

Abstract
TG-interacting factor 1 (TGIF1) is a transcriptional repressor that can modulate retinoic acid and transforming growth factor β signaling pathways. It is required for myeloid progenitor cell differentiation and survival, and mutations in the TGIF1 gene cause holoprosencephaly. Furthermore, we have previously observed that acute myelogenous leukemia (AML) patients with low TGIF1 levels had worse prognoses. Here, we explored the role of Tgif1 in murine hematopoietic stem cell (HSC) function. CFU assays showed that Tgif1−/− bone marrow cells produced more total colonies and had higher serial CFU potential. These effects were also observed in vivo, where Tgif1−/− bone marrow cells had higher repopulation potential in short- and long-term competitive repopulation assays than wild-type cells. Serial transplantation and replating studies showed that Tgif1−/− HSCs exhibited greater self-renewal and were less proliferative and more quiescent than wild-type cells, suggesting that Tgif1 is required for stem cells to enter the cell cycle. Furthermore, HSCs from Tgif1+/− mice had a phenotype similar to that of HSCs from Tgif1−/− mice, while bone marrow cells with overexpressing Tgif1 showed increased proliferation and lower survival in long-term transplant studies. Taken together, our data suggest that Tgif1 suppresses stem cell self-renewal and provide clues as to how reduced expression of TGIF1 may contribute to poor long-term survival in patients with AML.
INTRODUCTION
TG-interacting factor 1 (TGIF1) is a transcriptional repressor and a member of the three-amino-acid loop extension (TALE) class of homeodomain proteins (1). TGIF1 inhibits the transforming growth factor β (TGF-β) pathway by associating with Smad2 and recruiting corepressors, and it inhibits the downstream retinoic acid (RA) pathway by binding to the retinoid X receptor (RXR) response element and by interacting with RXR (2–7). In addition, it can bind to DNA directly through its own consensus binding site and impact the transcription of as yet undefined target genes (6). Mutations in TGIF1 are associated with holoprosencephaly (HPE), which is the most common structural abnormality of the forebrain in humans (8). The majority of these mutations would cause a loss of protein function and are hypothesized to alter signaling by TGF-β-related ligands (9–11). In mice, loss of both Tgif1 and Tgif2 is lethal, but epiblast-specific deletion of Tgif1 in combination with a null mutation in Tgif2 results in HPE, which is at least partly due to deregulation of Nodal signaling, suggesting that human TGIF1 mutations may cause HPE by affecting TGF-β signaling (12, 13).
There were several lines of data suggesting that TGIF1 could also have a role in hematopoiesis. As stated above, TGIF1 is a repressor of both TGF-β and RA signaling, and there is incontrovertible evidence that both of these pathways play an important role in hematopoiesis (14–16). Short hairpin RNA-mediated TGIF knockdown in the myeloid cell line HL60 (a well-characterized model for the study of committed myeloid progenitors) affected both proliferation and differentiation and induced a relative block in the cell cycle at the G0 stage (17). TGIF1 gene expression has been detected in murine hematopoietic stem cells (HSCs) (18) and in murine and human embryonic stem cells (19); TGIF1 is, in fact, represented on a short list of proteins proposed to mediate embryonic stem cell function (19). TGIF1 was also identified in a group of genes that are downregulated in fetal liver stem cells and upregulated in adult HSCs (20). Furthermore, and of possible clinical relevance, our unpublished data suggest that expression of TGIF1 is highly predictive of relapse-free and overall survival in patients with acute myelogenous leukemia (AML) (21). Patients whose blast cells expressed relatively lower levels of TGIF1 mRNA had a worse outcome than patients who had higher levels of expression.
HSCs are rare hematopoietic cells that reside in the bone marrow postnatally. These cells are capable of self-renewal (thus maintaining their own number) and can differentiate into any type of blood cell, losing their capacity of self-renewal in the process (22–24). The vast majority of HSCs in the bone marrow are quiescent; i.e., they are in the G0 phase of the cell cycle, which prevents their exhaustion and ensures a pool of self-renewing cells (25–27). When an HSC exits G0 to enter the cell cycle, it has the choice of self-renewal or differentiation. The balance between quiescence and growth, entry into and exit from the cell cycle, and self-renewal and differentiation is tightly controlled by a complex interplay between intrinsic and extrinsic factors, including transcription factors, cell surface receptors, and canonical signaling pathways (28–31). Regulation of stem cell function is still incompletely understood and, importantly, appears to be altered in acute leukemias.
Here we present data that suggest that Tgif1 modulates HSC biology by altering the exquisite balance between quiescence, self-renewal, and differentiation. We found that Tgif1 knockout resulted in increased HSC quiescence and self-renewal. Furthermore, our data show that this effect is associated with genes and pathways previously implicated in HSC function.
MATERIALS AND METHODS
Mice.
The generation, maintenance, and genotyping of Tgif1+/− mice (C57BL/6, CD45.2+) have been previously described (2, 32). Tgif1−/− and Tgif1+/+ mice were obtained by intercrossing Tgif1+/− mice, which ensured that Tgif1−/− and Tgif1+/+ mice had the same genetic background. B6-LY5.2/Cr (CD45.1+) mice were purchased from NCI/Charles River. Mice were housed in accordance with an approved protocol from Vanderbilt University's Institutional Animal Care and Use Committee.
Flow cytometry analysis.
A single-cell suspension of bone marrow cells was obtained by flushing the tibias and femurs of the euthanized mice. Following removal of the red blood cells, the remaining cells were stained with a cocktail of antibodies (CD3, Ter119, Gr1, Mac1, B220, streptavidin, Sca-1, c-Kit, CD45.1, CD45.2, Flt3, CD34, CD150, CD48, or CD16/CD32 [FcγR]) to discriminate between specific hematopoietic populations. Cells were designated as follows: lineage-negative (Lin−) Sca+ c-Kit+ (LSK); long-term hematopoietic stem cells (LT-HSCs), LSK/Flt3− CD34−; short-term hematopoietic stem cells (ST-HSCs), LSK/Flt3low CD34+; LT- and ST-HSCs (LT+ST-HSCs), LSK/Flt3low; signaling lymphocyte activation molecule-positive (SLAM) cells, LSK/CD48− CD150+; multipotent progenitors (MPPs), LSK/Flt3high; committed hematopoietic progenitor cells (HPCs), Lin− Sca− c-Kit+; common myeloid progenitors (CMPs), Lin− Sca− c-Kit+ CD34+ FcγRlow; granulocyte macrophage progenitors (GMPs), Lin− Sca− c-Kit+ CD34+ FcγRhigh; and megakaryocyte erythroid progenitors (MEPs), Lin− Sca− c-Kit+ CD34− FcγRlow. Flow cytometry analysis was performed on a Becton, Dickinson 5-laser LSRII instrument.
BrdU incorporation and apoptosis analysis.
For bromodeoxyuridine (BrdU) incorporation assays, mice were sacrificed 2 h after intraperitoneal injection of 1 mg BrdU (33). Bone marrow lineage-negative cells were isolated using a MACS lineage cell depletion kit (Miltenyi Biotech, Germany). These cells were then labeled with appropriate antibodies following the instructions of the BrdU flow kit (BD Pharmingen). Apoptosis was detected by staining the cells with antibody against annexin V.
Cell cycle analysis.
Bone marrow cells were lineage depleted using a MACS lineage cell depletion kit (Miltenyi Biotech), resuspended in RPMI 1640 with 10% fetal bovine serum, and incubated with 10 μg/ml of Hoechst 33342 (Invitrogen) and 50 μM verapamil (Sigma) for 45 min at 37°C. Cells were washed with HBS once and incubated with the indicated antibodies on ice for 20 min. Cells were then washed and resuspended in Cytofix buffer (BD Pharmingen) overnight. Cells were resuspended in staining buffer and incubated with 0.5 μM pyronin Y (Polysciences, Inc.) for 30 min on ice prior to flow cytometry analysis.
Stem cell and progenitor cell assays.
To evaluate the most primitive progenitors, we assessed their replating ability in methylcellulose CFU and serial replating assays. Briefly, 1.5 × 104 total bone marrow cells were plated in methylcellulose medium (Methocult GF M3434; Stem Cell Technologies). Colonies were counted between days 10 and 12. For the serial replating assay, colonies were first counted and then harvested, and then 1.5 × 104 cells were replated in fresh methylcellulose medium every 7 days for 4 weeks.
For competitive repopulation assays (CRAs), lethally irradiated B6-Ly5.2/Cr (CD45.1+) congenic mice were used as recipient mice. Tgif1+/+ or Tgif1−/− donor cells (CD45.2+) were mixed with B6-Ly5.2/Cr (CD45.1+) bone marrow cells (1:1), and the cell mixture was injected intravenously into the lateral tail vein of recipient mice. Reconstitution was evaluated by flow cytometry every 4 weeks after transplant by staining the peripheral blood with anti-CD45.2–fluorescein isothiocyanate and anti-CD45.1–phycoerythrin. For secondary CRAs, bone marrow cells were harvested from mice used in primary transplants, and 2 × 106 total bone marrow cells were injected into lethally irradiated recipient mice. To accurately reflect the actual difference in functional activities of donor HSCs relative to competitors, we calculated donor repopulation units (RU) as previously described (34).
Homing assay.
Bone marrow cells (1 × 107) isolated from Tgif1+/+ or Tgif−/− mice were labeled with the vital dye carboxyfluorescein diacetate succinimidyl ester (CFSE; Life Technologies, Grand Island, NY) and subsequently injected via the tail vein into lethally irradiated recipient C57BL/6 mice. Sixteen hours later, the recipient mice were euthanized and the bone marrow and spleens were isolated to determine cell homing to either organ.
Analysis of gene expression by quantitative real-time PCR (qRT-PCR).
TaqMan-based real-time PCR analyses to detect Tgif1 and Tgif2 were done on sorted cell populations according to the manufacturer's instructions using a Cells-to-CT kit (Ambion).
Western blot analysis.
Western blot analysis was done on nuclear extracts of bone marrow cells. Extracts were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membranes. Blots were probed with a rabbit anti-Tgif1 antibody (sc-9084; Santa Cruz Biotechnology) and a rabbit anti-histone H3 antibody (sc-10809; Santa Cruz Biotechnology).
Tgif1 overexpression.
Tgif1 cDNA was inserted into the vector pMSCV-IRES-GFP (MIG) to prepare MIG-Tgif1. Phoenix retroviral packaging cells were transfected with the parental vector (control) or MIG-Tgif1, and after 48 h, the retroviruses were harvested from the supernatants. Lin− cells isolated from Tgif1+/+ mouse bone marrow were then spinoculated with control retrovirus or retrovirus containing MIG-Tgif1 in the presence of 5 μg/ml Polybrene for 1 h at 1,350 × g and 32°C. Following spinoculation, cells were cultured in StemSpan serum-free expansion medium (Stemcell Technologies) supplemented with growth factors. BrdU incorporation and annexin V staining were performed at specific time points to evaluate proliferation and apoptosis of transduced Lin− cells (green fluorescent protein [GFP] positive).
To determine the reconstituting ability of Tgif1-overexpressing bone marrow cells. Lin− cells from Tgif1+/+ bone marrow were spinoculated with control MIG or MIG-Tgif1 retrovirus in the presence of 5 μg/ml Polybrene for 1 h at 1,350 × g and 32°C. After spinoculation, transformed Lin− cells were mixed with B6-Ly5.2/Cr (CD45.1+) Lin− cells and injected intravenously into the lateral tail vein of lethally irradiated B6-Ly5.2/Cr (CD45.1+) congenic mice. Reconstitution of transduced cells in recipient mice was evaluated by monitoring GFP expression by flow cytometry at 12 and 16 weeks after transplantation.
RNA sequencing.
The RNA sequencing library was prepared from approximately 200 ng RNA from LSK (Lin− Sca+ c-Kit+) cells isolated from the bone marrow of Tgif1−/− and Tgif1+/+ mice (3 replicates each) and sequenced on an Illumina HiSeq 2000 instrument. Sequencing analysis was performed by the Vanderbilt Technologies for Advanced Genomics (VANTAGE) shared resource. The mouse genome sequence was annotated using the UCSC annotation file (http://genome.ucsc.edu), and the TopHat and Cufflinks software packages were used for transcript alignment and for gene expression quantification (35).
GSEA and IPA.
We performed gene set enrichment analysis (GSEA) (36) to determine whether differentially expressed genes (between the Tgif1−/− LSK and Tgif1+/+ LSK cells) were randomly distributed throughout the gene expression data set or were enriched in the top or bottom of the gene expression list using the default weighted enrichment statistic. A false discovery rate of ≤0.25 was considered significant. Ingenuity pathway analysis (IPA) was performed to display closely related genes and identify biological pathways of relevance as previously described (17).
Statistics.
Data are presented as mean ± standard error of mean (SEM) and were analyzed with Student's t test. P values of <0.05 were considered statistically significant. Analyses were performed with Prism (version 5) software for Mac OS X (GraphPad Software Inc., La Jolla, CA).
RNA sequencing data accession number.
RNA sequencing data were submitted to the Gene Expression Omnibus (GEO) database under accession number GSE50739.
RESULTS
Tgif1 is expressed in bone marrow cell populations.
Since our goal was to elucidate the role of Tgif1 in hematopoiesis, we first determined whether Tgif1 was, in fact, expressed in bone marrow hematopoietic populations. Bone marrow cells from wild-type mice with the same genetic background as the knockout mice were sorted into various subpopulations and then analyzed for Tgif1 expression by real-time PCR analysis. We found that Tgif1 was ubiquitously expressed in the most primitive HSCs (SLAM cells) as well as various hematopoietic progenitor cell populations (Fig. 1A). As expected, Tgif1 was not expressed in Tgif1−/− bone marrow cells and expressed at reduced levels in the Tgif1+/− cells (see Fig. S1A in the supplemental material).
Fig 1.

(A) Tgif1 mRNA levels in hematopoietic bone marrow cells (BMCs) of wild-type (WT) mice; (B) total bone marrow cell counts in Tgif1−/− and wild-type mice enumerated by a hemocytometer; (C and D) flow cytometric analysis of the relative and absolute numbers of ST-HSC and SLAM cells in the bone marrow of Tgif1+/+ and Tgif −/− mice, respectively; (E and F) flow cytometric analysis of the relative and absolute numbers of CMPs and MEPs in the bone marrow of Tgif1+/+ and Tgif1−/− mice, respectively. Asterisks indicate P values, as follows: *, P = 0.05 to 0.01; **, P = 0.01 to 0.001; ***, P < 0.001. N.S., not significant.
Tgif1 knockout alters the bone marrow hematopoietic populations.
Since we found that Tgif1 was expressed in the hematopoietic compartments, we investigated whether Tgif1 deficiency had any effect on either the peripheral or bone marrow hematopoietic populations. Tgif1−/− mice lack exons 2 and 3 of the gene, which disrupts the Tgif1 open reading frame. Tgif1−/− mice thus do not express Tgif1, while Tgif1+/− mice express lower levels of Tgif1 than wild-type mice (see Fig. S1A in the supplemental material) (2). These mice, however, develop normally, allowing us to analyze their peripheral blood and bone marrow. At 8 and 12 weeks of life, complete blood counting (CBC) found no statistically significant differences in the number or types of peripheral blood cells between Tgif1+/+ and Tgif1−/− mice (see Table S1 in the supplemental material). Likewise, histological analysis of peripheral blood and bone marrow also did not show any obvious differences (data not shown). Tgif1−/− mice, however, had lower total bone marrow cells than wild-type mice (P = 0.0057) (Fig. 1B). Next, we enumerated the HSCs and progenitor cells in the bone marrow compartments of Tgif1+/+ and Tgif1−/− mice by flow cytometry (the flow schematic is shown in Fig. S1B in the supplemental material). The HSC population lacks lineage commitment markers and expresses stem cell antigen (Sca-1) and high levels of the stem cell factor receptor c-Kit; thus, it is referred to as the LSK population (a population enriched in stem and early progenitor cells). This LSK population can be further dissected into cells with the potential to reconstitute hematopoiesis on a long-term (SLAM cells) or a short-term (ST-HSCs) basis, as well as progenitors with no potential for self-renewal (MPPs). Tgif1−/− bone marrow had elevated percentages of ST-HSCs (P = 0.008) and the most primitive HSCs, the SLAM cells (LSK/CD48− CD150+) (P = 0.05) (Fig. 1C and D, left). Enumeration of the progenitor populations showed that Tgif1−/− bone marrow had a higher percentage of CMPs (P < 0.0001) (Fig. 1E, left) and a lower percentage of MEPs (P < 0.0001) than Tgif1+/+ bone marrow (Fig. 1F, left). We did not observe any differences in GMPs (P = 0.9719) (data not shown).
However, there was no significant difference in the absolute numbers of ST-HSCs and SLAM cells between Tgif1+/+ and Tgif1−/− mice (Fig. 1C and D, right), likely because Tgif1−/− mice had a decreased cellularity compared to Tgif1+/+ mice (Fig. 1B). Interestingly, however, the absolute numbers of CMPs (Fig. 1E, right), MEPs (Fig. 1F, right), and GMPs (data not shown) were in concordance with the relative numbers, which could suggest that the loss of Tgif1 had a proportionally greater significant effect on progenitor populations.
Since changes in apoptosis are a common cause of altered cell numbers or proportions, we evaluated apoptosis in the progenitor as well as the HSC populations using annexin V staining. These data showed that while CMP cells from Tgif1−/− mice were significantly less apoptotic than those from Tgif1+/+ mice (P = 0.004) (Fig. 2A), there were no significant differences in apoptosis between HSCs from Tgif1−/− and Tgif1+/+ mice (data not shown). Taken together, these data thus indicate that Tgif1 loss increases the primitive HSC and CMP cell populations without significantly changing peripheral blood hematologic parameters under steady-state conditions.
Fig 2.
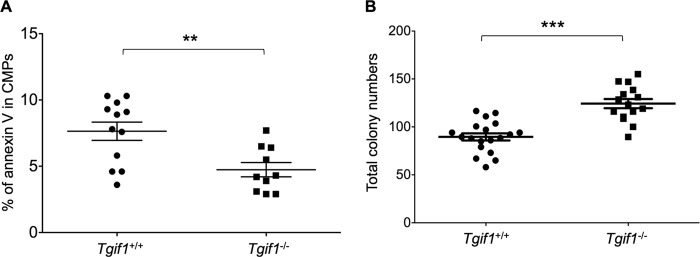
(A) CMPs isolated from Tgif1−/− mice were less apoptotic than wild-type CMPs, as determined by annexin V staining and flow cytometric analysis; (B) Tgif1−/− bone marrow cells produced more total colonies on methylcellulose medium than wild-type bone marrow cells. Each dot represents the total colonies from one plate. Colonies were scored at 10 to 12 days. Asterisks indicate the P values described in the legend to Fig. 1.
Tgif1 is required for hematopoietic progenitor cell differentiation and proliferation.
To test whether Tgif1 deletion affected myeloid progenitor cells' differentiation and proliferation, we performed in vitro CFU assays in semisolid medium supplemented with cytokines that allow the quantification of committed progenitor cells from mouse bone marrow. Fifteen thousand bone marrow cells were plated in methylcellulose medium, and myeloid, erythroid, and mixed-lineage colonies were counted after 10 to 12 days. We found that bone marrow cells from Tgif1−/− mice produced ∼40% more total colonies than bone marrow cells from healthy controls (P < 0.0001) (Fig. 2B). However, there was no significant difference in the percentages of specific types of progenitor colonies (data not shown).
Tgif1 loss disrupts HSC function.
To test whether Tgif1 loss affects HSC function, we performed competitive bone marrow transplantation studies. Lethally irradiated 8-week-old congenic recipient mice (CD45.1 allelotype) were transplanted with 106 Tgif1−/− or 106 Tgif1+/+ bone marrow cells (CD45.2 allelotype) in a 1:1 ratio with 106 wild-type competitor bone marrow cells (CD45.1 allelotype). We then examined peripheral blood for donor chimerism at 8 and 16 weeks after transplantation. Our data showed that Tgif1−/− bone marrow cells had a 1.9-fold higher reconstituting ability than Tgif1+/+ bone marrow cells (P = 0.0025) at 8 weeks, a measure of short-term reconstitution, and a 1.6-fold higher reconstituting ability than Tgif1+/+ bone marrow cells at 16 weeks, a measure of long-term reconstitution (P = 0.0096) (Fig. 3A). To ensure that the observed differences in reconstitution ability were not simply due to differences in homing, bone marrow cells (Tgif1+/+ and Tgif1−/−) were labeled with the vital dye CFSE and transplanted into donor mice. Sixteen hours later, the bone marrows and spleens of these mice were analyzed by flow cytometry and the number of cells positive for CFSE was quantified. These data showed that there were no differences in homing to bone marrow or spleen between Tgif1+/+ and Tgif1−/− bone marrow cells (Fig. 3B).
Fig 3.

Tgif1 loss disrupts HSC function and affects HSC self-renewal. (A) CRA of HSCs. A mixture of Tgif1+/+ or Tgif1−/− donor bone marrow cells (CD45.2+) and competitor cells (CD45.1+) was transplanted into recipient mice at a 1:1 ratio. The contribution of each population to the long-term reconstitution of the bone marrow was assessed by flow cytometry using anti-CD45.1 and anti-CD45.2 to enumerate cells in the peripheral blood. Donor repopulation units (Donor RU) were calculated at 8 and 16 weeks. (B) Tgif1−/− cells home to bone marrow. Bone marrow from Tgif1−/− and Tgif1+/+ mice were stained with CFSE and injected into irradiated recipient mice. The percentages of cells stained with CFSE in bone marrow and spleen are shown. (C) Number of bone marrow cell colonies formed in methylcellulose medium during 4 rounds of serial replating. (D) Secondary transplant experiment. Flow cytometry analysis for the presence of CD45.2 and CD45.1 was used to calculate donor RUs at 8 and 16 weeks after secondary bone marrow transplant from an initial CRA. Asterisks indicate the P values described in the legend to Fig. 1.
Tgif1 loss affects HSC self-renewal.
Tgif1−/− mice have increased reconstituting capacity in competitive bone marrow transplantation assays (Fig. 3A), suggesting that Tgif1 loss could increase stem cell self-renewal. A comparison of serially passaged Tgif1−/− and Tgif1+/+ bone marrow cells in methylcellulose showed that at each replating, there were consistently more colonies in Tgif1−/− plates than Tgif1+/+ control plates and that the differences were most pronounced at the fourth replating (Fig. 3C), when control bone marrow cells displayed a nearly 8-fold reduction in replating ability compared to Tgif1−/− bone marrow cells. Furthermore, Tgif1+/+ bone marrow cells could not be passaged beyond the third replating, while Tgif1−/− cells retained nearly 50% of their original replating capacity.
The most stringent functional test for stemness, or self-renewal capacity, of HSCs is the serial stem cell transplantation assay (37). To that end, bone marrow cells harvested from the primary transplant recipient mice were transplanted into irradiated secondary recipient mice and peripheral blood was examined for reconstitution at 8 and 16 weeks after transplant. Tgif1−/− bone marrow cells showed a significant (∼5-fold) improvement in reconstitution potential over the primary transplant relative to Tgif1+/+ bone marrow cells (P = 0.001 to 0.007) (Fig. 3D). Taken together, these data suggest that Tgif1−/− HSCs have a self-renewal advantage over Tgif1+/+ HSCs.
Tgif1−/− bone marrow cells are less proliferative and more quiescent than wild-type cells.
The long-term competitive repopulating ability of HSCs is associated with quiescence. HSCs are maintained in a quiescent state that is the G0 phase of the cell cycle, and this quiescence is thought to protect these cells against the loss of the self-renewal capacity needed for long-term maintenance of hematopoiesis. The more quiescent that the HSCs are, the better that their long-term repopulating potential will be. Thus, based on the results of our primary and secondary transplant experiments, we hypothesized that Tgif1−/− HSCs would be less proliferative and more quiescent than Tgif1+/+ cells. To investigate this hypothesis, we injected BrdU into Tgif1+/+ and Tgif1−/− mice to evaluate the number of cycling cells in their respective HSC populations. Two hours after BrdU injection, flow cytometry analysis revealed that both in the stem cell pools (LSK/Flt3low, LT+ST-HSCs) and in the most primitive HSCs, the SLAM cells (LSK/CD48− CD150+), the HSCs from Tgif1−/− mice incorporated significantly less BrdU than the HSCs from Tgif1+/+ mice (P = 0.0179 and P = 0.0208, respectively) (Fig. 4A and B). These data indicate that fewer Tgif1−/− HSCs entered the cell cycle.
Fig 4.

Tgif1−/− bone marrow has higher percentages of cells that do not enter the cell cycle and reside in G0 than Tgif1+/+ bone marrow. (A) Representative fluorescence-activated cell sorting profile of bone marrow HSCs from one Tgif1−/− mouse and one Tgif1+/+ mouse; (B) percentage of LT+ST-HSCs and SLAM cells that incorporate BrdU in Tgif1−/− and Tgif1+/+ bone marrow cells; (C) representative cell cycle profile from one Tgif1−/− mouse and one Tgif1+/+ mouse; (D) comparison of percentages of HSCs and progenitor cells in G0 and G1 phases of the cell cycle between Tgif1−/− and Tgif1+/+ mice (n = 6 in each case). Asterisks indicate the P values described in the legend to Fig. 1.
To further analyze the cell cycle in Tgif1−/− and Tgif1+/+ mice, we stained HSCs with Hoechst 33342 and pyronin Y, which distinguish G0 and G1 cells on the basis of RNA content. Our data showed that among different HSC and progenitor populations, the proportion of cells in G0 relative to G1 was significantly higher in Tgif1−/− mice than in Tgif1+/+ mice (Fig. 4C and D). However, the overall percentage of cells in G phase (G0 plus G1) did not differ between strains (Fig. 4D). There was, likewise, no difference in the percentage of cells in S or G2/M phase between Tgif1−/− and Tgif1+/+ cells (data not shown). Taken together, these data show that the HSCs of Tgif1−/− mice are less proliferative and more quiescent than the HSCs of Tgif1+/+ mice under steady-state conditions and suggest that Tgif1 could regulate the entrance of HSCs into the cell cycle.
Tgif1+/− heterozygotes also show altered progenitor and HSC function.
Since Tgif1−/− bone marrow had increased HSC pools and showed increased numbers of CFU and engraftment in primary and secondary transplant experiments and since TGIF1 abundance appears to impact survival in AML patients in a dose-related fashion, we investigated whether haploinsufficiency of Tgif1, which has previously been established to result in a ≥50% reduction in Tgif1 expression (2), would also have an effect on progenitor and/or HSC function. Analysis of progenitor and HSC populations showed that Tgif1+/− bone marrow had slightly increased LSK, ST-HSC, and CMP populations compared to Tgif1+/+ bone marrow, although the differences were not statistically significant (data not shown). However, compared to Tgif1+/+ bone marrow cells, Tgif1+/− bone marrow cells showed a significant enhancement of colony-forming capacity (P = 0.004) (Fig. 5A) and repopulating ability in both primary transplantation (P = 0.0005 at week 8 and P = 0.004 at week 16) (Fig. 5B) and secondary transplantation (P = 0.0001 at week 8 and P = 0.007 at week 16) (Fig. 5C). These data indicate that, like complete Tgif1 gene loss, decreased Tgif1 expression affects progenitor and HSC function and that the phenotypes observed in the Tgif1−/− mice are equally sensitive to Tgif1 heterozygosity.
Fig 5.
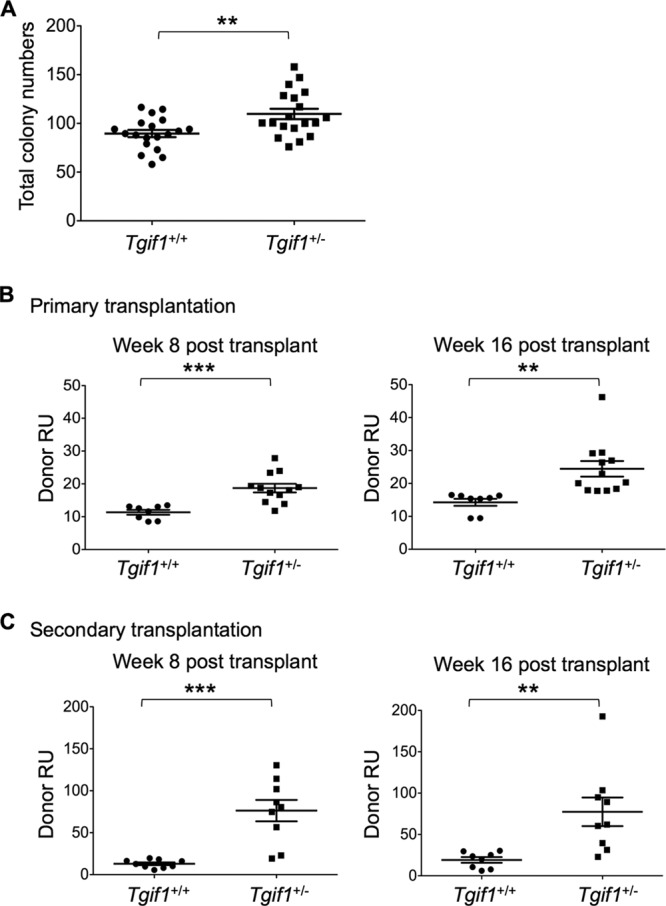
Analyses of Tgif1 heterozygotes. (A) CFU analysis of bone marrow cells derived from Tgif1+/+ and Tgif1+/− cells. (B) CRAs with a mixture of Tgif1+/+ or Tgif1+/− donor bone marrow cells (CD45.2+) and competitor cells (CD45.1+) transplanted in a 1:1 ratio in recipient mice. Donor repopulation units (Donor RU) were calculated at 8 and 16 weeks. (C) Secondary transplant experiments with bone marrow cells isolated from mice used in primary transplant experiments. Donor RU at week 8 and week 16 were calculated. Asterisks indicate the P values described in the legend to Fig. 1.
Bone marrow cells overexpressing Tgif1 show increased proliferation in culture and decreased hematopoietic repopulating activity in long-term transplant experiments.
Since Tgif1 deficiency altered HSC number and function, we investigated whether Tgif1 overexpression had an effect on bone marrow-derived HSC populations. HSCs isolated from wild-type mouse bone marrow were transduced with a Tgif1 retroviral expression vector or the empty MIG vector (see Fig. S2A in the supplemental material). Cells were then cultured in appropriate medium for 7 days, and BrdU incorporation was used to determine the rate of proliferation. Our data showed that HSC populations (Lin− Scahigh) transduced with the MIG-Tgif1 virus showed increased proliferation compared to HSCs infected with the MIG control (Fig. 6A). Furthermore, there was no difference in annexin V staining between the MIG-Tgif1-infected cells and the MIG-infected cells (Fig. 6B).
Fig 6.
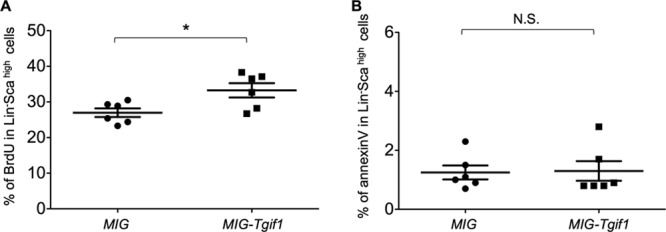
Tgif1 overexpression alters the proliferation of HSCs. Lin− cells isolated from Tgif1+/+ bone marrow cells were infected with MIG control or MIG-Tgif1 retrovirus to enforce the expression of Tgif1. (A) The percentage of cells that incorporated BrdU was compared between MIG control- and MIG-Tgif1-infected HSCs. (B) Control MIG- and MIG-Tigf1-infected HSCs were stained with the antibody to the apoptosis marker annexin V and analyzed by flow cytometry. Asterisks indicate the P values described in the legend to Fig. 1.
We next determined the proliferative potential of these cells in vivo. Lin− bone marrow cells from Tgif1+/+ CD45.2 mice were spinoculated with MIG control or MIG-Tgif1 retrovirus. Infected Lin− cells were then mixed with B6-Ly5.2/Cr (CD45.1+) Lin− cells and injected intravenously into the lateral tail vein of lethally irradiated B6-Ly5.2/Cr (CD45.1+) congenic mice. The long-term reconstitution ability of infected cells in the recipient mice was then evaluated by flow cytometry analysis for GFP expression at 12 and 16 weeks after transplantation. At 16 weeks posttransplant, cells infected with MIG-Tgif1 showed significantly decreased hematopoietic reconstituting function than MIG control-transduced cells (see Fig. S2B in the supplemental material). Taken together, these data reveal that Tgif1 overexpression increased the proliferation of HSC populations and reduced their long-term peripheral hematopoietic reconstitution potential, exactly opposite of the effects of Tgif1 deficiency.
A Tgif1-dependent transcriptional program implicates perturbation of multiple pathways involved in HSC quiescence and self-renewal.
TGIF1 is a transcriptional corepressor, and so to begin to determine the mechanisms by which it functions in bone marrow hematopoietic stem and progenitor cells, we compared gene expression in Tgif1−/− and Tgif1+/+ LSK bone marrow cells by RNA sequencing of biological triplicates (GEO accession number GSE50739). Our overall goal was to understand the signaling pathways by which Tgif1 regulates HSC biology. This analysis showed that the expression of 71 genes (31 upregulated and 40 downregulated) was significantly altered in Tgif1−/− cells compared to Tgif1+/+ cells (P ≤ 0.05) (see Table S2 in the supplemental material). A survey of the literature showed that 43% of these differentially expressed genes have either direct or indirect functions in hematopoietic cell biology, including proliferation, survival, and death. Of these, 6 genes were found to have functions in progenitor cells (including myeloid cells), 9 genes were found to have functions in cell cycle control and regulation of G0 populations, and 26 genes were found to have roles in the proliferation of hematopoietic cells (see Table S3 in the supplemental material).
We then used GSEA and IPA to further explore the biological implications of these data. We generated a rank-order list based on fold change expression differences such that the most upregulated genes in the Tgif1−/− cells were at the top of the ranked list, while the most upregulated genes in Tgif1+/+ cells (downregulated in Tgif1−/− cells) were at the bottom of the list. GSEA analysis showed a statistically significant enrichment (false discovery rate = 0.25) of gene sets involved in hemostasis, myeloid proliferation, self-renewal, and RA signaling (see Table S4 in the supplemental material). Enrichment of the last gene set in the upregulated genes in the transcriptome sequencing (RNA-seq) data is particularly interesting, given that all-trans RA is strongly implicated in many aspects of HSC function and TGIF1 has been recognized to function as a corepressor for the RAR-RXR-regulated transcription.
We next examined the functional categories of differentially expressed genes using IPA (see Table S5 in the supplemental material). Importantly, pathways known to be important in hematopoietic cells predominated, including pathways relevant to the HSC phenotype described in Tgif1 knockout mice: acute myeloid leukemia signaling, RA pathway signaling, Wnt/beta-catenin signaling, and mouse embryonic stem cell pluripotency (Fig. 7). To understand the intricate relationships of the genes defining the Tgif1−/− LSK cells, we employed IPA to identify gene networks (see Table S6 in the supplemental material). These networks contain many genes known to affect important functions of both normal and leukemic stem cells.
Fig 7.

Canonical pathways highlighted by IPA for the differentially expressed genes in the RNA-seq data. Numbers represent relative levels. GDNF, glial cell line-derived neurotropic factor.
DISCUSSION
In this study, we demonstrate that Tgif1 has a role in HSC maintenance, self-renewal, and quiescence. Importantly, Tgif1 affects HSC function in both the nullizygous state and the heterozygous state, while overexpression has opposite effects to loss of function in HSC populations. Tgif1 thus joins a very small list of genes whose knockdown has a positive effect and overexpression has a negative effect on HSC function (24). On the basis of its interactions with TGF-β and RA signaling pathways and its effects on myeloid cell functions, Tgif1 has been suggested to have a role in HSC regulation, and this study provides the first direct evidence by showing that Tgif1 has important actions in the most primitive hematopoietic stem and progenitor cells.
We showed that Tgif1 had an effect on both hematopoietic progenitor and HSC function. Tgif1−/− bone marrow produced more CFU and showed increased replating potential. Both short-term and long-term transplant studies further showed that Tgif1−/− bone marrow cells had significantly greater engraftment potential than wild-type bone marrow cells. Since long-term transplantation potential is derived from the contribution of the most primitive HSCs, these data suggest that Tgif1 knockout affects the function of the most undifferentiated populations of HSCs.
Quiescence and self-renewal are critical for pool preservation in adult HSCs and thereby long-term engraftment potential (28). One of the defining characteristics of adult HSCs is their quiescence and tendency to reside in a G0 state (27). Decreased quiescence can lead to defects in HSC self-renewal and eventual HSC exhaustion. Indeed, Tgif1−/− bone marrow had more HSCs in G0 phase than wild-type bone marrow, and secondary transplant data showed that Tgif1−/− bone marrow cells had enhanced self-renewal, as evidenced by significantly increased engraftment compared to the primary transplants. Our data are consistent with a previously reported finding of decreased proliferation and increased senescence in mouse embryonic fibroblasts carrying a Tgif1 deletion (38). Taken together, these data indicate that the enhanced engraftment ability in Tgif1−/− mice can likely be explained by their increased quiescence and potential for self-renewal.
While a number of gene knockouts have been found to alter HSC function, heterozygosity has been shown to affect HSC function in only a very small number of genes. By way of example, Gata2+/− bone marrow cells display poorly competitive repopulating ability, and their LSK population is more quiescent and displays an increased frequency of apoptosis. Likewise, haploinsufficiency of Shp2 resulted in decreased HSC repopulating capacity, with less quiescent LSK cells displaying reduced self-renewal ability. Finally, heterozygosity of Apc led to expansion of LT- and ST-HSC compartments and reduced HSC reconstituting capacity (39–41). Interestingly, our data show that a 50% reduction of Tgif1 can also affect both progenitor and HSC function. Tgif1+/− bone marrow showed multiple functional changes, including increased numbers of CFU and increased engraftment in primary as well as secondary transplantation.
While highly significant from a statistical standpoint, Tgif1 nullizygosity had only a mild effect on the HSC phenotype and no effect on the peripheral blood counts under steady-state conditions. One of the reasons for this might be gene redundancy. It has previously been hypothesized that the closely related homeobox gene Tgif2 might compensate for the reduced activity of Tgif1 in certain tissues or developmental contexts, since it shares many of the functional attributes of Tgif1 (12). In addition, Tgif1 and Tgif2 clearly perform overlapping functions during early embryonic development in mice (12, 13). Furthermore, our data also show that SLAM cells from Tgif1−/− mice express significantly higher levels of Tgif2 than SLAM cells from wild-type mice (see Fig. S3 in the supplemental material). Thus, our future plans involve investigating HSC function in mice with a combined knockout of Tgif1 and Tgif2.
Another important finding of our study was that Tgif1 overexpression in HSCs appeared to have the converse effect to Tgif1 knockdown. In this aspect, Tgif1 function resembles the effects of manipulating Myc expression in hematopoiesis, where conditional knockout of Myc in bone marrow cells resulted in accumulation of LT-HSCs, while its enforced expression resulted in lower HSC numbers (42).
The precise mechanism by which Tgif1 affects progenitor and HSC function remains to be defined. TGF-β and RA pathways were logical downstream candidates, since Tgif1 is an inhibitor of both pathways through its interactions with Smad2/3 proteins and RA receptor binding sites, respectively (2, 5–7), and there is evidence of effects of TGF-β on HSC quiescence (43, 44); furthermore, as discussed in the introduction, both have been shown to have important roles in hematopoiesis (14–16). Gene expression analysis did not show significant alterations of known TGF-β or RA target genes in the Tgif1−/− LSK cells. Although GSEA revealed activation of RA signaling, the RA gene set was not near the top of the list. However, the expression data identified multiple genes and pathways known to be involved in HSC quiescence and self-renewal that were perturbed in Tgif1−/− bone marrow cells. For example, Alox5 can block differentiation and division of leukemia stem cells and plays an essential role during chronic myeloid leukemia development (45). One interpretation of these data is that TGF-β and RA signaling pathways are not as important as direct regulation of transcription by Tgif1 in HSCs. It is also possible that one or both signaling pathways do, in fact, play a role but their role was masked by the heterogeneity of LSK cells; in comparison, the SLAM cell population is more primitive (and more homogeneous). Additionally, it should be noted that the gene responses to such signaling pathways are cell type specific, and the best-characterized target genes may not change in all cell types. Further, from our data on Tgif2 expression, it is possible that the effects were blunted by the increased Tgif2 expression in Tgif1-null cells (see Fig. S3 in the supplemental material). Ultimately, combined chromatin immunoprecipitation-sequencing analysis and expression profiling, potentially requiring a double-knockout (Tgif1 and Tgif2) mouse model, will be needed to resolve this issue.
TGIF1 is a TALE transcription factor, and other TALE proteins, including MEIS1 and PBX1, have been implicated in hematopoiesis and leukemogenesis (44, 46–48). For example, MEIS1 levels are important for leukemia stem cell function and chemotherapy resistance (49, 50), and PBX1 regulates HSC self-renewal and maintains HSC quiescence through multiple pathways (44). An increased HSC pool, enhanced quiescence, and an enhanced potential for self-renewal may make Tgif1−/− HSCs more prone to malignant transformation or increase leukemic stem cell or progenitor cell survival with chemotherapy. This would be analogous to what has been observed in MEF/ELF4 knockout mice, in which HSCs were increased in number, outcompeted wild-type cells in repopulation assays, and were less proliferative and more quiescent. MEF/ELF4−/− HSCs are also more prone to malignant transformation, although they have an otherwise normal life span without spontaneous tumor formation (51). TGIF1 could also impact AML cell biology through regulation of lymphoid enhancer-binding factor 1 (LEF1). High expression of LEF1 appears to be a favorable prognostic predictor in AML (52), and our data show that Lef1 levels are significantly lower in Tgif1−/− bone marrow cells than wild-type bone marrow cells.
Finally, we have found that a reduced TGIF1 RNA abundance in leukemic blasts is associated with poor long-term survival in patients with AML (21). In contrast to individuals with holoprosencephaly, these patients did not have a mutation in the TGIF1 coding or regulatory regions. Furthermore, we have shown that TGIF1 expression levels are a continuously variable trait, i.e., a quantitative genetic trait, in human lymphocyte cell lines (53). These data, when taken together with the data showing that Tgif1 heterozygosity affects HSC function and Tgif1 overexpression has the opposite phenotype to Tgif1 nullizygosity, suggest that variations in TGIF1 levels may be important for human HSC function and that TGIF1 may be a modifier of HSC and, potentially, leukemic stem cell function.
In summary, our data define Tgif1 to be a novel regulator of normal HSC function, and given the correlation between its abundance and prognosis in patients with AML, it may also have an important role in leukemic hematopoiesis.
Supplementary Material
ACKNOWLEDGMENTS
We thank Melissa Stauffer (Scientific Editing Solutions) for her editorial help.
The research presented in this report was supported by National Institutes of Health (K08 HL089903) and American Cancer Society (Scholar award LIB-118462) awards to R.H.
Footnotes
Published ahead of print 7 October 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01076-13.
REFERENCES
- 1. Burglin TR. 1997. Analysis of TALE superclass homeobox genes (MEIS, PBC, KNOX, Iroquois, TGIF) reveals a novel domain conserved between plants and animals. Nucleic Acids Res. 25:4173–4180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bartholin L, Powers SE, Melhuish TA, Lasse S, Weinstein M, Wotton D. 2006. TGIF inhibits retinoid signaling. Mol. Cell. Biol. 26:990–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Seo SR, Lallemand F, Ferrand N, Pessah M, L'Hoste S, Camonis J, Atfi A. 2004. The novel E3 ubiquitin ligase Tiul1 associates with TGIF to target Smad2 for degradation. EMBO J. 23:3780–3792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Seo SR, Ferrand N, Faresse N, Prunier C, Abecassis L, Pessah M, Bourgeade MF, Atfi A. 2006. Nuclear retention of the tumor suppressor cPML by the homeodomain protein TGIF restricts TGF-beta signaling. Mol. Cell 23:547–559 [DOI] [PubMed] [Google Scholar]
- 5. Wotton D, Lo RS, Lee S, Massague J. 1999. A Smad transcriptional corepressor. Cell 97:29–39 [DOI] [PubMed] [Google Scholar]
- 6. Wotton D, Lo RS, Swaby LA, Massague J. 1999. Multiple modes of repression by the Smad transcriptional corepressor TGIF. J. Biol. Chem. 274:37105–37110 [DOI] [PubMed] [Google Scholar]
- 7. Wotton D, Massague J. 2001. Smad transcriptional corepressors in TGF beta family signaling. Curr. Top. Microbiol. Immunol. 254:145–164 [PubMed] [Google Scholar]
- 8. Gripp KW, Wotton D, Edwards MC, Roessler E, Ades L, Meinecke P, Richieri-Costa A, Zackai EH, Massague J, Muenke M, Elledge SJ. 2000. Mutations in TGIF cause holoprosencephaly and link NODAL signalling to human neural axis determination. Nat. Genet. 25:205–208 [DOI] [PubMed] [Google Scholar]
- 9. Wallis DE, Muenke M. 1999. Molecular mechanisms of holoprosencephaly. Mol. Genet. Metab. 68:126–138 [DOI] [PubMed] [Google Scholar]
- 10. Wallis D, Muenke M. 2000. Mutations in holoprosencephaly. Hum. Mutat. 16:99–108 [DOI] [PubMed] [Google Scholar]
- 11. Ming JE, Muenke M. 2002. Multiple hits during early embryonic development: digenic diseases and holoprosencephaly. Am. J. Hum. Genet. 71:1017–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Powers SE, Taniguchi K, Yen W, Melhuish TA, Shen J, Walsh CA, Sutherland AE, Wotton D. 2010. Tgif1 and Tgif2 regulate Nodal signaling and are required for gastrulation. Development 137:249–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Taniguchi K, Anderson AE, Sutherland AE, Wotton D. 2012. Loss of Tgif function causes holoprosencephaly by disrupting the SHH signaling pathway. PLoS Genet. 8:e1002524. 10.1371/journal.pgen.1002524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Langer JC, Henckaerts E, Orenstein J, Snoeck HW. 2004. Quantitative trait analysis reveals transforming growth factor-beta2 as a positive regulator of early hematopoietic progenitor and stem cell function. J. Exp. Med. 199:5–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yamazaki S, Iwama A, Takayanagi S, Eto K, Ema H, Nakauchi H. 2009. TGF-beta as a candidate bone marrow niche signal to induce hematopoietic stem cell hibernation. Blood 113:1250–1256 [DOI] [PubMed] [Google Scholar]
- 16. Evans T. 2005. Regulation of hematopoiesis by retinoid signaling. Exp. Hematol. 33:1055–1061 [DOI] [PubMed] [Google Scholar]
- 17. Hamid R, Brandt SJ. 2009. Transforming growth-interacting factor (TGIF) regulates proliferation and differentiation of human myeloid leukemia cells. Mol. Oncol. 3:451–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Phillips RL, Ernst RE, Brunk B, Ivanova N, Mahan MA, Deanehan JK, Moore KA, Overton GC, Lemischka IR. 2000. The genetic program of hematopoietic stem cells. Science 288:1635–1640 [DOI] [PubMed] [Google Scholar]
- 19. Sato N, Sanjuan IM, Heke M, Uchida M, Naef F, Brivanlou AH. 2003. Molecular signature of human embryonic stem cells and its comparison with the mouse. Dev. Biol. 260:404–413 [DOI] [PubMed] [Google Scholar]
- 20. Venezia TA, Merchant AA, Ramos CA, Whitehouse NL, Young AS, Shaw CA, Goodell MA. 2004. Molecular signatures of proliferation and quiescence in hematopoietic stem cells. PLoS Biol. 2:e301. 10.1371/journal.pbio.0020301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Means-Powell JA, Kravtsov VD, Shyr Y, Levy SE, Greer JP, Koury MJ, Brandt SJ. 2003. Expression of homeobox gene TG-interaction factor is an independent predictor of survival in acute myelogenous leukemia, abstr 764, p 128a Platform presentation, 45th Am. Soc. Hematol. Meet. [Google Scholar]
- 22. Harrison DE, Astle CM. 1997. Short- and long-term multilineage repopulating hematopoietic stem cells in late fetal and newborn mice: models for human umbilical cord blood. Blood 90:174–181 [PubMed] [Google Scholar]
- 23. Zon LI. 2008. Self-renewal and differentiation at Cell Stem Cell. Cell Stem Cell 2:510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rossi L, Lin KK, Boles NC, Yang L, King KY, Jeong M, Mayle A, Goodell MA. 2012. Less is more: unveiling the functional core of hematopoietic stem cells through knockout mice. Cell Stem Cell 11:302–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M, Scadden DT. 2000. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science 287:1804–1808 [DOI] [PubMed] [Google Scholar]
- 26. Cheshier SH, Morrison SJ, Liao X, Weissman IL. 1999. In vivo proliferation and cell cycle kinetics of long-term self-renewing hematopoietic stem cells. Proc. Natl. Acad. Sci. U. S. A. 96:3120–3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pietras EM, Warr MR, Passegue E. 2011. Cell cycle regulation in hematopoietic stem cells. J. Cell Biol. 195:709–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zon LI. 2008. Intrinsic and extrinsic control of haematopoietic stem-cell self-renewal. Nature 453:306–313 [DOI] [PubMed] [Google Scholar]
- 29. Li L, Bhatia R. 2011. Stem cell quiescence. Clin. Cancer Res. 17:4936–4941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bianco P. 2011. Bone and the hematopoietic niche: a tale of two stem cells. Blood 117:5281–5288 [DOI] [PubMed] [Google Scholar]
- 31. Blank U, Karlsson G, Karlsson S. 2008. Signaling pathways governing stem-cell fate. Blood 111:492–503 [DOI] [PubMed] [Google Scholar]
- 32. Bartholin L, Melhuish TA, Powers SE, Goddard-Leon S, Treilleux I, Sutherland AE, Wotton D. 2008. Maternal Tgif is required for vascularization of the embryonic placenta. Dev. Biol. 319:285–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fischer MA, Moreno-Miralles I, Hunt A, Chyla BJ, Hiebert SW. 2012. Myeloid translocation gene 16 is required for maintenance of haematopoietic stem cell quiescence. EMBO J. 31:1494–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Harrison DE, Jordan CT, Zhong RK, Astle CM. 1993. Primitive hemopoietic stem cells: direct assay of most productive populations by competitive repopulation with simple binomial, correlation and covariance calculations. Exp. Hematol. 21:206–219 [PubMed] [Google Scholar]
- 35. Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7:562–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 102:15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lemischka IR, Raulet DH, Mulligan RC. 1986. Developmental potential and dynamic behavior of hematopoietic stem cells. Cell 45:917–927 [DOI] [PubMed] [Google Scholar]
- 38. Zerlanko BJ, Bartholin L, Melhuish TA, Wotton D. 2012. Premature senescence and increased TGFbeta signaling in the absence of Tgif1. PLoS One 7:e35460. 10.1371/journal.pone.0035460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rodrigues NP, Janzen V, Forkert R, Dombkowski DM, Boyd AS, Orkin SH, Enver T, Vyas P, Scadden DT. 2005. Haploinsufficiency of GATA-2 perturbs adult hematopoietic stem-cell homeostasis. Blood 106:477–484 [DOI] [PubMed] [Google Scholar]
- 40. Chan RJ, Li Y, Hass MN, Walter A, Voorhorst CS, Shelley WC, Yang Z, Orschell CM, Yoder MC. 2006. Shp-2 heterozygous hematopoietic stem cells have deficient repopulating ability due to diminished self-renewal. Exp. Hematol. 34:1230–1239 [DOI] [PubMed] [Google Scholar]
- 41. Wang J, Fernald AA, Anastasi J, Le Beau MM, Qian Z. 2010. Haploinsufficiency of Apc leads to ineffective hematopoiesis. Blood 115:3481–3488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wilson A, Murphy MJ, Oskarsson T, Kaloulis K, Bettess MD, Oser GM, Pasche AC, Knabenhans C, Macdonald HR, Trumpp A. 2004. c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev. 18:2747–2763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Brenet F, Kermani P, Spektor R, Rafii S, Scandura JM. 2013. TGFbeta restores hematopoietic homeostasis after myelosuppressive chemotherapy. J. Exp. Med. 210:623–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ficara F, Murphy MJ, Lin M, Cleary ML. 2008. Pbx1 regulates self-renewal of long-term hematopoietic stem cells by maintaining their quiescence. Cell Stem Cell 2:484–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen Y, Hu Y, Zhang H, Peng C, Li S. 2009. Loss of the Alox5 gene impairs leukemia stem cells and prevents chronic myeloid leukemia. Nat. Genet. 41:783–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kocabas F, Zheng J, Thet S, Copeland NG, Jenkins NA, Deberardinis RJ, Zhang C, Sadek HA. 2012. Meis1 regulates the metabolic phenotype and oxidant defense of hematopoietic stem cells. Blood 120:4963–4972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kumar AR, Li Q, Hudson WA, Chen W, Sam T, Yao Q, Lund EA, Wu B, Kowal BJ, Kersey JH. 2009. A role for MEIS1 in MLL-fusion gene leukemia. Blood 113:1756–1758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sanyal M, Tung JW, Karsunky H, Zeng H, Selleri L, Weissman IL, Herzenberg LA, Cleary ML. 2007. B-cell development fails in the absence of the Pbx1 proto-oncogene. Blood 109:4191–4199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rosales-Avina JA, Torres-Flores J, Aguilar-Lemarroy A, Gurrola-Diaz C, Hernandez-Flores G, Ortiz-Lazareno PC, Lerma-Diaz JM, de Celis R, Gonzalez-Ramella O, Barrera-Chaires E, Bravo-Cuellar A, Jave-Suarez LF. 2011. MEIS1, PREP1, and PBX4 are differentially expressed in acute lymphoblastic leukemia: association of MEIS1 expression with higher proliferation and chemotherapy resistance. J. Exp. Clin. Cancer Res. 30:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kumar AR, Sarver AL, Wu B, Kersey JH. 2010. Meis1 maintains stemness signature in MLL-AF9 leukemia. Blood 115:3642–3643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lacorazza HD, Yamada T, Liu Y, Miyata Y, Sivina M, Nunes J, Nimer SD. 2006. The transcription factor MEF/ELF4 regulates the quiescence of primitive hematopoietic cells. Cancer Cell 9:175–187 [DOI] [PubMed] [Google Scholar]
- 52. Metzeler KH, Heilmeier B, Edmaier KE, Rawat VP, Dufour A, Dohner K, Feuring-Buske M, Braess J, Spiekermann K, Buchner T, Sauerland MC, Dohner H, Hiddemann W, Bohlander SK, Schlenk RF, Bullinger L, Buske C. 2012. High expression of lymphoid enhancer-binding factor-1 (LEF1) is a novel favorable prognostic factor in cytogenetically normal acute myeloid leukemia. Blood 120:2118–2126 [DOI] [PubMed] [Google Scholar]
- 53. Means-Powell J, Martincic D, Brandt S, Hamid R. 2010. Genetic heterogeneity in Tg-Interacting Factor (TGIF) expression is a modifier of acute myeloid leukemia, abstr 523/T, p 127 Abstr. 60th Annu. Meet. Am. Soc. Hum. Genet., Washington, DC [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.