

Abstract
Normal cells secrete heat shock protein 90 alpha (Hsp90α) in response to tissue injury. Tumor cells have managed to constitutively secrete Hsp90α during invasion and metastasis. The sole function of extracellular Hsp90α (eHsp90α) is to promote cell motility, a critical event for both wound healing and tumor progression. The mechanism of promotility action by eHsp90α, however, has remained elusive. A key issue is whether eHsp90α still acts as a chaperone outside the cells or is a new and bona fide signaling molecule. Here, we have provided evidence that eHsp90α utilizes a unique transmembrane signaling mechanism to promote cell motility and wound healing. First, subdomain II in the extracellular part of low-density lipoprotein receptor-related protein 1 (LRP-1) receives the eHsp90α signal. Then, the NPVY but not the NPTY motif in the cytoplasmic tail of LRP-1 connects eHsp90α signaling to serine 473 but not threonine 308 phosphorylation in Akt kinases. Individual knockdown of Akt1, Akt2, or Akt3 revealed the importance of Akt1 and Akt2 in eHsp90α-induced cell motility. Akt gene rescue experiments suggest that Akt1 and Akt2 work in concert, rather than independently, to mediate eHsp90α promotility signaling. Finally, Akt1 and Akt2 knockout mice showed impaired wound healing that cannot be corrected by topical application with the eHsp90α protein.
INTRODUCTION
The cytosolic Hsp90 (heat shock protein 90) family proteins, including Hsp90α and Hsp90β, are among the most abundantly expressed proteins in almost all cells. Inside the cell, Hsp90 proteins act as an ATPase-dependent chaperone that binds and protects hundreds of client proteins from misfolding, structural damage, or protease degradation triggered by environmental insults or pathological stress (1–4). For the past 10 years, in particular, studies have convincingly made it clear that the same stress signals can also trigger the cells to secrete cytosolic Hsp90 proteins into the extracellular environment (5–8). Outside the cell, the sole function of extracellular Hsp90α (eHsp90α) is to promote cell motility. At least one physiological role for eHsp90α is promotion of tissue repair, such as skin wound healing (6, 9, 10). Certain tumor cells, such as those that maintain a steady-state level of hypoxia-inducible factor 1 alpha (HIF-1α), acquired the ability to constitutively secrete Hsp90α for invasion and metastasis (5, 6, 8, 11–14).
Why was eHsp90α chosen (by Mother Nature) for wound healing? For decades, the conventional wisdom dictated that growth factors were the driving force of skin wound healing (15). Despite extensive experimental studies and numerous clinical trials to determine if exogenous applications of growth factors, either alone or in combinations, stimulate wound healing in humans, only human recombinant platelet-derived growth factor BB (PDGF-BB) (becaplermin gel) received FDA approval for treatment of diabetic foot ulcers. However, PDGF-BB has since shown a modest efficacy, high cost, and risk of causing cancer in patients. Three potential obstacles for conventional growth factors in wound healing have been identified. (i) The action of a given growth factor is often restricted to limited cell types, while multiple cell types are involved in wound healing. (ii) The primary effects of a growth factor, such as promotion of cell proliferation and migration, can be nullified by transforming growth factor β3 (TGF-β3) present in human wounds. (iii) The effectiveness of a growth factor is greatly compromised by hyperglycemia, a hallmark of diabetic wounds (6, 9). In contrast, eHsp90α promotes motility of all types of skin cells, overrides the inhibition of TGF-β3, and remains fully functional even under hyperglycemia. In accordance with these in vitro features of eHsp90α, topical application of recombinant Hsp90α accelerated wound healing with an efficacy significantly stronger than that of becaplermin (PDGF-BB) gel (9).
While findings of studies for both wound healing and cancer agreed on the main function for eHsp90α, i.e., promoting cell motility, a fundamental debate centers on how eHsp90α carries out its extracellular function. Some studies suggested that even outside the cells, eHsp90α still acts as a chaperone protein by assisting proper folding and activation of other surface-bound or secreted proteins, such as HER2 tyrosine kinase receptor, matrix metalloproteinase 2 (MMP2), and extracellular matrix (ECM) proteins (16–18). Others argued that eHsp90α is no longer a chaperone and acts as a novel promotility factor via a cell surface receptor (19–21). However, neither of the studies in the past provided direct evidence for or against the two distinct hypotheses. In this study, we have established a novel eHsp90α cross-membrane signaling pathway that includes extracellular subdomain II and the cytoplasmic NPVY motif in the LRP-1 receptor and downstream activation of the Akt1 and Akt2 kinases. This pathway is essential for eHsp90α-stimulated skin cell migration in vitro and wound healing in vivo.
MATERIALS AND METHODS
Primary human dermal fibroblasts (HDFs) were purchased from Clonetics and cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (Gibco/Invitrogen Corp., Carlsbad, CA). The third or fourth passages of the cells were used for in vitro cell assays. Mouse monoclonal antibody against the p85 subunit of LRP-1/CD91 was purchased from Invitrogen Co. (Grand Island, NY) (37-7600). A proteome profiler human array kit was purchased from the R&D Systems (Minneapolis, MN). Rabbit anti-human Akt1 (C73H10), Akt2 (D6G4), and Akt3 (62AB) antibodies and anti-β-actin and anti-glyceraldehyde-3-phosphate dehydrogenase (anti-GAPDH) antibodies were purchased from Cell Signaling Technology (Danvers, MA). Antibody against the phosphorylated forms of p44/extracellular signal-regulated kinase 1 (ERK1) and p42/ERK2 (V8031) and the ProFection mammalian transfection system (E1200) were from Promega (Madison, WI). Rat type I collagen was purchased from BD Biosciences (San Jose, CA). Mini-LRP-1 receptor constructs (mLRP1-I to -IV) were gifts of Guojun Bu (Mayo Clinic, Jacksonville, FL). The QuikChange site-directed mutagenesis kit (200519) and XL-10 Gold Ultra competent cells were from Stratagene (La Jolla, CA). Akt1 and Akt2 knockout mice were provided by the laboratories of Nissim Hay and Bangyan Stiles, respectively. Carboxymethylcellulose (CMC) sodium salt (C5678) (pH 7.14) was from Sigma-Aldrich (St. Louis, MO). Stat Strips (adhesive bandages) were from Notro Max (Edgewood, MD). 3M Coban (self-adhesive wrap) was from 3M (St. Paul, MN). Other reagents were used as needed for experimental procedures.
Designing, testing, and cloning shRNAs against human LRP-1, Akt-1, Akt-2, and Akt-3 into lentiviral vector FG12.
Target sequences for small hairpin RNAs (shRNAs) against LRP-1, Akt-1, Akt-2, and Akt-3 were selected by using an RNA interference selection program, as previously described (20). Effectiveness of the synthetic double-stranded shRNAs for downregulation of their target genes was first tested by transfecting the shRNAs into 293T cells. Following 48 h of incubation, the cell lysates were blotted with antibodies against the corresponding gene products. Then, the most effective sequence of a given shRNA against a target gene was cloned into the lentiviral RNA interference (RNAi) delivery vector, FG-12. The selected shRNA (sense sequence) for human LRP-1 was GGAGTGGTATTCTGGTATA; for human Akt-1, it was TCACACCACCTGACCAAGA; for human Akt-2, it was GGGCTAAAGTGACCATGAA; and for Akt-3, it was GCAGGCACGTTAACTCGAA. Lentiviral production, titer determination, infection, and biochemical analyses were as previously described (20).
Cell migration assays.
The colloidal gold migration assay and the in vitro wound-healing (scratch) assay were conducted as described in detail previously (22). Data from independent experiments (n ≥ 3) were averaged and calculated (means ± standard deviations [SD]).
Production and purification of recombinant Hsp90α, F-5, and Hsp90β.
The cDNAs encoding the full-length human Hsp90α, F-5, or Hsp90β were generated by PCR and cloned into the pET15b vector (EMD Biosciences Inc., Darmstadt, Germany) at the BamHI site. The cloned mutation-free cDNA sequence was verified by DNA sequencing. The pET15b-Hsp90α construct was transformed into BL21-codonPlus (DE3)-RP competent cells following the manufacturer's instruction (Stratagene). Protein production and purification using the pET15b system were described previously (9). Briefly, Hsp90α protein synthesis was induced by addition of 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) to the bacterium culture (optical density [OD], 0.8 to 1), followed by an ∼5-h incubation at 25°C. The His-tagged Hsp90 protein was purified via a nickel-nitrilotriacetic acid (Ni-NTA) column containing HisBind resin according to the manufacturer's procedures (EMD Biosciences, Inc.). Partially purified protein was concentrated in Amicon Ultra 10K to 50K centrifugal filters (Millipore, Billerica, MA) to 1 to 2 ml, loaded onto a Superdex 200 HiLoad gel filtration column (GE Healthcare, Piscataway, NJ), and further purified by fast-protein liquid chromatography (FPLC) (AKTApurifier 10; GE Pharmacia). Proteins were eluted with Dulbecco's phosphate-buffered saline buffer (DPBS) at a flow rate of 1 ml/min. The fractions with Hsp90α were collected and concentrated in an Amicon Ultra 10K to 50K centrifugal filter to achieve a final concentration of 1 mg/ml. Proteins were stored in 10% glycerol–Dulbecco's phosphate-buffered saline at −80°C. We routinely obtained 10 to 20 mg of Hsp90α from 1 liter of bacterium culture.
Glutathione S-transferase (GST) fusion protein production and GST fusion protein pulldown assays were as described previously (20).
Site-directed mutagenesis on LRP-1 receptor.
Mutagenesis on the proximal NPXY motif from 4470NPTY to AATA was carried out using the sense and antisense primers CCATGAACGTGGAGATTGGAGCCGCCACCGCCAAGATGTACGAAGGCGGAG and CTCCGCCTTCGTACATCTTGGCGGTGGCGGCTCCAATCTCCACGTTCATGG, respectively. Mutagenesis on the distal NPXY motif from 4504NPVY to AAVA was carried out using the following primers: AAGCCCACCAACTTCACCGCCGCCGTGGCTGCCACACTCTACATGGG and CCCATGTAGAGTGTGGCAGCCACGGCGGCGGTGAAGTTGGTGGGCTT. The entire process followed the manufacturer's instructions (QuikChange site-directed mutagenesis kit). Selections for positive clones were carried out using XL-10 Gold Ultra competent cells.
Preparation of CMC gel with recombinant human Hsp90α proteins.
To prepare the wound-healing formula, CMC powder (viscosity, 50 to 200 cps; purity, 99.5%, sodium salt) was dissolved in double-distilled H2O in a tissue culture hood at a 10% (wt/vol) concentration. The mixture was incubated for 4 h at 37°C and then placed on a shaker for 24 h at 4°C. After being equilibrated back to room temperature, the CMC solution was mixed at a 1:1 ratio (vol/vol) with indicated concentrations of FPLC-purified, filtered (0.22 μm) recombinant Hsp90α or F-5 proteins. These mixtures were topically applied to skin wounds in the mouse models.
Wound healing in mice.
Thymic hairless (Foxn1) mice and BKS.Cg-m+/+Leprdb/J mice were obtained from The Jackson Laboratory. Akt-1−/− and Akt-2−/− mice were made available by laboratories of Nissim Hay and Bangyan Stiles, respectively. Mice were housed 4 per cage for maintenance and 1 mouse per cage during experiments at the animal facilities of the University of Southern California, under protocols approved by the University of Southern California Institutional Animal Use Committee. Mice were anesthetized with sodium pentobarbital (Nembutal; 50-mg/ml stock solution, murine dosage at 35 to 50 mg/kg of body weight) and isoflurane (as needed) prior to wound surgery. Full-thickness excision wounds (1 cm by 1 cm) were created by marking the area of the wound in the midback with a marker pen and a ruler, lifting the skin with a pair of forceps, and excising the full-thickness skin along the lines with a pair of surgical scissors. Each wound was dressed with a bandage and a self-adherent wrap (Coban) to prevent desiccation and infection for the duration of the experiments. Bandages and Coban wraps were changed every 3 days after an initial 4-day postsurgical recovery period. Standardized digital photographs were taken of the wounds, using a metric ruler. The photographs were examined using planimetry for objective evaluation for the degree of wound closure. The open-wound areas were determined using an image analyzer (AlphaEase FC, version 4.1.0; Alpha Innotech Corporation). The total pixels that cover the unhealed areas were drawn onto the digital photographs using a pattern overlay in the software program ImageJ (http://rsbweb.nih.gov/ij/). The number of pixels covering an open-wound area on a given day was divided by the number of pixels spreading over the initial day 0 wound to obtain the percentage of closure.
Statistical analyses.
Data are based on three independent experiments, in which three mutant and control animals were used for each stage. Colloidal gold salt migration assay quantification was achieved by measuring the individual tracks of 20 randomly selected individual cells per experimental condition, where each condition in an experiment was repeated at least three times. The data are presented as means ± SD. Statistical differences were evaluated using the two-tailed Student t test for comparisons of two groups or analysis of variance for comparisons of more than two groups. A P value of <0.05 was considered significant.
RESULTS
eHsp90α requires LRP-1 receptor to promote cell migration in vitro and wound closure in mice.
To investigate the mechanism of eHsp90α action, we focused on LRP-1, a suggested cell surface receptor for eHsp90α signaling (20, 21). We chose to use primary human dermal fibroblasts (HDFs) as the cell culture model for the investigation for the following reasons. (i) HDFs are essential for skin wound healing (15), (ii) HDFs secrete Hsp90α in response to skin wounding signals, such as hypoxia (10), and (iii) HDFs express LRP-1 and exhibit increased migration in response to recombinant eHsp90α stimulation (9, 20). To confirm the critical role of LRP-1 in eHsp90α signaling, we used the lentivirus system FG-12 to deliver shRNA against human LRP-1 to downregulate the endogenous LRP-1 protein in HDFs. As shown in Fig. 1A, infection with a control LacZ shRNA did not affect the LRP-1 protein levels in the cells (panel a, lane 1). In the shRNA-LRP-1-infected cells, LRP-1 expression was dramatically downregulated (panel a, lane 2) even with (deliberately) loading twice the amount of total lysates over that of the control (panel b, lane 2 versus lane 1). When these cells were subjected to colloidal gold migration assays, as shown in Fig. 1B, the LacZ shRNA-infected HDFs exhibited enhanced migration in response to both eHsp90α (panel b versus panel a) and PDGF-BB (panel c) (as a positive control) stimulation. However, eHsp90α was no longer able to promote any migration of the FG-12-shRNA-LRP-1-infected HDFs (panel e versus panel b). Interestingly, this defect was restricted only to eHsp90α signaling, since PDGF-BB was still able to stimulate migration in both control and LRP-1-downregulated cells (panel f versus panel c). These conclusions are supported by quantitation of the migration data as shown in Fig. 1C.
Fig 1.

LRP-1 is critical for eHsp90α to promote cell motility in vitro and wound healing in vivo. (A) The lentiviral RNAi system FG-12 mediated downregulation of endogenous LRP-1 in HDFs, confirmed by anti-LRP-1 antibody blotting of total cell lysates (panel a, lane 2 versus lane 1). We deliberately loaded twice the amount of the lysate from the shRNA-LRP-1-infected HDFs to emphasize the efficiency of downregulation. (B) Cells were subjected to a colloidal gold migration assay in response to either eHsp90α or PDGF-BB stimulation. A representative image of the “migration tracks” under each condition is shown, in which dotted circles point out the average size of the migration tracks. (C) Quantitation of the migration was presented as a migration index. (D) Full-thickness skin wounds (1 cm by 1 cm) in athymic nude mice (n = 3 mice per treatment per experiment) were treated with either the vehicle alone (−) or RAP (0.3 mM) (+). Images of a representative experiment are shown. Mean ± SD error of the percentage of the wound closure at days 0, 4, 8, and 12, RAP-treated wounds versus vehicle-treated wounds, is shown below the images. ∗, significant enhancement/inhibition (P ≤ 0.05) against results for vehicle-treated wounds.
To confirm the importance of LRP-1 in wound healing in vivo, we made use of a specific LRP-1 inhibitor, the receptor-associated protein (RAP) that blocks eHsp90α-stimulated cell migration in vitro (20). As clearly shown in Fig. 1D, topical application of recombinant RAP (only once on day 0) dramatically delayed wound closure in mice (right wound versus left wound from panels a to d). Quantitation of the data is shown below each of the wound images.
Extracellular subdomain II of LRP-1 mediates eHsp90α signaling.
The 13-kb cDNA for the human LRP-1 encodes a 515-kDa extracellular α subunit and a transmembrane 85-kDa β subunit that has a 100-amino-acid-long cytoplasmic tail. It is too large to be accommodated and expressed by any existing mammalian cDNA expression systems (23, 24). As schematically shown in Fig. 2A, LRP-1's extracellular domain is composed of four independent ligand-binding subdomains (I to IV). We therefore made use of four hemagglutinin (HA)-tagged mini-LRP-1 receptors (mLRP1-I to mLRP1-IV), in which each of four extracellular subdomains was fused to the p85 subunit gene (25, 26). Using these mini-LRP-1 receptors, we examined which subdomain(s) is sufficient to mediate eHsp90α signaling. Prior to expressing these mini-LRP-1 receptors in HDFs, we downregulated endogenous LRP-1 in the cells. As shown in Fig. 2B, we achieved an almost complete downregulation of endogenous LRP-1 (panel a, lane 2). We then used the lentivirus vector overexpressing system, pRRLsin, to express each of the mLRP1 cDNAs in these cells. Expression of the exogenous mLRP1 genes was verified by using anti-HA antibody Western blotting assays. As shown in Fig. 2C, the pRRLsin-mLRP1-infected HDFs showed comparable levels of mLRP1-I (lane 2), mLRP1-II (lane 3), mLRP1-III (lane 4), and mLRP1-IV (lane 5) expression.
Fig 2.

Subdomain II in LRP-1 mediates promotility signaling by eHsp90α. (A) A schematic representation of the four subdomains (I to IV) and cytoplasmic tail of human LRP-1. (B) Downregulation of endogenous LRP-1 in HDFs by FG-12 vector-delivered shRNA, shown by Western blotting. We loaded twice the amount of the lysate from shRNA-LRP-1-infected HDFs to demonstrate the efficiency of LRP-1 downregulation. (C) A lentiviral gene-overexpressing system, pRRL-sin-MCS-Deco, mediated expression of four mini-LRP-1 receptors (mLRP1-I to -IV) fused with the p85 subunit, confirmed by anti-HA antibody blotting. (D) GST-Hsp90α bead pulldown assays of the four mLRP1 receptors from lysates of the HDFs, as shown in panel C. (E) His-tagged full-length Hsp90α and F-5 on beads: pulldown assays of endogenous LRP-1 in HDFs. (F) LRP-1-downregulated HDFs expressing each of the four mLRP1 receptors were subjected to a colloidal gold migration assay in response to stimulation of either eHsp90α or PDGF-BB. The migration index of the experiments (n = 4) is shown. ∗, significant enhancement (P ≤ 0.05) compared to results with no treatment.
Using these cells, we first tested which subdomain in LRP-1 eHsp9α binds to. As shown in Fig. 2D, GST-Hsp90α did not bind subdomain I (lane 1), bound strongly to subdomain II (lane 2), bound to subdomain III slightly over the negative background (lane 3), and also bound significantly to subdomain IV (lane 4). Binding was apparently mediated by the F-5 fragment of Hsp90α, as previously reported by us (9, 20), since F-5 alone was able to pull down amounts of LRP-1 similar to those with full-length Hsp90α (Fig. 2E).
Then, we tested the hypothesis that one of the individual mLRP1 receptors should rescue the migratory defect of the LRP-1-downregulated HDFs in response to eHsp90α stimulation. As shown in Fig. 2F, downregulation of endogenous LRP-1 completely nullified the effect of eHsp90α (bar 3 versus bar 1). Expression of mLRP1-I (bar 6), mLRP1-III (bar 12), or mLRP1-IV (bar 15) was unable to rescue the defect. Interestingly, expression of mLRP1-II resumed migration of endogenous LRP-1-downregulated cells in response to eHsp90α stimulation (bar 9 versus bar 3). The reasons for eHsp90α binding to subdomain IV and not using it for signaling remain unclear. Again, the defect in migration in LRP-1-downregulated HDFs was specifically toward eHsp90α signaling, since PDGF-BB-stimulated migration remained unaffected (bars 2, 5, 8, 11, and 14). We conclude that eHsp90α binds to subdomain II of LRP-1 to execute its promotility signaling.
The intracellular NPVY motif in LRP-1 transmits eHsp90α's motility signal.
A critical question is whether eHsp90α can act as a bona fide signaling molecule that triggers cross-membrane signaling through the LRP-1 receptor. To provide answers to this question, we first focused on the two NPXY signaling motifs, 4470NPTY and 4504NPVY, in the cytoplasmic tail of LRP-1. As schematically shown in Fig. 3A, we replaced the three conserved amino acids in both NPXY motifs with alanines to create single (AAVA or AATA) and double (AAVA and AATA) mutants using mLRP1-II. The wild-type (wt) and mutant cDNAs of mLRP1-II were individually introduced into endogenous LRP-1-downregulated HDFs, as previously described. As shown in Fig. 3B, expression of the wt (lane 2) and NPXY mutant mLRP1-II proteins was confirmed by Western blotting with an antibody against the p85 subunit of LRP-1 (lanes 2 to 5 versus lane 1). When these cells were subjected to migration assays in response to eHsp90α, as shown in Fig. 3C, wt mLRP1-II was able to rescue the migratory defect in the endogenous LRP-1-downregulated HDFs in response to eHsp90α (bar 4 versus bar 2). Interestingly, mRLP1-II containing mutations in the proximal NPXY motif (AATA) also rescued the migration defect (bar 6). However, mLRP1-II that bears either the AAVA single or AATA/AAVA double mutations in the distal NxPY motif was unable to rescue the migratory defect of the cells in response to eHsp90α (bars 8 and 10). These data provide evidence for the first time that eHsp90α uses transmembrane signaling through LRP-1's NPVY motif to promote cell migration in the absence of any exogenously added factors, as schematically depicted in Fig. 3D.
Fig 3.

The NPVY motif in the cytoplasmic tail of LRP-1 transmits eHsp90α signaling. (A) Point mutations were introduced into two NPXY motifs, from NPTY to AATA and from NPVY to AAVA, using the mLRP1-II cDNA construct. (B) WT and mutant genes were expressed in LRP-1-downregulated HDFs, as detected by anti-p85 subunit of LRP-1 antibody blotting. (C) While the AATA mutation showed little effect (bar 6 versus bar 4), the mLRP1-II receptors with AAVY mutations completely blocked eHsp90α-stimulated cell migration (bars 8 and 10 versus bar 4). (D) A schematic representation of the finding shows the flow of the eHsp90α signaling. This experiment was repeated three times, and similar results were obtained.
Interestingly, however, we did not detect any tyrosine (Y) phosphorylation in LRP-1 in response to eHsp90α stimulation. As shown in Fig. 4A, PDGF-BB stimulated a strong tyrosine phosphorylation, including its 190-kDa receptor and several other protein species in the cells (lane 2 versus lane 1). Similarly, eHsp90α also triggered a weaker but significant, increase in global tyrosine phosphorylation in a time-dependent manner (lanes 3, 4, and 5, indicated by arrows). To study tyrosine phosphorylation of LRP-1, we first examined the efficiency of anti-LRP-1 antibody immunoprecipitation (IP). As shown in Fig. 4B, IP efficiency reached ∼50% based on the same volumes (TL versus IP) used and densitometry scanning data of the bands (B). Under these IP conditions, the immunoprecipitates of the same lysates (shown in Fig. 4A) were subjected to either anti-LRP-1 antibody blotting (10%) (C) or anti-PY blotting (90%) (D). As shown, we did not detect any significant tyrosine phosphorylation of LRP-1 (D).
Fig 4.
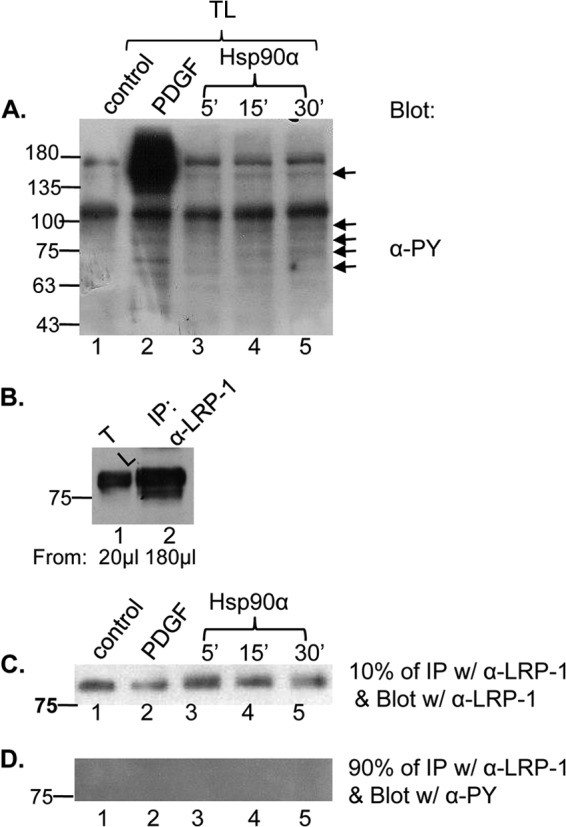
eHsp90α-induced increase in protein tyrosine phosphorylation does not include LRP-1. Parental HDFs were treated with PDGF-BB (50 ng/ml for 5 min) or with eHsp90α (0.1 μM) for the indicated time. (A) A fraction (20 μl) of the total lysates was analyzed by Western blotting using an anti-PY antibody. Arrows point out the protein species that showed increased tyrosine phosphorylation in a time-dependent fashion in response to eHsp90α. (B) Based on the differences in volumes of the same lysate used for total lysate (TL) and IP and densitometry scanning data, the calculated efficiency of IP by the anti-LRP-1 antibody was approximately 50 to 60% of the total cellular LRP-1 proteins. (C and D) The majority of the cell lysates shown in panel A was subjected to IP with the anti-LRP-1 antibody. Ten percent of the IP samples were subjected to Western blotting with the same anti-LRP-1 antibody (C). Ninety percent of the IP samples were blotted with the anti-PY antibody (D).
The NPVY motif connects eHsp90α signaling to Akt kinases.
To provide ultimate support for eHsp90α utilizing a cross-membrane signaling mechanism to execute its promotility function, we needed to prove that LRP-1 connects the eHsp90α signal to downstream cytoplasmic signaling networks. We undertook a “screening-then-focusing” approach. The parental and endogenous LRP-1-downregulated HDFs were treated without or with eHsp90α stimulation. The total lysates were subjected to a human phosphokinase antibody array that includes antibodies against the activated forms of 47 distinct protein kinases/pathways (see Materials and Methods). As shown in Fig. 5, in parental HDFs, eHsp90α stimulation caused increased phosphorylation of four pathways, including phospho-Stat5b, phospho-Stat5a, phospho-β-catenin, and phospho-Akt in serine 437 (S437) (panel A, b versus a) but not phospho-Akt in threonine-308 (T308), (panel b′ versus panel a′). Interestingly, the increased phosphorylation of all the four targets was undetectable in LRP-1-downregulated cells (panel B, d versus c). In addition, under a shorter period of exposure of the same blots (Fig. 5A and B), a dramatic increase in eHsp90α-stimulated phosphorylation of ERK1/2 became obvious (panel C, f versus panel e). ERK1/2 activation was also blocked in LRP-1-downregulated cells (panel D, h versus g).
Fig 5.

Pathway array detections of post-LRP-1 signaling by eHsp90α. The parental HDFs (A) and LRP-1-knocked-down HDFs (B) were serum starved for 16 h and either not treated (−) or treated (+) with recombinant Hsp90α (10 μg/ml or 0.1 μM for 10 min). The cell extracts were subjected to protein kinase array screening (see Materials and Methods). Activated pathways include phospho-Stat5b, phospho-Stat5a, phospho-β-catenin, phospho-Akt (S437), and phospho-ERK1/2. Akt(T-308), however, was negative. (C and D) When the same blots shown in panels A and B were underexposed (1 min instead of 3 min), they also revealed the activation of ERK1/2 in parent HDFs (C, panel f versus panel e) but not in LRP-1-knocked-down HDFs (D, panel h versus panel g).
Furthermore, we found that the NPVY motif of LRP-1 connects eHsp90α signaling to the Akt but not the ERK1/2 pathway. It is shown in Fig. 6A that in the endogenous LRP-1−/− background, eHsp90α-stimulated activation of both Akt (panel a, lane 2) and Erk1/2 (panel c, lane 2) was blocked, consistent with the array data. Reintroduction of the wt mLRP1-II or mLRP1-II-AATA mutant rescued eHsp90α-stimulated Akt activation in the cells (panel a, lanes 4 and 6 versus lane 2). In contrast, mLRP1-II-AAVA single and mLRP1-II-AATA/AAVA double mutants were unable to rescue eHsp90α-induced Akt phosphorylation (lanes 8 and 10). Interestingly, all mLRP1-II constructs, both wt and mutants, were able to rescue the eHsp90α-stimulated ERK1/2 activation (panel c, lanes 4, 6, 8, and 10 versus lane 2). These data indicate that the NPVY but not NPTY motif of LRP-1 connects eHsp90α signaling to the Akt pathway and eHsp90α stimulates ERK1/2 activation via some NPXY motif-independent mechanism. The relationships between NPXY motifs in LRP-1 and eHsp90α-stimulated phosphorylation of Stat5b, Stat5a, and β-catenin remain to be tested.
Fig 6.

The NPVY motif connects eHsp90α signaling to the Akt but not ERK1/2 pathway. (A) Based on the results of the protein kinase array, the endogenous LRP-1-downregulated HDFs expressing mLRP1-II with the AATA, AAVA, or AATA/AAVA mutations were serum starved and stimulated without or with eHsp90α (0.1 μM, 15 min.). Protein-equalized lysates of the cells were subjected to Western blotting with anti-phospho-Akt(S437), anti-phospho-ERK1/2, or control antibody. (B) Parental HDFs were serum starved and treated without or with increasing concentrations of eHsp90α (15 min) or PDGF-BB (50 ng/ml, 5 min). Total lysates were subjected to Western blotting with anti-phospho-Akt(S437) or anti-Akt protein antibody. (C) Parental HDFs were serum starved and treated without or with eHsp90α (10 μg/ml or 0.1 μM) for the indicated time. Total lysates were subjected to Western blotting with anti-phospho-Akt(S437), anti-phospho-ERK1/2, or anti-GAPDH antibody. (D) Parental HDFs were serum starved and stimulated with either full-length eHsp90α (0.1 μM) or F-5 (0.1 μM) for 15 min. Total lysates were immunoblotted with anti-phospho-Akt(S437) (a) or anti-GAPDH antibody (b). (E) Demonstration of the FPLC-purified human recombinant Hsp90β (lane 1) and Hsp90α (lane 2) proteins. (F) Parental HDFs were serum starved and stimulated with equal amounts of the Hsp90α or Hsp90β protein, and activation of Akt was measured by using anti-phospho-Akt antibody (a) or GAPDH antibody (b). (G) HDFs were infected with lentiviral vector carrying either the wt (lane 2) or dominant-negative (DN) mutant Akt1 gene. The inset shows a Western blot of the expressed Akt gene products. The bar graph shows that the cells then were subjected to a colloidal gold migration assay (see Materials and Methods). Only the migration index is shown.
To confirm the eHsp90α-Akt pathway, we carried out dose-response analysis and determined the time course of eHsp90α-stimulated Akt activation. We found that eHsp90α stimulation caused a dose-dependent activation of Akt (Fig. 6B), which was significantly weaker than PDGF-BB-stimulated Akt activation. While eHsp90α stimulated transient activation of ERK1/2 (Fig. 6C, panel b), it caused a sustained activation of Akt (Fig. 6C, panel a). As expected, both full-length eHsp90α and its F-5 fragment (amino acids (aa) 236 to aa 350) stimulation equally activated Akt (fig. 6D, panel a, lanes 2 and 4). Interestingly, stimulation with purified human recombinant eHsp90β (Fig. 6E, lane 1) caused significantly weaker Akt activation (Fig. 6F, lane 3) than eHsp90α (F, lane 2). We have previously reported that eHsp90β does not have any promotility effect (20).
More importantly, activation of the Akt pathway is critical for transmission of the eHsp90α promotility signal. We used lentivirus infection to overexpress a dominant-negative (DN) mutant of the Akt1 gene in HDFs. We found that Akt1-DN completely blocked eHsp90α-stimulated HDF migration (Fig. 6G).
Akt1 and Akt2 but not Akt3 mediate eHsp90α signaling.
We further investigated the individual roles of the three mammalian Akt isoforms, Akt1, Akt2, and Akt3. As shown in Fig. 7A, we introduced Akt isoform-specific shRNAs to effectively knock down endogenous Akt1 (panel a, lane 2), Akt2 (panel c, lane 2), and Akt3 (panel e, lane 2) in HDFs, respectively. Since the Western blot signal for Akt3 (panel e, lane 1) was significantly weaker than those of Akt1 and Akt2 (panels a and c, lanes 1), we asked the question of whether the weaker signal of Akt3 reflected its actual expression levels in HDFs or if it was due to a weaker affinity of the anti-Akt3 antibody used. As shown in Fig. 7B, the same anti-Akt3 antibody was able to detect strong expression of the Akt3 protein (panel a, lane 1 versus lane 2) in rat brain extract, even if the amount of loaded total proteins was just a fraction of those of HDF lysates (panel b, lane 1 versus lane 2). Therefore, Akt3 expression is intrinsically lower than those of Akt1 and Akt2 in HDFs.
Fig 7.

Akt1 and Akt2 are critical for eHsp90α signaling to promote HDF migration. (A) Downregulation of Akt1, Akt2, and Akt3 by the FG-12 lentiviral system was visualized by antibodies specific for each of the Akt isoforms. (B) The weaker detection of Akt3 was not due to lower affinity of the anti-Akt3 antibody, because it was able to detect strong Akt3 expression from a rat brain extract that was a fraction of the HDF lysate (based on GAPDH levels). (C) The selected shRNAs for Akt1, Akt2, and Akt3 did not cross-react with each other. (D) Effects of individual Akt downregulation on eHsp90α-stimulated HDF migration. Representative images of migration tracks are shown with dotted circles to indicate the average size of migration tracks under a given condition. (E) Quantitation of the migration data from three independent experiments.
Moreover, we demonstrated that the downregulation of Akt1, Akt2, and Akt3 was isoform specific and the three shRNAs did not cross-react among the Akt isoforms. As shown in Fig. 7C, Akt1 was knocked down only by shRNA against Akt1 (panel a, lane 2) but not by shRNA against either Akt2 (panel a, lane 3) or Akt3 (panel a, lane 4). Akt2 was knocked down only by shRNA against Akt2 (panel c, lane 3) but not by shRNA against Akt1 (panel c, lane 2) or Akt3 (panel c, lane 4). Similarly, Akt3 was selectively knocked down by shRNA against Akt3 (panel e, lane 4) but not by shRNA against either Akt1 (panel e, lane 2) or Akt2 (panel e, lane 3). As indicated (panels e and f), in order to visualize Akt3 expression, four times more total cellular protein was used for detecting Akt3 than for detecting Akt1 or Akt2.
Using colloidal gold migration assays, as shown in Fig. 7D, both eHsp90α and PDGF-BB stimulated migration in control shLacZ-infected HDFs, as expected (panels b and c versus panel a). However, downregulation of Akt1 alone (panels d, e, and f) completely blocked eHsp90α-stimulated migration (panel f versus panel c). Downregulation of Akt2 also showed a complete blockade of eHsp90α-stimulated cell migration (panel i versus panel c). In contrast, downregulation of Akt3 showed significantly less inhibition of eHsp90α-stimulated migration (panel l versus panel c). Interestingly, in all cases, PDGF-BB-stimulated cell migration remained unaffected, excluding the possibility that Akt downregulation affected the global physiology of the cells. Quantitation of the migration data is shown in Fig. 7E. We conclude that Akt1 and Akt2 work either independently or in concert to mediate eHsp90α signaling via the NPVY motif.
Akt1 and Akt2 work in concert to mediate eHsp90α signaling.
There are two proposed mechanisms of action by the Akt isoforms. First, each Akt isoform acts independently and does not compensate the other's absence. Second, the participating Akt isoforms, such as Akt1 and Akt2 in the case of eHsp90α signaling, act in concert to build a threshold Akt kinase activity that connect upstream signals to a common downstream pathway. To determine which mechanism mediates eHsp90α signaling, we carried out Akt1 versus Akt2 reciprocal rescue experiments. We reasoned that if Akt1 and Akt2 act independently, reexpression of Akt1 should rescue eHsp90α-stimulated cell migration only in Akt1-downregulated cells and not in Akt2-downregulated cells. However, if Akt1 and Akt2 work in concert, reexpression of Akt1 should rescue migration of both Akt1- and Akt2-downregulated cells. Similar predictions would apply to rescue with the Akt2 gene in Akt1- or Akt2-downregulated cells. As shown in Fig. 8A, in Akt1-downregulated cells (panel a, lane 2), we compensated the absence of Akt1 with exogenously expressed Akt2 (panel c, lane 2), implying that there was twice as much Akt2 in the cells. In reverse, as shown in Fig. 8C, in Akt2-downregulated cells (panel e, lane 2), we replaced the absence of Akt2 with exogenously expressed Akt1 (panel g, lane 2), i.e., making the cells have twice as much of Akt1 in comparison to the parental HDFs.
Fig 8.

Akt1 and Akt2 kinases work in concert to mediate eHsp90α signaling. (A) Akt1-downregulated HDFs (panel a, lane 2) were overexpressed with Akt2 (panel c, lane 2 versus lane 1) for testing any rescue effects. (B) Downregulation of Akt1 blocked eHsp90α-stimulated (bar 6) but not PDGF-BB-stimulated (bar 5) HDF migration, and expression of Akt2 replaced the absence of Akt1 (bar 9 versus bar 6) to rescue eHsp90α-stimulated HDF migration. (C) In reverse, Akt2-downregulated HDFs (panel e, lane 2) were overexpressed with Akt1 (panel g, lane 2 versus lane 1). (D) Downregulation of Akt2 blocked eHsp90α-stimulated but not PDGF-BB-stimulated HDF migration (bar 6 versus bar 5). Exogenous expression of Akt1 replaced the absence of Akt2 (bar 9 versus bar 6) to rescue eHsp90α-stimulated HDF migration.
When these cells were subjected to colloidal gold migration assays, we found that exogenous expression of Akt2 was sufficient to rescue the defect in eHsp90α-stimulated migration in Akt1-downregulated cells (Fig. 8B, bar 9 versus bar 6). The reverse was also true, since exogenous expression of Akt1 rescued the motility defect in the Akt2-downregulated cells in response to eHsp90α (Fig. 8D, bar 9 versus bar 6). These data suggest that Akt1 and Akt2 act in concert, instead of in parallel independent pathways, to mediate eHsp90α signaling. The absence of either Akt1 or Akt2, however, showed little inhibitory effect on PDGF-BB-stimulated migration of the same cells (Fig. 8B and D, bars 2, 5, and 8). Since we have previously shown that Akt is essential for PDGF-BB-stimulated HDF migration (22), we postulate that PDGF-BB activates the Akt isoforms so much more potently that removal of a single Akt isoform does not affect PDGF-BB signaling.
Akt1 and Akt2 are essential for eHsp90α to repair skin wounds in mice.
To ultimately prove a critical role for Akt1 and Akt2 in mediating eHsp90α signaling, we studied the effect of topically applied eHsp90α on skin wound healing in Akt1 and Akt2 knockout mice. We reasoned that if Akt1 and Akt2 are critical for eHsp90α signaling, topical application of the recombinant Hsp90α protein should no longer be able to accelerate wound closure in these mice. As shown in Fig. 9A, vehicle-alone treatment did not promote wound closure (panel b versus panel a). eHsp90α treatment significantly accelerated full-thickness wound closure in normal control mice (panel d versus panel c). In contrast, eHsp90α was unable to promote wound healing in Akt2 knockout mice (panel h versus panel g). Quantitation of three independent experiments, as shown in Fig. 9B, confirmed the observation (bar 8 versus bar 4). Similarly, in topical eHsp90α protein treatment greatly accelerated wound closure in control mice (Fig. 9C, panel d′ versus panel c′), but it failed to promote wound closure in Akt1 knockout mice (panel h′ versus panel g′). Quantitation of the experiments is shown in Fig. 9D (bar 8 versus bar 4).
Fig 9.

Akt1 and Akt2 are essential for both normal wound healing and efficacy of eHsp90α in mice. (A) Full-thickness excision wounds (1.0 cm by 1.0 cm) were created on the backs of Akt2 knockout mice. The wounds were treated with placebo (5% CMC, carboxymethycellulose gel) or CMC containing recombinant Hsp90α (30 μg/ml). The images of one representative experiment are shown between day 0 and the day 5. (B) Mean error of percentage of wound closure on day 5 ± SD, in reference to day 0 wounds. (C and D) Similar analyses were carried out using Akt1 knockout mice, except that 0.7-cm by 0.7-cm wounds (due to the smaller sizes of Akt1 knockout mice) were created. (E and F) H&E staining of day 14 full-thickness wound sections from either placebo (CMC)- or recombinant Hsp90α-treated wounds. A total of nine randomly selected images per condition from three independent experiments were analyzed. Representative images are shown. The unhealed wound gaps are indicated by red and yellow lines. Magnification bars, 7.5 μm. ReT, reepithelialization tongue (green lines), indicating the newly migrated keratinocytes at the wound edges.
Finally, in order to determine if the lack of eHsp90α-stimulated wound closure was due to a decrease in wound reepithelialization (i.e., keratinocyte migration) or in wound contraction, we performed hematoxylin and eosin (H&E) staining of the day 14 wounds. As shown in Fig. 9E and F, eHsp90α treatment caused increased reepithelialization and wound closure in the normal control mice (panel b versus panel a and panel f versus panel e). However, eHsp90α failed to accelerate reepithelialization and wound closure in either Akt2 knockout (panel d versus panel c) or Akt1 knockout (panel h versus panel g) mice.
DISCUSSION
It is clear now that the singular function of eHsp90α is to promote cell migration, a critical event in both tissue repair and tumor progression. During skin wound healing, eHsp90α drives migration of keratinocytes, dermal fibroblasts, and endothelial cells into the wound bed to promote wound closure (9, 10). In tumor progression, eHsp90α adds invasion and metastasis of the tumor cells (7, 16, 18, 27, 28). However, despite the understanding of eHsp90α function in both physiological and pathological processes, the mechanism by which eHsp90α promotes cell migration remained elusive. A line of studies suggested that eHsp90α still acts as a chaperone protein outside the cells, in which eHsp90α binds and maintains its extracellular client proteins in their active forms. For instance, eHsp90α was shown to promote cancer cell migration and invasion by binding to the cell surface receptor HER2, matrix metalloproteinase 2 (MMP2), and cochaperones such as Hsp70 (16, 18, 29). Another group of studies argued that eHsp90α no longer functions as a chaperone protein. Instead, it acts as a ligand, via its F-5 domain, that binds to the cell surface receptor, LRP-1, and triggers cross-membrane signaling to execute its promotility function (6, 7). The relationships between the “extracellular chaperone” and “extracellular signaling” mechanisms remain to be established. It is possible that these two mechanisms work in parallel to achieve the ultimate goal of eHsp90α: to promote cell motility. The key question was whether or not eHsp90α needs to go through the intracellular signaling networks to execute its promotility function. In this study, we have provided both in vitro and in vivo experimental evidence that eHsp90α uses a transmembrane signaling mechanism to promote skin cell migration and skin wound healing. The sequential steps of the “eHsp90α pathway” include the following: (i) eHsp90α engages subdomain II among four extracellular subdomains of LRP-1, (ii) eHsp90α signal crosses the membrane, bypasses the first NPTY motif, and then “exits” at the NPVY motif in the intracellular tail of LRP-1, and (iii) the NPVY motif connects eHsp90α signaling to Akt1 and Akt2 kinases. Akt1 and Akt2 knockout mice show defects in wound healing and do not respond to topically applied eHsp90α. A schematic representation of this finding is shown in Fig. 10.
Fig 10.
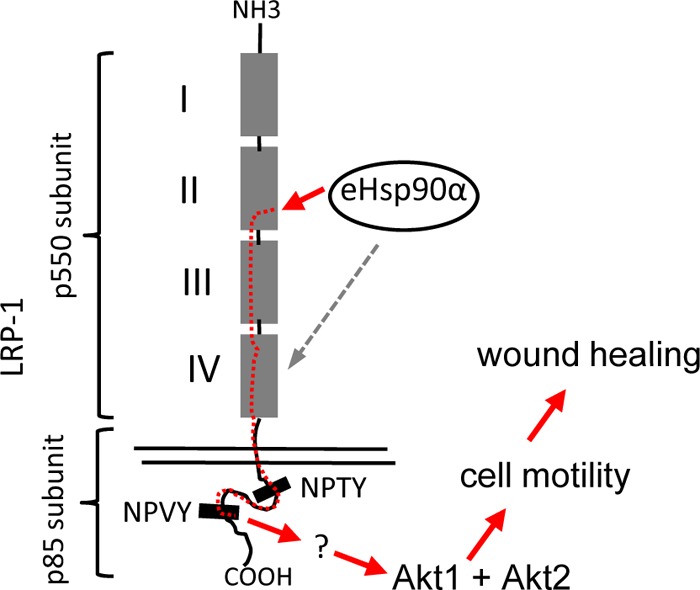
Schematic representation of eHsp90α signaling pathway. The red arrows depict the flow of eHsp90α signals from inside to outside the cell. The dotted line indicates the finding that eHsp90α can also bind to subdomain IV. Akt is essential for eHsp90α signaling to promote cell migration and wound healing. However, whether or not activation of Akt alone is sufficient to entirely replace eHsp90α is not clear. Other parallel pathways may also be involved in eHsp90α signaling. How LRP-1 mediates eHsp90α signaling to the other three pathways identified by the protein kinase array remains to be investigated. The question mark indicates intermediate activators whose identities remain unknown.
LRP-1 is a member of the low-density lipoprotein (LDL) receptor superfamily, which includes at least 11 structurally related members (LDL receptor, LRP-1/TβRV, LRP-1b, LRP-2/megalin, very low density lipoprotein (VLDL) receptor, LRP-3, LRP-4/MEGF7, LRP-5, LRP-6, ApoE receptor 2/LRP-8, and LRP-9) (23, 30). Mice with an LRP-1 gene knockout die at the embryonic stage of their development (31). The extracellular domains of all LRPs share a great degree of similarity, whereas their cytoplasmic tails vary in both length and amino acid sequence composition. All members of the LRP family contain one or more NPXY motifs conserved in their cytoplasmic tails. The NPXY motifs are important for cargo and scavenging functions via internalization or endocytosis of ligand-bound LRPs. In addition, they can also mediate cross-membrane signaling (23). Although results of our current study do not rule out the possibility that another membrane protein(s) also participates in eHsp90α–LRP-1 signaling, four lines of experimental data support the notion that LRP-1 is a major receptor that transmits eHsp90α signal. First, our cell migration assays were carried out under serum-free conditions, in which the only stimulus of cell migration present in the medium was recombinant Hsp90α proteins. Second, we ruled out the involvement of PDGF receptors in LRP-1 signaling, as suggested by others (24). Downregulation of either PDGF-Rα or PDGF-Rβ in HDFs showed little effect on eHsp90α-induced cell migration (10). And downregulation of LRP-1 in HDFs did not affect PDGF-BB-stimulated cell migration (this study). Third, identification of minireceptor II (subdomain II) as a fully functional receptor is distinct from another report that LRP-1 promotes tumor cell migration through subdomain IV of LRP-1 under a ligand-free condition (25). Finally, the identification of the NPVY motif at the cytoplasmic tail of LRP-1 and rapid activation of Akt kinases following eHsp90α stimulation provide direct evidence that LRP-1 mediates the transmembrane signaling of eHsp90α. Interestingly, our data also showed that none of the NPXY motifs was involved in eHsp90α signaling to activate the ERK1/2 pathway.
The three mammalian Akt isoforms (Akt1, -2, and -3) appear to have distinct functions, since homozygous knockout of individual Akt genes in mice resulted in distinct phenotypes (32). There are three possible explanations for these observations: (i) specific downstream targets/substrates for each of the Akt isoforms, (ii) distinct subcellular localizations for different Akt isoforms, and (iii) changes in threshold Akt activity in each Akt gene knockout (32). Our rescue experiments suggest that Akt1 and Akt2 work in concert, rather than individually, to mediate eHsp90α signaling. Knockdown of Akt1 or Akt2 alone completely inhibited eHsp90α-stimulated HDF migration. However, exogenous expression of Akt1 compensated for the absence of Akt2 in eHsp90α-stimulated cell migration and vice versa. More importantly, eHsp90α was no longer able to promote wound healing in Akt1 or Akt2 individual knockout mice. In contrast, in the same Akt-downregulated cells, the presence of either Akt1 or Akt2 alone was still sufficient to mediate PDGF-BB-stimulated cell migration. These data suggest that different extracellular stimuli require various degrees of threshold Akt kinase activity for signaling. Furthermore, our findings in this study also connect well with those in several recent reports showing that the downstream effect of Akt, the mTOR pathway, plays an essential role in wound healing (33–38). Therefore, taking all this together, it is becoming clear that the eHsp90α–subdomain II of LRP-1–NPVY motif–Akt1/2–mTOR pathway represents a new therapeutic target for skin wound healing.
Recent studies showed that Hsp90α knockout mice survived and developed otherwise normally with some specific tissue defects (39–41). One possible explanation is that Hsp90β was able to compensate for the absence of Hsp90α. However, the reverse was not the case, since Hsp90β gene knockout was reported to be embryonic lethal (42). These findings raised the question of what exactly Hsp90α does inside the cells, which stockpile Hsp90α in their cytoplasm. We argue that the purpose for all cells in preemptively storing Hsp90α is to be ready to promptly provide a steady supply for eHsp90α in response to environmental insults, such as tissue damage. A recent study from our laboratory showed that eHsp90α is better suited for tissue repair purposes, such as treatment of diabetic foot ulcers, than conventional growth factors (6). One unique property of eHsp90α, missing in conventional growth factors, is its ability to override inhibition by the TGF-β family of cytokines, abundantly present in injured tissues. In addition, eHsp90α but not conventional growth factors is able to override the inhibition of hyperglycemia in diabetic wounds (9, 43).
In summary, it is clear that Hsp90α has two distinct Mother Nature-assigned roles to play, an intracellular chaperone and an extracellular promotility factor. Both roles are designed for the cells to cope with environmental stress, such as a tissue injury, and both have been taken advantage of by tumor cells during invasion and metastasis. Recent recognition of the new function for eHsp90α has revealed a new line of therapeutic intervention. Evidence-based advantages of developing eHsp90α over conventional growth factors for wound healing have been shown in preclinical studies. Predicted advantages of targeting eHsp90α over intracellular eHsp90α in prevention of tumor progression remain to be tested upon availability of specific inhibitors.
ACKNOWLEDGMENTS
This work is supported by NIH grants GM066193 and GM067100 (to W.L.), AR46538 (to D.T.W.), and AR33625 (to M.C. and D.T.W.) and a VA Merit Award (to D.T.W.).
During this study, W.L. supervised all research. F.T., C.-F.C., and W.L. designed the experiments. F.T. performed most experiments and analyzed the results (with W.L.). A.B. performed all the experiments required for the revision. K.O. and M.C. provided further technical assistance. N.H. and B.S. provided the knockout mouse models. F.T., D.T.W., and W.L. wrote and edited the manuscript.
Footnotes
Published ahead of print 14 October 2013
REFERENCES
- 1. Csermely P, Schnaider T, Soti C, Prohászka Z, Nardai G. 1998. The 90-kDa molecular chaperone family: structure, function, and clinical applications. A comprehensive review. Pharmacol. Ther. 79:129–168 [DOI] [PubMed] [Google Scholar]
- 2. Horwich AL, Neupert W, Hartl FU. 1990. Protein-catalysed protein folding. Trends Biotechnol. 8:126–131 [DOI] [PubMed] [Google Scholar]
- 3. Whitesell L, Lindquist SL. 2005. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 5:761–772 [DOI] [PubMed] [Google Scholar]
- 4. Young JC, Moarefi I, Hartl FU. 2001. Hsp90: a specialized but essential protein-folding tool. J. Cell Biol. 154:267–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Eustace BK, Jay DG. 2004. Extracellular roles for the molecular chaperone, hsp90. Cell Cycle 3:1098–1100 [PubMed] [Google Scholar]
- 6. Li W, Sahu D, Tsen F. 2012. Secreted heat shock protein-90 (Hsp90) in wound healing and cancer. Biochim. Biophys. Acta 1823:730–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li W, Tsen F, Sahu D, Bhatia A, Chen M, Multhoff G, Woodley DT. 2013. Extracellular Hsp90 (eHsp90) as the actual target in clinical trials: intentionally or unintentionally. Int. Rev. Cell Mol. Biol. 303:203–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tsutsumi S, Neckers L. 2007. Extracellular heat shock protein 90: a role for a molecular chaperone in cell motility and cancer metastasis. Cancer Sci. 98:1536–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cheng CF, Sahu D, Tsen F, Zhao Z, Fan J, Kim R, Wang X, O'Brien K, Li Y, Kuang Y, Chen M, Woodley DT, Li W. 2011. A fragment of secreted Hsp90α carries properties that enable it to accelerate effectively both acute and diabetic wound healing in mice. J. Clin. Invest. 121:4348–4361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li W, Li Y, Guan S, Fan J, Cheng CF, Bright AM, Chinn C, Chen M, Woodley DT. 2007. Extracellular heat shock protein-90alpha: linking hypoxia to skin cell motility and wound healing. EMBO J. 26:1221–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dales JP, Garcia S, Meunier-Carpentier S, Andrac-Meyer L, Haddad O, Lavaut MN, Allasia C, Bonnier P, Charpin C. 2005. Overexpression of hypoxia-inducible factor HIF-1alpha predicts early relapse in breast cancer: retrospective study in a series of 745 patients. Int. J. Cancer 116:734–739 [DOI] [PubMed] [Google Scholar]
- 12. Kuroita T, Tachibana H, Ohashi H, Shirahata S, Murakami H. 1992. Growth stimulating activity of heat shock protein 90 alpha to lymphoid cell lines in serum-free medium. Cytotechnology 8:109–117 [DOI] [PubMed] [Google Scholar]
- 13. Sahu D, Zhao Z, Tsen F, Cheng CF, Park R, Situ AJ, Dai J, Eginli A, Shams S, Chen M, Ulmer TS, Conti P, Woodley DT, Li W. 2012. A potentially common peptide target in secreted heat shock protein-90α for hypoxia-inducible factor-1α-positive tumors. Mol. Biol. Cell 23:602–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Semenza GL. 2007. Evaluation of HIF-1 inhibitors as anticancer agents. Drug Discov. Today 12:853–859 [DOI] [PubMed] [Google Scholar]
- 15. Singer AJ, Clark RA. 1999. Cutaneous wound healing. N. Engl. J. Med. 341:738–746 [DOI] [PubMed] [Google Scholar]
- 16. Eustace BK, Sakurai T, Stewart JK, Yimlamai D, Unger C, Zehetmeier C, Lain B, Torella C, Henning SW, Beste G, Scroggins BT, Neckers L, Ilag LL, Jay DG. 2004. Functional proteomic screens reveal an essential extracellular role for hsp90 alpha in cancer cell invasiveness. Nat. Cell Biol. 6:507–514 [DOI] [PubMed] [Google Scholar]
- 17. Sims JD, McCready J, Jay DG. 2011. Extracellular heat shock protein (Hsp)70 and Hsp90α assist in matrix metalloproteinase-2 activation and breast cancer cell migration and invasion. PLoS One 6:e18848. 10.1371/journal.pone.0018848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stellas D, El Hamidieh A, Patsavoudi E. 2010. Monoclonal antibody 4C5 prevents activation of MMP2 and MMP9 by disrupting their interaction with extracellular HSP90 and inhibits formation of metastatic breast cancer cell deposits. BMC Cell Biol. 11:51. 10.1186/1471-2121-11-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen JS, Hsu YM, Chen CC, Chen LL, Lee CC, Huang TS. 2010. Secreted heat shock protein 90alpha induces colorectal cancer cell invasion through CD91/LRP-1 and NF-kappaB-mediated integrin alphaV expression. J. Biol. Chem. 285:25458–25466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cheng CF, Fan J, Fedesco M, Guan S, Li Y, Bandyopadhyay B, Bright AM, Yerushalmi D, Liang M, Chen M, Han YP, Woodley DT, Li W. 2008. Transforming growth factor alpha (TGFalpha)-stimulated secretion of HSP90alpha: using the receptor LRP-1/CD91 to promote human skin cell migration against a TGFbeta-rich environment during wound healing. Mol. Cell. Biol. 28:3344–3358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Woodley DT, Fan J, Cheng CF, Li Y, Chen M, Bu G, Li W. 2009. Participation of the lipoprotein receptor LRP1 in hypoxia-HSP90alpha autocrine signaling to promote keratinocyte migration. J. Cell Sci. 122:1495–1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li W, Fan J, Chen M, Guan S, Sawcer D, Bokoch GM, Woodley DT. 2004. Mechanism of human dermal fibroblast migration driven by type I collagen and platelet-derived growth factor-BB. Mol. Biol. Cell 15:294–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Herz J, Strickland DK. 2001. LRP: a multifunctional scavenger and signaling receptor. J. Clin. Invest. 108:779–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lillis AP, Mikhailenko I, Strickland DK. 2005. Beyond endocytosis: LRP function in cell migration, proliferation and vascular permeability. J. Thromb. Haemost. 3:1884–1893 [DOI] [PubMed] [Google Scholar]
- 25. Obermoeller-McCormick LM, Li Y, Osaka H, FitzGerald DJ, Schwartz AL, Bu G. 2001. Dissection of receptor folding and ligand-binding property with functional minireceptors of LDL receptor-related protein. J. Cell Sci. 114:899–908 [DOI] [PubMed] [Google Scholar]
- 26. Song H, Bu G. 2009. MicroRNA-205 inhibits tumor cell migration through down-regulating the expression of the LDL receptor-related protein 1. Biochem. Biophys. Res. Commun. 388:400–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gopal U, Bohonowych JE, Lema-Tome C, Liu A, Garrett-Mayer E, Wang B, Isaacs JS. 2011. A novel extracellular Hsp90 mediated co-receptor function for LRP1 regulates EphA2 dependent glioblastoma cell invasion. PLoS One 6:e17649. 10.1371/journal.pone.0017649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tsutsumi S, Scroggins B, Koga F, Lee MJ, Trepel J, Felts S, Carreras C, Neckers L. 2008. A small molecule cell-impermeant Hsp90 antagonist inhibits tumor cell motility and invasion. Oncogene 27:2478–2487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McCready J, Sims JD, Chan D, Jay DG. 2010. Secretion of extracellular hsp90alpha via exosomes increases cancer cell motility: a role for plasminogen activation. BMC Cancer 10:294. 10.1186/1471-2407-10-294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Willnow TE, Nykjaer A, Herz J. 1999. Lipoprotein receptors: new roles for ancient proteins. Nat. Cell Biol. 1:E157–E162. 10.1038/14109 [DOI] [PubMed] [Google Scholar]
- 31. Herz J, Clouthier DE, Hammer RE. 1992. LDL receptor-related protein internalizes and degrades uPA-PAI-1 complexes and is essential for embryo implantation. Cell 71:411–421 [DOI] [PubMed] [Google Scholar]
- 32. Dillon RL, Muller WJ. 2010. Distinct biological roles for the Akt family in mammary tumor progression. Cancer Res. 70:4260–4264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Castilho R, Squarize C, Gutkind J. 2013. Exploiting PI3K/mTOR signaling to accelerate epithelial wound healing. Oral Dis. 19:551–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Clark RA, Pavlis M. 2009. Dysregulation of the mTOR pathway secondary to mutations or a hostile microenvironment contributes to cancer and poor wound healing. J. Investig. Dermatol. 129:529–531 [DOI] [PubMed] [Google Scholar]
- 35. Feldmeyer L, Hofbauer GF, Böni T, French LE, Hafner J. 2012. Mammalian target of rapamycin (mTOR) inhibitors slow skin carcinogenesis, but impair wound healing. Br. J. Dermatol. 166:422–424 [DOI] [PubMed] [Google Scholar]
- 36. Jin Y, Tymen SD, Chen D, Fang ZJ, Zhao Y, Dragas D, Dai Y, Marucha PT, Zhou X. 2013. MicroRNA-99 family targets AKT/mTOR signaling pathway in dermal wound healing. PLoS One 8:e64434. 10.1371/journal.pone.0064434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kim YW, Lee WH, Choi SM, Seo YY, Ahn BO, Kim SH, Kim SG. 2012. DA6034 promotes gastric epithelial cell migration and wound-healing through the mTOR pathway. J. Gastroenterol. Hepatol. 27:397–405 [DOI] [PubMed] [Google Scholar]
- 38. Squarize CH, Castilho RM, Bugge TH, Gutkind JS. 2010. Accelerated wound healing by mTOR activation in genetically defined mouse models. PLoS One 5:e10643. 10.1371/journal.pone.0010643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Grad I, Cederroth CR, Walicki J, Grey C, Barluenga S, Winssinger N, De Massy B, Nef S, Picard D. 2010. The molecular chaperone Hsp90α is required for meiotic progression of spermatocytes beyond pachytene in the mouse. PLoS One 5:e15770. 10.1371/journal.pone.0015770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Imai T, Kato Y, Kajiwara C, Mizukami S, Ishige I, Ichiyanagi T, Hikida M, Wang JY, Udono H. 2011. Heat shock protein 90 (HSP90) contributes to cytosolic translocation of extracellular antigen for cross-presentation by dendritic cells. Proc. Natl. Acad. Sci. U. S. A. 108:16363–16368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kajiwara C, Kondo S, Uda S, Dai L, Ichiyanagi T, Chiba T, Ishido S, Koji T, Udono H. 2012. Spermatogenesis arrest caused by conditional deletion of Hsp90α in adult mice. Biol. Open 1:977–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Voss AK, Thomas T, Gruss P. 2000. Mice lacking HSP90beta fail to develop a placental labyrinth. Development 127:1–11 [DOI] [PubMed] [Google Scholar]
- 43. Suzuki S, Kulkarni AB. 2010. Extracellular heat shock protein HSP90beta secreted by MG63 osteosarcoma cells inhibits activation of latent TGF-beta1. Biochem. Biophys. Res. Commun. 398:525–531 [DOI] [PMC free article] [PubMed] [Google Scholar]