

Abstract
The unfolded protein response (UPR) is activated in response to hypoxia-induced stress such as in the tumor microenvironment. This study examined the role of CREB3L1 (cyclic AMP [cAMP]-responsive element-binding protein 3-like protein 1), a member of the UPR, in breast cancer development and metastasis. Initial experiments identified the loss of CREB3L1 expression in metastatic breast cancer cell lines compared to low-metastasis or nonmetastatic cell lines. When metastatic cells were transfected with CREB3L1, they demonstrated reduced invasion and migration in vitro, as well as a significantly decreased ability to survive under nonadherent or hypoxic conditions. Interestingly, in an in vivo rat mammary tumor model, not only did CREB3L1-expressing cells fail to form metastases compared to CREB3L1 null cells but regression of the primary tumors was seen in 70% of the animals as a result of impaired angiogenesis. Microarray and chromatin immunoprecipitation with microarray technology (ChIP on Chip) analyses identified changes in the expression of many genes involved in cancer development and metastasis, including a decrease in those involved in angiogenesis. These data suggest that CREB3L1 plays an important role in suppressing tumorigenesis and that loss of expression is required for the development of a metastatic phenotype.
INTRODUCTION
Homeostasis within the endoplasmic reticulum (ER) is essential for correct protein folding (1). Exposure of the ER to stresses such as glucose or nutrient depletion, expression of mutant or misfolded proteins, changes in calcium homeostasis, or hypoxia leads to the accumulation of unfolded proteins (2). In an attempt to compensate for this, the ER activates the unfolded protein response (UPR) (3–5). The UPR increases the folding capacity of the ER, reduces the translation of new proteins, and increases the degradation of misfolded proteins. If all else fails, the UPR signals for apoptosis. There are 3 major effector proteins of the UPR, inositol requiring 1 (IRE1), PKR-like ER kinase (PERK), and activating transcription factor 6 (ATF6) (6). Under nonstress conditions, all of these proteins are held in an inactive state in the ER membrane through binding to the chaperone GRP78/BIP1 (7–9). As unfolded proteins begin to accumulate in the ER, GRP78 detaches from these effectors in order to bind hydrophobic regions on the unfolded proteins, preventing their further transit and secretion. The unfolded proteins are then ubiquitinated and either refolded or degraded by the proteasome (6, 10). Once released from GRP78, IRE1, PERK, and ATF6 function to increase transcription of ER chaperones and members of the UPR. Proteins downstream of all 3 UPR receptor pathways have been identified as having proapoptotic roles; however, the point at which the “apoptotic switch” is activated is not known (11, 12).
A new member of the UPR, termed OASIS, was recently identified in mice (13). It is a transcription factor containing a basic region-leucine zipper (b/Zip) motif as well as a transmembrane sequence and is activated through regulated intramembrane proteolysis. Following activation of the UPR, OASIS is trafficked from the ER to the Golgi compartment, where it is cleaved to an active form. It then translocates into the nucleus and binds specifically to the cyclic AMP (cAMP) response element (CRE) consensus sequence and to ER stress response elements (ERSE) I and II, via its b/Zip domain (14). This transcriptional activation results in expression of many genes, including ER chaperones, such as GRP78, and OASIS itself (15).
A number of studies have suggested a role for the UPR in the development of cancer, more specifically, in regulating the balance between the senescence, proliferation, and apoptosis of cancer cells in the tumor microenvironment (16, 17). For example, XBP1 is a transcription factor activated downstream of IRE1, and small interfering RNA (siRNA)-mediated knockdown of XBP1 in cancer cells results in failure of the cells to form tumors in mice (18). Bi et al. showed that tumors derived from PERK-positive transformed cells grew much more rapidly than PERK-negative tumors in nude mice (19). Finally, several groups have shown that blocking the UPR makes tumor cells more sensitive to chemotherapy, both in vitro and in vivo (20). These data suggest that the UPR is essential for survival of cancer cells in the tumor microenvironment, possibly acting by increasing resistance to some of the stresses encountered in the microenvironment. Recently, our laboratory has identified the human and rat homologue to OASIS called CREB3L1 (CRE-binding protein 3-like protein 1) as a key protein expressed in nonmetastatic human and rat breast cancer cells whose expression is lost in metastatic cells.
In this report, we identify a clear relationship between loss of CREB3L1 expression and the development of a metastatic phenotype, providing strong evidence that CREB3L1 is a metastasis suppressor.
MATERIALS AND METHODS
Cell culture.
Our laboratory has established a rat mammary tumor model where highly (LN4D6) and poorly (CAbD5) metastatic subpopulations have been derived from the R3230Ac rat mammary adenocarcinoma (21). Stable LN4D6+pUB6-HA3CREB3L1 (LN4D6 CREB3L1) and LN4D6+pUB6-HA3 vector-only (LN4D6 vector) transfected cell lines were generated. The CREB3L1 sequence inserted into the vector was the complete coding sequence (amino acids 1 to 520 [NP_001005562.1]) with the addition of EcoRI and BamHI restriction sites for insertion. The same transfectants were generated with the human MDA-MB-435 cell line (ATCC HTB-129) using pcDNA 3.1(+) vector (amino acids 1 to 519 [NM_052854.3]). There have been conflicting reports in the literature about the origins of the MDA-MB-435 cells. Gene profiling of the MDA-MB-435 cells has suggested that it is of melanocytic rather than breast epithelial origin and is actually the M14 melanoma cell line (22, 23). However, it has also been suggested that, as the MDA-MB-435 cells secrete milk proteins (24) and have been shown to have two X chromosomes whereas the M14 cell line was derived from a man, they are in fact from a mammary origin (25). Knockdown of CREB3L1 in the CAbD5 cells was achieved using lentiviral small hairpin RNA (shRNA), according to the manufacturer's instructions (Santa Cruz Biotechnologies Inc.). Briefly, 1 × 105 infectious units of virus of CREB3L1 shRNA (sc-72995-V) or scrambled control shRNA (sc-108080) lentiviral particles were used to infect CAbD5 cells using 5 μg/ml Polybrene (sc-134220) prior to selection of stable clones in 50 μg/ml puromycin dihydrochloride (sc-108071). Knockdown was assessed using real-time PCR and Western blot analysis. LN4D6, MDA-MB-435, EA.hy926 (ATCC CRL-2922), and CAbD5 cells were maintained in complete RPMI 1640 (10% fetal bovine serum [FBS]) at 37°C with 5% CO2. The LN4D6 and MDA-MB-435 transfected cell lines were maintained in complete RPMI 1640 with 5 μg/ml blasticidin for selection at 37°C with 5% CO2. Hs578Bst (ATCC HTB-125), ZR-75-30 (ATCC CRL-1504), CAMA-1 (ATCC HTB-21), MDA-MB-361 (ATCC HTB-27), BT-20 (ATCC HTB-19), MDA-MB-157 (ATCC HTB-19), MDA-MB-435 (ATCC HTB-129), and HCC 1428 (ATCC CRL-2327) were obtained from the American Type Culture Collection and were maintained according to the supplier's instructions. All cell lines were routinely checked and negative for mycoplasma using a MycoSensor QPCR assay kit (Agilent Technologies).
Western blot analysis.
Western blot analyses were performed to determine the expression of hemagglutinin (HA)-CREB3L1 in the transfected cells as previously described (26). The quality of the available antibodies for CREB3L1 was deemed to be inadequate (many cross-reacting bands on Western blots), so we transfected the cells with human HA3-tagged CREB3L1, as described above, and probed for HA (Santa Cruz) (5 μg/ml), CREB3L1 (a gift from J. Ye, University of Texas [UT] Southwestern Medical Center), or GRP78 (Santa Cruz) (400 ng/ml) using either 25 μg or 75 μg of protein. Blots were also probed for GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (Santa Cruz) (5 μg/ml) as a loading control. Infrared 680-nm and 800-nm dye-tagged secondary antibodies (Odyssey) (200 ng/ml) were used, and proteins were visualized using a LiCOR Odyssey infrared imager.
Cell-based assays.
All cell lines were assayed for their ability to migrate and invade through an 8-μm-pore-size filter membrane toward medium containing 10% FBS or conditioned medium. Conditioned media were obtained from flasks of different cell types that had been growing in complete medium for 24 h. Negative controls were migrated toward 0.5% FBS-containing medium as previously described (27). Cells were serum starved for 24 h in RPMI 1640 containing 0.5% FBS prior to migration or invasion. Invasion assays were performed similarly with the exception that the filters were coated with 1 mg/ml Growth Factor Reduced Matrigel (BD Biosciences) and incubated for 24 h instead of 8 h. Data are expressed as numbers of migrated cells per field (10×).
Proliferation of cells was assessed using a Vybrant MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] cell proliferation assay kit (Invitrogen) according to the manufacturer's instructions. Briefly, 5 × 103 cells were plated in the wells of a 96-well plate in complete medium for 48 h at 37°C prior to analysis with MTT.
All cell lines were assessed for their ability to adhere to constituents of the extracellular matrix as previously described (28), with the wells of a 96-well plate coated with either 25 μg/ml fibronectin (Invitrogen) or 50 μg/ml rat tail collagen IV (Gibco).
Soft-agar assays were performed as previously described (29).
For wound-healing assays, cells were grown to confluence on 10-cm2 plates and 3 scratches were made using a 200-μl pipette tip. Media were replaced with either complete or conditioned RPMI 1640, and the cells were allowed to grow for 24 h, with photographs taken at specific time points.
Hypoxia and apoptosis assays.
Cells were plated at 1 × 105/well in 6-well plates and incubated for 24 h at 37°C with 5% CO2. The plates were then transferred to a hypoxia chamber and incubated at 37°C under hypoxic conditions (1% oxygen, 5% carbon dioxide, and 94% nitrogen) for 24 h and then allowed to recover under normoxic conditions for 24 h. Cells were resuspended at a concentration of 1 × 106 cells/ml in 200 μl of annexin binding buffer (Invitrogen). Fluorescein isothiocyanate (FITC)-conjugated annexin V (Invitrogen) was added at a concentration of 2 μg/ml. The cells were incubated for 15 min in the dark at 37°C. Following incubation, an additional 300 μl of binding buffer was added to dilute the annexin V. Immediately before analysis by flow cytometry, 12.5 μg/ml propidium iodide (Invitrogen) was added to each sample. The cells were then analyzed by flow cytometry using a Coulter XL flow cytometer.
Rat tumor model and immunohistochemistry.
In vivo breast cancer models were designed to assess the potential of the tumor cells to grow and subsequently metastasize to the draining lymph node. Fischer F334 rats (Charles River) were injected with wild-type LN4D6, LN4D6 pUB6 CREB3L1 transfected cells, or LN4D6 cells transfected with the empty vector as previously described (30). All animals used in this study were cared for and used in accordance with the guidelines of the Canadian Council on Animal Care and the regulations of the University of Saskatchewan Animal Care Committee (protocol number 20080071).
Immunohistochemical staining to detect angiogenesis was performed using a peroxidase detection reagent pack (Thermo Scientific) according to the manufacturer's instructions. Briefly, 5-μm-thick sections were incubated with a rat anti-CD34 primary antibody (R&D Systems) (10 μg/ml) overnight at 4°C followed by incubation with a horseradish peroxidase (HRP)–rabbit anti-goat secondary antibody (Invitrogen) (1:1,000) for 2 h at room temperature before detection with DAB (3,3′-diaminobenzidine tetrahydrochloride) for 10 min. Stained vessels were counted as either small (<2-mm diameter) or large (>2-mm diameter) in 9 random fields, and data were expressed as numbers of stained vessels per field (20×).
All images were taken at ×100 magnification on a Nikon Coolpix 990 camera using a Nikon Eclipse TE300 microscope, unless otherwise indicated.
Microarray.
Microarray analysis comparing LN4D6 wild-type cells and LN4D6 CREB3L1 transfected cells was performed. Briefly, total RNA was isolated from the cell lines using an RNeasy kit (Qiagen). RNA was analyzed in triplicate by the London Genomics Centre, Ontario, Canada, using the Rat 230 2.0 array. Data were analyzed by a qualified bioinformatician at the London Genomics Centre, with only genes that had a 1.4-fold increase or decrease in expression with a significance of P < 0.05 being included in the final results.
Real-time PCR.
Total RNA was isolated from cell lines and tumors using an RNeasy kit (Qiagen), and cDNA was generated using oligo(dT) primers with a Superscript II RNase reverse transcription system (Invitrogen). Real-time TaqMan PCR was performed according to Applied Biosystems protocols. All samples were loaded in triplicate wells, and fluorescence emission was detected by an ABI Prism 7000 sequence detection system (Applied Biosystems). Primers and probes were purchased from Applied Biosystems. All results were normalized to the expression of endogenous glyceraldehyde-3-phosphate dehydrogenase mRNA and are expressed as relative increases or decreases in expression compared to controls.
ChIP on Chip microarray.
Initially, a chromatin immunoprecipitation (ChIP) assay was performed using an EpiQuick chromatin immunoprecipitation kit (Epigentek) and an HA-X (F-7) antibody (Santa Cruz). All procedures were carried out according to the manufacturer's specifications. The ChIP DNA was then amplified using a GenomePLex whole-genome amplification 4 (WGA-4) kit (Sigma), according to the manufacturer's specifications. The amplified DNA was then purified using a QIAquick PCR purification kit (Qiagen). Samples were then sent to Roche Nimblegen (Reykjavik, Iceland) for analysis using microarray technology (Human ChIP 2.1 M Dlx Prom Arr Ser and Rat ChIP 385K Prom Set-2 Arr Ser). CREB3L1 was considered bound only to genes with a false-discovery-rate (FDR) value of <0.2, as the manufacturer recommended.
CAT activity.
Chloramphenicol acetyltransferase (CAT) assays were carried out to confirm selected ChIP on Chip data as previously described (31). Promoter sequences were generated for PTGES (5′-GACTCGAGGTCATTTGGGGTTAGGGGTGGTA and 3′-GAAGATCTGTCACACCGGGACTGAGAGTGAG) and fgfbp1 (5′-GACTCGAGGTCACACACTCTGGCTTTCCACACC and 3′-GAAGATCTGTCATCCAATGGCTTGAGACTGC) and inserted into the pCAT 3 basic vector using XhoI and BglII restriction enzymes (New England BioLabs).
Statistical analyses.
All results were expressed as means ± standard errors (SE) of the results from replicate samples. Unless otherwise stated, the statistical significance of changes was assessed by the application of either the Student t test or analysis of variance (ANOVA) using Microsoft Excel 2007, depending on which was more applicable.
Microarray data accession number.
Raw microarray data have been deposited in the NCBI Gene Expression Omnibus under accession no. GSE51857.
RESULTS
Our laboratory has established a rat mammary tumor model where highly and poorly metastatic subpopulations, termed R3230Ac-LN4 and R3230Ac-CAb, respectively, have been derived from the same R3230Ac rat mammary adenocarcinoma (21, 30, 32). In the experiments described in this paper, clonal isolates of these two cell populations, LN4D6 and CAbD5, were used. One gene that was found to be highly differentially expressed was CREB3L1, which was expressed in the poorly metastatic CAbD5 cell line but not expressed in the highly metastatic LN4D6 cell line (Fig. 1A). In human breast cancer cell lines, the expression of full-length CREB3L1 was also found in nonmetastatic or poorly metastatic cell lines but lost from metastatic cell lines (Fig. 1B). These results suggest that CREB3L1 expression is limited to cells of low metastatic potential.
Fig 1.
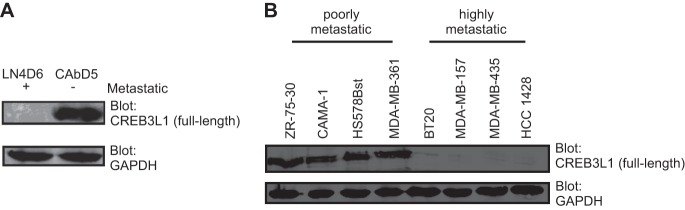
CREB3L1 is lost in highly metastatic cell lines. (A and B) Western blot analysis of (full-length) CREB3L1 expression in metastatic LN4D6 and poorly metastatic CAbD5 rat cell lines (A) and normal Hs578Bst, poorly metastatic ZR-75-30, CAMA-1, and MDA-MB-361, and highly metastatic BT-20, MDA-MB-157, MDA-MB-435, and HCC1428 human breast cancer cell lines (B). Briefly, Western blot analysis was performed using 75 μg of cell lysates, which were resolved by SDS-PAGE and immunoblotted with either anti-CREB3L1 or anti-GAPDH (loading control). The results are representative of 3 independent experiments.
We examined the effects of (HA-tagged) ectopically expressed CREB3L1 in the highly metastatic non-CREB3L1-expressing LN4D6 and MDA-MB-435 cell lines. Following transfection, CREB3L1 was expressed in the cells as 2 forms, the larger full-length form and the smaller cleaved form (Fig. 2A and B). The cleaved form, which is transcriptionally active, translocates to the nucleus, influencing transcription. We have included MDA-MB-435 cells in our analysis of CREB3L1 in breast cancer cell lines (see Materials and Methods for the rationale).
Fig 2.

Expression of CREB3L1 alters some of the tumorigenic properties of breast cancer cells. (A and B) LN4D6 and MDA-MB-435 cells were transfected with HA-tagged full-length CREB3L1. Briefly, 75 μg (for CREB3L1) or 25 μg (for HA and GAPDH) of cell lysates was resolved by SDS-PAGE and immunoblotted with either anti-CREB3L1 (A) or anti-HA (B) (HA-CREB3L1) in addition to anti-GAPDH as a loading control. (C to F) Determination of the metastatic properties of LN4D6 and MDA-MB-435 cells left untransfected (−) or transfected with a vector control or CREB3L1 on the migration (C) and invasion (D) of cells after 8 and 48 h, respectively, through an 8-μm-pore-size filter toward medium containing 10% FBS (+) or 0.5% FBS (−) and on the proliferation (E) and adhesion (F) of cells to fibronectin (filled bars) or collagen (open bars). Abs, absorption. (G) Fraction of apoptotic cells (1 × 105 cells plated) under normoxic conditions (open bars) or after 24 h of hypoxia followed by 24 h of growth under normoxic conditions (filled bars). (H) Numbers of colonies more than 32 cells in size formed in soft agar after 10 days (5 × 104 cells plated). The data are the means ± SE of the results of 3 independent experiments each with triplicate measurements. *, P < 0.01; **, P < 0.001.
Several cell-based assays were performed to determine the effects of CREB3L1 expression on the biological properties of the LN4D6 and MDA-MB-435 cells. CREB3L1 expression significantly reduced the migratory (Fig. 2C) and invasive (Fig. 2D) properties of the cells but had no effect on the proliferation (Fig. 2E) or adhesion to fibronectin or collagen (Fig. 2F) of any of the cell lines. When cells were grown under hypoxic conditions and then allowed to recover for 24 h, the cells expressing CREB3L1 showed a significant increase in apoptosis compared to the corresponding cells lacking CREB3L1 or vector controls, with no difference in apoptosis detected between the cell lines under normoxic conditions (Fig. 2G). Soft-agar experiments demonstrated a significantly decreased ability of the LN4D6 and MDA-MB-435 CREB3L1 transfected cells to form nonadherent colonies compared to the corresponding parental and vector control cell lines (Fig. 2H). These results indicate that cells lacking CREB3L1 expression are more migratory, invasive, and resistant to hypoxia-induced apoptosis and exhibit anchorage-independent growth, all properties associated with highly metastatic cells (33–35).
To evaluate the effects of CREB3L1 expression on primary tumor growth and metastasis, we employed a rat mammary tumor model of breast cancer metastasis. Rats (10/group for each cell type for each of 3 independent experiments) were injected with cells, including LN4D6 (Fig. 3A), LN4D6 vector (Fig. 3B), or LN4D6 CREB3L1 (Fig. 3C) cells. The majority of rats injected with LN4D6 as well as LN4D6 vector cells developed large primary tumors that metastasized to the popliteal lymph node (Fig. 3A and B and Table 1). In contrast, none of the animals injected with CREB3L1-expressing cells had metastases (Table 1), even among the 30% of animals that produced primary tumors > 1 cm in diameter. In addition, tumor regression was seen in 70% of the animals injected with the CREB3L1-expressing cells, with tumors reaching 0.4 to 0.7 cm in diameter before regression was observed (Fig. 3C).
Fig 3.
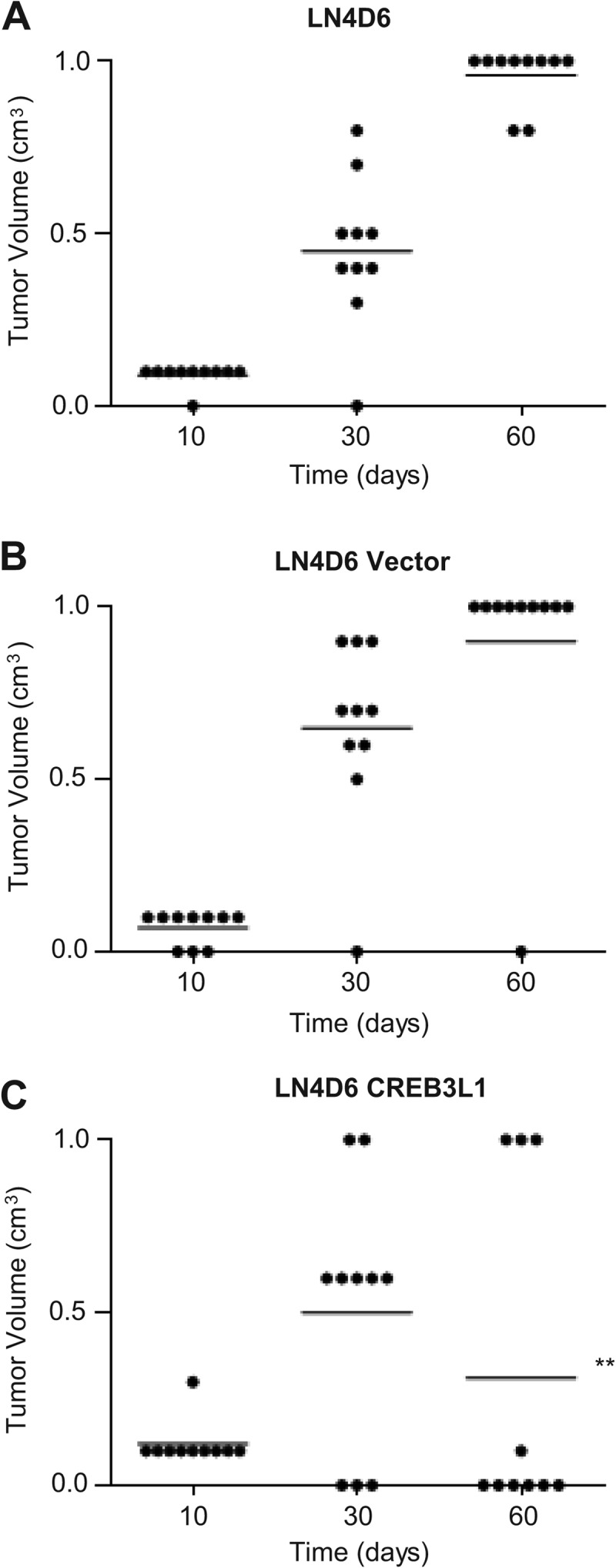
Expression of CREB3L1 prevents metastasis and results in tumor regression. Growth of tumors in a footpad model of cancer growth and metastasis in LN4D6 (A), LN4D6 vector control (B), and LN4D6 CREB3L1 (C) cells (1 × 106 cells) with 10 rats per cell line is shown. The results are representative of 1 of 3 independent experiments each giving similar results. Combined results from all 3 experiments are in Table 1. **, P < 0.001 (compared to LN4D6 at 60 days).
Table 1.
Summary of the numbers of animals with primary (>0.5-cm3) tumors and metastases from all 3 experiments
| Culture | No. of animals (n = 20 [10 per expt]): |
|
|---|---|---|
| With 0.5–1.0-cm3 primary tumor | With metastases | |
| LN4D6 | 29 | 26 |
| LN4D6 + vector | 28 | 24 |
| LN4D6 + CREB3L1 | 9 | 0 |
To determine if CREB3L1 expression had an impact on the formation of new vasculature, the experiment was repeated and tumors were removed at day 40 and stained for CD34, which is expressed on the surface of endothelial cells lining blood vessels (Fig. 4A to D). Tumors formed from cells expressing CREB3L1 had a significant (P < 0.001) reduction in the total number of blood vessels compared to the LN4D6 and vector control cell results. In particular, there was a significant (P < 0.001) decrease in the number of large (>2-mm-diameter) blood vessels in the CREB3L1-expressing tumors but no difference in the number of small (<2-mm-diameter) vessels (Fig. 4E). Altogether, these results strongly suggest that CREB3L1 plays a role in metastasis suppression and may also be involved in the suppression of hypoxic and/or angiogenic responses that allow large tumors to survive.
Fig 4.

CREB3L1 expression inhibits angiogenesis in tumors. (A to C) Immunohistochemistry for CD34 expression on blood vessels in tumors removed at day 40 from the footpad of rats injected with LN4D6 (A), LN4D6 vector control (B), and LN4D6 CREB3L1 (C) cells (1 × 106 cells). (D) A negative staining control assay was also performed on an LN4D6 vector section in which the primary antibody to CD34 was omitted. (E) The numbers of small vessels (<2 mm in diameter) and large vessels (>2 mm) in each field were qualified. A total of 9 fields from 3 different tumors in each group were assessed, and the results are expressed as vessels/field. The data are the means ± SE of the results of 2 independent experiments. **, P < 0.001.
To determine the potential impact of factors secreted by CREB3L1-expressing or -nonexpressing cells on neighboring cells, conditioned media from cells expressing or lacking CREB3L1 were added to endothelial cell migration and chemotaxis assays. Media conditioned by the LN4D6- and MDA-MB-435 CREB3L1-expressing cells showed a significantly reduced migration of EA.hy926 cells in a wound-healing assay compared to media conditioned by the corresponding cells transfected with vector only and complete medium (Fig. 5A and B). Chemotaxis assays of EA.hy926 cells across a membrane showed similar results, with the conditioned medium from cells expressing CREB3L1 inducing less cell migration than medium from nonexpressing cells (Fig. 5C). Together, these cell-based assays suggest that CREB3L1-expressing cells have reduced metastatic cell properties and may exert some of these effects through altered expression of secreted factors that influence neighboring cells.
Fig 5.

Conditioned media from CREB3L1-expressing cells induced less migration of endothelial cells. Data represent migration of the endothelial cell line EA.hy926 in wound-healing assays for 0, 6, 12, and 24 h (A and B) and chemotaxis assays for 8 h (C) in the presence of complete medium and conditioned media from LN4D6 and MDA-MB-435 cells transfected with vector only or CREB3L1 (×10 magnification). The results are representative of 3 independent experiments for the wound-healing assay data and the means ± SE of 3 independent experiments each with triplicate measurements for the chemotaxis data. **, P < 0.001.
To determine the effect of CREB3L1 on gene expression, two different approaches were taken. First, microarray analysis comparing LN4D6 CREB3L1 cells with LN4D6 cells was performed, identifying significant differences in the expression of a large number of genes (see Table S1 in the supplemental material). Some of the most significant (>2-fold) changes were in genes that influence tumor development and metastasis (Table 2). Selected differential gene expression results were further verified by real-time PCR analysis, as indicated (Table 2).
Table 2.
CREB3L1 regulates the expression of a number of genes involved in angiogenesis, metastasis, apoptosis, and growth in cancera
| Expression change category and gene | Process(es) | Function | Fold change in expression | Real-time fold change |
|---|---|---|---|---|
| Negative | ||||
| Prostaglandin E synthase | Angiogenesis | Overexpressed in colorectal and lung cancer; associated with disorganized vascular; deletion suppresses growth in intestinal cancer | −66 | −83 |
| Anp32A | Growth | Apoptosis; reduction causes cells to differentiate and decreases growth | −45 | −50 |
| Hdgfrp | Growth | Stimulates cell growth; downregulation inhibits lymphoma proliferation | −21 | |
| Igfbp5 | Metastasis/invasion | Overexpression increases invasion and progression in glioma; overexpression associated with poor outcome in breast cancer | −19 | |
| FGFbp1 | Angiogenesis | Increases angiogenesis | −19 | −20 |
| Hpgd | Growth | Downregulated in breast cancer; upregulation decreases growth and ability to form tumors | −7.3 | |
| TGFbi | Angiogenesis | Increases angiogenesis + inhibits immune system | −6.4 | |
| Fxyd3 | Apoptosis | Overexpressed in breast cancer; silencing decreases proliferation + increases apoptosis | −5.1 | |
| Adrenomedullin | Angiogenesis | Increases vascular growth + protection for hypoxic apoptosis | −5 | |
| Crystallin, alpha B | Apoptosis | Protects cancer cells from apoptosis; inhibition sensitizes cells to apoptosis | −4.9 | |
| Aquaporin 1 | Growth | Overexpressed in breast cancer and adenocarcinomas; induces proliferation + enhances anchorage-independent growth | −3.8 | |
| MMP13 | Metastasis/invasion | Increased in many cancers; involved in invasion | −3.6 | |
| Interleukin 13 receptor, alpha 2 | Growth/immune response | Acts as a decoy receptor for IL-13 + IL-4, preventing signaling; IL-13 + IL-4 suppress tumor growth | −2.6 | |
| CXCL10 | Angiogenesis | Angiostatic | −2.1 | |
| Positive | ||||
| JAM3 | Invasion | Enhances adhesion to ECM + invasion | 3.8 | |
| Apelin | Angiogenesis | Angiogenic factor; associated with earlier onset of tumor development | 3.4 | 3.98 |
| Angiopoietin 1 | Angiogenesis | Increases vascularization in early stages of cancer | 3.2 | |
| Cebpd | Growth | Induces cell cycle arrest; reduced in advanced breast cancer; possible tumor suppressor; STAT3 target gene and thus a mediator of proapoptotic gene expression | 3.1 | 3.3 |
| Legumain | Metastasis/invasion | Novel asparaginyl endopeptidase; increases migration + invasion in vitro + in vivo | 2.2 | |
| Calcium-activated chloride channel | Apoptosis | Downregulated in breast cancer cells resistant to apoptosis; overexpression in breast cancer reduces colony formation and increases apoptosis | 2.1 |
Data represent the top 20 significant negative and positive gene expression changes (>2-fold) from microarray analyses of LN4D6 with CREB3L1 compared to LN4D6 cells, with real-time expression validation for some of the genes. ECM, extracellular matrix; IL, interleukin.
Second, ChIP on Chip promoter analysis was performed on both LN4D6 and MDA-MB-435 cells expressing or lacking CREB3L1, identifying genes with promoters that are specifically bound by CREB3L1 (see Table S2A and B in the supplemental material), potentially regulating their transcription. Those genes that were identified in both cell lines in the ChIP on Chip experiments as targets for CREB3L1 regulation were compared with the genes whose expression was altered by CREB3L1 in the microarray data for the LN4D6 cell line. Those appearing in both lists were compiled (Table 3). Fragments of the promoters of two of these genes, pleiotrophin (Ptn) and FGFbp1, were used to confirm the ChIP on Chip data in CAT assays, showing decreased activity of the promoters in the presence of CREB3L1 (Table 3). These results demonstrate that CREB3L1 specifically associated with the promoter of these genes and regulates, in most instances negatively, their expression. A number of these CREB3L1 targets have known functions in cancer development, including cell growth, apoptosis, angiogenesis, invasion, and metastasis.
Table 3.
CREB3L1 specifically associates with the promoter of a number of genes involved in cancer, altering expressiona
| Expression change category and gene | Process | Function | Fold change in microarray | CAT activity fold change |
|---|---|---|---|---|
| Negative | ||||
| Banp_predicted | Tumor suppressor | Negatively regulates p53 transcription; functions as a cell cycle regulator and tumor suppressor | −37 | |
| FGFbp1 | Angiogenesis | Increases angiogenesis; elevated levels in many cancers | −19 | −11 |
| Chrnb4 | Unknown | Unknown | −18 | |
| Ptprf | Metastasis | Decreased in breast cancer, leading to prolonged activation of EGFR | −3.1 | |
| Gsta4 | Unknown | Unknown | −2.9 | |
| Pla2g2a | Metastasis | Prevents migration and invasion of cells in gastric cancer | −2.4 | |
| Mxi1 | Tumor suppressor | Antagonizes c-myc activity; mutated in some prostate cancers | −2.1 | |
| Rasl11b | Tumor suppressor | Ras-related GTPase; potential tumor suppressor | −2.1 | |
| Ptn | Angiogenesis | Increases angiogenesis; elevated levels in many cancers | −2 | −3.1 |
| Mt1a | Unknown | Unknown | −1.8 | |
| Rhebl1 | Tumor suppressor | Involved in the activation of mTor pathway; potential tumor suppressor | −1.8 | |
| Daf1 | Apoptosis | Complement-regulatory protein, protecting cells from apoptosis; upregulated in colorectal cancer and associated with a poor prognosis | −1.6 | |
| Blnk | Tumor suppressor | Regulates activation of JAK/STAT5 pathway in pre-B cells; loss of expression leads to development of pre-B cell leukemia | −1.6 | |
| Dlx1 | Growth/metastasis | Increased in ovarian cancer, promoting proliferation and migration | −1.5 | |
| Phyh | Unknown | Unknown | −1.5 | |
| Lrp1 | Invasion | Involved in the clearance of biological molecules from pericellular environment; increases the invasion of cancer cells | −1.5 | |
| Gstt1 | Protection | Involved in inactivation of carcinogenic substances; polymorphisms related to increased risk of many cancers | −1.5 | |
| Positive | ||||
| Sparc | Metastasis | Regulates cell-matrix interactions, cellular proliferation and migration; promotes metastasis | 2.7 | |
| Gpc4 | Unknown | Unknown | 1.7 | |
| Phf11 | Unknown | Unknown | 1.6 | |
| Hmgcr | Oncogene | Potential oncogene with increased expression promoting proliferation and transformation through Ras | 1.6 |
Genes that were present in both the ChIP on Chip data analysis of LN4D6 and MDA-MB-435 with CREB3L1 compared to LN4D6 and MDA-MB-435 cells and, importantly, were also identified in the microarray analysis as having significant negative and positive gene expression changes are shown. The ability of CREB3L1 to regulate selected promoters was confirmed using reporter constructs and CAT assays. EGFR, epidermal growth factor receptor.
Selected experiments performed in the LN4D6 and MDA-MB-435 cell lines were confirmed by knockdown of CREB3L1 expression (approximately 91%) in the poorly metastatic CREB3L1-expressing CAbD5 cells (Fig. 6A and B) to ensure that the effects being seen on the biological properties of the cells could be attributed to CREB3L1 expression. Knockdown of endogenous CREB3L1, but not a scrambled control, significantly increased the migratory (Fig. 6C) and invasive (Fig. 6D) properties of the cells but had no effect on the proliferation (Fig. 6E) or adhesion to fibronectin or collagen (Fig. 6F) of the cells. Knockdown of CREB3L1 expression also significantly increased the ability of the CAbD5 cells to form nonadherent colonies in soft-agar assays compared to the corresponding parental and scrambled control cell lines (Fig. 6G). When cells were grown under hypoxic conditions and then allowed to recover for 24 h, the knockdown of CREB3L1 resulted in a significant decrease in apoptosis compared to the corresponding cells expressing CREB3L1 (i.e., CAbD5) or scrambled controls, with no difference in apoptosis detected between the cell lines under normoxic conditions (Fig. 6H). These hypoxic conditions resulted in the induction of the UPR as demonstrated through induction of GRP78 in all cell lines with regulated intramembrane proteolysis of CREB3L1, when present, in the CAbD5 and scrambled control (Fig. 6I). Medium conditioned by the CAbD5 knockdown cells significantly stimulated the migration of EA.hy926 cells in a wound-healing assay at a level similar to that seen with complete medium compared to medium conditioned by the CAbD5 (CREB3L1-expressing) parental cells (Fig. 6J). These knockdown data are consistent with the ectopic studies showing that cancer cells lacking CREB3L1 have increased migration, invasion, resistance to hypoxia-induced apoptosis, and anchorage-independent growth, all properties associated with highly metastatic cells.
Fig 6.

Knockdown of CREB3L1 expression alters some of the tumorigenic properties of breast cancer cells. (A and B) CREB3L1 expression was knocked down using lentiviral delivery of CREB3L1 shRNA in CAbD5 cells. The level of knockdown was determined using real-time PCR analysis (A) and Western blot analysis (B). (C and D) Determination of the effect of the metastatic properties of CAbD5 with or without CREB3L1 shRNA knockdown (KD) or scrambled control shRNA (sc) cells on the migration (C) and invasion (D) of cells after 8 and 48 h, respectively, through an 8-μm-pore-size filter toward 10% FBS (+) or 0.5% FBS (−) and on proliferation (E) and adhesion (F) of cells to fibronectin (filled bars) or collagen (open bars). (G) Numbers of colonies more than 32 cells in size formed in soft agar after 10 days. (H) Fraction of apoptotic cells either grown under normoxic conditions (open bars) or after 24 h hypoxia followed by 24 h of growth under normoxic conditions (filled bars). (I) Induction of the UPR as demonstrated by expression of GRP78 and regulated intramembrane proteolysis of CREB3L1 (when present) following growth for 24 h under hypoxic conditions. (J) Migration of the EA.hy926 endothelial cell line in wound-healing assays for 0, 6, 12, and 24 h in the presence of complete medium and conditioned media from CAbD5 and CAbD5 CREB3L1 KD. The data are the means ± SE of the results of 3 independent experiments each with triplicate measurements. **, P < 0.001.
DISCUSSION
CREB3L1 is a member of the UPR pathway that is activated in response to cytotoxic conditions such as hypoxia (36, 37). The UPR has been shown to be cytoprotective in many situations, but prolonged activation leads to apoptosis (38). Numerous studies have identified a role for the UPR in regulating cancer development, both positively and negatively, but there is still a lack of understanding as to the specific role the UPR plays (17, 39). For example, increased expression of GRP78 is seen in breast and colon cancer cell lines (40) and XBP1 has been shown to be vital for cancer development (18).
However, studies in mouse models of prostate cancer demonstrated a decrease in the expression of UPR members in the progression from normal to high-grade prostatic intraepithelial neoplasia (39). So et al. hypothesized that, whereas upregulation of the UPR members may protect some cancers by increasing protein folding and prolonging survival, downregulation may prevent the activation of apoptotic events, allowing tumor progression (41). Our results support a model where loss of CREB3L1 expression may contribute to or be required for cancer progression and a metastatic phenotype, in a manner similar to that seen with the prostate models (39). This is consistent with recent findings that suggest a tumor suppressor role for CREB3L1, with chromosome fusion of the CREB3L1 gene resulting in the development of low-grade fibromyxoid sarcoma, and a potential role for CREB3L1 in preventing virus-induced tumorigenesis (42, 43).
This model for CREB3L1 function is supported by the rat studies in which cells expressing CREB3L1 initially formed primary tumors that subsequently regressed in 70% of animals, with no regression in the tumors derived from nonexpressing cells. Expression of CREB3L1 appears to limit not only the metastatic potential of the cancer cells, as seen in the migration and invasion assays, but also long-term survival. Sustained activation of the UPR can ultimately lead to apoptosis, and prolonged CREB3L1 activation under these stressful conditions appears to favor apoptosis of the cells rather than survival, as seen in the hypoxia experiments, making loss of CREB3L1 expression vital for tumor progression and cancer cell survival. This observation is in contrast to the behavior of some other members of the UPR family, such as ATF6 and XBP1, that have been shown to be critical for tumor development, especially under hypoxic conditions (18, 40). This highlights the need for further studies to more fully understand the complex roles of the various UPR family members in positively and negatively influencing cancer development.
The formation of new vasculature is essential for the delivery of oxygen and nutrients to a growing tumor (44, 45). Inhibition or retardation of this process exposes the tumor cells to hypoxic conditions that have been shown to prevent the growth of the tumor and also in some cases to result in tumor regression (46, 47). A number of studies have demonstrated that increased expression of both FGFbp1 and pleiotrophin promotes increased angiogenesis and tumor development (48, 49). We find that CREB3L1 specifically associated with the promoters of a number of genes, including FGFbp1 and pleiotrophin, decreasing their expression through its role as a transcription factor. This provides a possible mechanism by which loss of CREB3L1 expression results in enhanced expression of FGFbp1 and pleiotrophin to positively regulate angiogenesis and facilitate the growth of tumors. In addition, the decreased survival of CREB3L1-expressing cells under hypoxic conditions and the reduced ability to stimulate endothelial cell migration are two important factors that may influence the formation and survival of tumors from CREB3L1-expressing cells. This idea is supported by the findings indicating that the formation of new vasculature in tumors derived from CREB3L1-expressing cells was significantly impaired and is likely responsible for the regression seen in these tumors. The numbers of small vessels were similar for all of the tumors regardless of CREB3L1 expression and may explain why the tumors show an initial period of growth. However, the significant reduction in the number of large vessels in the tumors formed by CREB3L1-expressing cells may impair the formation and sustainability of larger tumors. These effects of CREB3L1 are quite novel, since previous studies have demonstrated that several other members of the UPR, including ATF6, increase the expression of VEGF-A and therefore promote angiogenesis under hypoxic conditions (50).
Our results clearly show that CREB3L1 has an important role in suppressing tumorigenesis. Through its role as a transcription factor, CREB3L1 suppresses the expression of genes involved in cancer cell survival and angiogenesis. Similar roles for CREB3L1 have been identified in other cell systems. The increase in protein production seen during cancer development and progression is mirrored in virus-infected cancer cells, where there is an excessive load on the ER as a result of synthesis of viral; both that increase and the viral protein synthesis can induce ER stress. CREB3L1 has been shown to prevent the spread of the hepatitis C virus by limiting the proliferation of virus-infected cells (43). In the case of the virus-infected cells, CREB3L1 was cleaved and translocated to the nucleus, where it activated the transcription of cell cycle inhibitors blocking the proliferation of these cells. Knockdown of CREB3L1 allowed increased viral replication (43). In addition, the same group demonstrated that CREB3L1 expression was required for the activation of p21 by the chemotherapy agent doxorubicin, preventing the proliferation of cancer cells. In cells lacking CREB3L1, the effectiveness of doxorubicin was decreased (51). In addition, CREB3L1 silencing through a chromosome fusion is associated with low-grade fibromyxoid sarcoma (42). Taken together, these studies clearly demonstrate a protective role for CREB3L1 in cells experiencing stressful or abnormal conditions through the regulation of transcriptional targets and the cell cycle.
In conclusion, our data support a model whereby normal cells express CREB3L1 and this represses the expression of genes for cell growth, cell survival, angiogenesis, migration, and invasion. Loss of CREB3L1 expression allows the expression of prosurvival and proangiogenic genes, promoting a phenotype that favors survival of cancer cells and progression to a metastatic phenotype, highlighting its importance as a tumor suppressor.
Supplementary Material
ACKNOWLEDGMENTS
We thank J. Ye (UT Southwestern Medical Center) for anti-CREB3L1 antibodies. We also thank J. Rideout, S. Junek, S. Smith, and A. Craven for expert technical assistance.
P.M. was supported by a postdoctoral fellowship from the Saskatchewan Health Research Foundation. Funding for the project was provided by the Canadian Breast Cancer Foundation-Prairies/NWT Region and the Saskatchewan Cancer Agency.
Footnotes
Published ahead of print 14 October 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00959-13.
REFERENCES
- 1. Zhang K, Kaufman RJ. 2004. Signaling the unfolded protein response from the endoplasmic reticulum. J. Biol. Chem. 279:25935–25938 [DOI] [PubMed] [Google Scholar]
- 2. Pahl HL. 1999. Signal transduction from the endoplasmic reticulum to the cell nucleus. Physiol. Rev. 79:683–701 [DOI] [PubMed] [Google Scholar]
- 3. Lee AS. 1992. Mammalian stress response: induction of the glucose-regulated protein family. Curr. Opin. Cell Biol. 4:267–273 [DOI] [PubMed] [Google Scholar]
- 4. Hampton RY. 2000. ER stress response: getting the UPR hand on misfolded proteins. Curr. Biol. 10:R518–R521 [DOI] [PubMed] [Google Scholar]
- 5. Kohno K. 2007. How transmembrane proteins sense endoplasmic reticulum stress. Antioxid. Redox Signal. 9:2295–2303 [DOI] [PubMed] [Google Scholar]
- 6. Ron D, Walter P. 2007. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8:519–529 [DOI] [PubMed] [Google Scholar]
- 7. Okamura K, Kimata Y, Higashio H, Tsuru A, Kohno K. 2000. Dissociation of Kar2p/BiP from an ER sensory molecule, Ire1p, triggers the unfolded protein response in yeast. Biochem. Biophys. Res. Commun. 279:445–450 [DOI] [PubMed] [Google Scholar]
- 8. Liu CY, Schroder M, Kaufman RJ. 2000. Ligand-independent dimerization activates the stress response kinases IRE1 and PERK in the lumen of the endoplasmic reticulum. J. Biol. Chem. 275:24881–24885 [DOI] [PubMed] [Google Scholar]
- 9. Shen J, Snapp EL, Lippincott-Schwartz J, Prywes R. 2005. Stable binding of ATF6 to BiP in the endoplasmic reticulum stress response. Mol. Cell. Biol. 25:921–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kim R, Emi M, Tanabe K, Murakami S. 2006. Role of the unfolded protein response in cell death. Apoptosis 11:5–13 [DOI] [PubMed] [Google Scholar]
- 11. Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, Lavail MM, Walter P. 2007. IRE1 signaling affects cell fate during the unfolded protein response. Science 318:944–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Szegezdi E, Logue SE, Gorman AM, Samali A. 2006. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 7:880–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kondo S, Murakami T, Tatsumi K, Ogata M, Kanemoto S, Otori K, Iseki K, Wanaka A, Imaizumi K. 2005. OASIS, a CREB/ATF-family member, modulates UPR signalling in astrocytes. Nat. Cell Biol. 7:186–194 [DOI] [PubMed] [Google Scholar]
- 14. Murakami T, Saito A, Hino S, Kondo S, Kanemoto S, Chihara K, Sekiya H, Tsumagari K, Ochiai K, Yoshinaga K, Saitoh M, Nishimura R, Yoneda T, Kou I, Furuichi T, Ikegawa S, Ikawa M, Okabe M, Wanaka A, Imaizumi K. 2009. Signalling mediated by the endoplasmic reticulum stress transducer OASIS is involved in bone formation. Nat. Cell Biol. 11:1205–1211 [DOI] [PubMed] [Google Scholar]
- 15. Murakami T, Kondo S, Ogata M, Kanemoto S, Saito A, Wanaka A, Imaizumi K. 2006. Cleavage of the membrane-bound transcription factor OASIS in response to endoplasmic reticulum stress. J. Neurochem. 96:1090–1100 [DOI] [PubMed] [Google Scholar]
- 16. Pyrko P, Schonthal AH, Hofman FM, Chen TC, Lee AS. 2007. The unfolded protein response regulator GRP78/BiP as a novel target for increasing chemosensitivity in malignant gliomas. Cancer Res. 67:9809–9816 [DOI] [PubMed] [Google Scholar]
- 17. Jamora C, Dennert G, Lee AS. 1996. Inhibition of tumor progression by suppression of stress protein GRP78/BiP induction in fibrosarcoma B/C10ME. Proc. Natl. Acad. Sci. U. S. A. 93:7690–7694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Romero-Ramirez L, Cao H, Nelson D, Hammond E, Lee AH, Yoshida H, Mori K, Glimcher LH, Denko NC, Giaccia AJ, Le QT, Koong AC. 2004. XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res. 64:5943–5947 [DOI] [PubMed] [Google Scholar]
- 19. Bi M, Naczki C, Koritzinsky M, Fels D, Blais J, Hu N, Harding H, Novoa I, Varia M, Raleigh J, Scheuner D, Kaufman RJ, Bell J, Ron D, Wouters BG, Koumenis C. 2005. ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. EMBO J. 24:3470–3481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Park HR, Tomida A, Sato S, Tsukumo Y, Yun J, Yamori T, Hayakawa Y, Tsuruo T, Shin-ya K. 2004. Effect on tumor cells of blocking survival response to glucose deprivation. J. Natl. Cancer Inst. 96:1300–1310 [DOI] [PubMed] [Google Scholar]
- 21. Carlsen SA, Barry M, Newton K. 1990. The identification of a neutral glycosphingolipid antigenic marker for metastatic cells in the R3230AC rat mammary adenocarcinoma. Clin. Exp. Metastasis 8:141–151 [DOI] [PubMed] [Google Scholar]
- 22. Ross DT, Scherf U, Eisen MB, Perou CM, Rees C, Spellman P, Iyer V, Jeffrey SS, Van de Rijn M, Waltham M, Pergamenschikov A, Lee JC, Lashkari D, Shalon D, Myers TG, Weinstein JN, Botstein D, Brown PO. 2000. Systematic variation in gene expression patterns in human cancer cell lines. Nat. Genet. 24:227–235 [DOI] [PubMed] [Google Scholar]
- 23. Rae JM, Creighton CJ, Meck JM, Haddad BR, Johnson MD. 2007. MDA-MB-435 cells are derived from M14 melanoma cells—a loss for breast cancer, but a boon for melanoma research. Breast Cancer Res. Treat 104:13–19 [DOI] [PubMed] [Google Scholar]
- 24. Sellappan S, Grijalva R, Zhou X, Yang W, Eli MB, Mills GB, Yu D. 2004. Lineage infidelity of MDA-MB-435 cells: expression of melanocyte proteins in a breast cancer cell line. Cancer Res. 64:3479–3485 [DOI] [PubMed] [Google Scholar]
- 25. Chambers AF. 2009. MDA-MB-435 and M14 cell lines: identical but not M14 melanoma? Cancer Res. 69:5292–5293 [DOI] [PubMed] [Google Scholar]
- 26. Anderson DH, Ismail PM. 1998. v-fps causes transformation by inducing tyrosine phosphorylation and activation of the PDGFbeta receptor. Oncogene 16:2321–2331 [DOI] [PubMed] [Google Scholar]
- 27. Mellor P, Harvey JR, Murphy KJ, Pye D, O'Boyle G, Lennard TW, Kirby JA, Ali S. 2007. Modulatory effects of heparin and short-length oligosaccharides of heparin on the metastasis and growth of LMD MDA-MB 231 breast cancer cells in vivo. Br. J. Cancer 97:761–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Maemura M, Akiyama SK, Woods VL, Jr, Dickson RB. 1995. Expression and ligand binding of alpha 2 beta 1 integrin on breast carcinoma cells. Clin. Exp. Metastasis 13:223–235 [DOI] [PubMed] [Google Scholar]
- 29. Chamberlain MD, Chan T, Oberg JC, Hawrysh AD, James KM, Saxena A, Xiang J, Anderson DH. 2008. Disrupted RabGAP function of the p85 subunit of phosphatidylinositol 3-kinase results in cell transformation. J. Biol. Chem. 283:15861–15868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Buckley ND, Carlsen SA. 1988. Involvement of soybean agglutinin binding cells in the lymphatic metastasis of the R3230AC rat mammary adenocarcinoma. Cancer Res. 48:1451–1455 [PubMed] [Google Scholar]
- 31. Ritchie S, Boyd FM, Wong J, Bonham K. 2000. Transcription of the human c-Src promoter is dependent on Sp1, a novel pyrimidine binding factor SPy, and can be inhibited by triplex-forming oligonucleotides. J. Biol. Chem. 275:847–854 [DOI] [PubMed] [Google Scholar]
- 32. Carlsen SA, Xie Y, Whitfield DM, Pang H, Krepinsky JJ. 1993. Isoglobotetraosylceramide is a marker for highly metastatic cells in rat mammary adenocarcinomas. Cancer Res. 53:2906–2911 [PubMed] [Google Scholar]
- 33. Mori S, Chang JT, Andrechek ER, Matsumura N, Baba T, Yao G, Kim JW, Gatza M, Murphy S, Nevins JR. 2009. Anchorage-independent cell growth signature identifies tumors with metastatic potential. Oncogene 28:2796–2805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nakanishi K, Sakamoto M, Yasuda J, Takamura M, Fujita N, Tsuruo T, Todo S, Hirohashi S. 2002. Critical involvement of the phosphatidylinositol 3-kinase/Akt pathway in anchorage-independent growth and hematogeneous intrahepatic metastasis of liver cancer. Cancer Res. 62:2971–2975 [PubMed] [Google Scholar]
- 35. Cifone MA, Fidler IJ. 1980. Correlation of patterns of anchorage-independent growth with in vivo behavior of cells from a murine fibrosarcoma. Proc. Natl. Acad. Sci. U. S. A. 77:1039–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Koumenis C, Wouters BG. 2006. “Translating” tumor hypoxia: unfolded protein response (UPR)-dependent and UPR-independent pathways. Mol. Cancer Res. 4:423–436 [DOI] [PubMed] [Google Scholar]
- 37. Mujcic H, Rzymski T, Rouschop KM, Koritzinsky M, Milani M, Harris AL, Wouters BG. 2009. Hypoxic activation of the unfolded protein response (UPR) induces expression of the metastasis-associated gene LAMP3. Radiother. Oncol. 92:450–459 [DOI] [PubMed] [Google Scholar]
- 38. Di Sano F, Ferraro E, Tufi R, Achsel T, Piacentini M, Cecconi F. 2006. Endoplasmic reticulum stress induces apoptosis by an apoptosome-dependent but caspase 12-independent mechanism. J. Biol. Chem. 281:2693–2700 [DOI] [PubMed] [Google Scholar]
- 39. Ouyang X, Jessen WJ, Al-Ahmadie H, Serio AM, Lin Y, Shih WJ, Reuter VE, Scardino PT, Shen MM, Aronow BJ, Vickers AJ, Gerald WL, Abate-Shen C. 2008. Activator protein-1 transcription factors are associated with progression and recurrence of prostate cancer. Cancer Res. 68:2132–2144 [DOI] [PubMed] [Google Scholar]
- 40. Scriven P, Brown NJ, Pockley AG, Wyld L. 2007. The unfolded protein response and cancer: a brighter future unfolding? J. Mol. Med. 85:331–341 [DOI] [PubMed] [Google Scholar]
- 41. So AY, de la Fuente E, Walter P, Shuman M, Bernales S. 2009. The unfolded protein response during prostate cancer development. Cancer Metastasis Rev. 28:219–223 [DOI] [PubMed] [Google Scholar]
- 42. Mertens F, Fletcher CD, Antonescu CR, Coindre JM, Colecchia M, Domanski HA, Downs-Kelly E, Fisher C, Goldblum JR, Guillou L, Reid R, Rosai J, Sciot R, Mandahl N, Panagopoulos I. 2005. Clinicopathologic and molecular genetic characterization of low-grade fibromyxoid sarcoma, and cloning of a novel FUS/CREB3L1 fusion gene. Lab. Invest. 85:408–415 [DOI] [PubMed] [Google Scholar]
- 43. Denard B, Seemann J, Chen Q, Gay A, Huang H, Chen Y, Ye J. 2011. The membrane-bound transcription factor CREB3L1 is activated in response to virus infection to inhibit proliferation of virus-infected cells. Cell Host Microbe 10:65–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Macchiarini P, Fontanini G, Dulmet E, de Montpreville V, Chapelier AR, Cerrina J, Ladurie FL, Dartevelle PG. 1994. Angiogenesis: an indicator of metastasis in non-small cell lung cancer invading the thoracic inlet. Ann. Thorac. Surg. 57:1534–1539 [DOI] [PubMed] [Google Scholar]
- 45. Hicklin DJ, Ellis LM. 2005. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J. Clin. Oncol. 23:1011–1027 [DOI] [PubMed] [Google Scholar]
- 46. Kedar D, Baker CH, Killion JJ, Dinney CP, Fidler IJ. 2002. Blockade of the epidermal growth factor receptor signaling inhibits angiogenesis leading to regression of human renal cell carcinoma growing orthotopically in nude mice. Clin. Cancer Res. 8:3592–3600 [PubMed] [Google Scholar]
- 47. Hood JD, Cheresh DA. 2002. Targeted delivery of mutant Raf kinase to neovessels causes tumor regression. Cold Spring Harbor Symp. Quant. Biol. 67:285–291 [DOI] [PubMed] [Google Scholar]
- 48. Aigner A, Brachmann P, Beyer J, Jager R, Raulais D, Vigny M, Neubauer A, Heidenreich A, Weinknecht S, Czubayko F, Zugmaier G. 2003. Marked increase of the growth factors pleiotrophin and fibroblast growth factor-2 in serum of testicular cancer patients. Ann. Oncol. 14:1525–1529 [DOI] [PubMed] [Google Scholar]
- 49. Chang Y, Zuka M, Perez-Pinera P, Astudillo A, Mortimer J, Berenson JR, Deuel TF. 2007. Secretion of pleiotrophin stimulates breast cancer progression through remodeling of the tumor microenvironment. Proc. Natl. Acad. Sci. U. S. A. 104:10888–10893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ghosh R, Lipson KL, Sargent KE, Mercurio AM, Hunt JS, Ron D, Urano F. 2010. Transcriptional regulation of VEGF-A by the unfolded protein response pathway. PLoS One 5:e9575. 10.1371/journal.pone.0009575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Denard B, Lee C, Ye J. 2012. Doxorubicin blocks proliferation of cancer cells through proteolytic activation of CREB3L1. eLife 1:e00090. 10.7554/eLife.00090 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.