1, releasing RIP1 from complex I and promoting the formation of complex II (25–27). However, additional factors regulating the balance between TNF-α-induced apoptosis and cell survival may exist.
In the present study, we showed that TRIM27 plays a role in the regulation of TNF-α-induced apoptosis by forming a complex with the Ub-specific protease USP7, leading to the removal of poly-Ub from RIP1.
MATERIALS AND METHODS
Generation of Trim-27−/− mice.
Trim27−/− mice were generated by homologous recombination in TT2 ES cells as described previously (28). The Trim27 gene was disrupted by replacing the 560-bp DNA fragment, which corresponds to the 430 bp upstream from the ATG initiation codon and the 130 bp of exon 1, with a neomycin cassette (see Fig. S1 in the supplemental material). This resulted in the deletion of the N-terminal initiator codon and the RING finger. Trim27−/− C57BL/6 congenic mice were generated by backcrossing onto a C57BL/6 genetic background for more than 10 generations. All mice used were 8- to 10-week-old males. Experiments were conducted in accordance with the guidelines of the Animal Care and Use Committee of the RIKEN Institute.
TNF-α–GalN-induced toxicity.
Mice were sensitized by intraperitoneal administration of GalN [d-(+)-galactosamine hydrochloride; Sigma] at 700 mg per kg body weight. After 15 min of GalN treatment, recombinant mouse TNF-α (20 μg/kg; Peprotech) was administered intraperitoneally, and the mortality rate was measured over the next 24 h.
Serum ALT and AST measurement.
Blood was collected from the saphenous vein at 4 h after TNF-α–GalN treatment, serum was prepared, and alanine aminotransferase (ALT) and aspartate transaminase (AST) activities were analyzed with a colorimetric assay kit according to the manufacturer's protocol (BioVision).
Assay of caspase 3 and caspase 8 activity.
Caspase-3 and -8 activities were measured using a caspase-3/8 colorimetric activity assay kit (Millipore). At 6 h after TNF-α–GalN treatment, livers were collected and homogenized in a Dounce homogenizer by extraction buffer (Millipore). After brief centrifugation, caspase-3 and -8 activities of the homogenate were measured according to the manufacturer's protocol.
TUNEL assay.
To examine apoptosis in hepatic cells, mouse livers were excised 6 h after treatment and fixed in 4% paraformaldehyde. Paraffin-embedded sections (5 μm) were analyzed by TUNEL (terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling) assay using an in situ cell death detection kit (Roche).
Histology and immunohistochemistry.
Histopathological analysis of paraffin-embedded liver tissue sections were performed by hematoxylin and eosin (H&E) staining. Cleaved caspase-3 immunohistochemistry was performed as described by Bellenger et al. (29) using anti-cleaved caspase-3 antibody (Cell Signaling Technology).
Cell culture and cell viability assay.
Mouse embryo fibroblasts (MEFs) were isolated from embryos at 14.5 days postcoitus. Spontaneously immortalized MEFs were isolated. MEFs, cells of the human hepatoma cell line HepG2, and cells of the human embryonic kidney cell line 293T were cultured in Dulbecco's modified Eagle's medium (Nissui Pharmaceuticals) with 10% bovine calf serum (HyClone) or fetal calf serum (Roche). To examine viability of MEFs, cells (1 × 104) were seeded into 96-well plates. At 24 h after incubation, cells were treated with various combinations of recombinant mouse TNF-α (Peprotech), cycloheximide (Sigma), anti-FAS antibody (BD Bioscience), TRAIL with enhancer antibody (Enzo Life Science), etoposide (Sigma), or the USP7 inhibitor HBX 41,108 (Calbiochem) for the times given in the figure legends, and cell viability was assessed colorimetrically using cell counting kit 8 (Dojindo, Tokyo, Japan).
Electrophoretic mobility shift assay (EMSA).
MEFs (1 × 106) were cultured for 24 h and treated with TNF-α (20 ng/ml) for times given in the figure legends. Nuclear extracts were prepared and subjected to EMSA using a 32P-radiolabeled double-stranded NF-κB probe (AGTTGAGGGGACTTTCCCAGGC) as described by Thapa et al. (30). For the supershift assay, nuclear extract was preincubated with anti-p65 antibody (Santa Cruz) on ice for 10 min before addition of radiolabeled NF-κB probe.
Flow cytometry.
One day after culture, MEFs were collected with trypsin, washed with phosphate-buffered saline (PBS) and fluorescence-activated cell sorting (FACS) buffer (1% bovine serum albumin [BSA]-PBS), blocked using Fc blocking reagent (BD Bioscience), and stained with phycoerythrin (PE)-conjugated anti-mouse TNFR antibody (Biolegend) for 30 min at 4°C. Cells were washed twice with FACS buffer and analyzed with a FACSCalibur flow cytometer (Becton, Dickinson).
Real-time RT-PCR.
Total RNA was isolated from MEFs using TRIzol (Invitrogen). Real-time reverse transcription-PCR (RT-PCR) was performed using an ABI 7500 real-time PCR instrument and the QuantiTect SYBR green one-step RT-PCR kit (Qiagen), according to the manufacturer's instructions. The PCR conditions were 50°C for 30 min, 95°C for 15 min, and 40 cycles of 94°C for 15 s, 60°C for 35 s, and 72°C for 35 s. The primer sequences are shown in Table S1 in the supplemental material. The relative level of mRNA expression was normalized to the amount of 18S rRNA using rRNA control reagents (Applied Biosystems).
Subcellular localization of TRIM27.
The subcellular localization of TRIM27 and USP7 was immunohistochemically examined as described previously (13). HepG2 cells or MEFs were transfected with 3 or 1.5 μg of the expression vector using Lipofectamine (Invitrogen). Forty-eight hours after transfection, cells were fixed and stained with anti-FLAG antibody M2 (Sigma) and MitoTracker red CMXRos (Molecular Probes) or rabbit polyclonal anti-USP7 antibody (Bethyl Laboratories). The signals were visualized by rhodamine- or fluorescein isothiocyanate-conjugated secondary antibodies (Jackson ImmunoResearch) and analyzed by confocal microscopy (Zeiss LSM510). For Nycodenz gradient fractionation, immortalized MEFs were transfected with the FLAG-TRIM27 or FLAG-RIP1 expression plasmid, harvested in ice-cold homogenizing buffer (10 mM Tris-HCl [pH 7.4], 250 mM sucrose, 5 mM EDTA, and protease inhibitor mixture), and passed 50 times through a Potter-Teflon homogenizer on ice. A postnuclear supernatant was obtained (3,000 rpm, 10 min, 4°C) and subjected to Nycodenz gradient fractionation as described previously (31).
Comparison of apoptosis-regulating factors by Western blotting.
MEFs were lysed by mild sonication in SDS sample buffer and boiled for 5 min. Protein concentration was determined by the Lowry method (Bio-Rad Laboratories), and 25 to 50 μg protein was typically subjected to SDS-PAGE. Anti-cleaved caspase-3 (Cell Signaling), anti-TNFR1 (R&D Systems), anti-TRADD (Santa Cruz), anti-TRAF2 (Santa Cruz), anti-RIP1 (BD Bioscience), anti-FADD (Santa Cruz), anti-CYLD (Santa Cruz), anti-USP7 (Bethyl Laboratories), anti-TRIM27 (IBL Chemicals), and anti-α-tubulin (Sigma) antibodies were utilized to check the respective endogenous protein levels by Western blotting.
In vivo ubiquitination assay.
For the ubiquitination assay of exogenously expressed proteins, HEK293T or HepG2 cells were transfected with a mixture of the plasmids indicated in the figure legends and empty vector using the calcium phosphate precipitation method. The plasmids used were pact-FLAG-RIP1, pact-FLAG-USP7, and pact-3xMyc-Ub to express various forms of Ub, pact-TRIM27 to express various forms of TRIM27, including FLAG-tagged or His-tagged TRIM27, a RING finger mutant, in which Cys-16, Cys-19, Cys-31, and His-33 were replaced with Ala, a B-box mutant, in which Cys-96, Cys-99, His-107, and Asp-110 were replaced by Ala, and a 4KR mutant, in which Lys-79, Lys-304, Lys-380, and Lys-382 were replaced with Arg. For immunoprecipitation of Flag-tagged proteins, at 40 h posttransfection, the cells were lysed in 1% SDS-containing radioimmunoprecipitation assay (RIPA) buffer by sonication. Lysates were then precleared and diluted with buffer lacking SDS to reduce the SDS concentration to 0.1% and immunoprecipitated overnight at 4°C with anti-FLAG M2 monoclonal antibody (Sigma). Immunocomplexes were captured with protein G-Sepharose beads (GE Health Care), washed three times with NETN buffer (20 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1% NP-40, 1 mM EDTA, 10% glycerol and protease inhibitor cocktail) containing 1 M NaCl and 2% NP-40, eluted with SDS sample buffer by boiling, and then separated by 7% SDS-PAGE and analyzed by immunoblotting. In the case of His-tagged TRIM27, transfected cells were scraped into urea buffer (8 M urea, 0.1 M sodium phosphate [pH 8.0], 0.3 M NaCl, 10 mM N-ethylmaleimide) and sonicated mildly on ice, and His-TRIM27 was purified using a His-selecting cobalt affinity gel (Sigma). Western blotting was performed as described above.
In the case of the endogenous ubiquitination assay, cells were stimulated with mouse TNF-α (10 ng/ml) and cycloheximide (1 μg/ml) and lysed with 1% SDS-containing RIPA buffer supplemented with 10 mM N-ethylmaleimide and protease inhibitors by vortexing and boiling for 5 min. Lysates were sonicated mildly on ice, diluted, precleared with Sepharose beads for 1 h at 4°C on a rotating wheel, and immunoprecipitated overnight at 4°C with anti-RIP-1 antibody (BD Biosciences) or anti-USP7 antibody (Bethyl Laboratories). Immunocomplex was captured, washed, eluted, and resolved as described above. Following transfer of proteins to nitrocellulose membranes, the membrane was autoclaved before incubation at denaturing buffer (6 M guanidine HCl, 20 mM Tris-HCl [pH 7.5], 5 mM beta-mercaptoethanol, 1 mM phenylmethylsulfonyl fluoride [PMSF]) for 30 min at 4°C. After extensive PBS washing, membrane was blocked at 20% heat-inactivated bovine calf serum and immunoblotting was performed with anti-Ub (Invitrogen) or anti-K63-linked Ub (Millipore) antibody using immunoreaction enhancer solution (Toyobo).
Purification and characterization of the TRIM27 complex.
The complex was purified as described previously (32). The HeLa S3 cell clone expressing FLAG–HA-TRIM27 was generated. Cells from a 4-liter culture were disrupted in hypotonic buffer, and the nuclear pellet was collected by centrifugation at 25,000 × g for 20 min. The pellet was extracted with buffer C (20 mM HEPES [pH 7.9], 25% glycerol, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM PMSF, and 0.5 mM dithiothreitol [DTT]), and lysates were collected by centrifugation. The TRIM27 complex was purified using anti-FLAG M2 monoclonal antibody (MAb)-conjugated agarose beads followed by anti-HA 12CA5 MAb-conjugated agarose beads in wash buffer. The purified proteins were separated by 4-to-20% gradient SDS-PAGE and silver stained. The protein bands were excised and analyzed by mass spectrometry. Anti-TRIM27 (IBL-America) or anti-USP7 (Bethyl Laboratories) antibody was used for immunoblotting.
Coimmunoprecipitation assays.
For coimmunoprecipitation experiments, 293T cells were directly used or transfected using the CaPO4 method with various plasmid mixtures, as described in the figure legends. Cells were collected and lysed by mild sonication in NETN buffer Lysates were then diluted, precleared, and subjected to immunoprecipitation by incubation with indicated antibodies for overnight at 4°C. The immunocomplexes were captured by protein G-Sepharose beads, washed three times with lysis buffer containing 0.2% NP-40, and separated by SDS-PAGE. Western blotting was performed using the antibodies described in the figure legends and the enhanced chemiluminescence (ECL) Western blotting system (Millipore). Aliquots of the lysates were also analyzed by Western blotting. For analysis of TNF-α-induced complex II, MEFs were treated with TNF-α (100 ng/ml) and cycloheximide (1 μg/ml) for the times indicated in the figure legends, and FADD/caspase-8/RIP1 complex was isolated and analyzed as reported by Ramakrishnan and Baltimore (33).
Knockdown of USP7.
The previously reported sequence of shRNA against human USP7 (34) was cloned into the pSuper.puro vector and transfected into 293T cells, which were selected with 1 μg/ml of puromycin (Sigma). Cell lysates from the isolated clones were used for Western blotting to examine the levels of USP7. For MEFs, the mouse Ufp7 gene was knocked down using small interfering RNA (siRNA) specific for mouse USP7 (target sequence, 5′-GACCCUGGAUUUGUGGUCACAUUAU-3′; Invitrogen). For gene-specific knockdown, immortalized MEFs were plated in 6-well plates and transfected with 10 μM control or USP7-specific siRNA using Lipofectamine RNAiMAX (Invitrogen). Forty-eight hours after transfection, cell lysates were prepared, and USP7 was detected by Western blotting.
RESULTS
TNF-α-induced apoptosis is impaired in Trim27-deficient mice.
To understand the physiological role of TRIM27, we generated Trim27-deficient (Trim27−/−) mice using homologous recombination in ES cells (see Fig. S1 in the supplemental material). Under pathogen-free conditions, Trim27−/− mice exhibited no obvious abnormality. We examined the abundance of CD4+ and/or CD8+ T cells, and also B220+ B cells, and there was no difference between wild-type (WT) and Trim27−/− mice (data not shown). As TRIM27 is a ubiquitin E3 ligase, and ubiquitination plays a role in the regulation of TNF-α signaling (17), we examined TNF-α signaling in Trim27−/− mice. We used a d-galactosamine (GalN)-sensitized mouse model of liver injury in which TNF-α induces hepatocyte apoptosis in the presence of GalN (35). GalN suppresses TNF-α survival signaling by inhibiting transcription in hepatocytes. WT mice treated with TNF-α and GalN died within 10 h, whereas 60% of Trim27−/− mice treated under the same conditions survived and lived even 24 h after injection (Fig. 1A). TNF-α–GalN-induced liver injury was observed preferentially in WT mice (Fig. 1B). The administration of TNF-α–GalN induced higher elevation of serum AST and ALT levels in WT than in Trim27−/− mice (Fig. 1C). Histological analysis of liver sections indicated that macrophages were infiltrated and hypostasis were evident in livers of TNF-α–GalN-treated WT mice (Fig. 1D). TUNEL staining of liver sections and immunostaining with anti-cleaved caspase 3 showed that the rate of apoptosis was higher in WT hepatocytes than in Trim27−/− cells (Fig. 1E and F). Furthermore, WT liver homogenates contained higher caspase-3 and caspase-8 activities than Trim27−/− liver homogenates (Fig. 1G). These results indicate that Trim27−/− hepatocytes are more resistant to the TNF-α–GalN-induced apoptosis than WT cells.
Fig 1.

TRIM27 positively regulates TNF-α-induced apoptosis. (A) Resistance of wild-type (WT) and Trim27−/− mice to TNF-α cytotoxicity. WT (n = 6) and Trim27−/− (n = 10) mice were treated with TNF-α (20 μg/kg) and GalN (700 mg/kg), and their mortality was monitored over next 24 h. (B) The liver of a mouse 6 h after TNF-α–GalN injection. (C) Serum AST and ALT levels were determined 4 h after TNF-α–GalN injection. Data are averages and standard errors of the means (SEM) (n = 3 for WT mice and 4 for Trim27−/− mice). *, P < 0.05. (D to F) Histological analysis (H&E staining) (D), TUNEL staining (E), and anti-cleaved caspase-3 immunostaining (F) were performed on liver sections 6 h after TNF-α–GalN injection. Infiltrated macrophages (arrows) and hypostasis are observed in the WT section. Scale bar, 10 μm. (G) Activities of caspase-3 and caspase-8 in liver homogenate of WT and Trim27−/− prepared at 6 h after TNF-α–GalN injection. Data are averages and SEM (n = 3). *, P < 0.05. (H) (Top) Immortalized MEFs from WT and Trim27−/− mice analyzed by Western blotting against TRIM27. The Trim27−/− immortalized MEF cell line expressing exogenous TRIM27 was established by infection with a TRIM27-expression retrovirus vector and was also used. α-Tubulin was examined as the control. (Bottom) WT and Trim27−/− MEFs (1 × 104 cells) were plated on 96-well plates and treated with TNF-α (10 ng/ml), or CHX alone (1 μg/ml), or in combination with TNF-α (10 ng/ml) and CHX (1 μg/ml). Six hours after treatment, cell viability was measured. Average values (n = 5) relative to the nontreated cells and standard deviations (SD) are shown. **, P < 0.01. The Trim27−/− MEF cell line expressing exogenous TRIM27 was used as the control. (I) Cleaved caspase-3 was determined by immunoblotting analysis of MEFs lysates prepared at 3 h after TNF-α–CHX addition.
TNF-α-induced apoptosis was further examined in primary MEFs in the presence of cycloheximide (CHX), which inhibits translation and thus inhibits TNF-α survival signaling. In WT MEFs, treatment with TNF-α or CHX alone did not inhibit cell viability, whereas the viability of MEFs treated with both TNF-α and CHX was significantly reduced (Fig. 1H). In contrast, TNF-α–CHX treatment only slightly reduced the cell viability of primary Trim27−/− MEFs. To verify that the lack of TRIM27 was responsible for this effect, a Trim27−/− MEF cell line expressing exogenous TRIM27 was generated by infection with the TRIM27-expression retrovirus vector (Fig. 1H, top). TNF-α–CHX reduced the cell viability of the Trim27−/− MEF cell line expressing TRIM27, similar to the effect on WT MEFs. When generation of cleaved caspase-3 was examined by Western blotting, TNF-α–CHX treatment induced higher level of cleaved caspase-3 in WT and Trim27−/− MEFs ectopically expressing TRIM27 than in Trim27−/− MEFs (Fig. 1I).
There was no difference in the expression level of TNF-α receptor between WT and Trim27−/− MEFs, as demonstrated by FACS and Western blotting (Fig. 2A and B). In addition, other key factors for TNF-α-induced apoptosis, TRADD, TRAF2, RIP1, caspase-8, FADD, and CYLD, were expressed at similar levels in WT and Trim27−/− MEFs (Fig. 2C).
Fig 2.

TRIM27 is not involved in TNF-α-induced survival signaling. (A) TNFR1 expression. TNFR1 expression in WT and Trim27−/− MEFs was examined by flow cytometry. (B) Expression of TNF-α signaling components. Expression levels of the indicated proteins were compared between WT and Trim27−/− MEFs by Western blotting. α-Tubulin was used as a control. (C) Analysis of the expression of survival-related genes. Primary MEFS from WT and Trim27−/− mice were treated with TNF-α (20 ng/ml), and the expression levels of four NF-κB-dependent survival-related genes were analyzed by qRT-PCR. Values are means ± SD (n = 3). (D) EMSA of NF-κB after treatment of MEFs with TNF-α. TNF-α (20 ng/ml) was added to the culture of WT and Trim27−/− MEFs, and nuclear extracts were prepared at the indicated times. EMSA was performed using the NF-κB DNA probe. Where indicated, anti-p65 antibodies were added, so that supershift of the NF-κB-DNA band was observed. To show the supershifted bands more clearly, a shorter exposure is shown on the right. (E) Analysis of complex II. Lysates of WT and Trim27−/− MEFs were prepared at the indicated times after addition of TNF-α (100 ng/ml)–CHX (1 μg/ml) and immunoprecipitated with anti-FADD, followed by Western blotting to detect RIP1, caspase-8, and FADD.
To determine whether TRIM27 plays a role in TNF-α survival signaling, such as through NF-κB target gene expression, which results in the inhibition of apoptosis, the expression of typical survival-related genes, including cFLIP, cIAP2, A20, and ICAM was assessed in the presence or absence of TNF-α. The results showed that the expression levels of these genes were similar between WT and Trim27−/− primary MEFs (Fig. 2C). The NF-κB activity in WT and Trim27−/− MEFs was examined using EMSA. At various times after TNF-α–CHX treatment, similar levels of NF-κB activities were detected in WT and Trim27−/− MEFs (Fig. 2D). These results indicate that TRIM27 does not affect TNF-α survival signaling. Trim27 mRNA level was also not significantly affected by TNF-α treatment (see Fig. S2A in the supplemental material).
To investigate the role of TRIM27 in the complex II-dependent apoptosis pathway, complex II formation was compared between WT and Trim27−/− MEFs. In WT cells, RIP1 and caspase-8 were coimmunoprecipitated with FADD 90 min after TNF-α–CHX treatment (Fig. 2E). However, the levels of RIP1 and caspase-8 coimmunoprecipitated with FADD were lower in Trim27−/− cells than in WT cells. These results suggest that TRIM27 is involved in the complex II-dependent apoptosis pathway but not in the complex I-dependent survival signaling pathway.
To test whether TRIM27 has a general role in the apoptosis induced by various stimuli, Fas-, TRAIL-, or etoposide-induced apoptosis was compared between WT and Trim27−/− MEFs. There was no difference in the Fas-, TRAIL-, or etoposide-induced apoptosis between WT and Trim27−/− MEFs (see Fig. S3A to C in the supplemental material), suggesting that TRIM27 does not have a general role in the various types of apoptosis.
Localization of TRIM27 to mitochondria.
Our previous studies suggested that TRIM27 is localized both in the nucleus and in the cytosol (13). As the data above suggest that TRIM27 is involved in TNF-α-induced apoptosis in hepatocytes and MEFs, we examined the subcellular localization of TRIM27 in the human hepatocyte-derived cell line HepG2 and in MEFs. Immunostaining for TRIM27 in HepG2 cells showed a predominantly cytoplasmic pattern, in addition to weak nuclear signals (Fig. 3A). Cytoplasmic TRIM27 signals partly overlapped with those of MitoTracker, a mitochondrion-selective fluorescent label, indicating the partial localization of TRIM27 to mitochondria. A similar pattern of overlapping of TRIM27 with MitoTracker was also observed in MEFs (see Fig. S4A in the supplemental material). Furthermore, TRIM27 subcellular localization was not affected by TNF-α or TNF-α–CHX treatment. These results were consistent with a prior study that showed that certain signaling molecules involved in TNF-α signaling, such as RIP1, localize to mitochondria (36). To confirm the mitochondrial localization of TRIM27, the cytosolic lysate was separated by Nycodenz gradient centrifugation, and the resulting fractions were analyzed by Western blotting with antibodies against TRIM27, mitochondrion-specific HSP70 (mtHSP70), and PER5. TRIM27 was detected in small amounts in the mitochondrial fraction, as shown with the anti-mtHSP70 antibody (Fig. 3B), and in large amounts in the peroxisome. Although the role of TRIM27 in the peroxisome is unknown, a recent report showed that the peroxisome is the signaling platform for antiviral innate immunity (37).
Fig 3.
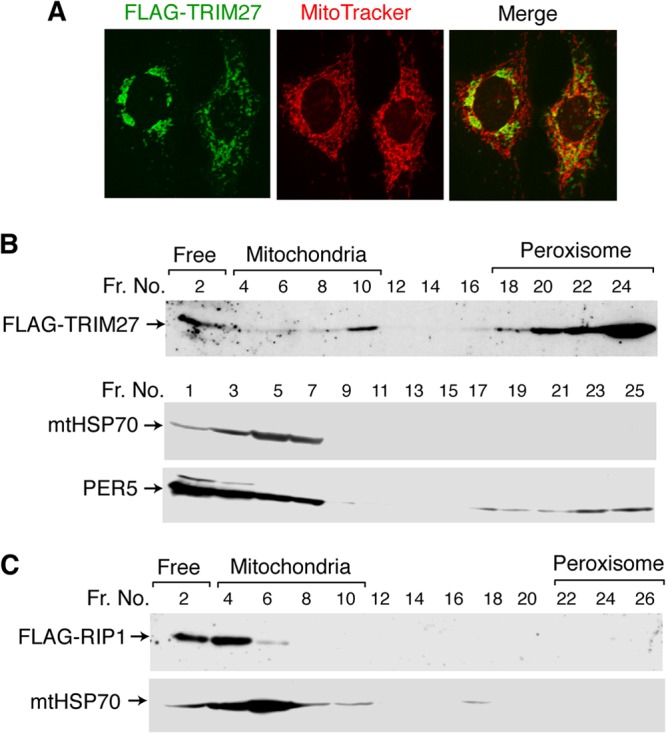
Localization of TRIM27 in mitochondria. (A) Subcellular localization of TRIM27. HepG2 cells were transfected with a FLAG-TRIM27 expression vector, incubated with Mitotracker, fixed with 1% paraformaldehyde, and incubated with an anti-FLAG antibody. After treatment with the corresponding secondary antibodies, fluorescence signals were visualized with a laser confocal microscope. The far right panel shows the merged signals for Mitotracker (red) and TRIM27 (green). (B) Subcellular fractionation experiments. Cytosolic fractions of immortalized WT MEFs transfected with FLAG-TRIM27 expression plasmids were subjected to Nycodenz gradient fractionation. The levels of FLAG-TRIM27, the mitochondrial marker mtHSP70, and the peroxisome marker PER5 in each fraction were analyzed by immunoblotting. (C) Localization of RIP1 in mitochondria. Cytosolic fractions of immortalized WT MEFs transfected with FLAG-RIP1 expression plasmids were subjected to Nycodenz gradient fractionation as described above. The levels of FLAG-RIP1 and mtHSP70 in each fraction were analyzed by immunoblotting.
To further examine the mitochondrial location of TRIM27, the mitochondrial fraction was isolated from HepG2 cells transfected with the FLAG-TRIM27 expression vector and analyzed by Western blotting (see Fig. S5A in the supplemental material), which showed that a significant amount of TRIM27 was present in mitochondria (see Fig. S5B in the supplemental material). The isolated mitochondrial fraction was then treated with digitonin to disrupt the mitochondrial outer membrane or proteinase K, which degrades proteins on the surface of the outer membrane. The pellet fraction after digitonin treatment (mitoplast) contained TRIM27, suggesting that TRIM27 is localized in the mitoplast or tightly associated with the inner membrane. Furthermore, proteinase K digestion of the mitochondrial fraction resulted in the loss of the FLAG tag containing the N-terminal portion, suggesting that the N-terminal portion of TRIM27 is located on the outer mitochondrial membrane (see Fig. S5C in the supplemental material).
TRIM27 induces RIP1 deubiquitination by forming a complex with USP7.
TRIM25, an RBCC motif-containing protein, directly ubiquitinates RIG-I and promotes RIG-I-dependent interferon production in response to dsRNA (6). We therefore hypothesized that the effect of TRIM27 on TNF-α-induced apoptosis may be mediated by ubiquitination of a regulatory factor. Of the several candidates examined, TRIM27 affected the ubiquitination status of only RIP1. However, contrary to its expected function as a Ub E3 ligase, TRIM27 deubiquitinated RIP1 (Fig. 4A). In the coimmunoprecipitation assay, RIP1 was coprecipitated with TRIM27 (Fig. 4B), suggesting the interaction between TRIM27 and RIP1. Isolation of the cytosolic fraction of HepG2 cells using Nycodenz gradient centrifugation indicated that RIP1 also localized to mitochondria (Fig. 3C), as reported previously (36). We previously showed that TRIM27 shuttles between the cytoplasm and the nucleus via its nuclear export signal (NES) (13). The TRM27 NES mutant, which is mainly localized in the nucleus, did not induce RIP1 deubiquitination (Fig. 4C), indicating that deubiquitination of RIP1 is mediated by cytosolic TRIM27.
Fig 4.

TRIM27 deubiquitinates RIP1. (A) Deubiquitination of RIP1 by TRIM27. HEK293T cells were transfected with the FLAG-RIP1 expression vector or control empty vector, together with the Myc-Ub expression vector and increasing amounts of the TRIM27 expression vector. (Top) Cell lysates were immunoprecipitated with an anti-FLAG antibody, and immunocomplexes were analyzed by Western blotting against anti-Myc. (Bottom) Cell lysates were probed against an anti-FLAG antibody. (B) Coimmunoprecipitation of TRIM27 with RIP1. HEK293T cells were transfected with vectors expressing FLAG-RIP1 and T7-TRIM27, and cell lysates were immunoprecipitated with anti-T7 antibody or control IgG, followed by Western blotting with anti-FLAG or anti-T7. (C) RIP1 deubiquitination is impaired with the TRIM27 NES mutant. Experiments were performed as described for panel A using 2 μg of WT and NES mutant of TRIM27, which cannot be exported from the nucleus. (D) Analysis of the ubiquitin chains removed from RIP1. Experiments were performed as described for panel A, except for the use of different vectors expressing the indicated ubiquitin mutants instead of WT ubiquitin.
A polyubiquitin (poly-Ub) chain is generated through the formation of a link between one of the seven Lys side chains of Ub and the C-terminal Gly of another Ub. To determine the type of poly-Ub chain on RIP1 that is removed by TRIM27, we used Ub mutants in which all but one Lys residue are replaced by Arg. Expression of Ub mutants with active K27, K29, or K48 linkages resulted in the TRIM27-induced deubiquitination of RIP1, whereas K11- and K63-linked poly-Ub was removed only slightly by TRIM27 (Fig. 4D). In contrast, K6- and K33-linked poly-Ub was not removed by TRIM27. These results suggest that TRIM27 removes K11-, K27-, K29-, K48-, or K63-linked poly-Ub from RIP1. However, the poly-Ub chains bound to RIP1 may have a complex structure with various Lys linkages; therefore, the specificity of TRIM27 for a particular chain remains unclear.
To understand the mechanism underlying the deubiquitination of RIP1 by TRIM27, TRIM27-interacting proteins were identified by purifying the complex formed. HeLa cells were infected with a retrovirus encoding FLAG- and HA-tagged TRIM27 (FH-TRIM27), and a cell line expressing FH-TRIM27 was isolated. The FH-TRIM27-containing complex was purified using anti-FLAG and anti-HA antibodies, and analyzed by SDS-PAGE. This complex contained a 120-kDa band in addition to FH-TRIM27 (Fig. 5A), which was identified by mass spectrometry as USP7 (also known as HAUSP [herpesvirus-associated ubiquitin-specific protease]), a ubiquitin-specific protease. USP7 colocalized with TRIM27 in the cytoplasm and in the nucleus of HepG2 cells (Fig. 5B) and MEFs (see Fig. S4B in the supplemental material), suggesting that these proteins interact. Furthermore, USP7 coimmunoprecipitated with TRIM27 in HepG2 cells transfected with the TRIM27 expression vector (Fig. 5C).
Fig 5.

USP7 is required for the TRIM27-induced deubiquitination of RIP1. (A) Formation of a complex between TRIM27 and USP7. The TRIM27 complex was purified from HeLa cells expressing FLAG- and HA-tagged TRIM27 (FH-TRIM27) or control HeLa cells (mock) using anti-FLAG and anti-HA antibodies and analyzed by SDS-PAGE followed by silver staining. Mass spectrometry analysis identified the two bands indicated by arrows as USP7 and FH-TRIM27. (B) Colocalization of TRIM27 and USP7. HepG2 cells were transfected with vectors expressing T7-tagged TRIM27 and FLAG-USP7 and immunostained with anti-T7 and anti-FLAG antibodies. After treatment with the corresponding secondary antibodies, fluorescence signals were visualized under a laser confocal microscope. Merged signals for T7-TRIM27 and FLAG-USP7 are shown on the far right panel. (C) Coimmunoprecipitation of TRIM27 and USP7. HEK 293T cells were transfected with the FLAG-TRIM27 expression vector or control empty vector, and cell lysates were immunoprecipitated with anti-FLAG antibody or control IgG. The immunocomplexes were subjected to Western blotting using anti-FLAG or anti-USP7 antibodies. (D) Generation of USP7 knockdown 293T cells. The 293T cell transfectants expressing the small hairpin RNA against the USP7 gene were isolated, and cell lysates were analyzed by Western blotting against anti-USP7. Equal amounts of total protein were loaded in each lane. (E) Knockdown of USP7 abrogates the TRIM27-induced deubiquitination of RIP1. Experiments were performed as described for Fig. 3A, using USP7 knockdown cells (clone 44) and control 293T cells. (F) Coimmunoprecipitation of endogenous TRIM27 with RIP1 and USP7. Whole-cell lysates from HEK 293T cells were immunoprecipitated with anti-TRIM27 or control IgG, and the immunocomplexes were analyzed by SDS-PAGE, followed by Western blotting with anti-RIP1, anti-USP7, or anti-TRIM27.
To examine whether USP7 is needed for the TRIM27-induced deubiquitination of RIP1, we generated USP7-knockdown 293T cell lines by introducing an siRNA expression vector. Among multiple cell lines isolated, one clone (clone 44) expressed USP7 at a significantly low level (Fig. 5D). When RIP1 was coexpressed with increasing amounts of TRIM27 in this cell line, RIP1 deubiquitination was not observed (Fig. 5E), indicating that USP7 is required for the TRIM27-induced deubiquitination of RIP1. These results suggest that the TRIM27-USP7 complex binds to RIP1, resulting in its deubiquitination. To further examine this interaction, we performed coimmunoprecipitation assays. Using 293T cell lysates, endogenous RIP1 and USP7 were coimmunoprecipitated with endogenous TRIM27 (Fig. 5F). However, in 293T cells ectopically expressing the three proteins, TRIM27 was coimmunoprecipitated with USP7 but not with RIP1 (see Fig. S6A in the supplemental material). When the USP7 mutant (C223A), in which the catalytic domain of the protease was disrupted by point mutation (38), was used, TRIM27 was coimmunoprecipitated with both USP7 and RIP1 (see Fig. S6B in the supplemental material). These results indicated that these three proteins interact, but RIP1 is immediately released after its deubiquitination by USP7. There was no difference in the USP7 mRNA level between WT and Trim27−/− MEFs (see Fig. S2B in the supplemental material), indicating that TRIM27 does not regulate TNF-α-induced apoptosis by regulating the USP7 level.
Ubiquitination of USP7 by TRIM27 is required for the TRIM27-induced deubiquitination of RIP1.
To examine the role of TRIM27 in the TRIM27/USP7 complex-induced deubiquitination of RIP1, we investigated whether TRIM27 ubiquitinates USP7. Ubiquitination assays in 293T cells showed that the WT and the RING finger TRIM27 mutant, in which four Zn-binding residues of the RING finger were replaced by Ala, ubiquitinated USP7, while the B-box mutant, in which four Zn-binding residues of the B box were replaced by Ala, did not (Fig. 6A and B). Similar results were obtained by using the USP7 catalytic domain mutant (C223A) (see Fig. S6C in the supplemental material). As both the RING finger and the B-box mutants interacted with USP7 (see Fig. S6D in the supplemental material), these results suggest that the B box of TRIM27 acts as the catalytic domain for the ubiquitination of USP7, or that the B box of TRIM27 recruits another Ub E3 ligase. As Lys-869 of USP7 is ubiquitinated (39), we examined the possible ubiquitination of USP7 at Lys-869. TRIM27 did not ubiquitinate the USP7-K869R mutant in which Lys-869 was mutated to Arg (USP7-K869R) (Fig. 6C). Expression of siRNA-resistant WT USP7 mRNA in USP7 knockdown 293T cells recovered the TRIM27-induced RIP1 deubiquitination (Fig. 6D, left, and Fig. 5E), whereas overexpression of siRNA-resistant USP7-K869R mRNA did not (Fig. 6D, right). These results indicate that the ubiquitination of USP7 by TRIM27 is required for the TRIM27-induced deubiquitination of RIP1.
Fig 6.

TRIM27 induces RIP1 deubiquitination by ubiquitinating and activating USP7. (A) Domain structure of TRIM27. (B) TRIM27 ubiquitinates USP7 via its B-box domain. HEK293T cells were cotransfected with FLAG-USP7 and Myc-ubiquitin expression vectors and a plasmid for the expression of the indicated form of TRIM27. (Top) Cell lysates were immunoprecipitated with anti-FLAG followed by immunoblotting against anti-Myc. (Bottom) Lysates were analyzed by Western blotting with anti-FLAG or anti-TRIM27 antibodies. (C) USP7 Lys-869 is ubiquitinated by TRIM27. HEK293T cells were transfected with an expression vector for FLAG-linked WT or the USP7 K869R mutant, in which Lys-869 is mutated to Arg, together with Myc-ubiquitin and TRIM27 expression vectors. (Top) Cell lysates were immunoprecipitated with anti-FLAG antibody, followed by immunoblotting against anti-Myc. (Bottom) Lysates were analyzed by Western blotting with anti-FLAG antibody. (D) TRIM27 ubiquitination of USP7 at Lys-869 is required for TRIM27-induced RIP1 deubiquitination. The USP7 knockdown HEK293T cells and the control 293T cells were transfected with the FLAG-RIP1 expression vector, together with vectors expressing Myc-ubiquitin and WT USP7 or the K869R USP7 mutant, which was resistant to USP7 shRNA, and increasing amounts of the TRIM27 expression vector. (Top) Cell lysates were immunoprecipitated with anti-FLAG antibody, and the immunocomplexes were subjected to Western blotting with anti-Myc antibody. (Bottom) Lysates were analyzed by Western blotting with anti-TRIM27, anti-FLAG, or anti-USP7 antibodies. (E) Analysis of polyubiquitin chains attached to USP7. Ubiquitination of the USP7 catalytic mutant C223A, which lacks ubiquitin protease activity, was examined as described for panel C, except for the use of the USP7-C223A mutant instead of WT USP7 and the indicated mutant ubiquitin instead of WT ubiquitin.
When TRIM27 was expressed with USP7, the TRIM27 level was increased (Fig. 6B). To further confirm this, we examined the effect of increasing amounts of USP7 on the TRIM27 level and its ubiquitination. TRIM27 level was increased by USP7 in a dose-dependent manner, and the degree of TRIM27 ubiquitination per TRIM27 molecule decreased (see Fig. S7A in the supplemental material). These results suggest that USP7 stabilizes TRIM27 by removal of poly-Ub from TRIM27.
As TRIM27 is a ubiquitin E3 ligase, we examined whether TRIM27 is autoubiquitinated via its RING finger domain. The degree of ubiquitination of the TRIM27 RING finger mutant was much lower than that of the WT (see Fig. S7B in the supplemental material). Next we determined the ubiquitinated sites of TRIM27 by mass spectrophotometry. Mutation of four ubiquitination sites (4KR) in TRIM27 dramatically decreased its ubiquitination (see Fig. S7C in the supplemental material). Two TRIM27 mutants, RING finger and 4KR mutants, were localized exclusively and mainly in the nuclei, respectively (see Fig. S7D in the supplemental material). Thus, autoubiquitination of TRIM27 may be required for its localization in the cytoplasm. Furthermore, the RING finger mutant of TRIM27 was colocalized with USP7 in the nucleus, while the 4KR mutant was not. This may be due to the decreased interaction between 4KR mutant and USP7 (see Fig. S7E in the supplemental material).
To analyze the nature of the TRIM27-generated poly-Ub linkage on USP7, various Ub mutants in which all but one Lys residue were mutated to Arg were used for ubiquitination assays. TRIM27 catalyzed the Lys-linked polyubiquitination of USP7 in all cases (Fig. 6E), suggesting that TRIM27 mediates the addition of poly-Ub chains to USP7 without selectivity for a particular linkage type.
Deubiquitination of RIP1 by the TRIM27-USP7 complex is required for TNF-α-induced apoptosis.
To further confirm that USP7 plays a role in TNF-α-induced apoptosis, USP7 mRNA was downregulated in immortalized MEF by using mouse siRNA. Western blot analysis showed that cells treated with siRNA sequence 407 expressed significantly lower levels of USP7 than control cells (Fig. 7A). Downregulation of USP7 made the cells resistant to TNF-α–CHX-induced apoptosis compared to control cells (Fig. 7B). Furthermore, when WT MEFs were treated with the USP7 inhibitor HBX (40), cells were also resistant to TNF-α–CHX-induced apoptosis (see Fig. S8A in the supplemental material). Downregulation of USP7 of HBX treatment also suppressed the generation of cleaved caspase-3 (Fig. 7C; also, see Fig. S8B in the supplemental material).
Fig 7.

TRIM27-USP7 regulation of RIP1 ubiquitination in the presence of TNF-α and CHX. (A) Knockdown of USP7 using siRNA in immortalized MEF. Three different sequences of mouse USP7 siRNA were transfected, and cell lysates were analyzed by Western blotting with anti-USP7 (top) or anti-tubulin (bottom) antibodies. (B) USP7 is involved in TNF-α–CHX-induced apoptosis. Cell viability was examined as described for Fig. 1H using control immortalized MEFs and USP7 knockdown cells. Experiments were repeated three times, and average values relative to the nontreated cells with SD are shown. **, P < 0.01. (C) Cleaved caspase-3 was determined by immunoblot analysis of MEFs lysates prepared at 2.5 h after TNF-α–CHX addition. (D and E) Ubiquitination of endogenous RIP1 after TNF-α–CHX treatment. Immortalized WT, Trim27−/− (D) or USP7 knockdown (E) MEFs were treated with TNF-α (10 ng/ml) and CHX (1 μg/ml) for the indicated times. Endogenous RIP1 was immunoprecipitated and analyzed by immunoblotting with anti-Ub (top) or anti-K63-linked Ub (bottom) antibody. Lysates were also analyzed by Western blotting with anti-RIP1 or anti-USP7 antibodies.
We next examined the ubiquitination of endogenous RIP1 in response to TNF-α–CHX treatment using WT, Trim27−/−, and USP7 knockdown MEF cell lines. The level of ubiquitinated RIP1 in Trim27−/− cells and USP7 knockdown cells was higher than in WT and control cells (Fig. 7D and E). The difference in the degree of RIP1 ubiquitination between WT and Trim27−/− MEFs and between WT and USP7 knockdown MEFs was more evident at 30 and 120 min after TNF-α–CHX treatment than at 0 min after treatment. This is consistent with the results that TRIM27 is involved in the TNF-α-induced apoptosis but not in the TNF-α-induced NF-κB activation, which occurs early after TNF-α stimulation. We also examined the K63-linked ubiquitination of RIP1. The level of K63-linked ubiquitination of RIP1 in Trim27−/− cells and USP7 knockdown cells was higher than in WT and control cells (Fig. 7D and E). However, the difference in the degree of K63-linked RIP1 ubiquitination between WT and Trim27−/− MEFs and between WT and USP7 knockdown MEFs was much less than the difference in the total ubiquitination of RIP1. As there was no significant difference in the NF-κB activity between WT and Trim27−/− MEFs (Fig. 2D), the K63-linked ubiquitinated RIP1 detected in Trim27−/− MEFs may contain the complex structure of poly-Ub together with K63-linked Ub, so that it may not function to activate the survival signaling. These results further support that the TRIM27/USP7 complex plays a role in TNF-α-induced apoptosis by mediating RIP1 deubiquitination.
We have also examined the degree of ubiquitination of endogenous USP7. There was no significant difference in the level of ubiquitinated USP7 between WT and Trim27−/− cells (see Fig. S9 in the supplemental material), suggesting that TRIM27 is a not a major ubiquitin E3 ligase for USP7, which affects the total level of ubiquitinated USP7 (see Discussion).
DISCUSSION
The results of the present study indicate that Trim27-deficient mice are resistant to TNF-α–GalN-induced apoptosis and that TRIM27 forms a complex with and polyubiquitinates USP7, which deubiquitinates RIP1 (Fig. 8). There may be multiple forms of ubiquitinated RIP1 which contain a complex structure of Ub in addition to K63-linked Ub. The deubiquitination of various forms of ubiquitinated RIP1 may trigger the formation of complex II, leading to the induction of apoptosis. In the signaling events downstream of complex I, K63-linked polyubiquitination of RIP1 is essential for the activation of NF-κB and thus the induction of cell survival-related genes, because this polyubiquitin chain binds to TAB2 and NEMO, activating TAK1 and IKK complexes (Fig. 8) (17, 20–22). The TRIM27/USP7 complex removed K27-, K29-, and K48-linked poly-Ub chains from RIP1 but had little effect on K11- and K63-linked poly-Ub. Consistent with this, TRIM27 knockdown did not affect the expression of survival-related genes. The addition of K11-linked poly-Ub to RIP1, which binds to NEMO, has been reported (41), but its role in the induction of survival genes is unknown. Although K48-linked ubiquitination is known to target proteins for proteasomal degradation, knockdown of TRIM27 did not increase RIP1 protein levels. These results suggest that RIP1 is modified by complex poly-Ub chains containing K11-, K27-, K29-, K48-, and K63-linked Ub. The Ub structure specificity of TRIM27 differs from that of CYLD and A20, which remove K63-linked poly-Ub chains from RIP1 and suppress the expression of survival-related genes (23–27). In the absence of TNF-α treatment, the degree of ubiquitination of RIP1 in Trim27−/− cells was higher than that in WT cells, and this difference became more pronounced 30 and 120 min after TNF-α–CHX treatment (Fig. 7E). These results suggest that TRIM27-dependent deubiquitination of RIP1 occurs constitutively in the absence of TNF-α, and its role in the generation of nonubiquitinated RIP1 becomes more evident in cells exposed to TNF-α, in which the expression of survival genes, including the RIP1-deubiquitinating enzymes A20 and CYLD, is suppressed. TRIM27 ubiquitination of USP7 at Lys-869 is required for USP7-induced deubiquitination of RIP1. The B-box mutant of TRIM27 was able to bind to USP7 but failed to ubiquitinate USP7, whereas the RING finger mutant of TRIM27 ubiquitinated USP7. TRIM family proteins have two types of B boxes, 1 and 2, and TRIM27 has only B-box 2. The two types of B box have a similar structure to that of the RING finger (3). Our results suggest that the B box functions as a Ub E3 ligase or that the B box recruits another Ub E3 ligase. The ability of TRIM27 to add K6-, K11-, K27-, K29-, K33-, K48-, and K63-linked poly-Ub to USP7 suggests that the structure of the poly-Ub chains attached to USP7 is complex. USP7 regulates the level of p53 and Mdm2 by inhibiting their degradation via removal of the K48-linked poly-Ub from these proteins (42). Nonubiquitinated USP7 actively removes poly-Ub from p53 and Mdm2, indicating that the polyubiquitination of USP7 is not required for its protease activity. Furthermore, nonubiquitinated USP7 forms a complex with TRIM27, indicating that the polyubiquitination of USP7 is not required for TRIM27/USP7 complex formation. Therefore, the Ub chain on USP7 may function to recruit RIP1 to the TRIM27/USP7 complex. The catalytic site mutant of USP7, but not WT USP7, forms a complex with RIP1, suggesting that RIP1 is released immediately after deubiquitination by USP7. Therefore, the attachment of poly-Ub to USP7 may stabilize the transient RIP1/USP7/TRIM27 complex.
Fig 8.

Model describing the role of TRIM27-USP7 in TNF-α-induced apoptosis. Upon binding to TNF-α, TNFR1 binds to TRADD and triggers the formation of complex I and II. Complex I, containing TRADD, TRAF2, TRAF5, RIP1, cIAP1, and cIAP2, activates NF-κB. The ubiquitin ligases cIAP1 and cIAP2 catalyze the K63-linked polyubiquitination of RIP1, which binds to TAB2 and NEMO. This induces activation of TAK1 and IKK complexes, followed by NF-κB activation to induce the transcription of survival genes. Activation of NF-κB triggers the dissociation of RIP1 from complex I and the formation of complex II containing FADD and procaspase-8, which induces apoptosis. Multiple forms of ubiquitinated RIP1, which contains not only K63-linked Ub but also other various forms of Ub, may exist. TRIM27 forms a complex with and ubiquitinates USP7, which deubiquitinates RIP1, upregulating complex II-dependent apoptosis.
There was no significant difference in the level of ubiquitinated USP7 between WT and Trim27−/− cells. TRIM27 ubiquitinates USP7 at Lys-869, which stimulates the USP7 activity. It is likely that USP7 is ubiquitinated at other sites by other ubiquitin E3 ligases, for instance, for proteasome-dependent degradation. The presence of similar levels of ubiquitinated USP7 in WT and Trim27−/− cells suggests that TRIM27 is a not a major ubiquitin E3 ligase for USP7, which affect the total level of ubiquitinated USP7.
TRIM27 and RIP1 were partly localized to mitochondria and peroxisomes. USP7 was also shown to localize to mitochondria (43). However, as the TRIM27/USP7 complex may have multiple functions, these data do not necessarily indicate that RIP1 deubiquitination occurs in mitochondria. Recently, the peroxisome was shown to function as the signaling platform for antiviral innate immunity (37); therefore, the TRIM27/USP7 complex could play a role in innate immunity. In fact, TRIM27 was suggested to inhibit the release of human immunodeficiency virus type 1 (HIV-1) and murine leukemia virus (MLV) and to inhibit MLV gene expression (44). TRIM27 and USP7 were also partly colocalized in the nucleus, suggesting their possible involvement in transcriptional regulation.
It was shown that USP7 binds to TRAF2 and TRAF6 and suppresses TNF-α-induced NF-κB activation (45). However, TRIM27 did not affect the TNF-α-dependent NF-κB target gene expression (Fig. 2C and D), and TRIM27 did not affect the TRF6 ubiquitination (data not shown). Furthermore, it is unlikely that all the USP7 proteins form the complex with TRIM27. Therefore, TRIM27 may not interact with TRAF2/6 together with USP7. Overexpression of TRIM27 suppresses interleukin 1-, virus infection- and TNF-α-induced cytokine production, and TRIM27 has been shown to bind to IKKα/IKKβ and suppress their activities (14). However, in the present study, TRIM27 knockdown did not affect the expression of survival-related genes. Overexpression of certain TRIM proteins may result in a specific phenotype that is not induced by loss of function. In particular, because the TRIM family of proteins can form heteromultimers via the coiled-coil region, the overexpression of certain TRIM proteins may affect the function of other members of the family. TRIM21 ubiquitinates TRIM5 and targets it for degradation (46), suggesting that the overexpression of TRIM27 could affect the level of another TRIM protein(s). TRIM27 has also been shown to activate Jun N-terminal kinase (JNK) and induce apoptosis (47), suggesting another possible function that needs to be investigated through loss-of-function assays.
A recent study that used Trim27-deficient mice, which were generated independently from ours, showed that TRIM27 catalyzes the K48-linked ubiquitination of the class II phosphatidylinositol 3 kinase C2β (PI3KC2β), promoting inhibition of its enzymatic activity, and negatively regulates CD4 T cells by inhibiting TCR-stimulated Ca2+ influx and cytokine production (48). TRIM27 also negatively regulates IgE receptor activation and downstream signaling by the same mechanism (49). Further analyses using Trim27−/− mice may help to elucidate the uncharacterized physiological roles of TRIM27.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to M. Takahashi for human TRIM27 (RFP) cDNA, T. Nagase for human USP7 cDNA, Y. Nakatani for the pOZ-FH-N vector, the staff of the Research Resources Center of the RIKEN Brain Science Institute for mass spectrometric analysis, and the members of the Experimental Animal Division of the RIKEN Tsukuba Institute for maintenance of the mice.
This work was supported in part by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Footnotes
Published ahead of print 21 October 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00465-13.
REFERENCES
- 1. Meroni G, Diez-Roux G. 2005. TRIM/RBCC, a novel class of ‘single protein RING finger' E3 ubiquitin ligases. Bioessays 27:1147–1157 [DOI] [PubMed] [Google Scholar]
- 2. Joazeiro CA, Weissman AM. 2000. RING finger proteins: mediators of ubiquitin ligase activity. Cell 102:549–552 [DOI] [PubMed] [Google Scholar]
- 3. Massiah MA, Simmons BN, Short KM, Cox TC. 2006. Solution structure of the RBCC/TRIM B-box1 domain of human MID1: B-box with a RING. J. Mol. Biol. 358:532–545 [DOI] [PubMed] [Google Scholar]
- 4. Reymond A, Meroni G, Fantozzi A, Merla G, Cairo S, Luzi L, Riganelli D, Zanaria E, Messali S, Cainarca S, Guffanti A, Minucci S, Pelicci PG, Ballabio A. 2001. The tripartite motif family identifies cell compartments. EMBO J. 20:2140–2151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ozato K, Shin DM, Chang TH, Morse HC., III 2008. TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 8:849–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, Jung JU. 2007. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446:916–920 [DOI] [PubMed] [Google Scholar]
- 7. Shi M, Deng W, Bi E, Mao K, Ji Y, Lin G, Wu X, Tao Z, Li Z, Cai X, Sun S, Xiang C, Sun B. 2008. TRIM30α negatively regulates TLR-mediated NF-κB activation by targeting TAB2 and TAB3 for degradation. Nat. Immunol. 9:369–377 [DOI] [PubMed] [Google Scholar]
- 8. Tsuchida T, Zou J, Saitoh T, Kumar H, Abe T, Matsuura Y, Kawai T, Akira S. 2010. The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity 33:765–776 [DOI] [PubMed] [Google Scholar]
- 9. Arimoto K, Funami K, Saeki Y, Tanaka K, Okawa K, Takeuchi O, Akira S, Murakami Y, Shimotohno K. 2010. Polyubiquitin conjugation to NEMO by tripartite motif protein 23 (TRIM23) is critical in antiviral defense. Proc. Natl. Acad. Sci. U. S. A. 107:15856–15861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Takahashi M, Ritz J, Cooper GM. 1985. Activation of a novel human transforming gene, ret, by DNA rearrangement. Cell 42:581–588 [DOI] [PubMed] [Google Scholar]
- 11. Shimono Y, Murakami H, Kawai K, Wade PA, Shimokata K, Takahashi M. 2003. Mi-2β associates with BRG1 and RET finger protein at the distinct regions with transcriptional activating and repressing abilities. J. Biol. Chem. 278:51638–51645 [DOI] [PubMed] [Google Scholar]
- 12. Krützfeldt M, Ellis M, Weekes DB, Bull JJ, Eilers M, Vivanco MD, Sellers WR, Mittnacht S. 2005. Selective ablation of retinoblastoma protein function by the RET finger protein. Mol. Cell 18:213–224 [DOI] [PubMed] [Google Scholar]
- 13. Harbers M, Nomura T, Ohno S, Ishii S. 2001. Intracellular localization of the Ret finger protein depends on a functional nuclear export signal and protein kinase C activation. J. Biol. Chem. 276:48596–48607 [DOI] [PubMed] [Google Scholar]
- 14. Zha J, Han KJ, Xu LG, He W, Zhou Q, Chen D, Zhai Z, Shu HB. 2006. The Ret finger protein inhibits signaling mediated by the noncanonical and canonical IκB kinase family members. J. Immunol. 176:1072–1080 [DOI] [PubMed] [Google Scholar]
- 15. Chen G, Goeddel DV. 2002. TNF-R1 signaling: a beautiful pathway. Science 296:1634–1635 [DOI] [PubMed] [Google Scholar]
- 16. Croft M. 2009. The role of TNF superfamily members in T-cell function and diseases. Nat. Rev. Immunol. 9:271–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vucic D, Dixit VM, Wertz IE. 2011. Ubiquitylation in apoptosis: a post-translational modification at the edge of life and death. Nat. Rev. Mol. Cell Biol. 12:439–452 [DOI] [PubMed] [Google Scholar]
- 18. Pobezinskaya YL, Liu Z. 2012. The role of TRADD in death receptor signaling. Cell Cycle 11:871–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Micheau O, Tschopp J. 2003. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114:181–190 [DOI] [PubMed] [Google Scholar]
- 20. Mahoney DJ, Cheung HH, Mrad RL, Plenchette S, Simard C, Enwere E, Arora V, Mak TW, Lacasse EC, Waring J, Korneluk RG. 2008. Both cIAP1 and cIAP2 regulate TNFα-mediated NF-κB activation. Proc. Natl. Acad. Sci. U. S. A. 105:11778–11783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, Gillard JW, Jaquith JB, Morris SJ, Barker PA. 2008. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 30:689–700 [DOI] [PubMed] [Google Scholar]
- 22. Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. 2006. Activation of IKK by TNFα requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol. Cell 22:245–257 [DOI] [PubMed] [Google Scholar]
- 23. Wertz IE, O'Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, Wu P, Wiesmann C, Baker R, Boone DL, Ma A, Koonin EV, Dixit VM. 2004. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-κB signalling. Nature 430:694–699 [DOI] [PubMed] [Google Scholar]
- 24. Shembade N, Ma A, Harhaj EW. 2010. Inhibition of NF-κB signaling by A20 through disruption of ubiquitin enzyme complexes. Science 327:1135–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Trompouki E, Hatzivassiliou E, Tsichritzis T, Farmer H, Ashworth A, Mosialos G. 2003. CYLD is a deubiquitinating enzyme that negatively regulates NF-κB activation by TNFR family members. Nature 424:793–796 [DOI] [PubMed] [Google Scholar]
- 26. Brummelkamp TR, Nijman SM, Dirac AM, Bernards R. 2003. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-κB. Nature 424:797–801 [DOI] [PubMed] [Google Scholar]
- 27. Kovalenko A, Chable-Bessia C, Cantarella G, Israël A, Wallach D, Courtois G. 2003. The tumour suppressor CYLD negatively regulates NF-κB signalling by deubiquitination. Nature 424:801–805 [DOI] [PubMed] [Google Scholar]
- 28. Takagi T, Harada J, Ishii S. 2001. Murine Schnurri-2 is required for positive selection of thymocytes. Nat. Immunol. 2:1048–1053 [DOI] [PubMed] [Google Scholar]
- 29. Bellenger J, Bellenger S, Bataille A, Massey KA, Nicolaou A, Rialland M, Tessier C, Kang JX, Narce M. 2011. High pancreatic n-3 fatty acids prevent STZ-induced diabetes in fat-1 mice: inflammatory pathway inhibition. Diabetes 60:1090–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Thapa RJ, Basagoudanavar S, Nogusa S, Irrinki K, Mallilankaraman K, Slifker MJ, Beg AA, Madesh M, Balachandran S. 2011. NF-κB protects cells from interferon-γ-induced RIP1-dependent necroptosis. Mol. Cell. Biol. 31:2934–2946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Khan MM, Nomura T, Kim H, Kaul SC, Wadhwa R, Zhong S, Pandolfi PP, Ishii S. 2004. The fusion oncoprotein PML-RARα induces endoplasmic reticulum (ER)-associated degradation of N-CoR and ER stress. J. Biol. Chem. 279:11814–11824 [DOI] [PubMed] [Google Scholar]
- 32. Yamauchi T, Ishidao T, Nomura T, Shinagawa T, Tanaka Y, Yonemura S, Ishii S. 2008. A B-Myb complex containing clathrin and filamin is required for mitotic spindle function. EMBO J. 27:1852–1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ramakrishnan P, Baltimore D. 2011. Sam68 is required for both NF-κB activation and apoptosis signaling by the TNF receptor. Mol. Cell 43:167–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Canning M, Boutell C, Parkinson J, Everett RD. 2004. A RING finger ubiquitin ligase is protected from autocatalyzed ubiquitination and degradation by binding to ubiquitin-specific protease USP7. J. Biol. Chem. 279:38160–38168 [DOI] [PubMed] [Google Scholar]
- 35. Lehmann V, Freudenberg MA, Galanos C. 1987. Lethal toxicity of lipopolysaccharide and tumor necrosis factor in normal and D-galactosamine-treated mice. J. Exp. Med. 165:657–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Temkin V, Huang Q, Liu H, Osada H, Pope RM. 2006. Inhibition of ADP/ATP exchange in receptor-interacting protein-mediated necrosis. Mol. Cell. Biol. 26:2215–2225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dixit E, Boulant S, Zhang Y, Lee AS, Odendall C, Shum B, Hacohen N, Chen ZJ, Whelan SP, Fransen M, Nibert ML, Superti-Furga G, Kagan JC. 2010. Peroxisomes are signaling platforms for antiviral innate immunity. Cell 141:668–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hu M, Li P, Li M, Li W, Yao T, Wu JW, Gu W, Cohen RE, Shi Y. 2002. Crystal structure of a UBP-family deubiquitinating enzyme in isolation and in complex with ubiquitin aldehyde. Cell 111:1041–1054 [DOI] [PubMed] [Google Scholar]
- 39. Fernández-Montalván A, Bouwmeester T, Joberty G, Mader R, Mahnke M, Pierrat B, Schlaeppi JM, Worpenberg S, Gerhartz B. 2007. Biochemical characterization of USP7 reveals post-translational modification sites and structural requirements for substrate processing and subcellular localization. FEBS J. 274:4256–4270 [DOI] [PubMed] [Google Scholar]
- 40. Colland F, Formstecher E, Jacq X, Reverdy C, Planquette C, Conrath S, Trouplin V, Bianchi J, Aushev VN, Camonis J, Calabrese A, Borg-Capra C, Sippl W, Collura V, Boissy G, Rain JC, Guedat P, Delansorne R, Daviet L. 2009. Small-molecule inhibitor of USP7/HAUSP ubiquitin protease stabilizes and activates p53 in cells. Mol. Cancer Ther. 8:2286–2295 [DOI] [PubMed] [Google Scholar]
- 41. Dynek JN, Goncharov T, Dueber EC, Fedorova AV, Izrael-Tomasevic A, Phu L, Helgason E, Fairbrother WJ, Deshayes K, Kirkpatrick DS, Vucic D. 2010. c-IAP1 and UbcH5 promote K11-linked polyubiquitination of RIP1 in TNF signalling. EMBO J. 29:4198–4209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li M, Brooks CL, Kon N, Gu W. 2004. A dynamic role of HAUSP in the p53-Mdm2 pathway. Mol. Cell 13:879–886 [DOI] [PubMed] [Google Scholar]
- 43. Marchenko ND, Wolff S, Erster S, Becker K, Moll UM. 2007. Monoubiquitylation promotes mitochondrial p53 translocation. EMBO J. 26:923–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Uchil PD, Quinlan BD, Chan WT, Luna JM, Mothes W. 2008. TRIM E3 ligases interfere with early and late stages of the retroviral life cycle. PLoS Pathog. 4:e16. 10.1371/journal.ppat.0040016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zapata JM, Pawlowski K, Haas E, Ware CF, Godzik A, Reed JC. 2001. A diverse family of proteins containing tumor necrosis factor receptor-associated factor domains. J. Biol. Chem. 276:24242–24252 [DOI] [PubMed] [Google Scholar]
- 46. Yamauchi K, Wada K, Tanji K, Tanaka M, Kamitani T. 2008. Ubiquitination of E3 ubiquitin ligase TRIM5 alpha and its potential role. FEBS J. 275:1540–1555 [DOI] [PubMed] [Google Scholar]
- 47. Dho SH, Kwon KS. 2003. The Ret finger protein induces apoptosis via its RING finger-B box-coiled-coil motif. J. Biol. Chem. 278:31902–31908 [DOI] [PubMed] [Google Scholar]
- 48. Cai X, Srivastava S, Sun Y, Li Z, Wu H, Zuvela-Jelaska L, Li J, Salamon RS, Backer JM, Skolnik EY. 2011. Tripartite motif containing protein 27 negatively regulates CD4 T cells by ubiquitinating and inhibiting the class II PI3K-C2β. Proc. Natl. Acad. Sci. U. S. A. 108:20072–20077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Srivastava S, Cai X, Li Z, Sun Y, Skolnik EY. 2012. Phosphatidylinositol-3-kinase C2β and TRIM27 function to positively and negatively regulate IgE receptor activation of mast cells. Mol. Cell. Biol. 32:3132–3139 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.